

Abstract
The Wnt/β-catenin pathway serves important roles in cancer development. The expression and function of Chibby (Cby), as a direct antagonist of β-catenin, in nasopharyngeal carcinoma (NPC) has not been fully investigated. The present study revealed that the mRNA and protein expression of Cby was significantly lower in NPC tissue than in the adjacent normal tissue. Low expression of Cby was significantly associated with the tumor and the clinical staging. Furthermore, Cby overexpression inhibited the proliferation of human NPC SUNE1 cells and induced cell cycle arrest. In addition, Cby overexpression also significantly enhanced the susceptibility of SUNE1 cells to apoptosis. These results indicated that Cby might serve as an anti-oncogenic gene in the development of NPC and could represent a potential therapeutic target for the human NPC therapy.
Keywords: nasopharyngeal carcinoma, Chibby, Wnt/β-catenin, cell cycle, apoptosis
Introduction
Nasopharyngeal carcinoma (NPC) is an endemic disease particularly prevalent among the people of Southern China (1). As one of the most common malignancies in this region, the incidence ratio of NPC ranges between 0.015–0.05%, which is 10- to 20-fold higher than for the population of China as a whole (2). A number of features contribute to this high incidence ratio, including ethnic, geographical and genetic factors (3). NPC arises from the epithelial lining of nasopharynx. At the early stage of disease, NPC is always asymptomatic and difficult to be identified by physical examination. Thus a large number of patients were diagnosed at advanced stages (1). Due to its relatively high radiosensitivity and inaccessible anatomical position, radiotherapy is the primary treatment for patients with NPC diagnosed in the early stage of disease (4,5). Nevertheless, the prognosis of the patient worsens significantly when the stage of the tumor increases or the patient undergoes distant metastasis (6). The overall and relapse-free survival times of the patients who underwent concurrent chemotherapy are significantly improved compared with those of the patients who received radiotherapy alone (7). However, the undesirable side effects of chemotherapy reduce the tolerance of patients to treatment. Therefore, investigating the underlying molecular mechanism of NPC may provide novel therapeutic targets or prognostic factors for this malignancy.
Chibby (Cby) is a small nuclear protein that consists of 126 amino acids; it is evolutionarily conserved from Drosophila to humans, and serves as an antagonist of β-Catenin, a central component of canonical Wnt signaling pathway (8). Cby physically interacts with the C terminus of β-Catenin and inhibits its transcriptional activation potential by competitive inhibition of the T-cell factor/lymphoid enhancer factor (TCF/LEF) family of transcription factors (9). Cby additionally cooperates with 14-3-3 adaptor proteins to transport β-Catenin out of the nucleus (10). In previous studies, overexpression of Cby promoted the differentiation of mouse cardiomyocytes from embryonic stem cells (11) and mice adipocyte differentiation from mouse embryo 3T3-L1 cells (12). In Cby-knockout mice, pulmonary malfunction was observed owing to reduced proliferation and the abnormal differentiation of lung epithelial cells (13). Cby is therefore involved in numerous cellular behaviors, including proliferation, survival and differentiation through the Wnt signaling pathway. The Wnt signaling pathway is essential to the genesis and development of cancer (14,15). The downregulated expression of Cby was validated in colon carcinoma cell lines by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) (16). In another study in pediatric ependymomas, methylation-specific PCR and bisulphite sequencing revealed the transcriptional inactivation of chromosome 22 open reading frame 2, where the gene encoding Cby is located (17). In addition, Cby has been demonstrated to suppress the growth of human colon adenocarcinoma SW480 cells through the inhibition of the canonical Wnt signaling pathway (18). In a recent study, Cby was found to activate endoplasmic reticulum (ER) stress and apoptosis in breakpoint cluster protein/Abl kinase 1-positive human myeloid leukemia cells by inducing the cytoplasmic accumulation of β-catenin (19). Thus, the downregulation of Cby in cancer cells may provide information regarding the use of Cby as therapeutic target and prognostic factor.
The present study first investigated the expression of Cby in the biopsy specimens of 45 patients with NPC and analyzed its association with disease development. Next, Cby was overexpressed in the NPC SUNE1 cell line and demonstrated that Cby overexpression significantly reduced the proliferation of SUNE1 cells by inducing cell cycle arrest and enhancing the susceptibility of SUNE1 cells to apoptosis.
Materials and methods
Patients and tissue samples
A total of 45 (32 male, 13 female; age range, 23–68 years; median age, 45 years) patients were diagnosed with NPC by biopsy between April 2010 to January 2011 in The First Affiliated Hospital of Xiamen University (Xiamen, China). No patients had undergone radiotherapy or chemotherapy. All patients were staged according to the International Union Against Cancer (UICC) Tumor-Node-Metastasis (TNM) Classification of Malignant Tumors, 7th edition (20). The pathological classification was determined according to World Health Organization Classification of Tumors Pathology and Genetics (21). Detailed clinical information of every patient was recorded. Written informed consent was acquired from every patient enrolled in the present study. The research program and experimental procedures were approved by the Ethics Board of The First Affiliated Hospital of Xiamen University.
Tumor samples and adjacent normal tissue were obtained by biopsy. Each specimen was divided into two parts: One was fixed in 10% pH-neutral formalin for 24 h at room temperature, embedded in paraffin for pathological examination; the other was snap-frozen and stored in liquid nitrogen for molecular biology analysis. All sample treatment procedures were finished within 30 min.
Immunohistochemistry
Immunohistochemistry was performed using the streptavidin-biotin complex technique. From the formalin-fixed, paraffin-embedded tissue, 4-µm-thick sections were cut and then dried at room temperature, deparaffinized in xylene for 15 min at room temperature, and then rehydrated in a descending ethanol series (100, 90, 80, 70, 50 and 10%) at room temperature. Tissue sections were submerged in 3% hydrogen peroxide for 10 min at room temperature to block endogenous peroxidase activity. Antigen retrieval was performed by immersing sections in 0.01 M sodium citrate buffer for 20 min at 95°C. Samples were cooled at room temperature for 1 h and then treated with rabbit serum blocking solution (Boster Biological Technology, Pleasanton, CA, USA) for a further 20 min at room temperature. Samples were incubated in primary rabbit polyclonal anti-Cby antibodies (cat. no., sc-393295; dilution, 1:500; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) for 1 h at room temperature, followed by biotin-labeled rabbit anti-mouse secondary antibody (cat. no., BA1005; dilution, 1:300; Boster Biological Technology) for 20 min. Next, streptavidin-coupled horseradish peroxidase (Boster Biological Technology) was added. Complexes were visualized in brown following treatment with 3,3′-diaminobenzidine (Boster Biological Technology) at room temperature. The slides were counterstained with hematoxylin for 1 min at room temperature, dehydrated, mounted and viewed under a light microscope (Olympus BH-2; Olympus Corporation, Tokyo, Japan) at a magnification of ×400. For every slide, PBS instead of primary antibody was used as negative control, and slides verified as positive prior to the experiment were used as positive control.
Evaluation of Cby histological expression
All histological slides were scored independently by two pathologists blinded for patient characteristics. Cby protein was present as brown granules in either the nucleus or cytoplasm. The intensity score of Cby staining was graded as follows: 0, no staining; 1, mild; 2, moderate; 3, intensive. To evaluate the percentage of Cby-positive cells, images of the tissue sections were generated using a ×40 objective lens, five high-magnification microscope fields (40,000 µm2) were randomly selected for each section and positive Cby staining cells were counted for every microscope field. The percentage score of Cby staining was generated as: 0, ≤10% cells were positively Cby stained; 1, 11–25% cells stained; 2, 26–50% cells stained; 3, >50% cells stained positively. The intensity and percentage score were summed to generate a composite with a maximum score of 6. A score of >2 represented a positive immunohistochemical identification of Cby (22). The Cby protein expression level was stratified as: 3, weak; 4/5, moderate; 6, strong.
Cell culture
The NPC SUNE1 cell line and an immortalized non-malignant nasopharyngeal epithelial NP69 cell line were purchased from Cell Bank of Xiangya School of Medicine (Central South University, Hunan, China). The SUNE1 cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) with 10% fetal bovine serum (FBS) and 100 U/ml penicillin-streptomycin at 37°C in 5% CO2, and the NP69 cells were cultured in defined keratinocyte-serum-free medium (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 0.2 ng/ml growth factors, 5% heat-inactivated FBS, and 100 U/ml penicillin-streptomycin.
Plasmid construction and electroporation
Total RNA was extracted using TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.) following the manufacturer's instructions. cDNA was produced from 1 µg total RNA using a RT Kit (Takara Bio, Inc., Otsu, Japan). The primer sets used were as follows: forward, 5′-GCTCTAGAATGGACTACAAAGACGATGACGACAAGATGCCTTTCTTTGGGAATACGG-3′ and reverse, 5′-CGACGCGTTCATTTTCTCTTCCGGCTGATC-3′, with XbaI and MluI enzyme digestion site incorporated into the forward and reverse primers, respectively; a FLAG sequence was inserted into the 5′ end of forward primer. The PCR products were enzyme digested and subcloned into plasmid plv-cs2.0 (Invitrogen; Thermo Fisher Scientific, Inc.), which contains the green fluorescent protein reporter gene. The recombination plasmid (named plv-cs2.0-Cby) DNA was identified by 1% agarose gel electrophoresis following cleavage with different restriction enzymes (XbaI/MluI and XbaI/KpnI) and DNA sequencing (performed by Shanghai Sangon Biotech Co., Ltd., Shanghai, China). A total of 1×106 SUNE1 cells were electroporated with 1 µg of either plv-cs2.0-Cby or plv-cs2.0 plasmid DNA in 100 µl DMEM media under 250 V square-wave pulse to reach a total electric capacity of 950 µF using a Gene PulserXcellEukarotic system (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Electroporated cells were seeded in 6-well plates. At 48 h after electroporation, cells were further cultured in selection media containing 1 µg/ml puromycin for 1 week, the plv-cs2.0-Cby SUNE1 cells and its control plv-cs2.0 SUNE1 cells were collected for subsequent experiments.
RNA extraction and RT-qPCR
Total RNA was extracted using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) following the manufacturer's instructions. cDNA was produced from 1 µg total RNA using RT Kit (Takara Bio, Inc.). qPCR was performed with SYBRGreen Real-Time PCR Master Mix (TAKARA, TOYOBO, Japan) and gene specific primer pairs for Cby. The qPCR reaction system was made up as: 10 µl PCR master mix, 2 µl cDNA, 1.2 µl primer sets (10 µmol) and 6.8 µl ddH2O to form a total volume of 20 µl. The primer pairs used in this experiment was as follows: Cby forward, 5′-TTTGGGAATACGTTCAGTCCG-3′ and reverse, 5′-TCAGCCGCAAGAGATTGTTC-3′; 18S RNA forward, 5′-GTCTGTGATGCCCTTAGATG-3′ and reverse, 5′-AGCTTATGACCCGCACTTAC-3′. The qPCRthermocycling conditions were: Initial denaturation at 95°C for 30 sec, followed by 45 cycles of denaturation at 90°C for 30 sec, annealing at 60°C for 20 sec, extension at 72°C for 25 sec, and termination at 72°C for 15 min. Each qPCR reaction was performed in triplicate. Relative gene expression was analyzed by using the 2−∆∆Cq method (23). 18S RNA was used as the internal reference gene. The relative n-fold ratio of tumor samples to the normal tissue was calculated; a ratio ≤0.66 was considered as under-expression and ≥1.5 as overexpression, as described previously (24).
Western blot analysis
Monolayer plv-cs2.0-Cby SUNE1 cells or control plv-cs2.0 SUNE1 cells in the exponential phase were lysed and total proteins were extracted using Radioimmunoprecipitation Assay buffer (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), following the manufacturer's protocol. The protein concentration was determined using a Bradford assay. A total of 80 µg protein per well were separated by 12% SDS-PAGE, then transferred onto a polyvinylidenedifluoride membrane (EMD Millipore, Billerica, MA, USA). Membranes were blocked in 7% bovine serum albumin (Boster Biological Technology) in TBS and 0.5% Tween-20 (TBST) for 1 h at room temperature, and then incubated with rabbit polyclonal anti-Cby antibody (cat. no., sc-393295; dilution 1:2,000) and anti-β-actin antibody (cat. no., sc-130300; 1:1,000; Santa Cruz Biotechnology, Inc.) for 2 h at room temperature and then washed in 0.5% TBST three times for 10 min. Horseradish peroxidase-conjugated rabbit anti-mouse secondary antibodies (cat. no., BA1005; dilution, 1:5,000; Boster Biological Technology) were then added for 1 h at room temperature. β-actin was used as an internal control. The signal was developed using the Western Lightning Plus-ECL reagent (Pierce; Thermo Fisher Scientific, Inc.). The density of the protein bands was analyzed using ImageJ 1.48 software (National Institutes of Health, Bethesda, MD, USA).
Dual-luciferase reporter assay
SUNE1 cells transfected with plv-cs2.0-Cby or plv-cs2.0 (1×105 cells/well) were seeded in 6-well plates for 24 h. Cells in each well were cotransfected with 100 ng pTOPFlash or pFOPFlash reporter plasmid, and 20 ng Renilla luciferase expression vector (Promega Corporation, Madison, WI, USA) using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) for a further 24 h. Then Wnt3a (30 nM; Sigma-Aldrich; Merck KGaA), an activator of the Wnt/β-catenin pathway, was added for 6 h. The luciferase activity was analyzed using a Dual-Luciferase Reporter Assay system (Promega Corporation), according to the manufacturer's instructions, with a Modulus single-tube-type multifunctional detector (Beijing Yuanpinghao Bio Co., Ltd., Beijing, China). The luciferase activity was normalized to renilla luciferase activity.
MTT assay
A total of 500 cells per well were transferred to 96-well plate and cultured at 37°C in 5% CO2. The MTT assay was performed every 24 h from day 0 to day 5. A total of 20 µl MTT reagent (5 mg/ml, ScienCell Research Laboratories, Inc., San Diego, CA, USA) were added to each well and further incubated in darkness for 4 h, after which the supernatant was aspirated and 150 µl dimethyl sulfoxide was added to each well. The plate was incubated at room temperature for 15 min and then the absorbance (A) was measured at 570 and 630 nm. The A630 value were subtracted from A570 value and plotted against the time of culture. Data were collected from three independent experiments at every time point.
Cell cycle and apoptosis determined by flow cytometry
A total of 1×106 cells following thymidine-synchronization at G1 phase prior to cell cycle analysis were harvested and washed in cold PBS, and fixed by adding 70% ethanol dropwise to the cell pellet. The cell suspension was stored at 4°C overnight. Fixed cells were centrifuged for 5 min at 4°C and 2,000 × g and resuspended in 400 µl cold PBS. Following this, 10 µl RNaseA (5 mg/ml) was added and incubated at 37°C for 1 h and then 50 µl propidium iodide (1 mg/ml; Sangon Biotech Co., Ltd.) was added then incubated in darkness at 37°C for 30 min. Data was acquired using a flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and analyzed using ModFit 3.3 (Verity Software House, Topsham, ME, USA) software.
The apoptosis of plv-cs2.0-Cby SUNE1 cells and control plv-cs2.0 SUNE1 cells treated with 20 µM 5-fluorouracil (5-FU) for 24 h (Sigma-Aldrich; Merck KGaA.) were detected by a FITC Annexin V/Dead Cell Apoptosis kit (Invitrogen, Thermo Fisher Scientific, Inc.), according to the manufacturer's protocol. Annexin V+, PI−+ cells were regarded as necrotic cells, whereas Annexin V+ but PI− cells were regarded as apoptotic cells. Data was acquired using a flow cytometer (BD Biosciences) and analyzed using ModFit 3.3 (Verity Software House.) software.
Hoechst staining assay
The plv-cs2.0-Cby SUNE1 cells and control plv-cs2.0 SUNE1 cells (5×105 cells/ml) were seeded in 35-mm culture dishes and cultured at 37°C in 5% CO2. Cells were fixed in 4% formalin at room temperature for 30 min when reached 90% confluence, then stained with Hoechst 33342 (5 µg/ml) for 10 min at room temperature. After washing with PBS, the apoptotic cells were counted under a fluorescence microscope (DMi8-M, Leica, Barcelona, Spain) at a magnification of ×400. Five microscope fields were randomly selected from every culture dish. Apoptotic rate (%)=apoptotic cells/total cells ×100.
Colony formation
A total of 500 cells of plv-cs2.0-Cby or plv-cs2.0 cells were cultured in six-well plates for 14 days, and then stained with 0.005% crystal violet for 30 min at room temperature. The number of foci containing more than 50 cells was counted.
Statistical analysis
All statistical analysis was performed with SPSS Statistics 17.0 (SPSS, Inc., Chicago, IL, USA). Data are expressed as the mean ± standard error of the mean. A paired t-test was used to analyze the difference in mRNA levels between NPC and adjacent normal tissue. The χ2 test was used to compare Cby protein expression status between NPC and adjacent normal tissue, as well as various clinical characteristic groups. Student's t-test was used to compare the differences of qPCR, western blot, MTT value, percentage of S phase cells and apoptosis rate in in vitro experiments. P<0.05 was consider to indicate a statistically significant difference.
Results
Patient characteristics
Of the 45 patients enrolled in this research, 32 (71.1%) were male while 13 were female. Patients' age ranged from 23–68 years old, with median age of 53. There were 24 (53.3%) patients with T1-T2 disease and 21 patients with T3-T4 patients. In total, 27 (60%) patients had regional lymph node metastases, whereas 18 patients did not; 19 (42.2%) patients had early-stage (I–II) disease and 26 patients had advanced stage (III–IV) disease. In total, 9 (20%) patients were diagnosed with keratinizing squamous cell carcinoma, 20 (44.4%) patients were diagnosed with undifferentiated non-keratinizing carcinoma, 16 (35.6%) patients were diagnosed with differentiated non-keratinizing carcinoma and none had basaloid squamous cell carcinoma.
mRNA expression of Cby is reduced in NPC tissue
NPC tissue from 45 NPC patients were isolated and analyzed by RT-qPCR. The 18S RNA was used as internal control. qPCR revealed that 68.9% (31/45) patients exhibited reduced expression of Cby in tumor tissue compared with matched non-cancerous tissue (≤0.66-fold), whereas 15.6% (7/45) patients exhibited overexpression of Cby (≥1.5-fold), with the other 15.6% (7/45) exhibiting a similar expression level; the difference had statistical significance (P<0.05). The relative level of Cby mRNA for each patient is presented in Fig. 1A. This result indicated that the expression of Cby was reduced in NPC tissue in majority of the NPC patients.
Figure 1.
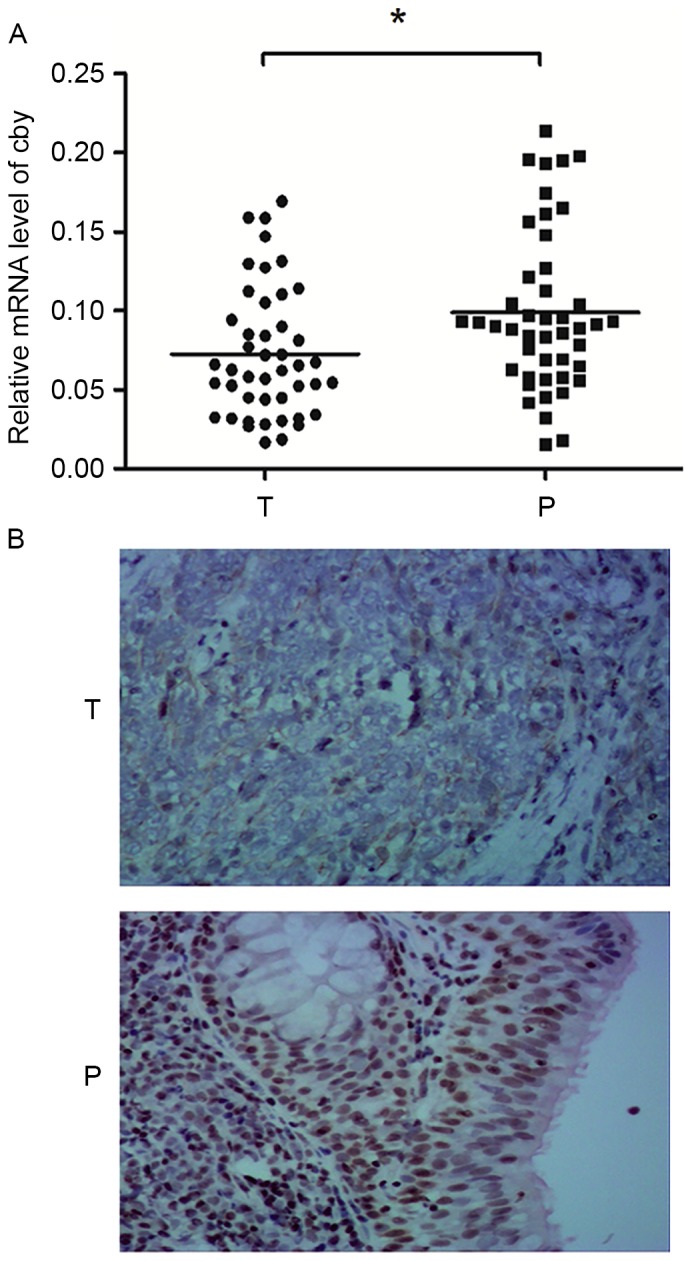
Expression of Cby in the clinical NPC tissues. (A) Scatter graph depicting the relative level of Cby mRNA of NPC tumor tissue (0.0725±0.0061) and adjacent normal tissue (0.0951±0.0509; Student's t-test, *P=0.0219, n=45). (B) The expression of Cby protein in NPC tissue and adjacent normal tissue. Cby expression is clearly observed in the nuclei of adjacent normal tissue cells, but expression is significant lower in NPC cells (original magnification, ×200). Cby, Chibby; NPC, nasopharyngeal cancer; T, NPC tissue; P, adjacent normal tissue.
When the Cby mRNA expression was analyzed based on different clinical groups, the difference between T1/T2 and T3/T4 groups was statistically significant (P<0.05), as was the difference between early (I/II) and advanced stage (III/IV) disease (P<0.05). The Cby mRNA expression level did not differ statistically between groups separated by sex, age, regional lymph node metastasis status and pathological classifications (Table I).
Table I.
The association of Cby mRNA expression with clinical features.
| Clinical features | Patients, n | RQ of Cby mRNA | 95% CI | P-value |
|---|---|---|---|---|
| Sex | >0.05 | |||
| Male | 32 | 0.0779 | 0.0697–0.0833 | |
| Female | 13 | 0.0775 | 0.0770–0.0803 | |
| Age, years | >0.05 | |||
| ≤50 | 27 | 0.0777 | 0.0696–0.0825 | |
| >50 | 18 | 0.0781 | 0.0752–0.0808 | |
| T staging | >0.05 | |||
| 1/2 | 24 | 0.0803 | 0.0770–0.0836 | |
| 3/4 | 21 | 0.0775 | 0.0685–0.0797 | |
| Lymph node metastasis | >0.05 | |||
| N0 | 18 | 0.0801 | 0.0752–0.0836 | |
| N1-3 | 27 | 0.0775 | 0.0696–0.0805 | |
| Clinical staging | <0.01 | |||
| I/II | 19 | 0.0810 | 0.0775–0.0896 | |
| III/IV | 26 | 0.0771 | 0.0687–0.0794 | |
| Pathological classification | >0.05 | |||
| WHO I | 9 | 0.0803 | 0.0728–0.0836 | |
| WHO II | 20 | 0.0773 | 0.0760–0.0821 | |
| WHO III | 16 | 0.0776 | 0.0696–0.0805 |
Cby, Chibby; RQ, relative quantity; CI, confidence interval; WHO I, keratinizing squamous cell carcinoma; WHO II, undifferentiated non-keratinizing carcinoma; WHO III, differentiated non-keratinizing carcinoma.
Cby protein expression is reduced in NPC tissue
Cby protein expression in NPC tissue and adjacent normal tissue were assessed by immunohistochemical staining. Scarlet-to-brown staining was observed in either the nucleus or the cytoplasm in Cby-positive cells (Fig. 1B). A total of 73.3% (33/45) patients exhibited a lower Cby expression score (the staining intensity and positive rate scores summed) in NPC tissue than normal tissue. The Cby-positive rates were 42.2% (19/45) and 91.1% (41/45) in NPC and adjacent normal tissues, respectively. The difference was statistically significant (χ2=24.2, P<0.01). This result suggested Cby protein expression was reduced in NPC tissue compared with adjacent normal tissue.
When Cby protein expression was analyzed based on different clinical groups, the differences between T1/T2 and T3/T4 groups (χ2=17.3, P<0.01), as well as early-stage (I/II) and advanced-stage (III/IV) groups (χ2=30.1, P<0.01) were statistically significant. The difference between groups separated based on gender, age, regional lymph node metastasis status and pathological classifications did not differ statistically (Table II).
Table II.
The association of Cby protein expression with clinical features.
| Clinical features | Patients, n | Cby protein PR, % (n) | χ2 value | P-value |
|---|---|---|---|---|
| Sex | 2.747 | >0.05 | ||
| Male | 32 | 50 (16/32) | ||
| Female | 13 | 23.1 (3/13) | ||
| Age, years | 0.137 | >0.05 | ||
| ≤50 | 27 | 44.4 (12/27) | ||
| >50 | 18 | 38.9 (7/18) | ||
| T staging | 17.257 | <0.01 | ||
| 1/2 | 24 | 70.8 (17/24) | ||
| 3/4 | 21 | 9.5 (2/21) | ||
| Lymph node metastasis | 2.186 | >0.05 | ||
| N0 | 18 | 55.6 (10/18) | ||
| N1-3 | 27 | 33.3 (9/27) | ||
| Clinical staging | 30.097 | <0.01 | ||
| I/II | 19 | 89.5 (17/19) | ||
| III/IV | 26 | 7.7 (2/26) | ||
| Pathological classification | 0.228 | >0.05 | ||
| WHO I | 9 | 44.4 (4/9) | ||
| WHO II | 20 | 45.0 (9/20) | ||
| WHO III | 16 | 37.5 (6/16) |
Cby, Chibby; PR, positive rate; LNM, lymph node metastases; WHO I, keratinizing squamous cell carcinoma; WHO II, undifferentiated non-keratinizing carcinoma; WHO III, differentiated non-keratinizing carcinoma.
Cby expression increases following plasmid electroporation
Human Cby DNA was transfected into NP69 and SUNE1 cell lines by electroporation. The results of western blot analysis indicated that the Cby protein level in SUNE1 cells was extremely low compared with NP69 cells (Fig. 2A), and was significantly different compared with the plv-cs2.0 control cells (7.16±0.58-fold of the control group; P<0.01) following electroporation (Fig. 2B). The results of western blot analysis indicated that the Cby protein level was increased. The results of the dual-luciferase reporter assay revealed that activation of the Wnt/β-catenin pathway was markedly reduced by Cby overexpression. In addition, activation of the Wnt/β-catenin pathway induced by Wnt3a was reduced by Cby overexpression (Fig. 2C). These results indicated that Cby expression was successfully upregulated and Wnt/β-catenin pathway was effectively inhibited following transfection with Cby.
Figure 2.

Establish of Cby overexpression SUNE1 cells model. (A and B) Expression of Cby in NP69 and SUNE1 cells detected by western blot analysisfollowing transfection withplv-cs2.0 (empty vector) and plv-cs2.0-Cby. (C) Activation of Wnt signaling assessed by β-catenin/T-cell factor-responsive luciferase reporter activity, normalized to β-Gal. Wnt3a, 30 nM. *P<0.05; **P<0.01; ***P<0.001. Cby, Chibby.
Cby overexpression inhibits the proliferation of SUNE1 cells
Cell survival was analyzed daily by an MTT assay between days 1 and 5. The MTT value did not differ significantly from day 1 to day 5 between the plv-cs2.0 and plv-cs2.0-Cby groups in NP69 cells; however, the growth rate of SUNE1 cells transfected with Cby decreased compared with those transfected with the blank vector from day 3 onwards, a difference that was statistically significant (P<0.01) following Cby overexpression (Fig. 3A).
Figure 3.

Effect of Cby overexpression on the proliferation of SUNE1 cells. (A) The growth of NP69 and SUNE1 cells transfected with plv-cs2.0 (empty vector) or plv-cs2.0-Cby were detected by MTT assay every 24 h from day 0 to day 5. (B) The anti-oncogenic potential of Cby, assessed by colony formation assays. The number of foci containing >50 cells was counted. **P<0.01; ***P<0.001. Cby, Chibby.
To investigate the anti-oncogenic potential of Cby further, colony formation assays were performed (Fig. 3B). The ability of plv-cs2.0-Cby-transfected SUNE1 cells to form foci (100±1.6%) was significantly impaired compared with that of plv-cs2.0 cells (57±5.2%) (P<0.01); however, no significant difference was observed in NP69 cells. This result demonstrated that overexpression of human Cby significantly inhibited the proliferation of SUNE1 cells.
Cby overexpression induces the cell cycle arrested and enhances the susceptibility of SUNE1 cells to apoptosis
To investigate the potential mechanism by which Cby overexpression inhibited the proliferation of SUNE1 cells, flow cytometry was performed to analyze the cell cycle distribution of cells. As depicted in Fig. 4A and B, the proportion of G1 phase cells for plv-cs2.0 and plv-cs2.0-Cby groups was 52.5±2.5 and 73.6±3.2%, respectively, which differed statistically (P<0.01). This result demonstrated that overexpression of human Cby significantly inhibited cell cycle progression in SUNE1 cells.
Figure 4.

Effect of Cby overexpression on the cell cycle distributions and apoptosis of SUNE1 cells. (A) Cell cycle analysis was performed by flow cytometry following plasmid transfection. The proportion of G1 phase cells for plv-cs2.0 (empty vector) and plv-cs2.0-Cby groups were 52.5±2.5 and 73.6±3.2%, respectively (P=0.0051). (B) Apoptosis analysis of the control group plv-cs2.0 and plv-cs2.0-Cby SUNE1 cells. Hoechst 33342 staining showed apoptotic cells with condensed chromatin or fragmented nuclei. Magnification, ×200. (C) Assessment of apoptosis of SUNE1 cells using flow cytometry with Annexin V-fluorescein isothiocyanate/phosphoinositide staining following treatment with 20 µM 5-fluorouracil (left) or 30 nM Wnt 3a (right). *P<0.05, **P<0.01. Cby, Chibby.
To detect the susceptibility of SUNE1 cells to apoptosis following Cby overexpression, the Hoechst staining assay was performed. Cells exhibiting condensed chromatin or fragmented nuclei were counted as apoptotic cells (Fig. 4B). The apoptosis rate in plv-cs2.0 and plv-c.s2.0-Cby was 3.55±0.54 and 9.85±1.46% (P<0.01), respectively, which differed statistically. Furthermore, the apoptosis rate induced by 5-FU was significantly increased from 1.82±1.2 to 2.75±1.7% following Cby overexpression (P<0.01). Down-regulation of 5-FU sensitivity induced by Wnt 3a could be significantly recued by Cby overexpression from 1.23±1.3 to 1.64±1.4% (P<0.05; Fig. 4C). This data indicated that overexpression of human Cby significantly facilitated the apoptosis of SUNE1 cells.
Discussion
The Wnt signaling pathway is an essential and fundamental signaling pathway that is highly evolutionally conserved. This pathway participates in a variety of processes, including development, tissue regeneration following injury and the maintenance of tissue homeostasis. Wnt signaling has versatile functions owing to its wide range of downstream target genes (25). The disordered expression of any Wnt signaling components will lead to abnormal activation of Wnt signaling and disruption of cell homeostasis, which in turn leads to cancer (26). The major alterations to Wnt signaling in cancer tissue are caused by the inactivation of negative regulators or the overexpression of positive regulators for different components of Wnt pathway, particularly β-catenin, which is a key component of the canonical Wnt pathway (27). A widely known example of the inactivation of negative regulators is the human tumor suppressor, adenomatous polyposis coli (APC). Genetic defects in APC are the cause of familial adenomatous polyposis, a heritable syndrome in which affected individuals develop hundreds of polyps in the large intestine at an early age and ultimately succumb to colorectal cancer (28). β-catenin phosphorylation at serines 33 and 37 creates a binding site for the E3 ubiquitin ligase β-Trcp, leading to β-catenin ubiquitination and degradation. Mutations of β-catenin at, and surrounding, these serine residues are frequently identified in cancer, generating mutant β-catenin that allows it to evade proteolytic degradation (29). These deficiencies all lead to the intracellular accumulation of β-catenin, thus enhancing its transcriptional activities (30). Evidence demonstrates that the inappropriate stabilization of β-catenin can cause surplus transcription of Wnt-downstream target genes, which can lead to cellular proliferation (31); such genes include MYC proto-oncogene, bHLH transcription factor (c-Myc) (32) and cyclin D1 (33,34).
Cby is a direct antagonist of β-catenin, and has been demonstrated to suppress colon tumor cell growth in vitro (18). The present study revealed that Cby expression was suppressed at the transcriptional and protein level in >60% of patients; however, these results were not completely consistent with previous reported studies (16), which may be due to differences in the biological characteristics of colon and head and neck cancer. Nonetheless, ~30% patients exhibited similar or even higher Cby expression levels in tumor tissue than the adjacent normal tissue (35). We hypothesize that there are two possible reasons for this result. First, it is widely accepted that cancer originates from and is developed under a series of cellular abnormalities (36). Cby truncation, where it is present, represents only one link in the chain of tumor development. Second, there is heterogeneity inside of a tumor, with β-catenin not uniformly distributed in the tumor (37), which means tumor cells located at the invasive front are exposed to growth factors and cytokines, which can further enhance β-catenin function; thus Cby expression in tumor will be affected by a number of unknown factors. As samples were acquired from a region of the tumor by biopsy, the Cby expression level may also vary between samples.
When taking account of clinical characteristics, it was found that Cby expression was associated with local NPC disease groups and stages. Cby mRNA and protein expression levels were declined more significantly in advanced diseases (T3/T4 disease and stage III/IV) than in preliminary diseases (T1/T2 diseases and stage I/II). The proliferation of SUNE1 cells was suppressed by Cby overexpression, which was consistent with the results of a previous study (18). In cell cycle analyses, an increased portion of cells arrested in the G0/G1 phase was observed, indicating that this effect is most likely mediated by cell cycle arrest caused by β-catenin dysfunction. Cyclin D1 is a direct target of β-catenin/TCF/LEF complex and is an important checkpoint regulator for the G1 to S progression (38). Cby may interfere with the transcriptional activation effect of β-catenin against cyclin D1. However, Fischer et al (18) observed that the major proportion of SW480 cells seemed to shift from G0/G1 phase to G2/M phase and then underwent cell cycle arrest. The exact mechanism of Cby activity remains unclear: One possible interpretation is Cby suppressed β-catenin's function in centrosomes, allowing mitosis to proceed by accumulating at G2/M phase induced by cyclin-dependent kinase 14/cyclin Y phosphorylates low-density lipoprotein receptor-related protein 6 (39). The difference observed in these two malignant cell lines may be attributable to the different time points at which Cby was expressed, thus leads to various dominant downstream effects. Further research, involving synchronizing the cell cycle prior to Cby administration, may be required.
The role of the Wnt/β-catenin signaling pathway in apoptosis is complicated, and the exact mechanism remains unclear. In the present study, overexpression of Cby increased apoptosis in SUNE1 cells to a certain extent, which may be caused by the transcriptional suppression of c-Myc (32), or by another Wnt/β-catenin-independent mechanism.
Although the association between the expression level of Cby and clinical prognostic factors has rarely been reported, β-catenin is considered to affect tumor invasion by cadherin-mediated cell-cell adhesion through direct binding to α-catenin (40). Since the extent of nasopharyngeal carcinoma, as elucidated by the TNM staging system, is the most import prognostic factor (41), the potential application of Cby for prognosis prediction is an attractive one. Also as an endogenous β-catenin antagonist, Cby may be utilized as a Wnt/β-catenin inhibitor for target therapy. However, the exact role of Cby in regulating the growth and proliferation of NPC requires elucidation by gain- and loss-of-function experiments in vitro and in vivo.
In summary, the data reported in the present study demonstrated that Cby expression was suppressed at the mRNA and protein levels in the NPC tissues of >60% patients. Overexpression of human Cby in SUNE1 cells significantly inhibited the proliferation and cell-cycle progression of SUNE1 cells, and significantly facilitated their apoptosis. These findings may serve as the basis for the use of Cby in future as a β-catenin inhibitor for targeted therapy, and can be used as the foundation for further investigation of the exact role of Cby in regulating NPC.
Acknowledgements
The present study was supported by grants from the National Natural Science Foundation of China (grant no. 81670936), the Project of Young and Middle-aged Backbone Talents Cultivation, Fujian, China (grant no. 2013-ZQN-JC-32) and the Project from Science and Technology Bureau of Xiamen, China (grant no. 3502Z20144004).
Competing interests
The authors declare that they have no competing interests.
References
- 1.Wu VW, Lam YN. Radiation-induced temporo-mandibular joint disorder in post-radiotherapy nasopharyngeal carcinoma patients: Assessment and treatment. J Med RadiatSci. 2016;63:124–132. doi: 10.1002/jmrs.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.He YQ, Xue WQ, Shen GP, Tang LL, Zeng YX, Jia WH. Household inhalants exposure and nasopharyngeal carcinoma risk: A large-scale case-control study in Guangdong, China. BMC Cancer. 2015;15:1022. doi: 10.1186/s12885-015-2035-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng WT, Choi CW, Lee MC, Chan SH, Yau TK, Lee AW. Familial nasopharyngeal carcinoma in Hong Kong: Epidemiology and implication in screening. Fam Cancer. 2009;8:103–108. doi: 10.1007/s10689-008-9213-9. [DOI] [PubMed] [Google Scholar]
- 4.Mesic JB, Fletcher GH, Goepfert H. Megavoltage irradiation of epithelial tumors of the nasopharynx. Int J Radiat Oncol Biol Phys. 1981;7:447–453. doi: 10.1016/0360-3016(81)90129-2. [DOI] [PubMed] [Google Scholar]
- 5.Lee AW, Poon YF, Foo W, Law SC, Cheung FK, Chan DK, Tung SY, Thaw M, Ho JH. Retrospective analysis of 5037 patients with nasopharyngeal carcinoma treated during 1976–1985: Overall survival and patterns of failure. Int J Radiat Oncol Biol Phys. 1992;23:261–270. doi: 10.1016/0360-3016(92)90740-9. [DOI] [PubMed] [Google Scholar]
- 6.Chua DT, Sham JS, Wei WI, Ho WK, Au GK. The predictive value of the 1997 American joint committee on cancer stage classification in determining failure patterns in nasopharyngeal carcinoma. Cancer. 2001;92:2845–2855. doi: 10.1002/1097-0142(20011201)92:11<2845::AID-CNCR10133>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 7.Wolden SL, Zelefsky MJ, Kraus DH, Rosenzweig KE, Chong LM, Shaha AR, Zhang H, Harrison LB, Shah JP, Pfister DG. Accelerated concomitant boost radiotherapy and chemotherapy for advanced nasopharyngeal carcinoma. J ClinOncol. 2001;19:1105–1110. doi: 10.1200/JCO.2001.19.4.1105. [DOI] [PubMed] [Google Scholar]
- 8.Takemaru K, Fischer V, Li FQ. Fine-tuning of nuclear-catenin by Chibby and 14-3-3. Cell cycle. 2009;8:210–213. doi: 10.4161/cc.8.2.7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takemaru K, Yamaguchi S, Lee YS, Zhang Y, Carthew RW, Moon RT. Chibby, a nuclear beta-catenin-associated antagonist of the Wnt/Wingless pathway. Nature. 2003;422:905–909. doi: 10.1038/nature01570. [DOI] [PubMed] [Google Scholar]
- 10.Li FQ, Mofunanya A, Harris K, Takemaru K. Chibby cooperates with 14-3-3 to regulate beta-catenin subcellular distribution and signaling activity. J Cell Biol. 2008;181:1141–1154. doi: 10.1083/jcb.200709091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh AM, Li FQ, Hamazaki T, Kasahara H, Takemaru K, Terada N. Chibby, an antagonist of the Wnt/beta-catenin pathway, facilitates cardiomyocyte differentiation of murine embryonic stem cells. Circulation. 2007;115:617–626. doi: 10.1161/CIRCULATIONAHA.106.642298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li FQ, Singh AM, Mofunanya A, Love D, Terada N, Moon RT, Takemaru K. Chibby promotes adipocyte differentiation through inhibition of beta-catenin signaling. Mol Cell Biol. 2007;27:4347–4354. doi: 10.1128/MCB.01640-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Love D, Li FQ, Burke MC, Cyge B, Ohmitsu M, Cabello J, Larson JE, Brody SL, Cohen JC, Takemaru K. Altered lung morphogenesis, epithelial cell differentiation and mechanics in mice deficient in the Wnt/β-catenin antagonist Chibby. PLoS One. 2010;5:e13600. doi: 10.1371/journal.pone.0013600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/-colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 15.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 16.Schuierer MM, Graf E, Takemaru K, Dietmaier W, Bosserhoff AK. Reduced expression of beta-catenin inhibitor Chibby in colon carcinoma cell lines. World J Gastroenterol. 2006;12:1529–1535. doi: 10.3748/wjg.v12.i10.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karakoula K, Suarez-Merino B, Ward S, Phipps KP, Harkness W, Hayward R, Thompson D, Jacques TS, Harding B, Beck J, et al. Real-time quantitative PCR analysis of pediatric ependymomas identifies novel candidate genes including TPR at 1q25 and CHIBBY at 22q12-q13. Genes Chromosomes Cancer. 2008;47:1005–1022. doi: 10.1002/gcc.20607. [DOI] [PubMed] [Google Scholar]
- 18.Fischer V, Brown-Grant DA, Li FQ. Chibby suppresses growth of human SW480 colon adenocarcinoma cells through inhibition of β-catenin signaling. J Mol Signal. 2012;7:6. doi: 10.1186/1750-2187-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mancini M, Leo E, Takemaru K, Campi V, Borsi E, Castagnetti F, Gugliotta G, Santucci MA, Martinelli G. Chibby drives β catenin cytoplasmic accumulation leading to activation of the unfolded protein response in BCR-ABL1+ cells. Cell Signal. 2013;25:1820–1827. doi: 10.1016/j.cellsig.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Zhu XZ. Introduction of WHO classification of tumours of soft tissue, the fourth edition. Zhonghua Bing Li XueZaZhi. 2013;42:363–365. doi: 10.3760/cma.j.issn.0529-5807.2013.06.002. (In Chinese) [DOI] [PubMed] [Google Scholar]
- 21.Jo VY, Fletcher CD. WHO classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology. 2014;46:95–104. doi: 10.1097/PAT.0000000000000050. [DOI] [PubMed] [Google Scholar]
- 22.Mattern J, Koomagi R, Volm M. Biological characterization of subgroups of squamous cell lung carcinomas. Clin Cancer Res. 1999;5:1459–1463. [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Gad S, Teboul D, Lièvre A, Goasguen N, Berger A, Beaune P, Laurent-Puig P. Is the gene encoding Chibby implicated as a tumour suppressor in colorectal cancer? BMC Cancer. 2004;4:31. doi: 10.1186/1471-2407-4-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duchartre Y, Kim YM, Kahn M. The Wnt signaling pathway in cancer. Crit Rev OncolHematol. 2016;99:141–149. doi: 10.1016/j.critrevonc.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: Can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16:3153–3162. doi: 10.1158/1078-0432.CCR-09-2943. [DOI] [PubMed] [Google Scholar]
- 28.Yang D, Zhang M, Gold B. Origin of Somatic mutations in β-catenin versus adenomatous polyposis coli in colon cancer: Random mutagenesis in animal models versus nonrandom mutagenesis in humans. Chem Res Toxicol. 2017;30:1369–1375. doi: 10.1021/acs.chemrestox.7b00092. [DOI] [PubMed] [Google Scholar]
- 29.Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat Rev. 2018;62:50–60. doi: 10.1016/j.ctrv.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Majidinia M, Aghazadeh J, Jahanban-Esfahlani R, Yousefi B. The roles of Wnt/β-catenin pathway in tissue development and regenerative medicine. J Cell Physiol. 2017 Nov 18; doi: 10.1002/jcp.26265. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 31.Polakis P. The many ways of Wnt in cancer. CurrOpin Genet Dev. 2007;17:45–51. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 32.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 33.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 34.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway; Proc Natl Acad Sci USA; 1999; pp. 5522–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren G, Zhao DA, Xu J, Li BA. Expression of CBY and methylation of CBY at promoter region in human laryngeal squamous cell carcinoma. Tumori. 2015;101:215–222. doi: 10.5301/tj.5000242. [DOI] [PubMed] [Google Scholar]
- 36.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-I. [DOI] [PubMed] [Google Scholar]
- 37.Brabletz T, Jung A, Hermann K, Günther K, Hohenberger W, Kirchner T. Nuclear overexpression of the oncoprotein beta-catenin in colorectal cancer is localized predominantly at the invasion front. Pathol Res Pract. 1998;194:701–704. doi: 10.1016/S0344-0338(98)80129-5. [DOI] [PubMed] [Google Scholar]
- 38.Monin MB, Krause P, Stelling R, Bocuk D, Niebert S, Klemm F, Pukrop T, Koenig S. The anthelmintic niclosamide inhibits colorectal cancer cell lines via modulation of the canonical and noncanonical Wnt signaling pathway. J Surg Res. 2016;203:193–205. doi: 10.1016/j.jss.2016.03.051. [DOI] [PubMed] [Google Scholar]
- 39.Davidson G, Shen J, Huang YL, Su Y, Karaulanov E, Bartscherer K, Hassler C, Stannek P, Boutros M, Niehrs C. Cell cycle control of wnt receptor activation. Dev Cell. 2009;17:788–799. doi: 10.1016/j.devcel.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 40.Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell. 2005;123:903–915. doi: 10.1016/j.cell.2005.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei WI, Sham JS. Nasopharyngeal carcinoma. Lancet. 2005;365:2041–2054. doi: 10.1016/S0140-6736(05)66698-6. [DOI] [PubMed] [Google Scholar]