

Abstract
Background
Despite improved survival following checkpoint inhibitors, there is still a potential role for anti-cancer therapeutic vaccines. Because of biological heterogeneity and neoantigens resulting from each patient’s mutanome, autologous tumor may be the best source of tumor-associated antigens (TAA) for vaccines. Ex vivo loading of autologous dendritic cells with TAA may be associated with superior clinical outcome compared to injecting irradiated autologous tumor cells. We conducted a randomized phase II trial to compare autologous tumor cell vaccines (TCV) and autologous dendritic cell vaccines (DCV) loaded with autologous TAA.
Methods
Short-term autologous tumor cell lines were established from metastatic tumor. Vaccines were admixed with 500 micrograms of GM-CSF and injected weekly for 3 weeks, then at weeks 8, 12,16, 20, and 24. The primary endpoint was overall survival. Secondary objectives were identification of adverse events, and results of delayed type hypersensitivity (DTH) reactions to intradermal tumor cell injections.
Results
Forty-two patients were randomized. All were followed from randomization until death or for five years; none were lost to follow-up. DCV was associated with longer survival: median 43.4 versus 20.5 months (95% CI, 18.6 to > 60 versus 9.3 to 32.3 months) and a 70% reduction in the risk of death (hazard ratio = 0.304, p = 0.0053, 95% CI, 0.131 to 0.702). Tumor DTH reactions were neither prognostic nor predictive. The most common treatment-related adverse events were mild to moderate local injection site reactions and flu-like symptoms; but grade 2 treatment-related adverse events were more frequent with TCV. Serum marker analyses at week-0 and week-4 showed that serum markers were similar at baseline in each arm, but differed after vaccination.
Conclusions
This is the only human clinical trial comparing DCV and TCV as platforms for autologous TAA presentation. DCV was associated with minimal toxicity and long-term survival in patients with metastatic melanoma. DTH to autologous tumor cells was neither prognostic for survival nor predictive of benefit for either vaccine.
Trial registration
Clinical trials.gov NCT00948480 retrospectively registered 28 July 2009.
Electronic supplementary material
The online version of this article (10.1186/s40425-018-0330-1) contains supplementary material, which is available to authorized users.
Keywords: Metastatic melanoma, Patient-specific vaccines, Dendritic-cell vaccines, Tumor-cell vaccines, Autologous tumor cell lines
Background
Relevance of therapeutic vaccine research
During the past decade introduction of new therapies has been associated with increased survival for patients with metastatic melanoma [1]. These have included oral enzyme inhibitors of signal-transduction driver pathways [2, 3], and monoclonal antibodies that block immune checkpoint inhibitors [4–6]. However, even with combination checkpoint inhibitors as initial therapy, the 3-year survival rates are less than 60% [7]. Therapeutic vaccines might add to the survival benefit of such patients. First, animal models suggest that anti-tumor vaccines and checkpoint inhibitors are complementary therapies [8, 9]. Second, the mechanism of action for the anti-programmed death-1 (PD1) checkpoint inhibitors is blocking the suppression of an existing immune response, but many patients show no evidence of an existing immune response in their tumors [10, 11]. Third, therapeutic vaccines have been associated with survival benefit in some patients [12, 13]. Fourth, dendritic cell vaccines can induce or enhance immune responses to patient-specific neoantigens [14]. For these and other reasons, vaccine research is still relevant for melanoma treatment [15].
Autologous tumor cells as sources of tumor-associated antigens
Because of biological heterogeneity and neoantigens resulting from each mutanome [16, 17], autologous tumor may be the best source of tumor-associated antigens (TAA) for vaccines. Short-term tumor cell lines derived from metastatic lesions is one source of TAA that could be used for patient-specific vaccines [18, 19]. They can express all TAA, including unique patient-specific TAA expressed only on self-renewing, proliferating autologous tumor cells that may represent tumor initiating stem cells and/or early progenitor cells. Using tumor cells from a short-term cell line assures lack of contamination with viable normal cells or immune suppressor cells such as regulatory T cells, or myeloid derived suppressor cells.
Whole tumor cell vaccines versus dendritic cell vaccines
Two potential therapeutic approaches with short-term tumor cell lines include (1) injecting irradiated tumor cells (ITC) in a manner similar to many anti-viral vaccines, and then relying on endogenous antigen-presenting cells to process TAA and induce anti-tumor immune responses; and (2) loading antigens from such cells ex vivo into autologous dendritic cells (DC) and vaccinating with these cells. Patient-specific tumor cell vaccines (TCV) derived from autologous short-term tumor cell lines were used to treat 74 metastatic melanoma patients. TCV was well-tolerated, associated with an objective response rate (ORR) of 9%, progression free survival (PFS) of 4.4 months, and when all patients were followed to death or for 5 years from enrollment with none lost to follow-up, median overall survival (OS) was 20.5 months, and 5-year OS 28% [20]. Subsequently 54 metastatic melanoma patients were treated with patient-specific dendritic cell vaccines (DCV) consisting of autologous dendritic cells (DC) loaded with TAA by incubating DC with 10 million ITC derived from autologous tumor cell lines. DCV was well-tolerated, associated with an ORR of 0%, PFS of 4.2 months, median OS greater than 5 years, and projected 5-year OS of 54% [21]. When all patients were followed to death or for 5 years from enrollment with none lost to follow-up, 5-year OS was 50%.
Significance of current report
A randomized trial (MACVAC) to compare these two approaches was initiated in October 2007, and is still the only human trial that has addressed whether a DCV is superior to a TCV. The trial was stopped prematurely due to discontinuation of financial support by the sponsoring hospital in April 2011. By then 42 patients had been randomized; all had initiated treatment per randomization assignment. Preliminary results were reported at a time when all patients had completed therapy; so minimum follow up was six months, median follow up was less than two years, maximum follow up less than four years, and 21 patients were deceased. OS was better in the DCV arm with projected 2-year survival rates of 72% versus 31% (p = 0.007) [22]. New information in the current report includes: (1) survival analysis performed after all patients had been followed for five years or until death, (2) details regarding patient characteristics and treatments administered before and after participation in this study, (3) results of a multivariate Cox regression analysis and hazards model, (4) survival results by treatment arm in various subsets of patients, (45) comparative results of delayed type hypersensitivity (DTH) reactions to autologous ITC, (6) comparison of adverse events by treatment arm, (7) comparison of serum cytokine results before and after vaccination, (8) and additional analyses regarding the feasibility of establishing tumor cell lines.
Methods
Design
This was designed as an open-label, randomized trial to compare autologous DCV to autologous TCV. The primary endpoint was overall survival. Secondary objectives were identification of adverse events, and results of delayed type hypersensitivity (DTH) reactions to intradermal tumor cell injections.
Autologous tumor cell lines
Surgically resected metastatic tumors were submitted on behalf of melanoma patients classified as recurrent stage 3 or distant stage 4. Tumors were mechanically and enzymatically dissociated into single-cell suspensions and grown in tissue culture as previously described [18, 22, 23]. The patient’s managing physician was notified once a cell line was successful. These cell cultures were the source of ITC used for autologous tumor DTH tests, for TCV, and for co-incubation with DC to create DCV.
Autologous dendritic cells
DC were derived from peripheral blood mononuclear cells obtained during a single leukapheresis procedure and monocytes were separated (Elutra® Cell Separation System, CaridianBCT, Lakewood, CO.). Monocytes were differentiated into DC over six days in the presence of granulocyte-macrophage colony stimulating factor (GM-CSF) and interleukin-4 as previously described [21, 22].
Investigational products
TCV consisted of ITC derived from the patient’s autologous tumor cell line as previously described [21–23]. Each dose contained about 10 million ITC.
DCV consisted of autologous DC that were incubated overnight with 10 million ITC for phagocytosis and antigen loading as previously described [21, 22]. The final product averaged 10 to 15 million cells per dose, but ranged from 3 to 30 million per dose among different patients and included residual ITC in many samples. There was substantial inter-patient variation in average dose, but little intra-patient variation in doses.
Patients
Eligibility criteria for the MACVAC trial were previously described [22]. Key eligibility criteria for randomization were: (1) availability of short-term autologous tumor cell line, (2) referral by the managing physician for vaccine therapy, (3) willingness to travel to Newport Beach, California for treatment, and (4) Karnofsky performance status ≥ 70. Patients with brain metastases were eligible if they had been treated with expectation of successful control.
Randomization
Patients were stratified by whether their most advanced stage of disease was 3 or 4 [24], and by whether they had measurable disease at the time of randomization [25]. They were then randomized 1:1 without blocking. Patients randomized to TCV were able to undergo DTH testing the next day and start treatment the following week. Patients randomized to DCV underwent a leukapheresis procedure the same or following day and could begin DTH testing and treatment about four weeks later.
Treatment schedule
At the time of each treatment, a cryopreserved vial of TCV or DCV was thawed and suspended in 500 micrograms of GM-CSF and injected within five hours of thawing. Subcutaneous injections were administered during weeks 1, 2, 3, 8, 12, 16, 20 and 24, as in earlier trials [21, 22]. Concurrent anti-cancer therapy was not allowed, but there was a provision by which patients could interrupt vaccine therapy in order to pursue another therapy because of progressive disease, and then when that therapy was completed, finish the remaining vaccine doses.
Follow up information
After completion of vaccine injections, patients were followed in person or by telecommunications every three months to collect information regarding administration of additional anti-cancer therapies, any new adverse events (AE) that might be attributable to vaccine treatment, approximate date of disease progression, or date and cause of death if deceased.
End points
Survival
The primary endpoint was OS from the date of randomization per intent-to treat to date of death. Managing physicians and clinical trial staff identified the first site of disease progression and a date of disease progression to estimate PFS.
Delayed type hypersensitivity (DTH) to autologous tumor cells
One week before starting either vaccine, patients were administered an intradermal skin test of 1 million ITC. Within 48 to 72 h the test was interpreted by nursing staff as negative (no induration), weakly positive (5 to 9 mm induration), or positive (≥ 1 cm induration). The test and interpretation was repeated one week after the three weekly injections had been administered.
Safety
AE were assessed at each visit for a vaccine injection, and four weeks after the last injection. AEs and serious AEs (SAEs) were classified and graded 0 to 5 per National Cancer Institute Common Toxicity Criteria for Adverse Events (NCI-CTCAE) version 3.0.
Serum markers
Cryopreserved serum samples (200 μl) from week-0 and week-4 were sent to Raybiotech, Inc. (Norcross, GA) for human cytokine protein array screening for the 110 different proteins then available in their Quantibody® Cytokine Array, which utilizes a validated, quantitative, multiplex enzyme-linked immunosorbent assay (ELISA). In order to normalize the results of various assays, week-0 and week-4 values for various assays were expressed as differences above or below the mean values for three normal controls.
Statistical analysis
Survival curves were generated using the method of Kaplan and Meier and compared using log rank tests (Mantel-Haenszel and Gehan’s Wilcoxon). The Cox regression model and the Wald test were used to estimate the hazard ratio associated with treatment and to identify the significance of potential prognostic factors and their impact on treatment differences. Variables associated with 3-year survival were explored by multiple univariate analyses. Proportions were compared using the Pearson Chi Square or Fisher’s Exact Test. Means were compared using the Student T-test. Nonparametric means were compared using the Mann-Whitney test.
Results
Establishing tumor cell lines
As previously reported median time from tissue harvest to successful cell line was 3.1 months [22], but the range was from 27 to 224 days. The sites of tumor collection were lymph nodes (n = 21), cutaneous or subcutaneous tissue (n = 11), lung (n = 3), breast (n = 2), and one each from brain, omentum, liver, spleen, and one with multiple sites. The median tumor weight was 2.3 g (range < 0.2 to 12.5). The cell line success rate was 46/79 (58%) for tumors ≥ 3 g compared to 30/108 (28%) for those < 3 g (p < 0.0001) (Additional file 1: Table S1). Among patients for whom cell lines were successful, 42/76 (55%) were referred for study enrollment. The median time to a successful cell culture was 81.0 days for both TCV and DCV; average time was 94.6 days (range17 to 237 days). There was no correlation between time to establish a cell line and survival.
Median time from tissue harvest to treatment was 3.0 months (range 3 to 23 months). While efforts were in progress to establish a cell line, disease progression occurred in 62% of patients who were eventually treated with DCV or TCV (Additional file 2: Table S2). Among patients with recurrent stage 3 at the time of tumor tissue acquisition, 9/18 were still NED at the time of randomization; another who had developed M1c disease after resection had a complete response to interleukin-2-based biochemotherapy. The other eight had progressed to detectable metastatic disease (7 measurable). Among 24 patients who had distant metastases at the time tissue was collected, only 5 were free of disease at randomization.
Treatment assignment, patient characteristics and other therapies
All patients were treated per randomization assignment (Additional file 3: Figure S1). Patient baseline characteristics at baseline are shown in Table 1. None of the specific individual characteristics differed significantly between the arms. There were no differences in therapies given previous or subsequent to DCV or TCV (Table 2). Patients were treated during November 2007 to August 2011 before widespread use of BRAF/MEK enzyme inhibitors, and the anti-cytotoxic T lymphocyte-4 (CTLA-4) and anti-programmed death-1 (PD-1) monoclonal antibody checkpoint inhibitors. The first FDA approvals of these agents were 2010 for ipilimumab, 2011 for vemurafinib, and 2014 for the anti-PD-1 inhibitors pembrolizumab and nivolumab.
Table 1.
Baseline characteristics of patients and by treatment arm
| Characteristic | All (n = 42) | TCV (n = 24) | DCV (N = 18) | P-value |
|---|---|---|---|---|
| Age ≥ 60 years | 20 (48%) | 12 (50%) | 8 (44%) | 0.72 |
| # Male | 27 (64%) | 16 (67%) | 11 (61%) | 0.71 |
| # from out of state | 16 (38%) | 9 (38%) | 7 (39%) | 0.93 |
| KPS = 100% | 21 (50%) | 12 (50%) | 9 (50%) | 1.00 |
| ↑LDH at randomization | 11 (26%) | 4 (17%) | 7 (39%) | 0.16 |
| Highest stage =4 | 33 (79%) | 17 (71%) | 16 (89%) | 0.16 |
| Prior brain metastases | 10 (24%) | 6 (25%) | 4 (22%) | 1.00 |
| Prior visceral metastases (non-CNS) | 22 (52%) | 14 (58%) | 8 (44%) | 0.45 |
| Measurable Disease | 17 (40%) | 9 (38%) | 8 (44%) | 0.65 |
| Detectable (not measurable) | 10 (24%) | 4 (17%) | 6 (33%) | 0.28 |
| NED at randomization | 15 (36%) | 11 (46%) | 4 (22%) | 0.19 |
| Stage 4 M1a at randomization | 3 (7%) | 1 (4%) | 2 (11%) | 0.57 |
| Stage 4 M1b at randomization | 9 (21%) | 6 (25%) | 3 (17%) | 0.71 |
| Stage 4 M1c at randomization | 15 (36%) | 6 (25%) | 9 (50%) | 0.094 |
TCV tumor cell vaccine, DCV dendritic cell vaccine, KPS Karnofsky Performance Status, LDH serum lactate dehydrogenase, CNS central nervous system, NED no evidence of disease, M1a metastatic disease soft tissue metastases only and normal LDH, M1b metastatic lung with or without soft tissue metastases, but no other visceral metastases and normal LDH, M1c metastases to visceral organs and/or elevated LDH
Table 2.
Anti-melanoma therapy prior and subsequent to participation in the MACVAC trial. Therapies: overall and by treatment arm
| All (n = 42) | TCV (n = 24) | DCV (N = 18) | P-value | |
|---|---|---|---|---|
| Previous therapy | ||||
| Surgeries Only | 7 (17%) | 3 (12%) | 4 (22%) | 0.44 |
| Radiation Therapy (not brain) | 17 (40%) | 8 (33%) | 9 (50%) | 0.28 |
| Brain Radiation Therapy | 10 (24%) | 6 (25%) | 4 (22%) | 1.00 |
| Chemotherapy | 23 (55%) | 15 (62%) | 8 (44%) | 0.24 |
| Interleukin-2 | 14 (33%) | 8 (33%) | 6 (33%) | 1.00 |
| IFN-α | 20 (48%) | 11 (46%) | 9 (50%) | 0.79 |
| GMCSF | 13 (31%) | 8 (33%) | 5 (28%) | 0.70 |
| Anti-VEGF | 8 (19%) | 4 (17%) | 4 (22%) | 0.71 |
| Vaccine | 5 (12%) | 4 (17%) | 1 (6%) | 0.37 |
| Anti-BRAF | 0 | 0 | 0 | – |
| Anti-CTLA4 | 1 (2%) | 1 (4%) | 0 | – |
| Anti-PD1 | 0 | 0 | 0 | – |
| Subsequent therapy | ||||
| Metastasectomy | 11 (26%) | 4 (17%) | 7 (39%) | 0.16 |
| Radiation Therapy (not brain) | 9 (21%) | 6 (25%) | 3 (17%) | 0.71 |
| Brain Radiation Therapy | 11 (26%) | 6 (25%) | 5 (28%) | 1.00 |
| Chemotherapy | 19 (45%) | 11 (46%) | 8 (44%) | 1.00 |
| Interleukin-2 | 7 (8% | 5 (21%) | 2 (11%) | 0.68 |
| IFN-α | 3 (7%) | 2 (8%) | 1 (6%) | 1.00 |
| GM-CSF | 7 (17%) | 4 (17%) | 3 (17%) | 1.00 |
| Anti-VEGF | 4 (10%) | 2 (8%) | 2 (11%) | 1.00 |
| Vaccine | 1 (2%) | 0 | 1 (6%) | – |
| Anti-BRAF | 7 (17%) | 3 (12%) | 4 (22%) | 0.44 |
| Anti-CTLA4 | 12 (29%) | 7 (29%) | 5 (28%) | 1.00 |
| Anti-PD1 | 1 (2%) | 0 | 1 (6%) | 0.43 |
| None | 9 (21%) | 7 (29%) | 2 (11%) | 0.26 |
MACVAC melanoma antigen cancer vaccine trial, TCV tumor cell vaccine, DCV dendritic cell vaccine, IFN-α interferon alpha, GM-CSF granulocyte macrophage colony stimulating factor, VEGF monoclonal antibody to vascular endothelial growth factor, BRAF enzyme endcoded by mutated BRAF gene, CTLA4 cytotoxic T Lymphocyte Antigen 4, PD1 programmed death molecule 1
Study objectives
Efficacy
DCV was associated with longer OS (Fig. 1a); there was no increase in PFS (Fig. 1b). A Cox regression analysis assessed the association between survival and other variables (Additional file 4: Table S3). Only DCV therapy and tumor burden (defined as measurable, detectable/equivocal but unmeasurable, or no evidence of disease) were strongly associated with survival. With the caveat that numbers of patients in subsets are quite small, for completeness survival by treatment arm for each of various clinical subsets associated with prognostic variables are shown in Table 3. In an earlier analysis at a time when minimal follow up was three years, and 17 patients were still in follow up, treatment in the DCV arm was the only variable associated with a survival difference. (Additional file 5: Table S4). As previously reported, there was one delayed complete response in a DCV-treated patient with progressing measurable disease when randomized [26]. At the time of 5-year follow up, she still had received no other anti-cancer therapy and was still in complete remission. Only one patient interrupted vaccine treatment to take another therapy, and then resumed the vaccine. That patient was in the TCV arm, stopped treatment to receive chemotherapy that he had received previously, and was still alive after 5 years.
Fig. 1.
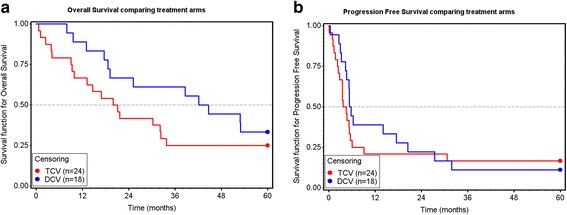
a Overall Survival by treatment arm. Median OS was 43.4 months versus 20.5 months for DCV and TCV respectively (18.6 to > 60 vs 9.3 to 32.3 months, 95% CI) (p = 0.194 Mantel-Haenzsel; p = 0.088 Gehan’s Wilcoxon). Adjusted Cox proportional hazard model revealed a 70% reduction in risk of death in the DCV arm (HR = 0.304, 95% CI 0.131 to 0.702, p = 0.0053, Wald test). Variables in multivariate analysis included age, stage, LDH, performance status, gender, M1 category, whether patient had measurable disease, treatment with dendritic cell vaccine, and whether patient lived outside California (see Additional file 4: Table S3. b Progression free survival (PFS) by treatment arm. Median PFS was 5.4 months in the DCV arm and 3.7 months in the TCV arm (4.0 to 8.0 vs 1.0 to 5.0 months, 95% CI p = 0.498)
Table 3.
Treatment effects in various subsets
| Subset | RX Arm |
# Pts |
Median OS Months |
3-year OS | P value |
|---|---|---|---|---|---|
| Measurable | DCV | 8 | 17.6 | 38% | 0.122 |
| TCV | 9 | 9.0 | 11% | ||
| Not Measurable | DCV | 10 | 44.6 | 80% | 0.332 |
| TCV | 15 | 32.2 | 33% | ||
| Not NED | DCV | 14 | 25.2 | 50% | 0.011 |
| TCV | 13 | 9.9 | 8% | ||
| NED | DCV | 4 | 53.0 | 75% | 0.561 |
| TCV | 11 | 33.7 | 40% | ||
| High LDH St 4 | DCV | 6 | 17.6 | 17% | 0.010 |
| TCV | 3 | 1.1 | 0% | ||
| WNL LDH | DCV | 11 | 53.0 | 82% | 0.079 |
| TCV | 20 | 21.1 | 25% | ||
| KPS =100 | DCV | 9 | 44.6 | 67% | 0.810 |
| TCV | 12 | 32.2 | 33% | ||
| KPS < 100 | DCV | 9 | 38.6 | 56% | 0.303 |
| TCV | 12 | 9.0 | 17% | ||
| Stage 4 | DCV | 16 | 38.6 | 56% | 0.292 |
| TCV | 17 | 16.9 | 24% | ||
| Stage 3 | DCV | 2 | > 60 | 100% | 0.141 |
| TCV | 7 | 32.2 | 29% |
RX arm treatment arm, Pts patients, OS overall survival, DCV dendritic cell vaccine, TCV = tumor cell vaccine, NED no evidence of disease, LDH serum lactate dehydrogenase, St 4 stage 4, KPS Karnofsky Performance Status
Delayed type hypersensitivity skin test reactivity
Tumor DTH tests were neither prognostic for survival nor predictive of therapeutic benefit in either arm. There was no difference between treatment arms in baseline DTH tests, nor in conversion rate at week-4 from a negative to positive or weakly positive tumor DTH test (TCV 5/20 vs DCV 1/17, p = 0.19) (Additional file 6: Table S5), nor in the rates of ever having a positive tumor DTH test (TCV 6/22 vs DCV 2/22, p = 0.26).
Safety
Both vaccines were well-tolerated. All eight planned doses were administered to 67% and 54% of patients in the DCV and TCV arms respectively. All early discontinuations were due to disease progression; no patients discontinued treatment because of toxicity. The frequencies of AEs attributed to study agents are summarized in Table 4. As in previous trials the most common AEs were injection site reactions and flu-like symptoms and nearly all treatment-related toxicity was mild to moderate in severity [20, 21]. There was only one grade 3 AE, a severe headache that occurred after the 8th and final DCV injection. Toxicity grade was higher in the TCV arm with 71% of TCV patients experiencing grade 2 or higher AEs as opposed to only 16% of DCV-treated patients (17/24 vs 3/18, p = 0.0007) (Table 5).
Table 4.
Summary by frequency of AEs of any severity, felt to possibly, likely, or almost certainly caused by injection of vaccines
| Adverse Event | All (N = 42) | TCV (n = 24) | DCV (n = 18) | P value |
|---|---|---|---|---|
| Injection site reactions | 28 (67%) | 16 (67%) | 12 (67%) | 1.00 |
| Flu-like symptoms | 14 (33%) | 9 (38%) | 5 (28%) | 0.742 |
| Nausea | 8 (19%) | 5 (21%) | 3 (17%) | 1.00 |
| Bone discomfort | 7 (17%) | 4 (17%) | 3 (17%) | 1.00 |
| Headache | 6 (14%) | 3 (12%) | 3 (17%) | 1.00 |
| Fatigue | 5 (12%) | 5 (21%) | 0 (0%) | 0.060 |
| Chills | 5 (12%) | 1 (4%) | 4 (22%) | 0.146 |
| Pruritus | 3 (7% | 2 (8%) | 1 (6%) | 1.00 |
| Arthralgias | 3 (7%) | 1 (4%) | 2 (11%) | 0.567 |
| Fever | 3 (7%) | 3 (12%) | 0 (0%) | 0.247 |
| Rash | 2 (5%) | 2 (8%) | 0 (0%) | 0.498 |
| Hives (urticarial) | 2 (5%) | 2 (8%) | 0 (0%) | 0.498 |
| Shingles | 2 (5%) | 2 (8%) | 0 (0%) | 0.498 |
| Myalgias | 1 (2%) | 0 (0%) | 1 (6%) | 0.429 |
TCV tumor cell vaccine, DCV dendritic cell vaccine
Table 5.
Highest grade of adverse events (AEs) felt possibly, likely or almost certainly caused by injection of vaccine
| Grade | All (N = 42) | TCV (n = 24) | DCV (n = 18) |
|---|---|---|---|
| Grade 0 | 6 (14%) | 2 (8%) | 4 (22%) |
| Grade 1 | 16 (38%) | 5 (21%) | 11 (61%) |
| Grade 2 | 19 (45%) | 17 (71%) | 2 (11%) |
| Grade 3 | 1 (2%) | 0 | 1 (5%) |
| Grade 4 | 0 | 0 | 0 |
TCV tumor cell vaccine, DCV dendritic cell vaccine
Serum markers
Paired week-0 and week-4 serum samples were available for 38 patients. Markers tested included 110 cytokines, growth factors, proteases, soluble receptors, and other proteins. Results were grouped together based on known associations with tumor growth, angiogenesis, and immune activation (Additional file 7: Table S6). At baseline serum levels of most markers were similar between the two arms, but tumor markers were higher in the DCV arm (Fig. 2), consistent with baseline tumor burden characteristics in that cohort (Table 1). The percent changes between week-0 and week-4 after the first three injections of DCV or TCV were quite different (Fig. 2b). TCV was associated with an increase in nearly all markers while DCV was associated with a decrease in several markers. In the TCV arm the tumor and inflammation markers increased from baseline. In the DCV arm tumor markers were only slightly increased from baseline, most inflammatory markers decreased from baseline, and some increased slightly. These results must be interpreted with caution because of the wide variability in assay results, and the use of only three donors for normalization.
Fig. 2.
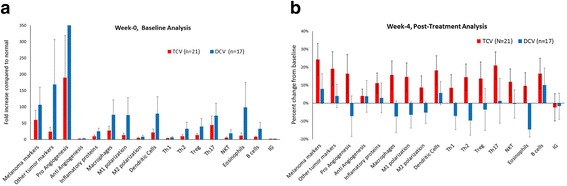
a Baseline analysis of serum cytokines. In order to summarize data for all tests, data is expressed as change in relation to values for set of assays compared to 3 normal control volunteers. There was substantial variation in markers among patients. At baseline most markers were elevated compared to normals, especially in the DCV arm. b The post treatment analysis of cytokines one week after the third weekly vaccine injection, showing changes compared to baseline. The changes associated with the two vaccines were quite different. Levels increased for nearly all markers in the TCV arm, but decreased for eight of the 17 groupings in the DCV arm
Discussion
To our knowledge, this is the first and only study conducted in cancer patients that addresses the question of whether survival differs between cancer patients treated with an autologous DCV versus an autologous TCV that both feature TAA from short-term autologous tumor cell lines. The most important observation in this study is that DCV was associated with a doubling of median OS and a 70% reduction in the risk of death compared to TCV. The median survival of 20.5 months in the TCV arm suggests that TCV may also have anti-tumor activity. The results in each arm of this randomized trial are quite similar to results previously reported for single arm trials of TCV [20], and DCV [21]. The lack of correlation between PFS and OS was also observed in the earlier trials, and has been observed for other immunotherapies that may provide long-lasting immune benefit [27]. Examples of FDA-approved agents that improved OS but had unimpressive ORR or PFS include sipuleucel-T in prostate cancer, [28] and ipilimumab in melanoma [29].
Strengths of the MACVAC trial include that it was a randomized trial, all patients received their assigned treatment, all patients were followed until death or for five years, and no patients were lost to follow up. Patients in both arms of the study were injected with vaccines that contained the patient-specific TAA from about 10 million tumor cells that were self-renewing in cell culture. The clinical study, including all leukapheresis procedures and treatment administration took place at a single institution, but patients were referred from all over the United States for this trial and remained under the clinical management of their referring physicians. Weaknesses of this trial are the small patient numbers, which was caused by the premature closure. Not only did this result in a much smaller population of subjects than originally planned, but also in an imbalance in the numbers of patients assigned to each treatment arm, and some imbalance in baseline prognostic factors. The imbalance in the treatment arms was due to the randomization of patients within each stratification, and the lack of blocking to assure approximately even randomization at any point in time. The imbalance in prognostic factors was slightly biased against the DCV arm. This also resulted in very small numbers of patients for exploratory subgroup analyses (Table 3), but this data was presented because of frequent questions from clinical other investigators regarding outcomes in these subgroups. Double-blinding was not used in this trial because it was a phase II trial, and because of the extra cost, inconvenience, and risks associated with leukapheresis procedures [30].
DTH tests to autologous tumor could be predictive of an effective anti-TAA immune response [31, 32]. We previously reported improved survival for 125 patients with various cancer histologies who had a positive DTH at any time during treatment with TCV [33], but this was not confirmed in TCV-treated metastatic melanoma patients [20]. There was no association between DTH reactivity and outcome in DCV-treated melanoma patients in the 54-patient single-arm trial [21]. The MACVAC trial confirmed that this tumor DTH test was not useful as a predictive or prognostic marker for either vaccine.
The MACVAC trial confirmed the limited toxicity associated with these vaccine products in previous trials [20, 21]. The most common AEs attributed to study injections were local injection site reactions and flu-like symptoms, which are well-known side effects of GM-CSF. Interestingly, there was somewhat higher grade toxicity (grade 2 as opposed to grade 1) in patients receiving TCV.
As part of this trial, serum was obtained at week-0 (baseline) and week-4 (after three weekly injections) and cryopreserved for later analysis. Comparisons of baseline samples and changes from week-0 to week-4 after the first three injections showed that the patterns of changes in cytokine levels differed between DCV and TCV. This data is included only to address the question raised by several investigators as to whether there was any evidence that there was a difference between the study arms in terms of changes in in any biological measurements after the first three weekly injections. Much more extensive evaluation would be needed to better understand the association between these changes and the clinical benefit observed in the DCV arm, especially in terms of immune effects on T cells and their recognition of tumor cells. Such experiments have not been performed because the cell samples obtained during this trial are no longer the property of the investigator.
The data regarding success in establishing tumor cell lines, and the time needed to establish them, is important for understanding the complex logistics involved in clinical trials of such patient-specific cellular products. Successful commercial application of autologous tumor cell lines would likely require a higher and faster rate of success in obtaining autologous tumor cells as a source of TAA in order to treat more patients, and to decrease potential differences in mutations between tumor cells growing in vitro and tumor cells growing in vivo. The objective of tissue culture was to increase the representation of self-renewing tumor cells (perhaps early progenitor cells) as the source of TAA rather than terminally differentiated tumor cells that predominate in a fresh tumor sample, However, this means that immune responses may be directed against only a small subset of cells in any tumor mass, which may explain why this approach is not associated with rapid regression of measurable tumor.
All attempts to establish dendritic cells were successful. In the DC arm no effort was made to standardize the number of DC injected, but the range of 3 million to 30 million cells among patients may be relatively narrow from a biological perspective. However, theoretically there could have been up to a 100-fold difference in the density of TAA per DC per patient. The potential impact of this is unclear given the autologous origin of the tumor cells and their unique neoantigens. Analyses failed to show any dose/survival relationship related to the number of dendritic cells injected among the 18 patients treated in this trial, and among the 54 reported previously [21].
Since MACVAC was conducted, enzyme-targeted BRAF inhibitors and monoclonal antibody immune checkpoint inhibitors, have become standard therapies for patients with metastatic melanoma, and are associated with improved survival [3, 7] Based on their complementary mechanisms of action, there is a good rationale for combining vaccines that present autologous TAA with these newer agents [34, 35]. There is also evidence that such patient-specific vaccines increase the survival benefit associated with the immune stimulating cytokine interleukin-2 [36]. Ex vivo loading of TAA onto DC outside of the immunosuppressive tumor environment may be especially advantageous in patients who have normal expression of major histocompatibility antigens, but have no infiltration of CD8+ T-lymphocytes in tumor biopsies. Pre-existing TAA recognition by the endogenous immune system is a prerequisite for possible benefit from anti-checkpoint therapy. DCV is worthy of further study combined or sequenced with these other agents. Other dendritic cell vaccines and patient-specific vaccine approaches already are showing promise in combination with checkpoint inhibitors [37, 38].
Conclusions
This is the only clinical trial that has compared dendritic cell and whole tumor cell vaccines as platforms for autologous TAA presentation. DCV was associated with superior survival compared to TCV and induced different changes in serum cytokines than did TCV. DCV was associated with minimal toxicity. As a potential biomarker, DTH to autologous tumor cells was neither prognostic for survival nor predictive of benefit from DCV.
Additional files
Table S1. Success rate for cell cultures started from metastatic melanoma samples obtained during 2006–2011 (era of MACVAC trial). (DOCX 14 kb)
Table S2. Relationship between stage at time of tumor collection for cell line and disease status at the time of randomization. (DOCX 13 kb)
Figure S1. Consort diagram for MACVAC trial. (DOC 31 kb)
Table S3. Mutlivariate Cox regression analysis and proportional hazards model for independent variables. (DOCX 14 kb)
Table S4. Multiple univariate analyses performed when all patients were either deceased or had been followed for a minimum of three years. (DOCX 15 kb)
Table S5. Results of delayed type hypersensitivity (DTH) skin test to autologous irradiated tumor cells, Week-0 (baseline) and Week-4 following three weekly vaccine injections. (DOCX 14 kb)
Table S6. Panel of markers used to investigate the serum of patients treated in randomized phase II trial, grouped by associated biologic activity. (DOCX 15 kb)
Acknowledgements
Linda D. Beutel and Denysha J. Carbonell of the Hoag Cancer Center Cell Biology Laboratory were instrumental in the production of the investigational products administered in this trial. Kelly Austin directed the collection of leukapheresis products. The following research associates employed in the Hoag Institute for Research and Education in Newport Beach, CA. collected the data for entry into case report forms for this clinical trial: Robin E. Ellis, Cristina deLeon, Cheryl Mayorga, and Alicia Bogardus. The statistical analyses, and in particular the Cox regression analysis, was performed by biostatistician Almut Hahn, who also reviewed the manuscript.
The following physicians referred two or more patients for this trial and provided copies of medical, radiology, and laboratory reports for data entered into case report forms: George B. Semeniuk, Orange Coast Oncology and Hematology, Newport Beach, CA., USA; Clark E. Haskins, New Mexico Cancer Center, Albuquerque, NM, USA; Robert W. Weber, St. Mary’s Medical Center and Sutter Health, San Francisco, CA, USA; David M Burtzo, Pacific Shores Medical Group, Huntington Beach, CA.
Funding
Support for the Hoag Cancer Center Cell Biology Laboratory was provided by the Hoag Hospital Foundation. Support for the Clinical Trials Office staff who collected data for this clinical trial in which these patients were treated was provided by Hoag Hospital. Funding support for the serum cytokine assays was provided by Caladrius Biosciences, Inc. Funding for statistical analysis was provided by AIVITA Biomedical, Inc. The funding bodies were not involved in the design of the study, the collection, analysis, and interpretation of data, nor in writing the manuscript.
Availability of data and materials
The clinical datasets used and/or analysed during the current study are available from the corresponding author on reasonable request. The cytokine data set is not available.
Abbreviations
- AE
Adverse event
- BRAF
Specific serine/threonine-protein kinase member of the Raf kinase family
- CTLA-4
Cytotoxic T lymphocyte–associated antigen 4
- DC
Dendritic cell
- DCV
Dendritic cell vaccine
- GM-CSF
Granulocyte-macrophage colony stimulating factor
- ITC
Irradiated tumor cells
- LDH
Lactate dehydrogenase
- MEK
Mitogen-activated protein kinase
- NED
No evidence of disease
- OS
Overall survival
- PD-1
Programmed cell death protein-1
- PFS
Progression-free Survival
- RECIST
Response criteria in solid tumors
- SAE
Severe adverse event
- TAA
Tumor associated antigens
- TCV
Tumor cell vaccine
Authors’ contributions
ROD conceived the study, drafted the clinical protocol, participated in the design, coordination, and execution of the clinical trial, participated in the statistical analysis, and drafted the manuscript. ANC participated in the study design, and coordination of the clinical trial, and directed the manufacturing of the tumor cell lines and production of the tumor cell and dendritic cell vaccines used in this trial, and provided content related to these activities. GIN coordinated the testing of serum samples and graphic presentation of the results and provided content related to this activity. EFM and TTA each enrolled and treated more than 10% of eligible, evaluable patients, and provided copies of medical, radiology, and laboratory reports for data entered into case report forms. CD participated in study design and writing the protocol, coordination and execution of the clinical trial, and coordinated data collection and compiled the data used for this analysis. All authors read and approved the manuscript.
Authors’ information
ROD is a former president of the Society for Immunotherapy of Cancer (2000–2002, then called the International Society for the Biotherapy of Cancer) after serving two previous terms on its Board of Directors. For more than a decade he was Chairman of the Cancer Biotherapy Research Group. From 1989 to 2011 he was Medical Director of the Hoag Cancer Center, where this trial was conducted, and Scientific Director for the Hoag Cell Biology Laboratory, where the investigational products used in this trial were manufactured.
Ethics approval and consent to participate
The MACVAC clinical trial was conducted per the Declaration of Helsinki after approval by the Western Institutional Review Board (Seattle, WA), and in accord with assurances filed with and approved by the Department of Health and Human Services. All patients gave written informed consent prior to randomization. In addition to Institutional Review Board oversight, conduct of the trial was monitored by an external consultant, and Hoag Hospital’s institutional clinical research oversight committee. The protocol and manufacturing procedures were reviewed by the US Food and Drug Administration in association with BB-IND 5838 for the tumor cell vaccine, 8554 for the dendritic cell vaccine. The trial was registered on ClinicalTrials.gov (NCT00436930) on 28 July 2009.
Consent for publication
Not applicable
Competing interests
ROD is an employ of AIVITA Biomedical, Inc. ANC is an employee of TCR2. GIN is an employ of AVITIA Biomedical, Inc. ROD, ANC, and GIN are former employees of Caladrius Biosciences, Inc. and retain stock in that company. None of the other authors declared any conflicts as related to this trial.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s40425-018-0330-1) contains supplementary material, which is available to authorized users.
Contributor Information
Robert O. Dillman, Phone: 949-735-7229, Email: robert.dillman55@gmail.com
Andrew N. Cornforth, Email: acorn4th@gmail.com
Gabriel I. Nistor, Email: gabriel@aivitabiomedical.com
Edward F. McClay, Email: emcclay@pacificoncology.com
Thomas T. Amatruda, Email: thomas.amatruda@usoncology.com
Carol Depriest, Email: carol.depriest5@gmail.com.
References
- 1.Ugurel S, Röhmel J, Ascierto PA, Flaherty KT, Grob JJ, Hasuchild A, et al. Survival of patients with advanced metastatic melanoma: the impact of novel therapies. Eur J Cancer. 2016;53:125–134. doi: 10.1016/j.ejca.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 2.Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurfenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;13(37):1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 3.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–39. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 4.Schadendorf D, Hodi S, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33(17):1889–1894. doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 6.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377(14):1345–1356. doi: 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony- stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190(3):355–366. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li B, VanRoey M, Wang C, Chen TH, Korman A, Jooss K. Anti-programmed death-1 synergies with granulocytes macrophage colony-stimulating factor-secreting tumor cell immunotherapy providing therapeutic benefit to mice with established tumors. Clin Cancer Res. 2009;15(5):1623–1634. doi: 10.1158/1078-0432.CCR-08-1825. [DOI] [PubMed] [Google Scholar]
- 10.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 11.Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016;6(8):827–837. doi: 10.1158/2159-8290.CD-15-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 13.de Rosa F, Ridolfi L, Fiammenghi L, Petrini M, Granato AM, Ancarani V, et al. Dendritic cell vaccination for metastatic melanoma: a 14 year monoinstitutional experience. Melanoma Res. 2017;27(4):351–357. doi: 10.1097/CMR.0000000000000356. [DOI] [PubMed] [Google Scholar]
- 14.Carreno BM, Magrini V, Becker-Hapak, Kaabinejadian S, Hundal J, Petti AA, et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803–808. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dillman RO. An update on the relevance of vaccine research for the treatment of metastatic melanoma. Melanoma Manage. 2017;4:203–15. [DOI] [PMC free article] [PubMed]
- 16.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 17.Türeci Ö, Vormehr M, Diken M, Kreiter S, Huber C, Sahin U. Mutanome directed cancer immunotherapy. Curr Opin Immunol. 2016;39:14–22. doi: 10.1016/j.coi.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 18.Dillman RO, Nayak SK, Beutel L. Establishing in vitro cultures of autologous tumor cells for use in active specific immunotherapy. J Immunother Emphasis Tumor Immunol. 1993;14(1):65–69. doi: 10.1097/00002371-199307000-00009. [DOI] [PubMed] [Google Scholar]
- 19.Dillman RO, Cornforth AN, Nistor G. Cancer stem cell antigen-based vaccines: the preferred strategy for active specific immunotherapy of metastatic melanoma? Expert Opin Biol Ther. 2013;13(5):643–656. doi: 10.1517/14712598.2013.759556. [DOI] [PubMed] [Google Scholar]
- 20.Dillman RO, DePriest C, DeLeon C, Barth NM, Schwartzberg LS, Beutel LD, et al. Patient-specific vaccines derived from autologous tumor cell lines as active specific immunotherapy: results of exploratory phase I/II trials in patients with metastatic melanoma. Cancer Biother Radiopharm. 2007;22(3):309–321. doi: 10.1089/cbr.2007.345. [DOI] [PubMed] [Google Scholar]
- 21.Dillman RO, Selvan SR, Schiltz PM, McClay EF, Barth NM, Depriest C, et al. Phase II trial of dendritic cells loaded with antigens from self-renewing, proliferating autologous tumor cells as patient-specific anti-tumor vaccines in patients with metastatic melanoma: final report. Cancer Biother Radiopharm. 2009;24(3):311–319. doi: 10.1089/cbr.2008.0599. [DOI] [PubMed] [Google Scholar]
- 22.Dillman RO, Cornforth AN, Depriest C, McClay EF, Amatruda TT, de Leon C, et al. Tumor stem cell antigens as consolidative active specific immunotherapy: a randomized phase II trial of dendritic cells versus tumor cells in patients with metastatic melanoma. J Immunother. 2012;35(8):641–649. doi: 10.1097/CJI.0b013e31826f79c8. [DOI] [PubMed] [Google Scholar]
- 23.Selvan SR, Carbonell DJ, Fowler AW, Beatty AR, Ravindranath MH, Dillman RO. Establishment of stable cell lines for personalized melanoma cell vaccine. Melanoma Res. 2010;20(4):280–292. doi: 10.1097/CMR.0b013e3283390696. [DOI] [PubMed] [Google Scholar]
- 24.Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 26.Dillman RO, Nanci AA, Williams ST, Kim RB, Hafer RL, Coleman CL, et al. Durable complete response of refractory, progressing metastatic melanoma after treatment with a patient-specific vaccine. Cancer Biother Radiopharm. 2010;25(5):553–557. doi: 10.1089/cbr.2010.0819. [DOI] [PubMed] [Google Scholar]
- 27.Dillman RO. Cancer vaccines: can they improve survival? Cancer Biother Radiopharm. 2015;30(4):147–151. doi: 10.1089/cbr.2014.1805. [DOI] [PubMed] [Google Scholar]
- 28.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 29.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flanigan RC, Polcari AJ, Shore ND, Price TH, Sims RB, Maher JC, et al. An analysis of leukapheresis and central venous catheter use in the randomized, placebo controlled, phase 3 IMPACT trial of sipuleucel-T for metastatic castrate resistant prostate cancer. J Urol. 2013;189(2):521–526. doi: 10.1016/j.juro.2012.09.029. [DOI] [PubMed] [Google Scholar]
- 31.Berd D, Maguire HC. Ur, McCue P, Mastrangelo MJ. Treatment of metastatic melanoma with an autologous tumor-cell vaccine: clinical and immunologic results in 64 patients. J Clin Oncol. 1990;8(11):1858–1867. doi: 10.1200/JCO.1990.8.11.1858. [DOI] [PubMed] [Google Scholar]
- 32.Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, et al. Vaccination of melanoma patients with peptide-or tumor lysate-pulsed cells. Nat Med. 1998;4(3):328–332. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 33.Dillman RO, Beutel LD, Barth NM, de Leon C, O’Connor AA, Depriest C, Nayak SK. Irradiated cells from autologous tumor cell lines as patient-specific vaccine therapy in 125 patients with metastatic cancer: induction of delayed-type hypersensitivity to autologous tumor is associated with improved survival. Cancer Biother Radiopharm. 2002;17(1):51–66. doi: 10.1089/10849780252824073. [DOI] [PubMed] [Google Scholar]
- 34.Dillman RO, Cornforth AN, Nestor GI. Dendritic cell vaccines for melanoma: past, present, and future. Melanoma Manag. 2016;3:267–283. doi: 10.2217/mmt-2016-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dillman RO. Is there a role for therapeutic cancer vaccines in the age of checkpoint inhibitors? Human Vaccine Immunother. 2017;13(3):528–532. doi: 10.1080/21645515.2016.1244149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dillman RO, Depriest C, McClure SE. High-dose IL-2 in metastatic melanoma: better survival in patients immunized with antigens from autologous tumor cell lines. Cancer Biother Radiopharm. 2014;29(2):53–57. doi: 10.1089/cbr.2013.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol. 2016;34:2619–2626. doi: 10.1200/JCO.2016.67.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vreeland TJ, Clifton GT, Herbert GS, Hale DF, Jackson DO, Berry JS, Peoples GE. Gaining ground on a cure through synergy: combining checkpoint inhibitors with cancer vaccines. Expert Rev Clin Immunol. 2016;12:1347–1357. doi: 10.1080/1744666X.2016.1202114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Success rate for cell cultures started from metastatic melanoma samples obtained during 2006–2011 (era of MACVAC trial). (DOCX 14 kb)
Table S2. Relationship between stage at time of tumor collection for cell line and disease status at the time of randomization. (DOCX 13 kb)
Figure S1. Consort diagram for MACVAC trial. (DOC 31 kb)
Table S3. Mutlivariate Cox regression analysis and proportional hazards model for independent variables. (DOCX 14 kb)
Table S4. Multiple univariate analyses performed when all patients were either deceased or had been followed for a minimum of three years. (DOCX 15 kb)
Table S5. Results of delayed type hypersensitivity (DTH) skin test to autologous irradiated tumor cells, Week-0 (baseline) and Week-4 following three weekly vaccine injections. (DOCX 14 kb)
Table S6. Panel of markers used to investigate the serum of patients treated in randomized phase II trial, grouped by associated biologic activity. (DOCX 15 kb)
Data Availability Statement
The clinical datasets used and/or analysed during the current study are available from the corresponding author on reasonable request. The cytokine data set is not available.