

Abstract
Background
Animals living at high altitude must adapt to environments with hypoxia and low temperatures, but relatively little is known about underlying genetic changes. Toad-headed lizards of the genus Phrynocephalus cover a broad altitudinal gradient of over 4000 m and are useful models for studies of such adaptive responses. In one of the first studies to have considered selection on mitochondrial protein-coding regions in an ectothermic group distributed over such a wide range of environments, we analysed nineteen complete mitochondrial genomes from all Chinese Phrynocephalus (including eight genomes sequenced for the first time). Initial analyses used site and branch-site model (program: PAML) approaches to examine nonsynonymous: synonymous substitution rates across the mtDNA tree.
Results
Ten positively selected sites were discovered, nine of which corresponded to subunits ND2, ND3, ND4, ND5, and ND6 within the respiratory chain enzyme mitochondrial Complex I (NADH Coenzyme Q oxidoreductase). Four of these sites showed evidence of general long-term selection across the group while the remainder showed evidence of episodic selection across different branches of the tree. Some of these branches corresponded to increases in altitude and/or latitude. Analyses of physicochemical changes in protein structures revealed that residue changes at sites that were under selection corresponded to major functional differences. Analyses of coevolution point to coevolution of selected sites within the ND4 subunit, with key sites associated with proton translocation across the mitochondrial membrane.
Conclusions
Our results identify mitochondrial Complex I as a target for environment-mediated selection in this group of lizards, a complex that frequently appears to be under selection in other organisms. This makes these lizards good candidates for more detailed future studies of molecular evolution.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4569-1) contains supplementary material, which is available to authorized users.
Keywords: Phrynocephalus, Mitochondrial genes, Complex I, Selective evolution, Altitude
Background
Altitudinal gradients can produce environmental variation that has a major influence on the survival and evolution of organisms [1]. Animal populations commonly found at extremely high altitudes are expected to adapt their phenotypes, including physiology and metabolism, to local environmental conditions. Changes in the structure and function of proteins associated with these adaptive responses are therefore predicted.
The metabolic impacts of low temperature and hypoxia at high altitude suggest that proteins encoded by the mitochondrial genome could be integral to adaptive responses due to their association with oxidative phosphorylation of ADP to ATP. Five protein complexes participate in this process, which produces 95% of the energy used by eukaryotes [2]. Four of these complexes (I, III, IV, and V) contain polypeptides encoded by mitochondrial genes. Several studies have reported evidence of positive Darwinian selection on mitochondrial proteins, although purifying selection is more commonly detected [3, 4].
Previous studies of the effects of selection on mitochondrial evolution have primarily focused on mammals [5–13] and/or focused on single genes or complexes that are expected to have experienced positive selection [14, 15]. Nonetheless, some research has linked altitudinal/latitudinal gradients to selection-mediated mitochondrial evolution. For example, adaptive responses to northern latitudes or high altitudes have been linked to the evolution of mitochondrial-encoded proteins in modern humans [16–18]. Previous mammalian studies have also examined possible altitude-related selection on genes in the oxidative phosphorylation pathway. Positive selection on three amino acid residues in the ND2 and ND6 genes were found for one of two high altitude Chinese Rhinopithecus monkeys [17], while analyses of the Caprini, which are adapted to a high altitude existence, detected greatest selection on ATPase and cytochrome b genes, and least selection on ND and COX genes [19]. Nevertheless, there do not seem to have been any studies of ectotherms occupying a wide altitudinal range to date.
The altitudinal range of Chinese Phrynocephalus lizards spans over 4200 m, with some species such as P. theobaldi inhabiting regions up to approximately 5000 m. The latter represents one of the world’s highest altitude lizard species. Viviparous Phrynocephalus are restricted to the Qinghai-Tibetan Plateau (QTP), while oviparous Phrynocephalus are only found at lower altitudes outside the QTP. Viviparous and oviparous Chinese Phrynocephalus split approximately 10 Ma ago, with subsequent diversification occurring at very different altitudes [20]. Different viviparous species can also occupy very different environments, e.g., P. erythrurus and P. forsythii which show a 4200 m altitudinal difference. Intraspecific altitudinal ranges can also be substantial, exceeding 1500 m in some cases [21].
The distributions of Chinese Phrynocephalus make them excellent models for the study of ectotherm adaptation to cold environments. Previous studies have highlighted the impact of altitude on geographic variation of intra- and interspecific life history traits in Phrynocephalus [21–24], as well as its influence on physiological and metabolic processes [25]. Comparative transcriptome analyses of P. przewalskii (oviparous) and P. vlangalii (viviparous) have detected several nuclear genes that are likely to have experienced selection as part of the shift to a high-altitude existence [26]. To date, no research has investigated altitude-related evolution of mitochondrial genes in Phrynocephalus, despite the importance of these genes for metabolic processes that could be affected by the low temperatures and oxygen concentrations found at high altitudes.
In this study, we examined mitochondrial protein evolution among all species and major evolutionary lineages within Phrynocephalus with the aim of: (1) detecting genes and sites that have experienced significant selection during the diversification of the group, (2) analysing whether selected sites have important functions and/or have undergone significant co-evolution with other key functional sites.
Methods
Mitochondrial genomes
All experimental protocols were performed in accordance with guidelines from the China Council on Animal Care and approved by the Ethics Committee for Animal Experiments at China Jiliang University.
A total of 19 complete mitogenomes representing all Chinese Phrynocephalus species were analysed in our study. The genomes provided a comprehensive coverage of deep mitochondrial lineages in the group. Seven of these mitogenomes have been previously published by our group (GenBank: KJ551842, KJ885621, KJ830752, KJ749841, KJ630904, KF572032, KM093859), and four mitogenomes had been published by other researchers (GenBank KC119493, KC578685, KM093858, KP126516). The origins of all samples are shown in Fig. 1 and more detailed information is provided in the Additional file 1.
Fig. 1.

Map showing sites from which Phrynocephalus were sampled. All species/specimen numbers shown on the map correspond to Additional file 1 (map data were provided by the Scientific Data Centre of the Chinese Academy of Sciences and the final map produced by the authors using the software ArcGis 10.2)
Eight mitochondrial genomes represented by individuals primarily from Tibetan plateau were newly-sequenced for this study (GenBank MF039058–65) from tissues of voucher specimens stored at China Jiliang University and Lanzhou University. These mitogenomes represented the species/subspecies P. vlangalii vlangalii, P. v. pylzowi, P. v. nanschanica, P. theobaldi theobaldi, P. t. orientalis, P. erythrurus erythrurus, and P. frontalis were obtained using PCR. LA Taq Hot Start Version Polymerase (TaKaRa Bio, Osaka, Japan) was used with four pairs of designed primers (Additional file 2) and each portion was sequenced using a primer-walking method. The TA-rich region of the Control Region, which is difficult to sequence directly, was amplified from genomic DNA using primers containing a 5′ EcoRI and a 3′ X-hol site, cloned into the pGex-4 t-1 vector and sequenced. Full details of the mitogenome sequencing protocol are available in our previous work [27].
Phylogenetic tree construction
Phylogenetic relationships among all of the taxa have been inferred by previous studies, however, given the availability of such a large mitochondrial dataset, we applied a maximum likelihood (ML) approach to re-examine these relationships and ensure a robust topology for analyses of selection. The genomes were divided into six partitions: the three codon positions, rRNA, tRNA and control region sequences. Small areas of potentially ambiguous alignment were trimmed from the rRNAs, tRNAs and control regions leaving a 15,422 bp alignment. Control region sequence was missing for P. axillaris. ML analyses were carried out using RAxML ver. v8.2.10 [28] on a Linux Debian operating system. The raxmlHPC-PTHREADS command was used for the analyses and the GTR + G model was specified for each partition (−m command). Oviparous and viviparous species are unequivocally monophyletic [20] and this was specified as a topological constraint (backbone option). An ML tree was identified for the original sequence and additional trees were determined for 2000 bootstrap-resampled datasets to indicate levels of support for branches (−# option). No outgroup was specified and so the ML tree was rooted with a midpoint root for display purposes.
Tests of selection
Statistical analyses of selection are based on nonsynonymous: synonymous ratios (ω) within protein-coding sequence, across the (unrooted) mtDNA tree that we inferred here. We used the CODEML program in PAML ver. 4.8 [29], to detect signatures of selection using both more general site models and also branch-site models (where ω can vary both among amino acid sites in the protein and across branches on the tree). Specific details of the analyses are described below.
Likelihoods were obtained under the following site models: i) one ratio, i.e., the same ω for all sites (M0), ii) two classes of neutral sites in which either ω = 1 or ω = 0 (M1a), iii) positive selection, i.e., as for M1a but with a third class of sites under positive selection (ω > 1) (M2a), iv) sites follow a general discrete distribution (M3), v) ω follows a beta distribution (M7), vi) as for M7 but with an additional class of sites under positive selection (ω > 1) (M8). A complete description of the models is available in the PAML manual.
The six models were applied to concatenated mtDNA coding sequences. Likelihood ratio tests (LRT) were used to compare pairs of models. M0-M3 LRT was used to test for variable ω between sites. M1a-M2a LRT and M7-M8 LRT were both used to determine the existence of positively selected sites. Sites were considered to have experienced positive selection when at least one codon model showed ω > 1 and one LRT was significant for either the M1a-M2a or M7-M8 comparisons.
Posterior probabilities for site classes were obtained using a Bayes Empirical Bayesian (BEB) approach [30]. This allowed identification of positively selected sites under the M2a and M8 models. We considered that there was strong evidence for a coding site being under positive selection when the BEB posterior probability was greater than 0.9 and ω > 1. This represents a stringent requirement given that ω is unlikely to average more than 1 across the entire group.
In the widely-used branch-site model known as model A, the branches are divided into foreground and background groups [30]. (This model provided more conservative LRT results than those of the branch-based test model [31]). In the foreground lineages a proportion of the sites are under positive selection (ω > 1), while the background lineages feature a class of conserved sites with ω between 0 and 1, and a class of neutral sites with ω fixed at 1 [30]. Because it was difficult to select foreground branches a priori, we defined each possible branch as a foreground branch and then tested whether positive selection was present. Selection was tested by comparison of two models: null Model A, in which the sites in the foreground lineages are set at ω = 1, against the alternative Model A, in which ω > 1. We used the BEB to obtain posterior probabilities.
We conducted additional analyses to evaluate the robustness of the signal of positive selection in these data. The instantaneous rate change of codons i to j depends on parameter πj (equilibrium frequency of codon j), which can be estimated in several ways: i) using nucleotide frequencies observed at each of the three codon positions within the gene (F3 × 4 model), ii) using the empirical nucleotide frequencies observed at the gene irrespective of codon position (F1 × 4) and iii) with all nucleotide frequencies are set equal (Fequal). Hence, we repeated the M7-M8 and Model A LRTs under the F1 × 4 and Fequal models for comparison with the F3 × 4 model.
Analyses of physicochemical changes
Additional analyses of selection on amino acids were carried out using the program TreeSAAP ver. 3.2 software [32] which analyses significant changes in up to 31 physicochemical amino acid properties during evolution. The basis of this approach is comparison of changes in physicochemical properties inferred for the specified topology with an expected distribution of changes in under the condition that any amino acid is equally likely to be replaced by any other (as is the case under the neutral model). Congruent findings with the analyses of non-synonymous: synonymous ratios provide stronger evidence for selection. The mtDNA tree was used to infer amino acid replacement events and these were then assigned to one of eight physicochemical ‘magnitude of change’ categories (one being the smallest change and eight the highest). We focused only on physicochemical properties in categories 6, 7 and 8 as these represent the strongest positive selection. A sliding window size of 15 sites was used. Significance was assessed by comparing observed and expected mean changes in amino acid properties, χ2 goodness-of-fit tests were used to compare observed with expected distributions and deemed notably significant where P < 0.0001.
Structure and co-evolutionary analyses of protein residues
Protein alignments containing amino acid residues were used to determine whether positively selected sites that were related to protein functions or located in important protein functional domains. Co-evolution between sites under positive selection and other sites within each protein was analysed using the online system at http://coevolution.gersteinlab.org/coevolution/ using Statistical Coupling Analysis (SCA). A total of 800 sequences were aligned (the maximum number allowed). These included the reference sequence and known structure (PDB ID 3RKO) of the ND protein within Complex I (only ND was analysed following the results of the selection analyses), one P. putjatia sequence from our study and 798 sequences from different genera from Uniprot online (mostly from the SWISS-PROT database). The sequence alignments corresponding to the five important ND subunits are given in the Additional file 3. Chimera X software [33] was used to portray the three-dimensional structure of proteins. The locations of Phrynocephalus residues found to be under selection within the PDB ID 3RKO structural model were displayed. In addition, 25 ND protein structures which encompassed the Phrynocephalus sites under selection were independently estimated using the Phyre2 online platform [34]. The residue side chains (for loci under selection) were obtained for all 19 individuals. To determine possible coevolutionary roles for a given selective residue in proton channel or other residues, Z-tests were computed on: i) SCA scores (s) (which do not incorporate distance between loci) ii) distances between loci (d) and iii) s/d [35–37].
Results
Mitochondrial genomes and phylogenetic analyses
Eight mitogenomes for the species/subspecies P. v. pylzowi, P. v. vlangalii, P. t. theobaldi, P. frontalis, P. t. orientalis, P. e. erythrurus, and P. v. nanschanica were obtained using PCR-based sequencing. Genome lengths varied from 16,251 bp (P. v. nanschanica) to 17,129 bp (P. t. orientalis). All sequences have been deposited in Genbank (GenBank accessions: MF039058–65).
The ML Phrynocephalus tree inferred from the mitogenomes was similar, but not identical, to that inferred previously [20] (Additional file 4). Topological differences involved nodes that were not strongly supported. In the current analysis, bootstrap support for the split between P. forsythi and the remaining Phrynocephalus was weak showing uncertainty over this relationship. Jin & Brown (2013) found strong (Bayesian) support for P. forsythi originating from the most basal viviparous node in both nuclear and mtDNA trees. Hence we favoured the latter P. forsythii position within the viviparous clade. The position of P. helioscopus within the oviparous group also differed from our previous Bayesian analysis. However, posterior support for the corresponding node was weak in that analysis, so the current ML topology for oviparous species was favoured. In sum, one adjustment was made to the topology of the ML tree for use in selection analyses.
Random-sites analyses of selection
Values of ω much greater than 1 were detected for models M2A and M8 (both allow a proportion of sites to be under positive selection), while M3 did not reveal any positively selected sites (Table 1). The LRT that compared M0-M3 was highly significant (P < 0.001), which indicated a strong signal of variable ω among sites (Table 2). The M1a-M2a LRT was not significant (P > 0.9996) (Table 2). Nevertheless, the M7-M8 LRT was highly significant (P < 0.0001). These results are not unexpected as the M1a-M2a LRT has very low power compared with the M7-M8 LRT, particularly when the proportion of positively selected sites is very small [38]. Posterior probabilities obtained using BEB methods under M8 showed strong evidence of long-term positive selection at four sites within three ND subunits (Table 3): one site in ND3 (#9), two sites in ND4 (#187 and #454) and one site in ND6 (#100).
Table 1.
Site-models, log-likelihood values, transition: transversion ratio (κ) and parameter estimates for Phrynocephalus mitochondrial gene sequences. ω and ωi are nonsynonymous: synonymous ratios, pi refers to the proportion of sites in class i (see PAML manual for more details on these site models)
| Model Number | Log Likelihood | κ | Parameter Estimates |
|---|---|---|---|
| M0 (one ratio) | −53,947.45 | 8.02 | ω = 0.0755 |
| M1A (nearly neutral) | −53,215.56 | 8.69 | p0 = 0.9173, p1 = 0.0827 ω0 = 0.0410, ω1 = 1.0000 |
| M2A (positive selection) | −53,215.56 | 8.69 | p0 = 0.9173, p1 = 0.0827, p2 = 0.0000 ω0 = 0.0410, ω1 = 1.0000, ω2 = 30.1188 |
| M3 (discrete) | −52,976.30 | 8.20 | p0 = 0.7920, p1 = 0.0003, p2 = 0.2077 ω0 = 0.0149, ω1 = 0.0533, ω2 = 0.3429 |
| M7 (beta) | −52,936.28 | 8.27 | p = 0.1797, q = 1.7626 |
| M8 (beta & ω) | −52,926.55 | 8.29 | p0 = 0.9981, p1 = 0.0020, ωs = 2.2162, p = 0.1863, q = 1.9069 |
Table 2.
Results of log-likelihood ratio tests that compare different site models
| Test for | Null Model | Experimental Model | df | P-value |
|---|---|---|---|---|
| Difference in ω among sites | Model 0 (one ratio) | Model 1A (nearly neutral) | 2 | < 0.0001 |
| Positive selection | Model 1A (nearly neutral) | Model 2A (positive selection) | 2 | > 0.9996 |
| Difference in ω among sites | Model 0 (one ratio) | Model 3 (discrete) | 2 | < 0.0001 |
| Positive selection | Model 7 (beta) | Model 8 (beta & ω) | 2 | < 0.0001 |
Table 3.
Sites found to be under long-term positive selection, as determined from posterior probabilities obtained using the Bayes empirical Bayes (BEB) approach
| Gene Name | Amino Acid Position | BEB |
|---|---|---|
| ND3 | 9 | 0.988 |
| ND4 | 187 | 0.987 |
| ND4 | 454 | 0.988 |
| ND6 | 100 | 0.994 |
Branch-site analysis revealed several sites under episodes of positive selection on just six branches of the tree (Branches a-f in Fig. 2). Both the oviparous and viviparous groups contained three of these branches. Perhaps the most notable of these was the ancestral branch of all P. theobaldi, which reaches the highest elevation of all species. These corresponded to genes (site positions): ND2 (#10), COXIII (#171), ND4 (#109 and 448), ND5 (#365) and ND6 (#137) (Table 4). Multiple sites from different branches (> 90%) experienced purifying selection, approximately 8% of sites were neutral, while less than 1% of sites had undergone positive selection (Table 4).
Fig. 2.

Phylogenetic tree that was used for selection analyses and showing branches on which there was evidence of selection: branches a, b, c, d, e, and f (bold lines) were found to have highly significant values of ω (non-synonymous: synonymous ratios), provided as branch labels. Altitudes of capture sites are given on the left of dashed lines, while the altitudinal ranges that have been described for the oviparous species are given on the right of these lines. References for these descriptions are denoted as *unpublished personal observation, a [51], b [52], c [53], d [54], e [55]
Table 4.
Parameter estimates (to 3 d.p.) under null and alternative hypotheses for branch-site Model A (four parameter version), for six significant branch/site combinations. Background site classes are: 0 (0 < ω0 < 1), 1 (ω1 = 1), 2a (0 < ω0 < 1) and 2b (ω1 = 1). Foreground site classes are: 0 (0 < ω0 < 1), 1 (ω1 = 1), 2a (ω2 ≥ 1) and 2b (ω2 ≥ 1). The six branches (a, b, c, d, e, f) are shown in Fig. 1
| Branch | Amino Acid Position (gene) | Model | Parameter | 0 | 1 | 2a | 2b |
|---|---|---|---|---|---|---|---|
| a | 109 (ND4) | Model A (Null) | Proportion | 0.913 | 0.082 | 0.004 | 0.000 |
| Background ω | 0.040 | 1.000 | 0.041 | 1.000 | |||
| Foreground ω | 0.041 | 1.000 | 1.000 | 1.000 | |||
| Model A (Alternative) | Proportion | 0.915 | 0.082 | 0.003 | 0.000 | ||
| Background ω | 0.041 | 1.000 | 0.041 | 1.000 | |||
| Foreground ω | 0.041 | 1.000 | 998.999 | 998.999 | |||
| b | 137 (ND6) | Model A (Null) | Proportion | 0.917 | 0.083 | 0.001 | 0.000 |
| Background ω | 0.041 | 1.000 | 0.041 | 1.000 | |||
| Foreground ω | 0.041 | 1.000 | 1.000 | 1.000 | |||
| Model A (Alternative) | Proportion | 0.917 | 0.083 | 0.001 | 0.000 | ||
| Background ω | 0.041 | 1.000 | 0.041 | 1.000 | |||
| Foreground ω | 0.041 | 1.000 | 352.529 | 352.529 | |||
| c | 10 (ND2) | Model A (Null) | Proportion | 0.908 | 0.082 | 0.010 | 0.001 |
| Background ω | 0.041 | 1.000 | 0.041 | 1.000 | |||
| Foreground ω | 0.041 | 1.000 | 1.000 | 1.000 | |||
| Model A (Alternative) | Proportion | 0.917 | 0.082 | 0.000 | 0.000 | ||
| Background ω | 0.041 | 1.000 | 0.041 | 1.000 | |||
| Foreground ω | 0.041 | 1.000 | 70.079 | 70.079 | |||
| d | 448 (ND4) | Model A (Null) | Proportion | 0.914 | 0.083 | 0.003 | 0.000 |
| Background ω | 0.041 | 1.000 | 0.041 | 1.000 | |||
| Foreground ω | 0.041 | 1.000 | 1.000 | 1.000 | |||
| Model A (Alternative) | Proportion | 0.917 | 0.083 | 0.000 | 0.000 | ||
| Background ω | 0.041 | 1.000 | 0.041 | 1.000 | |||
| Foreground ω | 0.041 | 1.000 | 682.538 | 682.538 | |||
| e | 171 (COXIII) | Model A (Null) | Proportion | 0.884 | 0.080 | 0.034 | 0.003 |
| Background ω | 0.041 | 1.000 | 0.041 | 1.000 | |||
| Foreground ω | 0.041 | 1.000 | 1.000 | 1.000 | |||
| Model A (Alternative) | Proportion | 0.911 | 0.082 | 0.006 | 0.001 | ||
| Background ω | 0.041 | 1.000 | 0.041 | 1.000 | |||
| Foreground ω | 0.041 | 1.000 | 6.409 | 6.409 | |||
| f | 365 (ND5) | Model A (Null) | Proportion | 0.909 | 0.082 | 0.008 | 0.001 |
| Background ω | 0.041 | 1.000 | 0.041 | 1.000 | |||
| Foreground ω | 0.041 | 1.000 | 1.000 | 1.000 | |||
| Model A (Alternative) | Proportion | 0.916 | 0.083 | 0.001 | 0.000 | ||
| Background ω | 0.041 | 1.000 | 0.041 | 1.000 | |||
| Foreground ω | 0.041 | 1.000 | 10.593 | 10.593 |
Re-analyses of the M7-M8 LRT and Model A LRT using the F1 × 4 and Fequal models on individual coding genes rather than the concatenated sequence revealed the same positively selected sites and similar posterior probabilities. This supported the robustness of both the site and branch-site analyses.
Analyses of physicochemical changes
More than half (sixteen) of the eigenvalues for physicochemical characteristics changed in selective sites within ND3 (#9) and ND6 (#100), both of which had experienced long-term positive selection. Moreover, all other residues except COXIII (#171) that showed evidence of selection (in PAML analyses) corresponded to the greatest observed changes in physicochemical characteristics within each subunit (see Fig. 3). One ND1 site showed changes in physicochemical properties, eleven of which represented changes to basic characteristics of the subunit, despite significant positive selection not previously being detected. ND subunits under selection, as detected by analyses of ω, therefore also tended to show important physicochemical changes.
Fig. 3.

Summary of selection in the mitogenome of Phrynocephalus. The 13 mitochondrial coding genes are represented and labelled (figure does not include tRNAs). Filled black circles represent positively selected sites (p < 0.001) identified by TreeSAAP analysis. The dataset was analysed for significant changes in each of the 31 physicochemical properties. The y-axis represents the number of properties for which selection was inferred (for each site). Open triangles are positively selected sites identified by independent analyses of ω
Structural and co-evolutionary analyses of protein amino acid residues
Nine out of the ten sites that were detected by the site and branch-site model analyses were in ND subunits and therefore provided strong evidence of selection primarily affecting the hydrophobic region of Complex I of the mitochondrial respiratory chain (Fig. 4), the remaining site was in Complex III. All selected sites in ND subunits were located in the helices of these subunits in the Complex I crystal structure (Fig. 4), while two residues (#187 ND4, #100 ND6) appeared in random coils but quite near the residues of helices in the simulated 3D structure (high confidence was obtained for all, i.e., 100%). The final residue (#454) of ND4 was absent in the template and could not be included. The simulated three-dimensional structure (Fig. 4) shows that changes at all of the selected residues caused structural changes in residue side chains (Fig. 4).
Fig. 4.
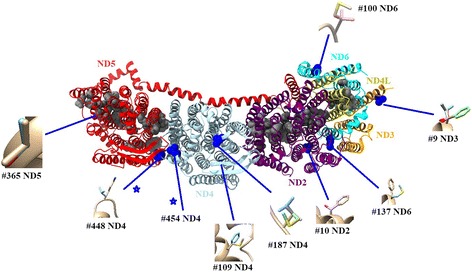
The three dimensional structure of Complex I showing the locations of amino acids under selection as determined by predictive models for individual Phrynocephalus ND subunits. Residues marked as blue circles are the selected residues within the reference crystal structure (PDB ID 3RKO). Diagrams of residue side chains are given for each. Each ND subunit is coloured individually, protein names correspond to the protein subunits in Complex I. Blue stars indicate the two residues that show significant co-evolutionary relationship with key residues (in grey sphere) in the chain of proton translocation
SCA analyses of co-evolution of protein spatial structure (the hydrophobic region of Complex I) provided probabilities close to the 0.05 level (P = 0.049 and P = 0.060, respectively) for each of the two selected residues in ND4 (#454 and #448). This provided some evidence for co-evolutionary relationships with residues that played key or important roles in proton transport as well as other residues not responsible for proton translocation (Table 5).
Table 5.
Results of SCA co-evolutionary analyses for each selective site. The t and corresponding P-values are given for the SCA scores (s), distances between sites (d), and the ratios of these measures (s/d)
| Selective sites | n | s | d | s/d | ||||
|---|---|---|---|---|---|---|---|---|
| Gene | t | P | t | P | t | P | ||
| ND2 | 10 | 346 | 0.430 | 0.670 | −2.065 | 0.045* | 1.140 | 0.255 |
| ND3 | 9 | 91 | 0.774 | 0.513 | 0.171 | 0.877 | 1.058 | 0.293 |
| ND4 | 109 | 421 | 1.344 | 0.192 | −0.554 | 0.584 | 0.137 | 0.891 |
| ND4 | 187 | 421 | 0.285 | 0.778 | 1.042 | 0.305 | 0.973 | 0.331 |
| ND4 | 448 | 421 | 1.422 | 0.168 | −1.618 | 0.118 | 1.851 | 0.062 |
| ND4 | 454 | 421 | −1.743 | 0.093 | −0.949 | 0.351 | −1.967 | 0.049* |
| ND5 | 365 | 537 | 1.004 | 0.333 | −4.434 | < 0.001* | −0.015 | 0.988 |
| ND6 | 100 | 97 | −0.552 | 0.598 | −2.795 | 0.027* | 0.715 | 0.496 |
| ND6 | 137 | 97 | 1.715 | 0.128 | −0.341 | 0.743 | 1.488 | 0.143 |
*P < 0.05
Discussion
The hydrophobic region of the respiratory Complex I comprises mitochondrial proteins: one piston arm of its sub-element NuoL (equates to ND5) combines with three proton pumps including NuoL (ND5), NuoM (ND4) and NuoN (ND2). This structure plays an important role in proton transport for ATP synthesis [39]. A total of nine out of ten selective sites were related to ND proteins within Complex I and indicated that there was stronger selection on ND genes than on mitochondrial genes that coded for other respiratory chain complexes. The positively selected sites were not key sites directly responsible for proton translocation in the channel, but at least two selected sites in NuoM (ND4) do appear to have a coevolutionary relationship with proton transport channel residues.
In addition to detection of Complex I selection through non-synonymous: synonymous ratios, we found notable changes in the selected residues’ physicochemical properties. Sites with highest nonsynonymous: synonymous ratios tended to show some of the greatest changes in physicochemical properties suggesting that selection was acting at sites associated with important structures/functions.
Several studies have used variation in non-synonymous: synonymous ratios to infer variable selection pressures across mitochondrial genes [40] including ND subunits [7, 41–45]. The degree of hydrophobicity has been found to differ among animal groups for the ND5 subunit, and to a lesser extent the ND1, ND2 and ND4 subunits, pointing towards possible functions associated with selection [44]. Four out of nine selected sites in Complex I were found in ND4, indicating that this subunit might experience particularly strong selection in response to environmental differences. While several studies have found selection on Complex I, e.g., [7, 17], others have found selection on other complexes, e.g., [19]. For example, research on primates has indicated that electron transport chain-related proteins of mitochondrial Complex III and Complex IV underwent distinct selection pressures [9].
Adaptive responses to high-altitude in vertebrates are likely to involve numerous genetic interactions. How differences in elevation might shape selection on the ND complex in ectotherms remains speculative. Phrynocephalus lizards living at high altitudes/latitudes need to store and release energy to fuel metabolism under low oxygen concentrations and/or at lower temperatures than species from warmer regions. Several temperature-dependent traits, such as locomotor performance, are likely to be under quite strong selection pressures in lizards [46] and are linked to oxidative phosphorylation. Colonization of lower temperature environments might therefore be expected to lead to selection on this process, involving changes in the associated proteins. The lack of clear statistical evidence to support positive selection in the ancestor of all viviparous lineage therefore runs counter to this expectation because this lineage is potentially associated with the largest change in altitude. Nevertheless, there are some more subtle indications of altitude-related selection on Complex I, which are discussed below.
Proton transport is closely related to mitochondrial metabolism. Previous analyses of positive selection on protein-coding genes have often been linked to external factors, such as climatic variation [16, 47, 48], high temperature and physical activity [49], extreme temperatures and oxygen levels [50]. Mitochondrial respiratory rate appears to differ in P. erythrurus relative to a congeneric oviparous species, providing potential advantages at high altitude [25]. Our branch site analyses indicated that three lineages within the high-elevation viviparous group, namely P. vlangalii, P. forsythii and P. theobaldi, had undergone significant positive selection during the evolutionary history of the group. Phrynocephalus theobaldi reaches some of the highest elevations within this group which could explain why selection was detected in the lineage ancestral to the both of the subspecies analysed. Phrynocephalus forsythii inhabits a wide elevational range and extends down to the lowest ranges within the group (in fact, the capture site for P. forsythii here was much lower than the known elevational ranges of other viviparous Phrynocephalus lizards, by approximately 2000-4000 m). Similarly we detected positive selection in the P. vlangalii group, which was found at lower elevations than the sister taxa. Hence the latter two species do not exhibit selection for high altitudes, but potentially the opposite (selection for low altitudes). In the oviparous group, the branch that is ancestral to P. versicolor, P. przewalskii, and P. frontalis, and a subsequent branch ancestral to the latter two species, both show evidence of selection. These species are found at higher altitudes than other oviparous species and are distributed in the cooler eastern highlands, suggesting temperature related positive selection. The branch leading to P. helioscopus also show evidence of very strong selection. While this is found at lower altitudes than the P. versicolor, P. przewalskii, and P. frontalis group, it has the northernmost distribution of all Phrynocephalus lizards which again might suggest temperature-related selection.
Our research overcame some limitations of previous studies. For example, it is often relatively difficult to specifically detect sites that have experienced selection and are of functional significance because: i) most previous studies only analysed subsets of codons/mitochondrial protein-coding regions, ii) species groups used in some studies were incomplete, and so branches represented longer and potentially more varied histories; iii) unlike many previous studies, we were able to analyse mitochondrial protein-coding regions that corresponded to well-known structures and functional sites (specifically Complex I).
Conclusions
Taken together, we found strong evidence for selection on subunits associated with the hydrophobic region of mitochondrial complex I and we infer that this is at least partially associated with historical altitude and latitude range shifts that individual species within the group have undergone. The decrease in environmental temperatures with increasing altitude and latitude may have led to selection on oxidative phosphorylation, but consideration of the adaptive nature of these molecular changes remains speculative. Future studies of climate-related selection on the protein complexes associated with oxidative phosphorylation in ectotherms will help to determine general trends and allow more detailed insights into the processes involved.
Additional files
Voucher specimen and mitogenome used in selection analyses. (DOC 49 kb)
Primer pairs used in amplification of fragments of mitogenomes. (DOC 63 kb)
Multiple sequence alignment was used in statistical coupling analyses for ND proteins. (XLS 224 kb)
ML tree for Phrynocephalus inferred from 15,417 bp of mtDNA (6 partitions; GTRGAMMA model for each). Values on nodes are bootstrap proportions (2000 bootstraps). (DOCX 172 kb)
Acknowledgements
We thank Xiaolong Tang for sample collection, and Qian Wang, Liufang Zhu, Kailong Zhang and many others for lab work and/or analyses.
Funding
This work was supported by the National Natural Science Foundation of China (31372183, 31772447).
Availability of data and materials
Sequence data were deposited in GenBank with accession numbers: MF039058–65, KJ551842, KJ885621, KJ830752, KJ749841, KJ630904, KF572032, KM093858–59, KC119493, KC578685, KP126516, listed in Additional file 1. Phylogenetic data were submitted to TreeBASE (http://purl.org/phylo/treebase/phylows/study/TB2:S22322).
Authors’ contributions
YTJ, YBW, HJT, SS, LXZ contributed sampling and/or lab work, YTJ, HJT, RPB contributed to statistical analyses and production of the paper. All authors read and approved the final manuscript.
Ethics approval
All experimental protocols were performed in accordance with guidelines from the China Council on Animal Care and approved by the Ethics Committee of Animal Experiments at China Jiliang University. No live animals were used in the study.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4569-1) contains supplementary material, which is available to authorized users.
Contributor Information
Yuanting Jin, Email: jinyuanting@126.com.
Yubin Wo, Email: wybhpp@163.com.
Haojie Tong, Email: hj_tong2012@163.com.
Sen Song, Email: hj_tong2012@163.com.
Lixun Zhang, Email: zhanglixun@lzu.edu.cn.
Richard P. Brown, Email: r.p.brown@ljmu.ac.uk
References
- 1.Malhotra A, Thorpe RS. Experimental detection of rapid evolutionary response in natural lizard populations. Nature. 1991;353(6342):347–348. doi: 10.1038/353347a0. [DOI] [Google Scholar]
- 2.Soltoff SP. ATP and the regulation of renal cell function. Annu Rev Physiol. 1986;48(1):9–31. doi: 10.1146/annurev.ph.48.030186.000301. [DOI] [PubMed] [Google Scholar]
- 3.Garvin MR, Bielawski JP, Gharrett AJ. Positive Darwinian selection in the piston that powers proton pumps in complex I of the mitochondria of Pacific salmon. PLoS One. 2011;6(9) [DOI] [PMC free article] [PubMed]
- 4.Subramanian S, Mohandesan E, Millar CD, Lambert DM. Distance-dependent patterns of molecular divergences in tuatara mitogenomes. Sci Rep. 2015;5:8703. doi: 10.1038/srep08703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elson JL, Turnbull DM, Howell N. Comparative genomics and the evolution of human mitochondrial DNA: assessing the effects of selection. Am J Hum Genet. 2004;74(2):229–238. doi: 10.1086/381505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kivisild T, Shen P, Wall DP, Do B, Sung R, Davis K, Passarino G, Underhill PA, Scharfe C, Torroni A. The role of selection in the evolution of human mitochondrial genomes. Genetics. 2006;172(1):373–387. doi: 10.1534/genetics.105.043901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mishmar D, Ruizpesini E, Mondragonpalomino M, Procaccio V, Gaut B, Wallace DC. Adaptive selection of mitochondrial complex I subunits during primate radiation. Gene. 2006;378(3):11–18. doi: 10.1016/j.gene.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 8.Ruizpesini E, Dan M, Brandon M, Procaccio V, Wallace DC. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science. 2004;303(5655):223. doi: 10.1126/science.1088434. [DOI] [PubMed] [Google Scholar]
- 9.Grossman LI, Wildman DE, Schmidt TR, Goodman M. Accelerated evolution of the electron transport chain in anthropoid primates. Trends Genet. 2004;20(11):578–585. doi: 10.1016/j.tig.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Finch TM, Zhao N, Korkin D, Frederick KH, Eggert LS. Evidence of positive selection in mitochondrial complexes I and V of the African elephant. PLoS One. 2014;9(4):e92587. doi: 10.1371/journal.pone.0092587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garvin MR, Bielawski JP, Sazanov LA, Gharrett AJ. Review and meta-analysis of natural selection in mitochondrial complex I in metazoans. J Zool Syst Evol Res. 2015;53(1):1–17. doi: 10.1111/jzs.12079. [DOI] [Google Scholar]
- 12.Luo Y, Gao W, Gao Y, Tang S, Huang Q, Tan X, Chen J, Huang T. Mitochondrial genome analysis of Ochotona curzoniae and implication of cytochrome c oxidase in hypoxic adaptation. Mitochondrion. 2008;8(5):352–357. doi: 10.1016/j.mito.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 13.Melo-Ferreira J, Vilela J, Fonseca MM, da Fonseca RR, Boursot P, Alves PC. The elusive nature of adaptive mitochondrial DNA evolution of an arctic lineage prone to frequent introgression. Genome biology and evolution. 2014;6(4):886–896. doi: 10.1093/gbe/evu059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wise CA, Sraml M, Easteal S. Departure from neutrality at the mitochondrial NADH dehydrogenase subunit 2 gene in humans, but not in chimpanzees. Genetics. 1998;148(148):409–421. doi: 10.1093/genetics/148.1.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zink RM. Natural selection on mitochondrial DNA in Parus and its relevance for phylogeographic studies. Proceedings Biological Sci. 2005;272(1558):71. doi: 10.1098/rspb.2004.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dan M, Ruizpesini E, Golik P, Macaulay V, Clark AG, Hosseini S, Brandon M, Easley K, Chen E, Brown MD. Natural selection shaped regional mtDNA variation in humans. Proc Natl Acad Sci U S A. 2003;100(1):171–176. doi: 10.1073/pnas.0136972100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu L, Wang X, Ting N, Zhang Y. Mitogenomic analysis of Chinese snub-nosed monkeys: evidence of positive selection in NADH dehydrogenase genes in high-altitude adaptation. Mitochondrion. 2011;11(3):497–503. doi: 10.1016/j.mito.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Zhou T, Shen X, Irwin DM, Shen Y, Zhang Y. Mitogenomic analyses propose positive selection in mitochondrial genes for high-altitude adaptation in galliform birds. Mitochondrion. 2014;18:70–75. doi: 10.1016/j.mito.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 19.Hassanin A, Ropiquet A, Couloux A, Cruaud C. Evolution of the mitochondrial genome in mammals living at high altitude: new insights from a study of the tribe Caprini (Bovidae, Antilopinae) J Mol Evol. 2009;68(4):293–310. doi: 10.1007/s00239-009-9208-7. [DOI] [PubMed] [Google Scholar]
- 20.Jin YT, Brown RP. Species history and divergence times of viviparous and oviparous Chinese toad-headed sand lizards (Phrynocephalus) on the Qinghai-Tibetan plateau. Molecular Phylogenetics & Evolution. 2013;68(2):259–268. doi: 10.1016/j.ympev.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 21.Jin YT, Li JQ, Liu NF. Elevation-related variation in life history traits among Phrynocephalus lineages on the Tibetan plateau: do they follow typical squamate ecogeographic patterns? J Zool. 2013;290(4):293–301. doi: 10.1111/jzo.12042. [DOI] [Google Scholar]
- 22.Jin YT, Liao PH. An elevational trend of body size variation in a cold-climate agamid lizard, Phrynocephalus theobaldi. Curr Zool. 2015;61(3):444–453. doi: 10.1093/czoolo/61.3.444. [DOI] [Google Scholar]
- 23.Jin YT, Liu NF. Altitudinal variation in reproductive strategy of the toad-headed lizard, Phrynocephalus vlangalii in North Tibet plateau (Qinghai) Amphibia-Reptilia. 2007;28(4):509–515. doi: 10.1163/156853807782152507. [DOI] [Google Scholar]
- 24.Jin YT, Liu NF, Li JL. Elevational variation in body size of Phrynocephalus vlangalii in the North Qinghai-Xizang (Tibetan) plateau. Belg J Zool. 2007;137:197–202. [Google Scholar]
- 25.Tang X, Xin Y, Wang H, Li W, Zhang Y, Liang S, He J, Wang N, Ma M, Chen Q. Metabolic characteristics and response to high altitude in Phrynocephalus erythrurus (Lacertilia: Agamidae), a lizard dwell at altitudes higher than any other living lizards in the world. PLoS One. 2013;8(8):e71976. doi: 10.1371/journal.pone.0071976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang W, Qi Y, Fu J. Exploring the genetic basis of adaptation to high elevations in reptiles: a comparative transcriptome analysis of two toad-headed agamas (genus Phrynocephalus). PLoS One. 2014;9(11) [DOI] [PMC free article] [PubMed]
- 27.Liao P, Jin Y. The complete mitochondrial genome of the toad-headed lizard subspecies, Phrynocephalus theobaldi orientalis (Reptilia, Squamata, Agamidae) Mitochondrial DNA. 2014;27(1):559. doi: 10.3109/19401736.2014.905857. [DOI] [PubMed] [Google Scholar]
- 28.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 29.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 30.Yang Z, Wong WSW, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22(4):1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- 31.Zhang J, Kumar S, Nei M. Small-sample tests of episodic adaptive evolution: a case study of primate lysozymes. Mol Biol Evol. 1997;14(12):1335–1338. doi: 10.1093/oxfordjournals.molbev.a025743. [DOI] [PubMed] [Google Scholar]
- 32.Woolley S, Johnson J, Smith MJ, Crandall KA, Mcclellan DA. TreeSAAP: selection on amino acid properties using phylogenetic trees. Bioinformatics. 2003;19(5):671–672. doi: 10.1093/bioinformatics/btg043. [DOI] [PubMed] [Google Scholar]
- 33.Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE. UCSF chimerax: meeting modern challenges in visualization and analysis. Protein Sci. 2017; [DOI] [PMC free article] [PubMed]
- 34.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286(5438):295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 36.Suel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nat Struct Mol Biol. 2003;10(1):59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 37.Dekker JP, Fodor AA, Aldrich RW, Yellen G. A perturbation-based method for calculating explicit likelihood of evolutionary co-variance in multiple sequence alignments. Bioinformatics. 2004;20(10):1565–1572. doi: 10.1093/bioinformatics/bth128. [DOI] [PubMed] [Google Scholar]
- 38.Yang ZH. Computational molecular evolution. Oxford: Oxford University Press; 2006. [Google Scholar]
- 39.Sazanov LA, Hinchliffe P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science. 2006;311(5766):1430. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- 40.Meiklejohn CD, Montooth KL, Rand DM. Positive and negative selection on the mitochondrial genome. Trends Genet. 2007;23(6):259–263. doi: 10.1016/j.tig.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 41.Marshall HD, Baker AJ, Grant AR. Complete mitochondrial genomes from four subspecies of common chaffinch (Fringilla coelebs): new inferences about mitochondrial rate heterogeneity, neutral theory, and phylogenetic relationships within the order Passeriformes. Gene. 2013;517(1):37–45. doi: 10.1016/j.gene.2012.12.093. [DOI] [PubMed] [Google Scholar]
- 42.Morales HE, Pavlova A, Joseph L, Sunnucks P. Positive and purifying selection in mitochondrial genomes of a bird with mitonuclear discordance. Mol Ecol. 2015;24(11):2820–2837. doi: 10.1111/mec.13203. [DOI] [PubMed] [Google Scholar]
- 43.Da FR, Johnson WE, O'Brien SJ, Ramos MJ, Antunes A. The adaptive evolution of the mammalian mitochondrial genome. BMC Genomics. 2008;9(1):119. doi: 10.1186/1471-2164-9-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liò P. Phylogenetic and structural analysis of mitochondrial complex I proteins. Gene. 2005;345(1):55. doi: 10.1016/j.gene.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 45.Pabijan M, Spolsky C, Uzzell T, Szymura JM. Comparative analysis of mitochondrial genomes in Bombina (Anura; Bombinatoridae) J Mol Evol. 2008;67(3):246–256. doi: 10.1007/s00239-008-9123-3. [DOI] [PubMed] [Google Scholar]
- 46.Bennett AF. Thermal dependence of locomotor capacity. Am J Phys Regul Integr Comp Phys. 1990;259(2):R253–R258. doi: 10.1152/ajpregu.1990.259.2.R253. [DOI] [PubMed] [Google Scholar]
- 47.Moilanen JS, Finnila S, Majamaa K. Lineage-specific selection in human mtDNA: lack of polymorphisms in a segment of MTND5 gene in haplogroup J. Molecular Biology & Evolution. 2003;20(20):2132–2142. doi: 10.1093/molbev/msg230. [DOI] [PubMed] [Google Scholar]
- 48.Wilson CC, Bernatchez L. The ghost of hybrids past: fixation of Arctic charr (Salvelinus alpinus) mitochondrial DNA in introgressed population of lake trout (S. namaycush) Mol Ecol. 1998;7(1):127–132. doi: 10.1046/j.1365-294x.1998.00302.x. [DOI] [Google Scholar]
- 49.Dalziel AC, Moyes CD, Fredriksson E, Lougheed SC. Molecular evolution of cytochrome c oxidase in high-performance fish (teleostei: Scombroidei) J Mol Evol. 2006;62(3):319. doi: 10.1007/s00239-005-0110-7. [DOI] [PubMed] [Google Scholar]
- 50.Rocco FD, Parisi G, Zambelli A, Vida-Rioja L. Rapid evolution of cytochrome c oxidase subunit II in camelids (Tylopoda, Camelidae) J Bioenerg Biomembr. 2006;38(5):293–297. doi: 10.1007/s10863-006-9048-8. [DOI] [PubMed] [Google Scholar]
- 51.Zhao EM, Zhao KT, Zhou KY: Fauna Sinica, Reptilia Vol. 2, Squamata, Lacertilia. Beijing: Science Press (in Chinese); 1999.
- 52.Jin YT, Yang ZS, Brown RP, Liao PH, Liu NF: Intraspecific lineages of the lizard Phrynocephalus putjatia from the Qinghai-Tibetan plateau: impact of physical events on divergence and discordance between morphology and molecular markers. Mol Phylogenet Evol 2014, 71:288–297. [DOI] [PubMed]
- 53.Jin Y, Liu N, Brown RP. The geography and timing of genetic divergence in the lizard Phrynocephalus theobaldi on the Qinghai-Tibetan plateau. Sci Rep. 2017;7 [DOI] [PMC free article] [PubMed]
- 54.Jin YT, Liu NF. Phylogeography of Phrynocephalus erythrurus from the Qiangtang plateau of the Tibetan plateau. Molecular Phylogenetics & Evolution. 2010;54(3):933–940. doi: 10.1016/j.ympev.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 55.Jin YT, Brown RP, Liu NF. Cladogenesis and phylogeography of the lizard Phrynocephalus vlangalii (Agamidae) on the Tibetan plateau. Mol Ecol. 2008;17(8):1971–1982. doi: 10.1111/j.1365-294X.2008.03721.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Voucher specimen and mitogenome used in selection analyses. (DOC 49 kb)
Primer pairs used in amplification of fragments of mitogenomes. (DOC 63 kb)
Multiple sequence alignment was used in statistical coupling analyses for ND proteins. (XLS 224 kb)
ML tree for Phrynocephalus inferred from 15,417 bp of mtDNA (6 partitions; GTRGAMMA model for each). Values on nodes are bootstrap proportions (2000 bootstraps). (DOCX 172 kb)
Data Availability Statement
Sequence data were deposited in GenBank with accession numbers: MF039058–65, KJ551842, KJ885621, KJ830752, KJ749841, KJ630904, KF572032, KM093858–59, KC119493, KC578685, KP126516, listed in Additional file 1. Phylogenetic data were submitted to TreeBASE (http://purl.org/phylo/treebase/phylows/study/TB2:S22322).