

Abstract
The present study aimed to explore the potential clinical significance of microRNA (miR)-124-3p expression in the hepatocarcinogenesis and development of hepatocellular carcinoma (HCC), as well as the potential target genes of functional HCC pathways. Reverse transcription-quantitative polymerase chain reaction was performed to evaluate the expression of miR-124-3p in 101 HCC and adjacent non-cancerous tissue samples. Additionally, the association between miR-124-3p expression and clinical parameters was also analyzed. Differentially expressed genes identified following miR-124-3p transfection, the prospective target genes predicted in silico and the key genes of HCC obtained from Natural Language Processing (NLP) were integrated to obtain potential target genes of miR-124-3p in HCC. Relevant signaling pathways were assessed with protein-protein interaction (PPI) networks, Gene Ontology (GO) enrichment analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) and Protein Annotation Through Evolutionary Relationships (PANTHER) pathway enrichment analysis. miR-124-3p expression was significantly reduced in HCC tissues compared with expression in adjacent non-cancerous liver tissues. In HCC, miR-124-3p was demonstrated to be associated with clinical stage. The mean survival time of the low miR-124-3p expression group was reduced compared with that of the high expression group. A total of 132 genes overlapped from differentially expressed genes, miR-124-3p predicted target genes and NLP identified genes. PPI network construction revealed a total of 109 nodes and 386 edges, and 20 key genes were identified. The major enriched terms of three GO categories included regulation of cell proliferation, positive regulation of cellular biosynthetic processes, cell leading edge, cytosol and cell projection, protein kinase activity, transcription activator activity and enzyme binding. KEGG analysis revealed pancreatic cancer, prostate cancer and non-small cell lung cancer as the top three terms. Angiogenesis, the endothelial growth factor receptor signaling pathway and the fibroblast growth factor signaling pathway were identified as the most significant terms in the PANTHER pathway analysis. The present study confirmed that miR-124-3p acts as a tumor suppressor in HCC. miR-124-3p may target multiple genes, exerting its effect spatiotemporally, or in combination with a diverse range of processes in HCC. Functional characterization of miR-124-3p targets will offer novel insight into the molecular changes that occur in HCC progression.
Keywords: hepatocellular carcinoma, microRNA-124-3p, reverse transcription-quantitative polymerase chain reaction, gene expression omnibus, natural language processing, functional analysis
Introduction
MicroRNAs (miRNAs/miRs) are a class of short endogenous non-coding RNAs that suppress gene expression by either degrading mRNA or repressing mRNA translation through complementary binding to their specific target mRNAs in the 3′-untranslated region (1,2). Mature miRNAs are small single-stranded RNAs 19–22 nucleotides in length. Increasing evidence suggests that miRNAs are involved in diverse basic biological processes, including cell proliferation, apoptosis and differentiation (3,4). Studies have also verified that miRNAs are ectopically expressed in tumors and operate as tumor oncogenes or suppressors during tumor progression, indicating the potential for miRNAs as biomarkers for cancer diagnosis and therapy (2).
Hepatocellular carcinoma (HCC) is the third leading cause of global cancer-associated mortality, with the highest incidence occurring in Asia (5). The World Health Organization (WHO) estimates that there are ~56,400 newly diagnosed cases of HCC worldwide per year (6), with a significantly higher occurrence in males. Notably, more than half of all HCC cases are diagnosed in China (7). Multiple risk factors contribute to the occurrence of HCC, including chronic viral hepatitis and alcohol abuse. HCC development typically involves several steps, progressing from chronic hepatitis, cirrhosis and dysplastic nodules to HCC (8). It has been revealed that miRNAs serve a pivotal role in HCC progression and may provide novel insight for diagnostic and therapeutic HCC strategies (9). miR-124-3p has been implicated in HCC and its expression has been demonstrated to be downregulated in HCC tissues (2,10). Additionally, several target genes of miR-124-3p have been discovered (2,10,11). However, to the best of our knowledge, previous studies combining gene expression omnibus (GEO) data and bioinformatics analysis to explore the mechanisms underlying the role of miR-124-3p in HCC have not been performed. Additionally, previous studies have only focused on a specific target gene of miR-124-3p. Therefore, the potential mechanisms by which miR-124 suppresses tumorigenesis in HCC remain unclear; miR-124-3p may be involved in the regulation of several other undiscovered mRNAs and signaling pathways. The present study investigated miR-124-3p expression and its potential target genes in HCC by combining reverse transcription-quantitative polymerase chain reaction (RT-qPCR) with GEO data, using prediction software and natural language processing (NLP).
The clinical significance of miR-124-3p expression in hepatocarcinogenesis and development and the potential target genes of functional pathways in HCC patients was analyzed. Gene expression profiles were examined from primary HCC samples, literature searches using PubMed were performed and microarray data was downloaded from GEO. miR-124-3p was transfected into the HepG2 cell line to confirm the prospective targeted genes predicted in silico (relevant data were downloaded from GSE6207, GEO Profiles) and key genes of HCC were identified from NLP. The identified differentially expressed genes (DEGs) were subsequently integrated to confirm the potential target genes of miR-124-3p in HCC. Finally, the relevant signaling pathways in HCC were assessed to determine the key genes involved in the maintenance of these pathways, in order to further identify novel therapeutic targets in HCC.
Materials and methods
Tissue specimens
A total of 101 formalin-fixed, paraffin-embedded (FFPE) HCC tissues (80 males and 21 females) with clear pathology diagnoses were collected (10% neutral formalin-fixed for 24 h at room temperature). These HCC patients underwent primary surgical resection at the First Affiliated Hospital of Guangxi Medical University (Nanning, China) between January 2012 and February 2014 and did not receive any additional treatment. The age of the HCC patients ranged from 29 to 82 years old, with a mean age of 52 years. Among the 101 cases, 70 were successfully followed up. Clinical stages were classified according to the WHO Tumor-Node-Metastasis (TNM) criteria (12). Recurrence-free survival (RFS) was defined as the length of time between surgery and recurrence. Additionally, 101 adjacent non-HCC tissues were obtained to serve as internal controls. The present study was approved by the Research Ethics Committee of the First Affiliated Hospital of Guangxi Medical University (Nanning, China), and written informed consent was obtained from all patients.
RNA extraction and RT-qPCR
The total RNA, including miRNA, was isolated from tumor sections using the miRNeasy FFPE kit (Qiagen China Co., Ltd., Shanghai, China), as previously described (13–15). The miScript Reverse Transcription and the miScript SYBR-Green PCR kits (218073 and 218161 respectively; Qiagen China Co., Ltd.) were used to evaluate miR-124-3p expression levels. Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was performed in triplicate using the 7900HT PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA), in order to determine miRNA expression. The following temperature protocol was applied: 16°C for 30 min, 42°C for 30 min and 85°C for 5 min. LightCycler 480 SYBR-Green I Master (Roche Diagnostics GmbH) was used for qPCR, with the following thermocycling conditions: Initial pre-denaturation for 5 min at 95°C, followed by 40 cycles of 95°C with 10 sec, 60°C for 10 sec and 72°C for 10 sec; evaluation of the solubility curve was performed at 95°C for 5 sec and 65°C for 1 min, which was followed by cooling at 40°C for 30 sec. Each experiment was repeated in triplicate. The abundance of miR-124-3p in each sample was normalized to references genes RNU6B and RNU48, which was demonstrated to be a stable internal control in previous studies (16). The sequences purchased from Applied Biosystems (Thermo Fisher Scientific, Inc.) were as follows: miR-124-3p, 5′-UCGGGGAUCAUCAUGUCACGAG-3′ (cat. no. 4427975-001288), RNU6B, 5′-CGCAAGGAUGACACGCAAAUUCGUGAAGCGUUCCAUAUUUUU-3′ (cat. no. 4427975-001093); and RNU48, 5′-GAUGACCCCAGGUAACUCUGAGUGUGUCGCUGAUGCCAUCACCGCAGCGCUCUGACC-3′ (cat. no. 4427975-001006). The relevant expression of miR-124-3p was quantified using the formula 2−∆Cq (17).
Literature review search
PubMed was searched to identify previous studies regarding miR-124-3p expression, and clinical parameters were also collected if available. The search terms were as follows: ‘malignant* OR cancer OR tumor OR neoplas* OR carcinoma’, ‘hepatocellular OR liver OR hepatic OR HCC’ and ‘miR-124 OR miRNA-124 OR microRNA-124 OR ‘miR 124’ OR ‘miRNA 124’ OR ‘microRNA 124’ OR miR-124-3p OR miRNA-124-3p OR microRNA-124-3p’. A meta-analysis with P-values alone was subsequently conducted to pool the collected data if no relative expression of miR-124-3p was provided. To allow the combination of the P-values from each study, two-tailed P-values were converted into one-tailed P-values by dividing by two. A meta-analysis using Fisher's method was subsequently performed (18).
Public microarray data analysis of miR-124-3p
To verify the difference in miR-124-3p expression between HCC and normal liver tissues, the GEO database of the National Center for Biotechnology Information (NCBI) of the National Institute of Health (NIH; http://www.ncbi.nlm.nih.gov/geo/) and the ArrayExpress data of the European Bioinformatics Institute (EBI; http://www.ebi.ac.uk/arrayexpress/) were searched. The following terms were searched: ‘malignant* OR cancer OR tumor OR neoplas* OR carcinoma’, ‘hepatocellular OR liver OR hepatic OR HCC’ and ‘miR-124 OR miRNA-124 OR microRNA-124 OR ‘miR 124’ OR ‘miRNA 124’ OR ‘microRNA 124’ OR miR-124-3p OR miRNA-124-3p OR microRNA-124-3p’. Following screening, seven datasets [GSE57555 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE57555) (19), GSE41874 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE41874), GSE40744 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE40744) (20), GSE21362 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE21362) (21), GSE20077 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE20077) (22), GSE12717 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE12717) (23) and GSE31383 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE31383) (24)] were included. Among them, dataset GSE57555 contained 11 cholangiocarcinoma samples, 5 HCC samples and 16 normal controls; GSE41874 contained 3 primary HCC samples, 3 metastatic hepatocellular carcinoma (metastatic HCC) samples and 4 normal controls; GSE40744 contained 9 HCC, 17 HCC surrounding non-tumorous tissue affected by cirrhosis (HCC-CIR), 18 hepatitis C virus-associated cirrhosis without HCC (CIR), 13 hepatitis B virus-associated acute liver failure (ALF), 7 surrounding normal liver of liver angioma (NLA) and 12 non-cancerous liver tissues; GSE21362 contained 73 HCC and 73 normal control samples; GSE20077 was a cell line dataset, which contained 7 HCC cell lines and 3 normal primary human hepatocytes; GSE12717 contained 10 HCC and 6 normal control samples; GSE31383 contained 9 HCC and 10 normal control samples. In addition, a dataset GSE64989 contained 8 recurrent HCC and 10 non-recurrent HCC samples, and two datasets [GSE67138 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE67138) and GSE67139 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE67139)] about vascular invasion were also included. GSE67138 contained 23 vascular invasion and 34 non-vascular invasions; GSE67139 contained 58 vascular invasion and 57 non-vascular invasions. The significant differences between the case groups and normal controls for the included datasets were calculated using Student's t-test.
To explore the DEGs associated with miR-124-3p in HCC, GEO profiles were searched with the terms ‘HCC AND miR-124-3p’ and a gene set (GSE6207) investigated miR-124-3p transfection in the HepG2 cell line was identified. The effects in the miR-124 overexpression group with the negative control group were subsequently compared by assessing fold change (FC), to identify downregulated genes and potential miR-124 targets (FC <0.95 were selected). The GSE6207 dataset (25) was retrieved to explore the mechanism by which miR-124-3p may be involved in hepatocarcinogenesis.
In silico analysis of target genes of miR-124-3p
Predicted targets of miR-124-3p and its target sites were analyzed using miRWalk2.0 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/) (26), which combines 12 existing miRNA-target prediction programs (miRWalk, Microt4, miRanda, miRbridge, miRDB, miRMap, miRNAMap, Pictar2, PITA, RNA22, RNAhybrid and TargetScan) to provide comprehensive potential targets for miR-124-3p. The genes contained in >5 prediction software programs were selected in order to obtain more reliable targets.
NLP
NLP is a novel computerized approach to analyze text in order to achieve human-like language processing. Under this approach, programmers create software to ‘read’ the text and extract key pieces of information from clinical notes, procedures, radiology or pathology reports and laboratory results (27,28). A literature search was performed in PubMed to obtain all associated electronic records. The literature search queries were as follows: (hepatocellular carcinoma) AND (resistance OR prognosis OR metastasis OR recurrence OR survival OR carcinogenesis OR sorafenib OR bevacizumab) and [‘1980/01/01’ (PDAT): ‘2015/05/25’ (PDAT)]. Subsequently, all pertinent molecules were retrieved and a list was generated, primarily comprising proteins and genes. Gene mention tagging was conducted with A Biomedical Named Entity Recognizer (ABNER; http://pages.cs.wisc.edu/~bsettles/abner/). ABNER also assisted in conjunction resolution. Gene name normalization conformed to the standard names in the Entrez database provided by NCBI (https://www.ncbi.nlm.nih.gov/gene/). Finally, statistical analysis was performed; P<0.01 was considered to indicate a statistically significant difference, as previously described (29,30). The P-value of a certain HCC-related gene was calculated with the following formula:
Where N indicates all the eligible studies, m is the frequency of the related genes, n is the frequency of HCC and k is the co-occurrence of HCC and a certain gene.
Construction of protein-protein interaction (PPI) networks
Overlapping genes were placed into the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING; version 10.0; http://string-db.org/) to construct the PPI network. The direct (physical) and indirect (functional) associations between the proteins were identified using four methods: Literature-reported protein interactions, high-throughput experiments, genome analysis and prediction, and co-expression studies. A node with a degree of high connectivity was perceived as a hub node. By scrutinizing the connectivity degrees of the nodes in the PPI networks, the hub genes were determined. Subsequently, the expression of hub genes identified from PPI networks were calculated with TCGA data. Firstly, the publicly available RNA-seq data of the mRNA level of Liver hepatocellular carcinoma (LIHC) samples were downloaded directly from the TCGA data portal (https://tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp) via bulk download mode [LIHC (cancer type), RNASeqV2 (data type), level 3 (data level)]. Then the relative expression of these hub genes were extracted and calculated on a log2 scale to investigate the potential targets of miR-124-3p in HCC. The immunohistochemical data of most potential targets of miR-124-3p were downloaded from The Human Protein Atlas (http://www.proteinatlas.org/).
Functional and signaling pathway analyses
Further functional and signaling pathway analyses were performed on a set of condition-specific genes, those that overlapped in the GEO database, target prediction software and NLP. The functional and signaling pathway analyses of the selected DEGs was performed on a public database platform; the Database for Annotation, Visualization and Integrated Discovery (DAVID; https://david.ncifcrf.gov/), which provides a functional interpretation of large lists of genes derived from genomic studies. The analyses also included Gene Ontology (GO) function analysis (http://www.geneontology.org/), Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg/) analysis and Protein Annotation Through Evolutionary Relationships (PANTHER) pathway analysis (http://www.pantherdb.org/). GO function analysis categorized the selected genes into groups, in accordance with the following three independent classification standards: Biological process (BP), cellular component (CC), and molecular function (MF). A Benjamini P-value of <0.05 was used to indicate a statistically significant difference in the above pathway enrichment analyses. The results were visualized as three GO maps using Cytoscape version 3.4.0 (http://cytoscape.org/). Subsequently, the pathway with the top priority was selected for further evaluation and the most significant aberrantly expressed genes were examined for their prospective diagnostic and prognostic value. The top 30 enriched pathways of KEGG were visualized using the ggplot2 (version 2.2.1) package of R/Bioconductor (version 3.4.2) Project for Statistical Computing (https://www.r-project.org).
Statistical analysis
SPSS 22.0 (IBM Corp., Armonk, NY, USA) was used for statistical analysis. All data are presented as the mean ± standard deviation. The receiver operating characteristic (ROC) curve was drawn to identify the diagnostic significance of miR-124-3p and clinical parameters. The criteria for assessing the area under the ROC curve (AUC) were as follows: 0.5–0.7, poor evidence for diagnosis; 0.7–0.9, moderate evidence for diagnosis; 0.9–1.0, high evidence for diagnosis. The correlations between miR-124-3p expression and clinicopathological parameters were determined using Spearman's rank correlation. Significant differences between HCC and non-cancerous liver tissues were analyzed using the Student's t-test. The significant differences amongst three groups was examined using one-way analysis of variance. Since no significant differences were identified, no post-hoc test was performed. A box-whisker plot was generated to demonstrate miR-124-3p expression from all GEO datasets using GraphPad Prism (version 5.0; GraphPad Software, Inc., La Jolla, CA, USA). RFS was assessed using the Kaplan-Meier method as well as Cox Regression and the log-rank test was performed to compare survival times. Kaplan-Meier survival curves were drawn to evaluate the association between miR-124-3p expression and the survival rate of 101 patients with HCC. P<0.05 was considered to indicate a statistically significant difference.
Results
Expression of miR-124-3p in HCC tissues and its association with clinical parameters
RT-qPCR analysis of miR-124-3p expression in 101 HCC and adjacent non-cancerous tissues revealed that the relative expression of miR-124-3p in HCC tissues was 2.4439±1.54599, which was significantly reduced compared with expression in the adjacent non-cancerous liver tissues (3.5279±1.82462; P<0.001). The AUC of the low miR-124-3p levels for HCC diagnosis was 0.691 (95% CI, 0.618–0.763; P<0.001; Fig. 1), with a cut-off value of 3.30, which indicated that miR-124-3p is a poor diagnostic marker for HCC. The expression of miR-124-3p in HCC samples with a single tumor node was significantly increased (2.7356±1.73799) compared with those with multiple tumor nodes (2.0659±1.16857; P=0.030). Compared with advanced stage HCC, the relative expression of miR-124-3p in early stage patients was markedly increased (stage I and II, 3.4600±1.97104; stage III and IV, 2.1096±1.21910; P=0.003). Spearman's rank correlation analysis revealed a correlation between miR-124-3p and clinical stage (r=−0.305; P=0.002). The clinicopathological parameters of the 101 HCC patients are presented in Table I.
Figure 1.
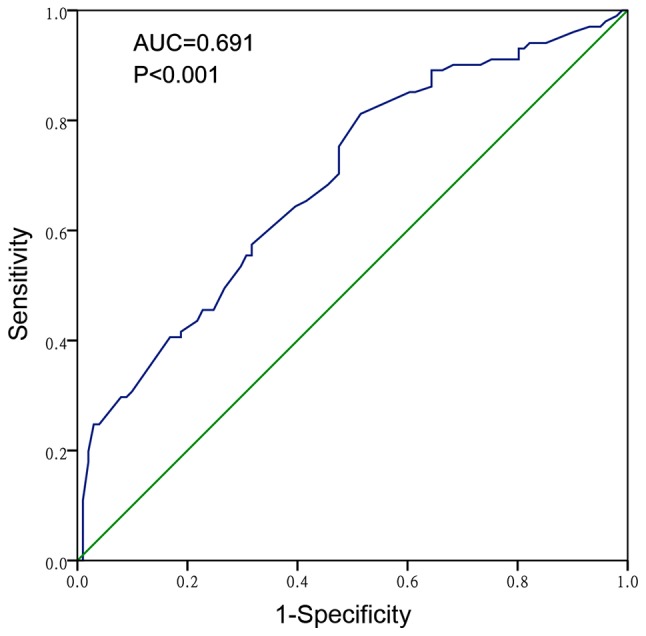
Receiver operating characteristic curve of the diagnostic value of miR-124-3p expression in HCC. The AUC of miR-124-3p was calculated to be 0.691 (95% CI, 0.618–0.763; P<0.001). AUC, area under curve; miR-124-3p, microRNA-124-3p.
Table I.
Association between miR-124-3p and clinicopathological parameters in hepatocellular carcinoma.
| Relative miR-124-3p expression (2−∆Cq) | Spearman's rank correlation | |||||
|---|---|---|---|---|---|---|
| Clinicopathological feature | n | Mean ± standard deviation | t | P-value | r | P-value |
| Tissue type | ||||||
| Adjacent non-cancerous | 101 | 3.5279±1.82462 | −4.556 | <0.001a | − | − |
| HCC | 101 | 2.4439±1.54599 | ||||
| Age, years | ||||||
| ≥50 | 51 | 2.2182±1.17362 | −1.484 | 0.142 | 0.081 | 0.423 |
| <50 | 50 | 2.6740±1.83446 | ||||
| Sex | ||||||
| Male | 80 | 2.4642±1.62180 | 0.257 | 0.797 | 0.026 | 0.800 |
| Female | 21 | 2.3662±1.24620 | ||||
| Differentiation | ||||||
| High | 7 | 1.6857±0.74482 | 0.985 | 0.377 | 0.038 | 0.704 |
| Moderate | 64 | 2.5442±1.60274 | ||||
| Low | 30 | 2.4067±1.54405 | ||||
| Size | ||||||
| <5 cm | 21 | 2.4381±1.60171 | −0.019 | 0.985 | 0.028 | 0.777 |
| ≥5 cm | 80 | 2.4454±1.54141 | ||||
| Tumor nodes | ||||||
| Single | 57 | 2.7356±1.73799 | 2.200 | 0.030a | −0.193 | 0.053 |
| Multi | 44 | 2.0659±1.16857 | ||||
| Metastasis | ||||||
| No | 49 | 2.5980±1.85646 | 0.960 | 0.340 | 0.014 | 0.890 |
| Yes | 52 | 2.2987±1.18255 | ||||
| Clinical TNM stage | ||||||
| I–II | 25 | 3.4600±1.97104 | 3.228 | 0.003a | −0.305 | 0.002a |
| III–IV | 76 | 2.1096±1.21910 | ||||
| Portal vein tumor embolus | ||||||
| − | 69 | 2.5571±1.62674 | 1.082 | 0.282 | −0.082 | 0.414 |
| + | 32 | 2.1997±1.34728 | ||||
| Vaso-invasion | ||||||
| − | 63 | 2.5705±1.66786 | 1.060 | 0.292 | −0.070 | 0.485 |
| + | 38 | 2.2339±1.31371 | ||||
| Tumor capsular infiltration | ||||||
| With complete capsule | 49 | 2.6039±1.71438 | 1.010 | 0.315 | −0.060 | 0.552 |
| No capsule or infiltration | 52 | 2.2931±1.36838 | ||||
| AFP | ||||||
| − | 46 | 2.3150±1.43969 | 0.314 | 0.755 | −0.008 | 0.944 |
| + | 39 | 2.4205±1.66341 | ||||
| Cirrhosis | ||||||
| − | 54 | 2.4148±1.33719 | 0.198 | 0.844 | 0.066 | 0.509 |
| + | 47 | 2.4772±1.77018 | ||||
t, Student's t-test
P<0.05; miR-124-3p, microRNA-124-3p; TNM, tumor-node-metastasis; AFP, α-fetoprotein.
Expression of miR-124-3p is correlated with the prognosis of patients with HCC
Kaplan-Meier analysis and Cox Regression were performed in order to determine the prognostic role of miR-124-3p. The follow-up information of 70 patients was obtained. In total, 44 patients exhibited low miR-124-3p expression (mean, <2.9859), while 26 patients exhibited high expression. The median survival time of the low miR-124-3p expression group was 57.00 months which was significantly reduced compared with that of the high expression group (the survival rate at each time point was >50%). The RFS rate was significantly reduced in the low miR-124-3p expression group compared with the high expression group (P=0.001; Fig. 2). The Cox Regression also demonstrated a hazard ratio of 0.069 (95%CI, 0.009–0.552; P=0.012).
Figure 2.
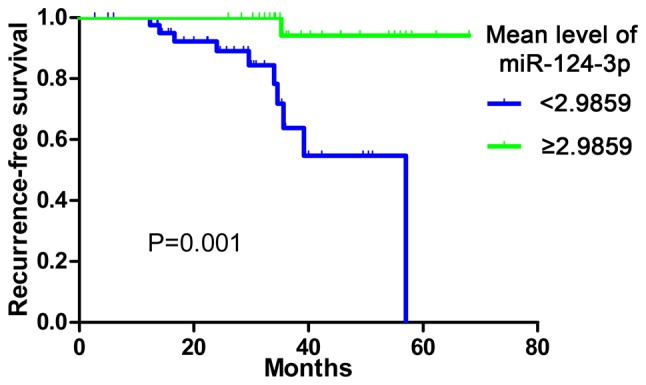
miR-124-3p expression and hepatocellular carcinoma survival. The Kaplan-Meier survival curve demonstrated that the median survival of the low miR-124-3p expression group was reduced compared with that of the high expression group. P=0.001. miR-124-3p, microRNA-124-3p; censored, patients lost to follow-up or succumbed to other causes (not HCC).
Expression of miR-124-3p in eight GEO datasets and previous reports
To confirm the RT-qPCR results of the present study, GEO datasets were searched to compare the expression of miR-124-3p between HCC and non-cancerous tissues. miR-124-3p expression levels in all seven microarray chips [GSE57555 (19), GSE41874 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE41874), GSE40744 (20), GSE21362 (21) GSE20077 (22), GSE12717 (23) and GSE31383 (24)], including 119 HCC and 124 non-cancerous tissues, were presented in Fig. 3. A recurrence [GSE64989 (31)] and two invasive datasets (GSE67139 and GSE67138) were also presented. Student's t-test revealed that miR-124-3p was significantly reduced in HCC compared to normal control samples with GSE 40744 data. While no significant difference could be found in other datasets. The seven datasets and RT-qPCR data from the present study were further analyzed by a meta-analysis, in which standardized mean differences (SMD) with a 95% CI [SMD and 95% CI, 0.01 (−0.38–0.40)] from selected datasets were pooled. However, no significant differences were observed between the HCC and non-cancerous liver groups (data not shown). Literatures review searching obtained four reliable studies. The PubMed literature search revealed that miR-124-3p expression in HCC was reduced compared with that in adjacent non-cancerous liver tissues, which was consistent with the results obtained in the present study (pooled P-value of four studies, P=1.05×10−10).
Figure 3.

Scatter diagram presenting the miR-124-3p expression of 10 microarray chips. (A) miR-124-3p expression in CCC, HCC and adjacent non-cancerous tissues. P=0.1306. (B) miR-124-3p expression in primary HCC, metastatic HCC and normal tissues. P=0.1965. (C) miR-124-3p expression in HCC tissues and normal liver tissues. P=0.2522. (D) miR-124-3p expression in HCC tissues and non-cancerous tissues. P=0.0849. (E) miR-124-3p expression in a human liver cancer cell line and normal primary human hepatocytes. P=0.7174. (F) miR-124-3p expression in HCC, HCC-CIR, CIR, ALF, NLA and non-cancerous liver tissues. P<0.001. (G) miR-124-3p expression in HCC tissues and human healthy liver tissues. P=0.8708. (H) miR-124-3p expression in HCC and adjacent non-tumor tissues. P<0.001. (I) miR-124-3p expression in recurrent HCC tissues and non-recurrent HCC tissues. P=0.1165. (J) miR-124-3p expression in tumor vascular invasion tissues and non-tumor vascular invasion tissues. P=0.2450. (K) miR-124-3p expression in tumor vascular invasion tissues and non-tumor vascular invasion tissues. P=0.2071. miR-124-3p, microRNA-124-3p; HCC, hepatocellular carcinoma; CCC, cholangiocarcinoma; HCC-CIR, HCC surrounding non-tumorous tissue affected by cirrhosis; CIR, hepatitis C virus-associated cirrhosis without HCC; ALF, hepatitis B virus-associated acute liver failure; NLA, surrounding normal liver of liver angioma.
Potential targets of miR-124-3p in HCC
To identify potential targets of miR-124-3p, GEO profiles were searched and gene chip data were downloaded such that the targets and functions of miR-124-3p could be analyzed. The fold-change (FC) between the miR-124 transfection group and the negative control group was subsequently calculated and FC values <0.95 were selected; a total of 4,261 mRNAs were attained following duplicate exclusion. The prediction process was then performed online using the miRWalk 2.0, which contained 12 computational algorithms. To increase the reliability of the present study, 3,902 mRNAs that appeared in ≥5 prediction software programs were screened, and any duplicates were excluded. Additionally, the titles and abstracts of 64,577 studies were included through the use of NLP data and text mining tools and a total of 1,800 HCC-associated genes were subsequently identified. The potential target mRNAs of miR-124-3p in HCC were determined by combining the predicted targets obtained using the three methods described earlier. In total, 132 mRNAs (Table II) were selected for PPI network analysis, GO analysis, KEGG pathway annotation and PANTHER pathway analysis, in order to identify the genes largely representative of the prospective molecular mechanism of miR-124-3p in HCC. A flow chart demonstrating the aforementioned process is presented in Fig. 4.
Table II.
Identified potential target genes of microRNA-124-3p in hepatocellular carcinoma.
| ZNF148 | ZDHHC2 | ZBTB20 | YAP1 | WSB1 | WHSC1 | WASF2 | VIM | VASH1 | UBE3C | TNFRSF10B | TLR7 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TLN2 | TJP1 | TGFA | TFRC | TFPI | TET1 | TCF4 | TCF3 | SULF1 | ST8SIA2 | SREBF1 | SQSTM1 |
| SPTBN1 | SPRY2 | SPRY1 | SPHK1 | SPARC | SPAG9 | SP1 | SOX9 | SOS1 | SORT1 | SOD2 | SMYD3 |
| SMAD5 | SMAD4 | SEC62 | SEC13 | SART3 | ROCK1 | RELA | RAB27A | PVRL2 | PTPN12 | PTP4A1 | PSEN1 |
| PRRX1 | PRLR | PPP1R13L | PPARA | PIK3CA | PEA15 | PAQR3 | NAAA | MYB | MTR | MTAP | MPZL1 |
| MAPRE1 | MAPK14 | MAPK10 | MAPK1 | LRP6 | LRP1 | LOX | KLF4 | KIF14 | JAG1 | ITGB1 | IQGAP1 |
| ING3 | IL7R | IL11 | IGFBP3 | HTATIP2 | HNMT | HLA-E | HIPK2 | HDLBP | HDAC4 | GYPA | GSN |
| GSC | GRIN1 | GGPS1 | G3BP1 | FMNL2 | FGFR2 | FGFR1 | ETS1 | ERN1 | ERBB3 | ERBB2 | EPHA7 |
| EPHA3 | EGR1 | E2F5 | DTL | DPP4 | DLGAP5 | DAB2IP | DAB2 | CTGF | CRKL | CHEK1 | CEACAM1 |
| CDK6 | CDK4 | CDK2 | CDC25B | CD44 | CCNG2 | CAPN2 | C1GALT1 | BMP6 | BCL2L2 | BCL2L11 | AURKA |
| ASPH | ARHGDIA | ARHGAP1 | ARAF | ANXA7 | ANGPT2 | AKT2 | AHR | ABI1 | ABCC4 | ABCC12 | ABCA2 |
Figure 4.

Illustration of the workflow pipeline. The genes selected for bioinformatics analysis overlapped in the GEO database, prediction software and NLP analysis, resulting in the identification of 132 genes. miR-124-3p, microRNA-124-3p; GEO, gene expression omnibus; FC, fold change; NLP, natural language processing; PPI, protein-protein interaction; GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PANTHER, Protein Annotation Through Evolutionary Relationships.
Analysis of PPI networks
The influence of multiple genes, interacting through a signaling pathway, on cellular function is more significant than the influence of a single gene. Therefore, a PPI network was constructed with 132 genes imported into the STRING software. Consequently, a total of 109 nodes and 386 edges were involved in the constructed network. Furthermore, 20 key genes with >10 nodes were identified, including mitogen-activated protein kinase 1 (MAPK1), MAPK14, early growth response 1 (EGR1), phosphatidylinositol-4,5-biphosphate 3-kinase catalytic subunit α (PIK3CA), RELA proto-oncogene, nuclear factor κ B subunit (RELA), SOS Ras/Rac guanine nucleotide exchange factor 1 (SOS1), SMAD family member 4 (SMAD4), erb-b2 tyrosine kinase 2, cyclin dependent kinase 2 (CDK2), ETS proto-oncogene 1, transcription factor (ETS1), Sp1 transcription factor (SP1), MYB proto-oncogene, transcription factor, fibroblast growth factor receptor 1 (FGFR1), AKT serine/threonine kinase 2 (AKT2), aurora kinase A, CDK4, FGFR2, SMAD5, MAPK10 and cell division cycle 25B. A total of 23 genes and proteins acted independently. PPI networks are presented in Fig. 5.
Figure 5.

Protein-protein interaction network. Interactions among the prospective 132 target genes were illustrated using the Search Tool for the Retrieval of Interacting Genes/Proteins online database and Cytoscape v3.4.0. Network nodes represent proteins and edges represent protein-protein associations. The blue nodes indicate the identified key genes.
In order to further study the potential role of miR-124-3p and the 20 key genes identified in HCC, the literature was reviewed and validated prediction software was searched. The target genes that had been validated by a luciferase report assay were subsequently determined. The results revealed that MAPK14, EGR1, RELA, CDK2, CDK4, ETS1, SOS1 and SP1 were upregulated following miR-124-3p knockdown. GSE22058 data were also mined to examine the genome-wide expression profiles of miRNAs and mRNAs of 96 HCC samples and their adjacent non-cancerous liver tissues. This revealed that EGR1 and ETS1 were negatively correlated with miR-124-3p expression (Fig. 6). Additionally, validation of the association between miR-124-3p and the 20 identified key genes based on TCGA data was attempted, but insufficient data was available.
Figure 6.

Correlation between microRNA 124-3p and (A) EGR1 or (B) ETS1 expression based on the GSE22058 dataset. EGR1, early growth response protein 1; ETS1, ETS proto-oncogene 1.
Validation of potential target gene expression based on TCGA with data from The Human Protein Atlas
Based on the literature review and prediction software search, 11/20 key genes identified by the PPI network were selected for further analysis, namely MAPK14, EGR1, RELA, PIK3CA, SOS1, SMAD4, CDK2, CDK4, ETS1, SP1 and AKT2. The relative expression of these 11 genes calculated with TCGA data revealed that no significant differences were detected in the expression of PIK3CA, SMAD4 and AKT2 between the HCC and adjacent non-cancerous liver tissues. EGR1 and ETS1 were downregulated in the HCC tissues when compared with adjacent non-cancerous tissues (data not shown). Notably, MAPK14, RELA, SOS1, CDK2, CDK4 and SP1 were significantly overexpressed in liver cancer tissues, compared with expression in adjacent liver tissues (P<0.01).
The protein expression of the six aforementioned genes was also collected, as they were more likely to be direct miR-124-3p targets. The immunohistochemical data downloaded from The Human Protein Atlas revealed that all these genes were overexpressed in HCC tissues, with the exception of SOS1, which was also positively stained in normal tissues. Therefore, MAPK14, RELA, CDK2, CDK4 and SP1 were considered direct targets of miR-124-3p in HCC. Expression profiles of the five mRNAs with their corresponding immunohistochemical results are presented in Fig. 7.
Figure 7.

Overexpression of five potential target genes of microRNA 124-3p in HCC, based on TCGA and The Human Protein Atlas data. The TCGA RNAseq profiles of HCC were extracted to explore the relative expression of (A) MAPK14, (B) RELA, (C) CDK2, (D) CDK4 and (E) SP1 in HCC and adjacent non-cancerous liver tissues. Immunohistochemical data was downloaded from The Human Protein Atlas. TCGA, The Cancer Genome Atlas; HCC, hepatocellular carcinoma; MAPK14, mitogen-activated protein kinase 14; RELA, RELA proto-oncogene, nuclear factor κB subunit; CDK2, cycle dependent kinase 2; CDK4, cycle dependent kinase 4; SP1, SP1 transcription factor.
Functional and signaling pathway analyses
The 132 identified genes were classified into three GO categories using the online analysis tool DAVID. The BP genes exhibited significant enrichment in the regulation of cell proliferation (GO, 0042127; P=1.71×10−10), positive regulation of cellular biosynthetic processes (GO, 0031328; P=1.86×10−10) and positive regulation of biosynthetic processes (GO, 0009891; P=2.57×10−10). Among the CC genes, the top three functional items were cell leading edge (GO, 0031252; P=4.06×10−7), cytosol (GO, 0005829; P=1.12×10−5) and cell projection (GO, 0042995; P=1.18×10−5). Among the MF genes, the most clustered GO terms were protein kinase activity (GO, 0004672; P=4.92×10−7), transcription activator activity (GO, 0016563; P=8.83×10−5) and enzyme binding (GO, 0019899; P=2.77×10−4). Three visualized GO maps (BP, CC and MF) were drawn by EnrichmentMap, a Cytoscape v3.4.0 plug-in. In the enrichment map, the number of nodes and edges of the three GO categories was: BP, 72 and 1366; CC, 41 and 461; MF, 18 and 92, respectively (Fig. 8). The top 10 categories for 3 GO terms are presented in Table III.
Figure 8.

GO enrichment maps were drawn using the Cytoscape v3.4.0 EnrichmentMap plug-in. The rhombus represents different terms of biological processes. The associations among terms are represented by arrows. The number of nodes and edges of the three GO categories was: biological process, 72 and 1366; cellular component, 41 and 461; and molecular function, 18 and 92; respectively. GO, gene ontology.
Table III.
GO functional annotation for most significantly associated targets of microRNA-124-3p.
| GO ID | GO term | Count (%) | P-value | Benjamini | FDR | Gene symbol |
|---|---|---|---|---|---|---|
| Biological process | ||||||
| GO:0042127 | Regulation of cell proliferation | 30 (1.7) | 1.71×10−10 | 2.83×10−7 | 2.88×10−7 | FGFR2, FGFR1, ERBB3, ERBB2, PRRX1, CHEK1, ABI1, JAG1, SOX9, ITGB1, IL11, TGFA, ASPH, TCF3, RELA, etc.. |
| GO:0031328 | Positive regulation of cellular biosynthetic process | 28 (1.6) | 1.86×10−10 | 1.54×10−7 | 3.13×10−7 | PPARA, SOX9, TLR7, IL11, ZNF148, SQSTM1, YAP1, TCF4, TCF3, AKT2, EGR1, SREBF1, RELA, GRIN1, SMAD5, etc. |
| GO:0009891 | Positive regulation of biosynthetic process | 28 (1.6) | 2.57×10−10 | 1.42×10−7 | 4.32×10−7 | PPARA, SOX9, TLR7, IL11, ZNF148, SQSTM1, YAP1, TCF4, TCF3, AKT2, EGR1, SREBF1, RELA, GRIN1, SMAD5, etc. |
| GO:0045944 | Positive regulation of transcription from RNA polymerase II promoter | 21 (1.2) | 2.78×10−10 | 1.15×10−7 | 4.68×10−7 | SREBF1, EGR1, PPARA, RELA, SMAD5, GRIN1, SMAD4, SOX9, AHR, IL11, HDAC4, SP1, ZNF148, SQSTM1, ETS1, etc. |
| GO:0010557 | Positive regulation of macromolecule biosynthetic process | 27 (1.5) | 3.57×10−10 | 1.18×10−7 | 6.00×10−7 | PPARA, SOX9, TLR7, IL11, ZNF148, SQSTM1, YAP1, TCF4, TCF3, AKT2, EGR1, SREBF1, RELA, GRIN1, SMAD5, etc. |
| GO:0006468 | Protein amino acid phosphorylation | 27 (1.5) | 5.45×10−10 | 1.50×10−7 | 9.16×10−7 | FGFR2, FGFR1, ERBB3, ERBB2, ABI1, AURKA, CHEK1, TGFA, PIK3CA, AKT2, ROCK1, CDK6, MAPK10, CDK4, CDK2, etc. |
| GO:0042981 | Regulation of apoptosis | 29 (1.7) | 1.35×10−9 | 3.20×10−7 | 2.28×10−6 | ING3, HTATIP2, ERBB3, ERBB2, BCL2L2, SOX9, PEA15, CD44, SQSTM1, SOS1, PIK3CA, ARHGDIA, RAB27A, ROCK1, RELA, etc. |
| GO:0043067 | Regulation of programmed cell death | 29 (1.7) | 1.69×10−9 | 3.49×10−7 | 2.84×10−6 | ING3, HTATIP2, ERBB3, ERBB2, BCL2L2, SOX9, PEA15, CD44, SQSTM1, SOS1, PIK3CA, ARHGDIA, RAB27A, ROCK1, RELA, etc. |
| GO:0010941 | Regulation of cell death | 29 (1.7) | 1.84×10−9 | 3.37×10−7 | 3.08×10−6 | ING3, HTATIP2, ERBB3, ERBB2, BCL2L2, SOX9, PEA15, CD44, SQSTM1, SOS1, PIK3CA, ARHGDIA, RAB27A, ROCK1, RELA, etc. |
| GO:0045941 | Positive regulation of transcription | 24 (1.4) | 2.67×10−9 | 4.42×10−7 | 4.49×10−6 | SREBF1, EGR1, PPARA, RELA, SMAD5, GRIN1, SMAD4, SOX9, AHR, CDK2, IL11, HDAC4, MAPK1, SP1, ZNF148, etc. |
| Cellular component | ||||||
| GO:0031252 | Cell leading edge | 11 (0.6) | 4.06×10−7 | 9.92×10−5 | 5.26×10−4 | SPRY1, TLN2, GSN, WASF2, ARHGAP1, PIK3CA, ABI1, CDK6, ITGB1, IQGAP1, AKT2 |
| GO:0005829 | Cytosol | 29 (1.7) | 1.12×10−5 | 0.00136 | 0.01446 | VIM, BCL2L2, ABI1, ANXA7, SPRY2, SPRY1, GSN, SQSTM1, SOS1, PIK3CA, ARHGDIA, AKT2, ROCK1, RELA, SMAD5, etc. |
| GO:0042995 | Cell projection | 20 (1.1) | 1.18×10−5 | 9.61×10−4 | 0.01530 | TLN2, ERBB2, VIM, GRIN1, WASF2, CDK6, ABI1, ITGB1, IQGAP1, MAPK1, EPHA7, SPRY1, LRP1, PSEN1, GSN, etc. |
| GO:0005667 | Transcription factor complex | 11 (0.6) | 1.78×10−5 | 0.00109 | 0.02304 | GSC, E2F5, RELA, SMAD5, SMAD4, YAP1, TCF4, CDK4, TCF3, KLF4, CDK2 |
| GO:0000267 | Cell fraction | 23 (1.3) | 2.00×10−4 | 0.00970 | 0.25820 | FGFR1, GYPA, GRIN1, SPHK1, ABI1, CAPN2, ITGB1, BCL2L11, PTPN12, SOD2, ANXA7, MAPK1, LRP1, PSEN1, ARAF, etc. |
| GO:0009986 | Cell surface | 12 (0.7) | 2.74×10−4 | 0.01108 | 0.35385 | FGFR2, LRP1, PSEN1, PRLR, CD44, SULF1, PVRL2, TGFA, SORT1, IL7R, ITGB1, DPP4 |
| GO:0005654 | Nucleoplasm | 20 (1.1) | 2.79×10−4 | 0.00967 | 0.35999 | ING3, GSC, E2F5, RELA, SMAD5, SMAD4, CHEK1, CDK4, SART3, CDK2, CDC25B, HDAC4, MAPK1, SQSTM1, etc. |
| GO:0031981 | Nuclear lumen | 27 (1.5) | 3.55×10−4 | 0.01078 | 0.45888 | ING3, E2F5, CHEK1, SOX9, SART3, IQGAP1, ZNF148, SQSTM1, YAP1, TCF4, MYB, TCF3, ZBTB20, GSC, RELA, etc. |
| GO:0030027 | Lamellipodium | 6 (0.3) | 3.94×10−4 | 0.01063 | 0.50892 | SPRY1, GSN, WASF2, PIK3CA, ABI1, AKT2 |
| GO:0005856 | Cytoskeleton | 26 (1.5) | 4.10×10−4 | 0.00996 | 0.52974 | TLN2, VIM, WASF2, ABI1, AURKA, ABCA2, CHEK1, SEC62, IQGAP1, SPRY2, PEA15, GSN, SOS1, ARHGDIA, KIF14, etc. |
| Molecular function | ||||||
| GO:0004672 | Protein kinase activity | 21 (1.2) | 4.92×10−7 | 1.73×10−4 | 6.73×10−4 | FGFR2, FGFR1, ROCK1, ERBB3, ERBB2, CDK6, CHEK1, AURKA, MAPK10, CDK4, CDK2, EPHA3, MAPK1, EPHA7, CRKL, etc. |
| GO:0016563 | Transcription activator activity | 14 (0.8) | 8.83×10−5 | 0.01542 | 0.12078 | EGR1, PPARA, HTATIP2, SMAD5, SMAD4, PRRX1, SOX9, HDAC4, SP1, ZNF148, YAP1, MYB, TCF3, KLF4 |
| GO:0019899 | Enzyme binding | 15 (0.9) | 2.77×10−4 | 0.03193 | 0.37791 | FMNL2, ROCK1, ERBB2, RELA, VIM, CHEK1, AURKA, IQGAP1, HDAC4, SPAG9, MAPK1, LRP1, SP1, SQSTM1, SORT1 |
| GO:0004714 | Transmembrane receptor protein tyrosine kinase activity | 6 (0.3) | 3.51×10−4 | 0.03042 | 0.47936 | FGFR2, FGFR1, EPHA7, ERBB3, ERBB2, EPHA3 |
| GO:0004674 | Protein serine/threonine kinase activity | 13 (0.7) | 5.34×10−4 | 0.03689 | 0.72816 | ROCK1, AURKA, CHEK1, CDK6, MAPK10, CDK4, CDK2, MAPK1, MAPK14, ARAF, HIPK2, ERN1, AKT2 |
| GO:0005524 | ATP binding | 26 (1.5) | 0.00148 | 0.08344 | 2.01226 | FGFR2, FGFR1, ERBB3, ERBB2, ABCA2, AURKA, CHEK1, PIK3CA, AKT2, KIF14, ROCK1, G3BP1, SPHK1, CDK6, etc. |
| GO:0003702 | RNA polymerase II transcription factor activity | 9 (0.5) | 0.00172 | 0.08284 | 2.32651 | SREBF1, PPARA, HTATIP2, SP1, ETS1, ZNF148, RELA, SOX9, KLF4 |
| GO:0010843 | Promoter binding | 5 (0.3) | 0.00177 | 0.07487 | 2.39182 | SREBF1, HDAC4, SP1, SMAD4, TCF3 |
| GO:0032559 | Adenyl ribonucleotide binding | 26 (1.5) | 0.00179 | 0.06771 | 2.42412 | FGFR2, FGFR1, ERBB3, ERBB2, ABCA2, AURKA, CHEK1, PIK3CA, AKT2, KIF14, ROCK1, G3BP1, SPHK1, CDK6, etc. |
| GO:0019838 | Growth factor binding | 6 (0.3) | 0.00267 | 0.08983 | 3.59394 | ERBB3, CTGF, ERBB2, SORT1, IL7R, IGFBP3 |
GO, gene ontology.
KEGG pathway enrichment analysis revealed that pancreatic cancer (hsa05212; P=6.80×10−8), prostate cancer (hsa05215; P=5.28×10−7), non-small cell lung cancer (hsa05223; P=9.25×10−7), chronic myeloid leukemia (hsa05220; P=1.16×10−6) and pathways in cancer (hsa05200; P=3.64×10−6) had the most significant enrichment. The top 30 enriched pathways were presented using the ggplot2 (version 2.2.1) package of R/Bioconductor (version 3.4.2) Project for Statistical Computing (https://www.r-project.org/), a free software environment for statistical computing and graphics (32) (Fig. 9).
Figure 9.

KEGG pathway analysis of microRNA 124-3p predicted target genes in hepatocellular carcinoma. Enrichment analysis was performed to identify pathways enriched in olfactory transduction using the ggplot2 (version 2.2.1) package of R/Bioconductor (version 3.4.2) Project for Statistical Computing (https://www.r-project.org/). KEGG, Kyoto Encyclopedia of Genes and Genomes.
Regarding the PANTHER pathway, the following three terms were identified as the most significant: Angiogenesis (P00005; P=1.78×10−7), epidermal growth factor receptor signaling pathway (P00018; P=1.54×10−6), and FGF signaling pathway (P00021; P=4.32×10−5). The top 10 categories for KEGG and the PANTHER pathway are presented in Table IV.
Table IV.
KEGG and PANTHER functional annotation for most significantly associated targets of miR-124-3p.
| Database ID | Database term | Count (%) | P-value | Benjamini | FDR | Gene symbol |
|---|---|---|---|---|---|---|
| KEGG | ||||||
| hsa05212 | Pancreatic cancer | 11 (0.6) | 6.80×10−8 | 6.39×10−6 | 7.46×10−5 | MAPK1, RELA, ERBB2, ARAF, SMAD4, TGFA, PIK3CA, CDK6, MAPK10, CDK4, AKT2 |
| hsa05215 | Prostate cancer | 11 (0.6) | 5.28×10−7 | 2.48×10−5 | 5.79×10−4 | FGFR2, FGFR1, MAPK1, RELA, ERBB2, SOS1, ARAF, TGFA, PIK3CA, CDK2, AKT2 |
| hsa05223 | Non-small cell lung cancer | 9 (0.5) | 9.25×10−7 | 2.90×10−5 | 0.00101 | MAPK1, ERBB2, SOS1, ARAF, TGFA, PIK3CA, CDK6, CDK4, AKT2 |
| hsa05220 | Chronic myeloid leukemia | 10 (0.6) | 1.16×10−6 | 2.74×10−5 | 0.00128 | MAPK1, CRKL, RELA, SOS1, ARAF, SMAD4, PIK3CA, CDK6, CDK4, AKT2 |
| hsa05200 | Pathways in cancer | 18 (1.0) | 3.64×10−6 | 6.84×10−5 | 0.00399 | FGFR2, FGFR1, ERBB2, RELA, SMAD4, CDK6, MAPK10, CDK4, ITGB1, CDK2, MAPK1, CRKL, ETS1, SOS1, ARAF, TGFA, PIK3CA, AKT2 |
| hsa04012 | ErbB signaling pathway | 10 (0.6) | 4.13×10−6 | 6.47×10−5 | 0.00453 | MAPK1, CRKL, ERBB3, ERBB2, SOS1, ARAF, TGFA, PIK3CA, MAPK10, AKT2 |
| hsa04722 | Neurotrophin signaling pathway | 11 (0.6) | 1.12×10−5 | 1.51×10−4 | 0.01230 | MAPK1, CRKL, PSEN1, RELA, MAPK14, SOS1, SORT1, PIK3CA, MAPK10, ARHGDIA, AKT2 |
| hsa05214 | Glioma | 8 (0.5) | 3.27×10−5 | 3.84×10−4 | 0.03585 | MAPK1, SOS1, ARAF, TGFA, PIK3CA, CDK6, CDK4, AKT2 |
| hsa05211 | Renal cell carcinoma | 8 (0.5) | 6.52×10−5 | 6.81×10−4 | 0.07153 | MAPK1, CRKL, ETS1, SOS1, ARAF, TGFA, PIK3CA, AKT2 |
| hsa04520 | Adherens junction | 8 (0.5) | 1.21×10−4 | 0.00113 | 0.13211 | FGFR1, MAPK1, TJP1, ERBB2, WASF2, PVRL2, SMAD4, IQGAP1 |
| PANTHER | ||||||
| P00005 | Angiogenesis | 17 (1.0) | 1.78×10−7 | 8.17×10−6 | 1.68×10−4 | FGFR2, FGFR1, SPHK1, JAG1, MAPK10, EPHA3, MAPK1, EPHA7, CRKL, ETS1, MAPK14, SOS1, ARAF, ARHGAP1, etc. |
| P00018 | EGF receptor signaling pathway | 13 (0.7) | 1.54×10−6 | 3.55×10−5 | 0.00146 | MAPK1, SPRY2, DAB2IP, SPRY1, ERBB3, MAPK14, ERBB2, SOS1, ARAF, PIK3CA, TGFA, MAPK10, AKT2 |
| P00021 | FGF signaling pathway | 11 (0.6) | 4.32×10−5 | 6.61×10−4 | 0.04078 | FGFR2, SPRY2, FGFR1, MAPK1, SPRY1, MAPK14, SOS1, ARAF, PIK3CA, MAPK10, AKT2 |
| P00056 | VEGF signaling pathway | 8 (0.5) | 2.25×10−4 | 0.00259 | 0.21257 | MAPK1, ETS1, MAPK14, ARAF, ARHGAP1, SPHK1, PIK3CA, AKT2 |
| P04393 | Ras Pathway | 8 (0.5) | 5.48×10−4 | 0.00503 | 0.51707 | MAPK1, ETS1, MAPK14, SOS1, ARAF, PIK3CA, MAPK10, AKT2 |
| P00010 | B cell activation | 6 (0.3) | 0.01190 | 0.08766 | 10.69454 | MAPK1, MAPK14, SOS1, ARAF, PIK3CA, MAPK10 |
| P00006 | Apoptosis signaling pathway | 7 (0.4) | 0.01767 | 0.11053 | 15.50490 | MAPK1, TNFRSF10B, RELA, PIK3CA, BCL2L2, MAPK10, AKT2 |
| P04398 | p53 pathway feedback loops 2 | 5 (0.3) | 0.01786 | 0.09846 | 15.66488 | MAPK14, PIK3CA, CCNG2, CDK2, AKT2 |
| P00054 | Toll receptor signaling pathway | 5 (0.3) | 0.01987 | 0.09748 | 17.27652 | MAPK1, RELA, MAPK14, MAPK10, TLR7 |
| P00034 | Integrin signaling pathway | 9 (0.5) | 0.02426 | 0.10682 | 20.71556 | MAPK1, CRKL, MAPK14, SOS1, ARAF, PIK3CA, MAPK10, ITGB1, PTPN12 |
| P00047 | PDGF signaling pathway | 8 (0.5) | 0.02650 | 0.10625 | 22.42248 | MAPK1, ETS1, SOS1, ARAF, ARHGAP1, PIK3CA, MAPK10, AKT2 |
| P00053 | T cell activation | 6 (0.3) | 0.04509 | 0.16213 | 35.34827 | MAPK1, SOS1, ARAF, PIK3CA, MAPK10, AKT2 |
| P00059 | p53 pathway | 6 (0.3) | 0.04510 | 0.16213 | 35.34827 | TNFRSF10B, PIK3CA, IGFBP3, CCNG2, CDK2, AKT2 |
KEGG, Kyoto encyclopedia of genes and genomes; PANTHER, protein annotation through evolutionary relationship.
Discussion
The occurrence and development of HCC is a multistep process that involves the activation of tumor oncogenes and/or the inactivation of tumor suppressor genes. These factors are of vital importance for the design of effective therapeutic strategies for the treatment of HCC. Given that the prognosis of advanced HCC remains poor regardless of improvements in innovative therapeutic approaches, the development of a more effective method of diagnosis and therapy and/or a novel biomarker for early stage HCC is urgently required. Previous studies have demonstrated the crucial involvement of miRNAs in carcinogenesis (33–35). The function of miRNAs in hepatocarcinogenesis has also been assessed. The ectopic expression of several miRNAs has been detected in HCC, including miR-21, miR-542-5p, miR-29, miR-140-5p, miR-221 and miR-490-3p. Previous studies have also reported low miR-124 expression in HCC (2,10,36,37), prostate cancer (38), breast cancer (39), gastric cancer (40) and pancreatic cancer (41). In line with the results of previous studies, the present study demonstrated that miR-124-3p was downregulated in HCC and that its downregulation was significantly associated with certain clinical characteristics, including the TNM stage. Additionally, the data indicated that miR-124-3p expression is positively associated with RFS in patients with HCC (P=0.012). Meta-analysis of GEO data only, and meta-analysis of GEO data plus in-house PCR revealed no significant difference of miR-124-3p between HCC and non-cancerous liver groups. Certain chips contained fewer HCC samples than non-cancerous samples, while other chips contained more HCC samples than non-cancerous samples. Additionally, technology may be another cause of this difference. For example, amongst the data included here, certain parts were detected using gene chip technology, while a number were detected by PCR. Conversely, seven microarrays come from different platforms, which is another reason to cause the difference. TCGA was used in order to validate the downregulation of miR-124-3p, but this was unsuccessful due to the lack of available data.
miRNAs are a class of short endogenous non-coding RNAs that inhibit gene expression by either degrading mRNA or repressing mRNA translation. Theoretically, there are hundreds of potential targets for a single miRNA. Previous studies have demonstrated that miRNAs modulate a wide variety of targets, indicating that a single miRNA may regulate cancer progression in multiple steps by targeting numerous genes. Zheng et al (37) reported that miR-124 inhibits epithelial-mesenchymal transition by inhibiting the mRNA and protein expression of ROCK2 and EZH2 in HCC. Lang and Ling (10) demonstrated that miR-124 inhibits cell proliferation by targeting PIK3CA in HCC. The literature search of the present study revealed that miR-124-3p acts via the regulation of CTGF, ITGB1, RhoG and ROCK1 expression. Recently, with the progression of high-throughput sequencing and bioinformatics, circular RNAs (circRNAs) have received increasing attention (42,43). Increasing evidence indicates that circRNAs may serve a major role in various types of cancer (44–46), including HCC. Shang et al (47) identified hsa_circ_0005075 as a potential HCC biomarker involved in HCC development. Qin et al (48) and Xu et al (49) also demonstrated that circRNAs may serve as potential novel biomarkers in HCC. Notably, Zheng et al (50) performed RNA immunoprecipitation and luciferase screening, demonstrating that circHIPK3 may function as a sponge for miRNAs, including miR-12, as circHIPK3 is directly adsorbed to miR-124 and suppresses its activation. In the present study, the target genes of miR-124-3p were explored by assessing the overlapping genes retrieved from GEO profiles, prediction software and NLP. A total of 132 potential target mRNAs of miR-124-3p in HCC were identified. Based on the prospective roles of circRNAs in human cancer, future studies should aim to further investigate the association between circRNA and miR-124-3p in HCC.
As the diagnosis of HCC at early stages is difficult, when patients were diagnosed with HCC, it is generally too late to remove the tumor with surgical excision. Various novel therapies for advanced HCC are being actively pursued, including molecular targeted therapy, systemic chemotherapy, immunotherapy and arginine deprivation therapy (51). These treatment therapies are all involved in the molecular pathways of HCC development. Furthermore, the majority of the molecular targeted drugs currently being investigated are multi-targeted drugs. Therefore, it is crucial to elucidate the molecular pathogenesis of HCC. Following the prediction of potential target genes, functional and signaling pathway analyses were conducted. Additionally, enriched GO term, KEGG pathway and PANTHER pathway analyses of miR-124-3p target genes were performed. The results revealed that the predicted target genes of miR-124-3p involved 396 GO terms. The top-ranking terms all exhibited a large number of gene clusters, particularly in BP terms. KEGG pathway enrichment analysis demonstrated that the predicted target genes of miR-124-3p were concentrated in 41 signaling pathways, and the top ten signaling pathways were involved in the occurrence and development of several types of cancer. PANTHER pathway analysis indicated that the predicted target genes of miR-124-3p were concentrated in 14 terms, which were all involved in various molecular signaling pathways. Therefore, taken together with the results of previous studies, these observations suggested that miR-124-3p may target multiple genes and may function spatiotemporally or in combination with several diverse processes. For example, among the 14 terms of the PANTHER pathway, ‘Angiogenesis’, ‘EGF receptor signaling pathway’, ‘FGF signaling pathway’, ‘VEGF signaling pathway’ and ‘PDGF signaling pathway’ are all involved in angiogenesis. However, other target genes involved in ‘B cell activation’, ‘Toll receptor signaling pathway’ and ‘T cell activation’ indicated that immunological mechanisms may be the other factor that affected the genesis and the development of HCC. In addition, ‘Ras pathway’, ‘Apoptosis signaling pathway’ and ‘P53 pathway’ have also been predicted. Further research on the mechanisms of above pathway may be beneficial in the development of novel therapeutic agents against HCC.
In conclusion, it was confirmed that miR-124-3p was downregulated in HCC. Assessing the overlaps in GEO data, miR-124-3p predicted target genes and NLP, 132 reliable potential target genes of miR-124-3p were identified that may serve key functions in HCC. The pathway analysis results suggested that the functional characterization of the identified miR-124-3p targets will offer novel insight into the underlying molecular mechanisms that occur in HCC. Given the complexity of molecular pathway involvement, the mechanism of hepatocarcinogenesis requires further study with powerful analysis tools, as well as through the study of in vitro and in vivo models. The identified miR-124-3p target genes have the potential to be employed as innovative prognosticators and/or therapeutic targets for HCC.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Huang DH, Wang GY, Zhang JW, Li Y, Zeng XC, Jiang N. MiR-501-5p regulates CYLD expression and promotes cell proliferation in human hepatocellular carcinoma. Jpn J Clin Oncol. 2015;45:738–744. doi: 10.1093/jjco/hyv063. [DOI] [PubMed] [Google Scholar]
- 2.Lu Y, Yue X, Cui Y, Zhang J, Wang K. MicroRNA-124 suppresses growth of human hepatocellular carcinoma by targeting STAT3. Biochem Biophys Res Commun. 2013;441:873–879. doi: 10.1016/j.bbrc.2013.10.157. [DOI] [PubMed] [Google Scholar]
- 3.Otsuka M, Kishikawa T, Yoshikawa T, Yamagami M, Ohno M, Takata A, Shibata C, Ishibashi R, Koike K. MicroRNAs and liver disease. J Hum Genet. 2017;62:75–80. doi: 10.1038/jhg.2016.53. [DOI] [PubMed] [Google Scholar]
- 4.Huang Y, Shen XJ, Zou Q, Wang SP, Tang SM, Zhang GZ. Biological functions of microRNAs: A review. J Physiol Biochem. 2011;67:129–139. doi: 10.1007/s13105-010-0050-6. [DOI] [PubMed] [Google Scholar]
- 5.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 6.Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: Worldwide incidence and trends. Gastroenterology. 2004;127(5 Suppl 1):S5–S16. doi: 10.1053/j.gastro.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 7.Sherman M. Hepatocellular carcinoma: Epidemiology, surveillance, and diagnosis. Semin Liver Dis. 2010;30:3–16. doi: 10.1055/s-0030-1247128. [DOI] [PubMed] [Google Scholar]
- 8.Shangguan H, Tan SY, Zhang JR. Bioinformatics analysis of gene expression profiles in hepatocellular carcinoma. Eur Rev Med Pharmacol Sci. 2015;19:2054–2061. [PubMed] [Google Scholar]
- 9.Huang S, He X. The role of microRNAs in liver cancer progression. Br J Cancer. 2011;104:235–240. doi: 10.1038/sj.bjc.6606010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lang Q, Ling C. MiR-124 suppresses cell proliferation in hepatocellular carcinoma by targeting PIK3CA. Biochem Biophys Res Commun. 2012;426:247–252. doi: 10.1016/j.bbrc.2012.08.075. [DOI] [PubMed] [Google Scholar]
- 11.Furuta M, Kozaki KI, Tanaka S, Arii S, Imoto I, Inazawa J. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis. 2010;31:766–776. doi: 10.1093/carcin/bgp250. [DOI] [PubMed] [Google Scholar]
- 12.World Health Organization (WHO), corp-author WHO Handbook for Reporting Results of Cancer Treatment. WHO; Geneva: 1979. [Google Scholar]
- 13.Rong M, He R, Dang Y, Chen G. Expression and clinicopathological significance of miR-146a in hepatocellular carcinoma tissues. Ups J Med Sci. 2014;119:19–24. doi: 10.3109/03009734.2013.856970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rong M, Chen G, Dang Y. Increased miR-221 expression in hepatocellular carcinoma tissues and its role in enhancing cell growth and inhibiting apoptosis in vitro. BMC Cancer. 2013;13:21. doi: 10.1186/1471-2407-13-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen G, Umelo IA, Lv S, Teugels E, Fostier K, Kronenberger P, Dewaele A, Sadones J, Geers C, De Grève J. miR-146a inhibits cell growth, cell migration and induces apoptosis in non-small cell lung cancer cells. PLoS One. 2013;8:e60317. doi: 10.1371/journal.pone.0060317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Ren F, Luo Y, Rong M, Chen G, Dang Y. Down-regulation of miR-193a-3p dictates deterioration of HCC: A clinical real-time qRT-PCR study. Med Sci Monit. 2015;21:2352–2360. doi: 10.12659/MSM.894077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Egger M, Smith GD, Altman DG. Systematic Reviews in Health Care. Meta-analysis in Context. 2nd. BMJ Publishing Group; London: 2001. [DOI] [Google Scholar]
- 19.Murakami Y, Kubo S, Tamori A, Itami S, Kawamura E, Iwaisako K, Ikeda K, Kawada N, Ochiya T, Taguchi YH. Comprehensive analysis of transcriptome and metabolome analysis in Intrahepatic Cholangiocarcinoma and Hepatocellular Carcinoma. Sci Rep. 2015;5:16294. doi: 10.1038/srep16294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diaz G, Melis M, Tice A, Kleiner DE, Mishra L, Zamboni F, Farci P. Identification of microRNAs specifically expressed in hepatitis C virus-associated hepatocellular carcinoma. Int J Cancer. 2013;133:816–824. doi: 10.1002/ijc.28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato F, Hatano E, Kitamura K, Myomoto A, Fujiwara T, Takizawa S, Tsuchiya S, Tsujimoto G, Uemoto S, Shimizu K. MicroRNA profile predicts recurrence after resection in patients with hepatocellular carcinoma within the Milan criteria. PLoS One. 2011;6:e16435. doi: 10.1371/journal.pone.0016435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He XX, Chang Y, Meng FY, Wang MY, Xie QH, Tang F, Li PY, Song YH, Lin JS. MicroRNA-375 targets AEG-1 in hepatocellular carcinoma and suppresses liver cancer cell growth in vitro and in vivo. Oncogene. 2012;31:3357–3369. doi: 10.1038/onc.2011.500. [DOI] [PubMed] [Google Scholar]
- 23.Su H, Yang JR, Xu T, Huang J, Xu L, Yuan Y, Zhuang SM. MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res. 2009;69:1135–1142. doi: 10.1158/0008-5472.CAN-08-2886. [DOI] [PubMed] [Google Scholar]
- 24.Wang PR, Xu M, Toffanin S, Li Y, Llovet JM, Russell DW. Induction of hepatocellular carcinoma by in vivo gene targeting; Proc Natl Acad Sci USA; 2012; pp. 11264–11269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Wang X. Systematic identification of microRNA functions by combining target prediction and expression profiling. Nucleic Acids Res. 2006;34:1646–1652. doi: 10.1093/nar/gkl068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dweep H, Gretz N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat Methods. 2015;12:697. doi: 10.1038/nmeth.3485. [DOI] [PubMed] [Google Scholar]
- 27.Chang EK, Yu CY, Clarke R, Hackbarth A, Sanders T, Esrailian E, Hommes DW, Runyon BA. Defining a patient population with cirrhosis: An automated algorithm with natural language processing. J Clin Gastroenterol. 2016;50:889–894. doi: 10.1097/MCG.0000000000000583. [DOI] [PubMed] [Google Scholar]
- 28.Alsawas M, Alahdab F, Asi N, Li DC, Wang Z, Murad MH. Natural language processing: Use in EBM and a guide for appraisal. Evid Based Med. 2016;21:136–138. doi: 10.1136/ebmed-2016-110437. [DOI] [PubMed] [Google Scholar]
- 29.Zhang X, Tang W, Chen G, Ren F, Liang H, Dang Y, Rong M. An encapsulation of gene signatures for hepatocellular carcinoma, microRNA-132 predicted target genes and the corresponding overlaps. PLoS One. 2016;11:e0159498. doi: 10.1371/journal.pone.0159498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang WT, Wang HL, Yang H, Ren FH, Luo YH, Huang CQ, Liang YY, Liang HW, Chen G, Dang YW. Lower expressed miR-198 and its potential targets in hepatocellular carcinoma: A clinicopathological and in silico study. Onco Targets Ther. 2016;9:5163–5180. doi: 10.2147/OTT.S108828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liese J, Peveling-Oberhag J, Doering C, Schnitzbauer AA, Herrmann E, Zangos S, Hansmann ML, Moench C, Welker MW, Zeuzem S, et al. A possible role of microRNAs as predictive markers for the recurrence of hepatocellular carcinoma after liver transplantation. Transpl Int. 2016;29:369–380. doi: 10.1111/tri.12733. [DOI] [PubMed] [Google Scholar]
- 32.Ito K, Murphy D. Application of ggplot2 to Pharmacometric Graphics. CPT Pharmacometrics Syst Pharmacol. 2013;2:e79. doi: 10.1038/psp.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu C, Zeng Q, Xu W, Jiao L, Chen Y, Zhang Z, Wu C, Jin T, Pan A, Wei R, et al. miRNA-100 inhibits human bladder urothelial carcinogenesis by directly targeting mTOR. Mol Cancer Ther. 2013;12:207–219. doi: 10.1158/1535-7163.MCT-12-0273. [DOI] [PubMed] [Google Scholar]
- 34.Ma Y, Han W, Yang L, He L, Wang H. The regulation of miRNAs in inflammation-related carcinogenesis. Curr Pharm Des. 2015;21:3023–3031. doi: 10.2174/1381612821666150514105606. [DOI] [PubMed] [Google Scholar]
- 35.Wan X, Ding X, Chen S, Song H, Jiang H, Fang Y, Li P, Guo J. The functional sites of miRNAs and lncRNAs in gastric carcinogenesis. Tumour Biol. 2015;36:521–532. doi: 10.1007/s13277-015-3136-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu L, Dai W, Li J, He LI, Wang F, Xia Y, Chen K, Li S, Liu T, Lu J, et al. Methylation-regulated miR-124-1 suppresses tumorigenesis in hepatocellular carcinoma by targeting CASC3. Oncotarget. 2016;7:26027–26041. doi: 10.18632/oncotarget.8266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng F, Liao YJ, Cai MY, Liu YH, Liu TH, Chen SP, Bian XW, Guan XY, Lin MC, Zeng YX, et al. The putative tumour suppressor microRNA-124 modulates hepatocellular carcinoma cell aggressiveness by repressing ROCK2 and EZH2. Gut. 2012;61:278–289. doi: 10.1136/gut.2011.239145. [DOI] [PubMed] [Google Scholar]
- 38.Shi XB, Xue L, Ma AH, Tepper CG, Gandour-Edwards R, Kung HJ, deVere White RW. Tumor suppressive miR-124 targets androgen receptor and inhibits proliferation of prostate cancer cells. Oncogene. 2013;32:4130–4138. doi: 10.1038/onc.2012.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lv XB, Jiao Y, Qing Y, Hu H, Cui X, Lin T, Song E, Yu F. miR-124 suppresses multiple steps of breast cancer metastasis by targeting a cohort of pro-metastatic genes in vitro. Chin J Cancer. 2011;30:821–830. doi: 10.5732/cjc.011.10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Xie S, Liu M, Chen Z, Liu X, Wang L, Li D, Zhou Y. The clinical significance of downregulation of mir-124-3p, mir-146a-5p, mir-155-5p and mir-335-5p in gastric cancer tumorigenesis. Int J Oncol. 2014;45:197–208. doi: 10.3892/ijo.2014.2415. [DOI] [PubMed] [Google Scholar]
- 41.Wang P, Chen L, Zhang J, Chen H, Fan J, Wang K, Luo J, Chen Z, Meng Z, Liu L. Methylation-mediated silencing of the miR-124 genes facilitates pancreatic cancer progression and metastasis by targeting Rac1. Oncogene. 2014;33:514–524. doi: 10.1038/onc.2012.598. [DOI] [PubMed] [Google Scholar]
- 42.Zhao W, Cheng Y, Zhang C, You Q, Shen X, Guo W, Jiao Y. Genome-wide identification and characterization of circular RNAs by high throughput sequencing in soybean. Sci Rep. 2017;7:5636. doi: 10.1038/s41598-017-05922-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Becker HF, Heliou A, Djaout K, Lestini R, Regnier M, Myllykallio H. High-throughput sequencing reveals circular substrates for an archaeal RNA ligase. RNA Biology. 2017;14:1075–1085. doi: 10.1080/15476286.2017.1302640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nair AA, Niu N, Tang X, Thompson KJ, Wang L, Kocher JP, Subramanian S, Kalari KR. Circular RNAs and their associations with breast cancer subtypes. Oncotarget. 2016;7:80967–80979. doi: 10.18632/oncotarget.13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, Zhang Y, Huang L, Zhang J, Pan F, Li B, Yan Y, Jia B, Liu H, Li S, Zheng W. Decreased expression of hsa_circ_001988 in colorectal cancer and its clinical significances. Int J Clin Exp Pathol. 2015;8:16020–16025. [PMC free article] [PubMed] [Google Scholar]
- 46.Li P, Chen S, Chen H, Mo X, Li T, Shao Y, Xiao B, Guo J. Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin Chim Acta. 2015;444:132–136. doi: 10.1016/j.cca.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 47.Shang X, Li G, Liu H, Li T, Liu J, Zhao Q, Wang C. Comprehensive circular RNA profiling reveals that hsa_circ_0005075, a new circular RNA biomarker, is involved in hepatocellular crcinoma development. Medicine (Baltimore) 2016;95:e3811. doi: 10.1097/MD.0000000000003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qin M, Liu G, Huo X, Tao X, Sun X, Ge Z, Yang J, Fan J, Liu L, Qin W. Hsa_circ_0001649: A circular RNA and potential novel biomarker for hepatocellular carcinoma. Cancer Biomark. 2016;16:161–169. doi: 10.3233/CBM-150552. [DOI] [PubMed] [Google Scholar]
- 49.Xu L, Zhang M, Zheng X, Yi P, Lan C, Xu M. The circular RNA ciRS-7 (Cdr1as) acts as a risk factor of hepatic microvascular invasion in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2017;143:17–27. doi: 10.1007/s00432-016-2256-7. [DOI] [PubMed] [Google Scholar]
- 50.Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, Luo Y, Lyu D, Li Y, Shi G, et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun. 2016;7:11215. doi: 10.1038/ncomms11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gong XL, Qin SK. Progress in systemic therapy of advanced hepatocellular carcinoma. World J Gastroenterol. 2016;22:6582–6594. doi: 10.3748/wjg.v22.i29.6582. [DOI] [PMC free article] [PubMed] [Google Scholar]