

Abstract
Background
Cerebral microhemorrhages (CMH) are commonly found in the aging brain. CMH are also the neuropathological substrate of cerebral microbleeds (CMB), demonstrated on brain MRI. Recent studies demonstrate the importance of systemic inflammation in CMH development, but the relationships among inflammation, aging, and CMH development are not well-defined. In the current study, we hypothesized that the pathogenesis of inflammation-induced CMH in mice differs by age.
Methods
We studied young (3 months, n = 20) and old (18 months, n = 25) C57BL/6 mice injected with low-dose lipopolysaccharide (LPS; 1 mg/kg, i.p.) or saline at 0, 6, and 24 h. Seven days after the first LPS/saline injection, brains were harvested, sectioned, and stained with hematoxylin and eosin (H&E) and Prussian blue (PB) to estimate acute/fresh and sub-acute CMH development, respectively. The relationships between microglial/macrophage activation (ionized calcium-binding adapter molecule-1), astrocyte activation (glial fibrillary acidic protein), blood-brain barrier (BBB) disruption (brain immunoglobulin G), aging, and CMH development were examined using immunohistochemistry.
Results
Aging alone did not increase spontaneous H&E-positive CMH development but significantly increased the number, size, and total area of LPS-induced H&E-positive CMH in mice. LPS- and saline-treated aged mice had significantly larger PB-positive CMH compared with young mice, but the total area of PB-positive CMH was increased only in LPS-treated aged mice. Aged mice had significantly increased microglial/macrophage activation, which correlated with H&E- and PB-positive CMH development. Aged mice treated with LPS had significantly increased astrocyte activation and BBB disruption compared with young LPS-treated mice.
Conclusions
Aging makes the brain more susceptible to inflammation-induced CMH in mice, and this increase in CMH with aging is associated with microglial/macrophage activation.
Keywords: Animal models, Cerebral microhemorrhage, Cerebral microbleeds, Inflammation, Hemosiderin, Aging
Background
Age is the most significant independent risk factor for cerebral microbleeds (CMB), which are identified by brain MRI and have as their pathologic substrate cerebral microhemorrhages (CMH) [1, 2]. In addition to aging, CMB are associated with hypertension, cerebral amyloid angiopathy (CAA), Alzheimer’s disease, cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), and chronic kidney disease, among other diseases [3–7]. Despite their high prevalence and clinical importance, the mechanisms underlying age-related CMB increase are not well-understood. One mechanism suggested by clinical findings is the activation of inflammatory cascades followed by blood-brain barrier (BBB) damage resulting in CMH development [8–11]. In fact, a pro-inflammatory systemic state is a common factor observed in normal aging and diseases with a high prevalence of CMB [12–15].
Recently, we showed that systemic inflammation induced by lipopolysaccharide (LPS) administration results in CMH development in mice and that CMH development was associated with levels of systemic and neuroinflammatory markers [2, 16]. However, the relationships among aging, inflammation, and CMH development are not well-defined [3]. The current study aimed to determine if aging exacerbates inflammation-induced CMH, and we hypothesized that the pathogenesis of inflammation-induced CMH in mice differs by age. We utilized the inflammation-induced CMH mouse model which results in MRI-demonstrable CMB [2]. LPS was used as the inflammatory stimulus to induce CMH development in young (3-month old) and aged (18-month old) mice. Age-related changes in both hematoxylin and eosin (H&E)-positive (acute) and Prussian blue-positive (sub-acute) CMH were examined in relationship to markers of neuroinflammation and BBB damage.
Methods
Mouse treatment
All animal procedures were approved by the University of California, Irvine, Institutional Animal Care and Use Committee, and followed the ARRIVE Guidelines for animal experiments reporting. Young (3-month old) and aged (18-month old) C57BL/6 male and female mice (National Institute of Aging, Bethesda, MD) were randomly assigned to two treatment groups: one treated with a 1 mg/kg dose of LPS derived from gram-negative bacterium Salmonella typhimurium (Sigma-Aldrich, St. Louis, MO) and the other treated with saline intraperitoneally (i.p.) at three times at 0, 6, and 24 h. Seven days after the first LPS/saline injection, mice were anesthetized with a lethal dose of Nembutal (150 mg/kg, i.p.), cardiac perfusions were performed using ice-cold PBS, and brains were processed for CMH detection as described in our previous work [16]. The sample size for each group was as follows: young-saline = 10, aged-saline = 10, young-LPS = 10, and aged-LPS = 15. The average weight of the mice was 28.1 ± 0.9 g at the beginning of the study and 27.1 ± 0.8 at the end of the study.
Microhemorrhage detection
Right hemispheres were fixed in 4% paraformaldehyde (Boston BioProducts, Ashland, MA) at 4 °C, examined for surface CMH, and sectioned into 40-μm coronal sections using a vibratome (Technical Products International, Inc., St. Louis, MO). Every fourth, fifth, sixth, and seventh section was collected. Every sixth section was stained with H&E by Research Services Core offered by the Department of Pathology and Laboratory Medicine at UCI Medical Center to detect fresh (acute) CMH. Every seventh section was stained with Prussian blue (PB) to detect hemosiderin (a marker of sub-acute CMH) [16]. Briefly, sections were stained using 5% potassium hexacyanoferrate trihydrate (Sigma, St. Louis, MO) and 5% hydrochloric acid (Sigma, St. Louis, MO) for 30 min, rinsed in water and counterstained with Nuclear Fast Red (Sigma, St. Louis, MO), dehydrated, and cover slipped. Remaining sections were used for immunohistochemistry. CMH were counted at a × 20 magnification by a blinded observer, and digitized images were used to determine CMH size (μm2) and positive area (expressed as a percent of total area analyzed), by an observer blinded to the experiment using RGB CMH analyzer program and NIH Image J software 1.62, respectively [16].
IgG, Iba-1, and GFAP immunohistochemistry
Parenchymal IgG (BBB damage marker), Iba-1 (microglial/macrophage marker), and GFAP (astrocyte marker) immunohistochemistry were performed [16]. Briefly, 40-μm sections were incubated in 0.5% hydrogen peroxide in 0.1 M PBS (pH 7.4) containing 0.3% Triton-X100 (phosphate-buffered saline with triton-X100 (PBST)) for 30 min at room temperature (RT), washed with PBST, and blocked with PBST containing 2% bovine serum albumin for 30 min at RT. Sections were then incubated overnight at 4 °C with a rabbit anti-mouse IgG antibody (1:200 dilution; Jackson ImmunoResearch, West Grove, PA), rabbit antibody against Iba-1 (1:200 dilution, Wako Chemicals USA, Richmond, VA), or rabbit antibody against GFAP (1:2000 dilution, Abcam, Cambridge, MA). After washing with PBST, sections were incubated at RT for 1 h with biotinylated anti-rabbit IgG (1:500 dilution; Jackson ImmunoResearch, West Grove, PA), followed by 1 h incubation at RT with ABC complex, and developed with 3,3′-diaminobenzidine (DAB) (Vector Laboratories, Burlingame, CA) as per manufacturer’s instructions. Sixteen images per brain section were acquired at × 20 magnification, and the total positive immunoreactive area (expressed as percent of total analyzed area) was quantified using NIH Image J software 1.62 by an observer blinded to the experimental groups [16].
Statistical analysis
Based on our previous work showing a mean number of CMH per brain section of 1.3 ± 0.8 in the LPS group and 0.03 ± 0.05 in the saline group, we estimated that at least 10 mice per group were needed to detect a significant difference between LPS and control mice at 80% power. One-way ANOVA with Bonferroni’s post hoc test for normal data and Kruskal-Wallis with Dunn’s multiple comparison test for non-normal data were used to compare more than two groups. Two-way ANOVA with Tukey’s post hoc test was used for sex comparisons. Pearson r correlation was used for correlation analysis. One-sample t test was used to compare group means with a hypothesized mean = 0 when the values in the control group were zero (e.g., surface microhemorrhages). A two-tailed p value of < 0.05 was considered statistically significant.
Results
Survival
All the saline-treated young and aged mice and LPS-treated young mice survived the duration of the study (n = 10 per group). Four aged mice treated with LPS died prematurely reducing the number included in the analysis to 15.
Cerebral microhemorrhages
Aging exacerbated the development of LPS-induced surface CMH, and the number of surface CMH in LPS-treated aged mice increased 23-fold compared with LPS-treated young mice (Fig. 1a, e). No surface CMH were observed in saline-treated mice, young or old.
Fig. 1.
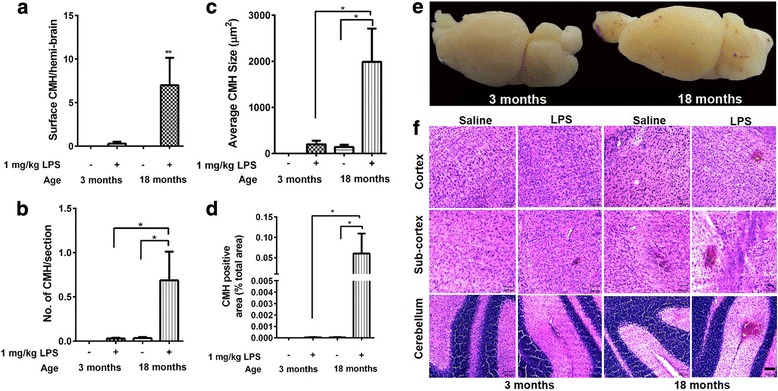
Surface and acute H&E-positive cerebral microhemorrhages (CMH). No development of surface CMH in young and aged saline-treated mice (n = 10 per group). Significant development of surface CMH in aged LPS-treated mice (n = 15) compared with young LPS-treated (n = 10) and aged saline-treated mice (n = 10) (a). No development of acute (H&E-positive) parenchymal CMH in saline-treated young mice. Significantly greater number, size, and total area of acute parenchymal CMH in LPS-treated aged mice compared with LPS-treated young mice and saline-treated aged mice (b–d). Brain images showing development of surface CMH in LPS-treated aged mice compared with LPS-treated young mice (e). Representative images showing acute CMH in different brain regions (f). Data are presented as mean ± SEM. One-sample t test for surface CMH, one-way ANOVA with Bonferroni’s post-test, or Kruskal-Wallis test with Dunn’s post-test. *p < 0.05 and **p < 0.01. Scale bar = 100 μm
In young mice, no spontaneous (in the absence of LPS) H&E-positive acute CMH were observed in saline-treated mice and a 1 mg/kg triple dose of LPS did not result in significant development of H&E-positive acute CMH (Fig. 1b–d, f). Aging did not cause a significant increase in spontaneous acute CMH development in saline-treated mice; however, similar to the effect on surface CMH development, aging significantly increased the development of H&E-positive acute parenchymal CMH in LPS-treated mice (Fig. 1b–d, f). All three parameters (number, size, and total area) of H&E-positive acute parenchymal CMH were significantly increased with age in LPS-treated mice (Fig. 1b–d). H&E-positive lesions were significantly higher in the cerebellum compared with the cortex and the sub-cortex (data not shown).
The number of PB-positive sub-acute parenchymal CMH was significantly higher in the LPS-treated young and LPS-treated aged mice compared with their respective saline controls (Fig. 2a–c). The number of PB-positive CMH lesions increased twofold in LPS-treated aged mice compared with LPS-treated young mice, although this increase did not reach statistical significance. Aging independently (with and without LPS) caused a significant increase in PB-positive lesion size; average size increased approximately threefold in both saline- and LPS-treated aged mice compared with their respective young controls (Fig. 2b). Total PB-positive lesion area, which is a function of both lesion number and size, was significantly increased with age in LPS-treated mice (Fig. 2c). The number of H&E-positive acute and PB-positive sub-acute lesions were significantly correlated (Pearson r = 0.92, p < 0.0001) in aged mice (Fig. 2d) but not in young mice (data not shown).
Fig. 2.
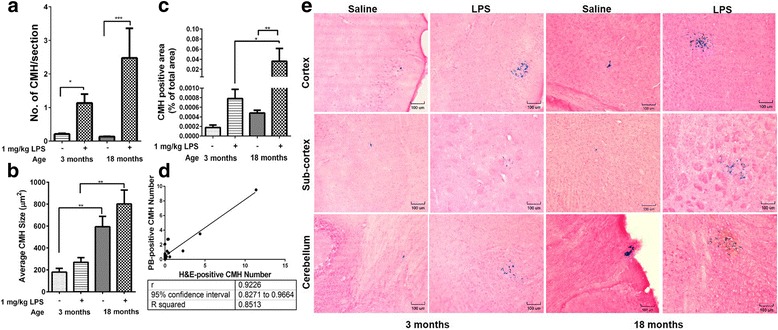
Sub-acute Prussian Blue (PB)-positive cerebral microhemorrhages (CMH). Significantly higher number of PB-positive lesions in LPS-treated young (n = 10) and LPS-treated aged mice (n = 15) compared with their respective saline controls (n = 10 per group) (a). Significant increase in PB-positive lesion size with aging (b). Significant increase in PB-positive total lesion area in LPS-treated aged mice compared with LPS-treated young mice and saline-treated aged mice (c). PB-positive and H&E-positive lesion number are significantly correlated in aged mice (d). Representative images showing PB-positive stains in different brain regions (e). Scale bar = 100 μm. Data are presented as mean ± SEM. One-way ANOVA with Bonferroni’s post-test or Kruskal-Wallis test with Dunn’s post-test; *p < 0.05, **p < 0.01, and ***p < 0.001
Relationship between CMH development, neuroinflammation, BBB damage, and aging
Aging was associated with a significant increase in brain Iba-1-positive immunoreactivity, independent of LPS treatment (Fig. 3a). Immunoreactive area of the astrocyte activation marker (GFAP) and BBB damage marker (brain IgG) on the other hand were significantly increased with age only in LPS-treated mice (Fig. 3b, c). Overall, both the H&E-positive and PB-positive lesion numbers were significantly correlated with Iba-1-positive immunoreactivity (Fig. 3d, e).
Fig. 3.
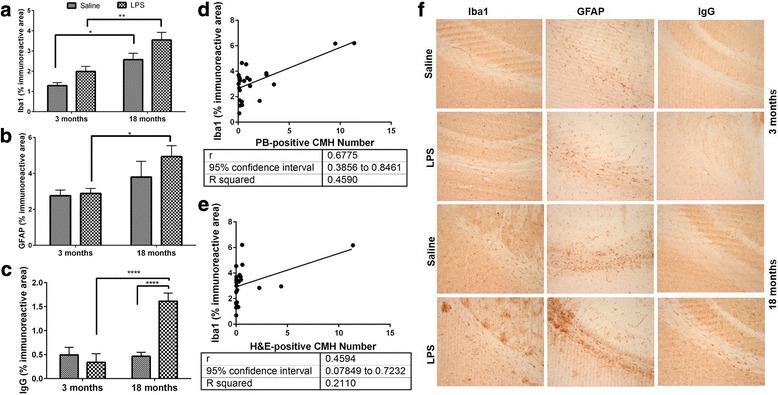
Neuroinflammation, BBB disruption, and CMH development. Significantly higher Iba-1-positive immunoreactivity with aging (a), and GFAP- (b) and IgG- (c) positive immunoreactivity in LPS-treated aged mice compared with LPS-treated young mice. PB- and H&E-positive CMH numbers are significantly correlated with Iba-1 positive immunoreactivity in aged mice (d, e). Representative images of Iba-1, GFAP, and IgG immunohistochemistry (f). Data are presented as mean ± SEM. One-way ANOVA with Bonferroni’s post-test or Kruskal-Wallis test with Dunn’s post-test and Pearson correlation; *p < 0.05, **p < 0.01, and ****p < 0.0001
Sex and CMH development
In an exploratory analysis of the relationship between sex and CMH development, the number (p < 0.01), size (p < 0.0001), and total area (p < 0.01) of H&E-positive acute parenchymal CMH were increased with age in LPS-treated male mice compared with LPS-treated female aged mice. No sex differences were observed in PB-positive CMH and in markers of neuroinflammation and BBB injury among young or aged mice (data not shown).
Discussion
In the current study, we investigated the effect of aging on inflammation-induced CMH development in a mouse model. We demonstrated negligible spontaneous acute CMH development in saline-treated young and aged mice. Exacerbation of acute CMH development was observed in LPS-treated aged mice and not in LPS-treated young mice. Since H&E staining enables the detection of fresh or acute microhemorrhages in the brain [2], the presence of fresh microhemorrhages in aged LPS-treated mice suggests that inflammation-induced CMH development is an ongoing process that lasts days after the last LPS injection in aged mice but not young mice (last LPS injection at day 2 and sacrifice at day 7 in the current study). PB on the other hand detects hemosiderin that remains at the bleeding site for a prolonged period of time, thus enabling the detection of cumulative CMH load at the time of sacrifice.
LPS treatment resulted in significant development of PB-positive sub-acute CMH in both young and aged mice. Consistent with other findings and our own work, we found low numbers of spontaneous PB-positive CMH in young and aged mice [2, 17]. Total PB-positive CMH load was significantly higher in LPS-treated aged mice and not in LPS-treated young mice. Spontaneous PB-positive CMH were significantly larger in aged mice, but this effect was independent of LPS treatment. The number of PB-positive lesions was highly associated with the number of H&E-positive lesions (r2 = 0.85) further suggesting that hemosiderin-positive sub-acute microhemorrhages detected by PB may help predict the susceptibility of the aging brain to develop acute or fresh microhemorrhages. Taken together, these results show that aging and inflammation together make the brain more vulnerable to CMH development and further corroborate previous studies that support the role of inflammation in the pathogenesis of CMH [8, 9, 18].
Though the exact molecular mechanisms underlying inflammation-induced CMH development are not completely understood, loss of microvascular integrity associated with inflammatory changes is associated with CMH development [19]. In the current study, we observed an increase in BBB disruption in LPS-treated aged mice consistent with our previous work [16]. Systemic administration of LPS is known to result in BBB injury and inflammation by altering tight junction protein expression, enlarging intercellular clefts, and increasing cytokine and chemokine release [20, 21]. These effects of LPS are mediated by its binding to toll-like receptor 4 (TLR4), which activates signaling pathways implicated in BBB disruption [22]. The contribution of TLR4 in intracerebral hemorrhage (ICH)-induced brain injury and inflammation has been documented [23], and a recent study showed that endothelial TLR4 activation by its canonical ligand LPS accelerates cerebral cavernous malformations (CCM) [24]. Given the role of TLR4 activation in ICH and CCM development and increased expression of TLR4 with the LPS dose used in the current study [25], TLR4 activation via LPS may mediate at least some of the effects observed in the current study. Overall, increased LPS-mediated BBB disruption in the current study further supports the role of the inflammation-induced loss of BBB integrity in CMH development [11]; the underlying role of TLR4 in CMH development needs further investigation. Loss of BBB integrity detected at the time of sacrifice in the aged mice in the current study may further explain the presence of acute CMH several days after the last LPS injection in aged mice and not LPS-treated young mice.
Studies show that hemosiderin deposits in the brain are often surrounded by macrophages which can further initiate an inflammatory response [26]. Consistent with this and our previous work, we found a significant increase in two different markers of neuroinflammation, Iba-1 (microglial/macrophage activation marker) and GFAP (astrocyte activation marker), in aged mice [2]. Iba-1 immunoreactivity was elevated with aging in the current study and was independent of LPS treatment. Further, we found that microglia/macrophage activation was significantly associated with CMH development in aged mice. We also found an increase in astrocyte activation in the aged mice treated with LPS, further indicating the increased susceptibility of the aging brain to neuroinflammation.
The role of microglia/macrophages in the pathogenesis of CMH is not well-defined. Apart from exhibiting a cytotoxic phenotype (M1 phenotype), microglia/macrophages can also mediate repair mechanisms in the brain depending on the brain microenvironment (M2 phenotype) [27, 28]. M2 macrophages are associated with hematoma resolution after intracerebral hemorrhage in a mouse model [29]. Whether the macrophage/microglial response to CMH observed in the current study is protective (clearing the CMH) or detrimental (triggering local inflammation) needs further investigation.
Sex differences in CMB development have been reported in clinical settings and CMB are more prevalent in males [30]. An exploratory analysis of our data showed that LPS-treated aged male mice were more susceptible to acute (H&E-positive) CMH development compared to LPS-treated aged female mice. We observed no sex differences in subacute (PB-positive) CMH development in aged mice. These sex differences in LPS-induced CMH development are consistent with prior observations on sex differences in response to LPS [31]. We speculate that inflammation-induced CMH development is an ongoing process in aged male mice at the time of sacrifice (7 days), compared with aged female mice. Future experimental studies are needed to fully define this process.
Conclusions
The current study highlights the relationships among aging, inflammation, and CMH development. Aging increases the susceptibility of the brain to inflammation-induced CMH development in mice, and this increase in CMH development is significantly associated with microglial/macrophage activation. The exact role of microglial activation in CMH pathogenesis needs further investigation.
Acknowledgements
We would like to thank Tuan Ngo, Gurjit Pannu, Jiwei Cheng, and Masood Akram for their assistance.
Funding
Funding for this study was provided by a grant from NINDS: RO1 NS20989 (MJF and DHC).
Availability of data and materials
Community-established norms of data sharing are fully respected.
Abbreviations
- BBB
Blood-brain barrier
- CAA
Cerebral amyloid angiopathy
- CADASIL
Cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- CCM
Cerebral cavernous malformations
- CMB
Cerebral microbleeds
- CMH
Cerebral microhemorrhages
- DAB
3,3′-Diaminobenzidine
- GFAP
Glial fibrillary acidic protein
- H&E
Hematoxylin and eosin
- Iba-1
Ionized calcium-binding adapter molecule-1
- ICH
Intracerebral hemorrhage
- IgG
Immunoglobulin G
- LPS
Lipopolysaccharide
- PB
Prussian blue
Authors’ contributions
RKS designed and performed experiments, analyzed data, prepared the figures, and wrote the paper. MMG performed experiments and collected data. VV and KK performed the Prussian blue and immunohistochemical staining. APH helped with data analysis. RK provided expertise with mouse brain histology. DHC participated in the conception of the study and was involved in revising the manuscript critically for important intellectual content. MJF conceived the study, designed and coordinated the experiments, and helped in the drafting and editing of the manuscript. All authors read and approved the final manuscript.
Ethics approval
All animal procedures were approved by UCI Institutional Animal Care and Use Committee and were carried out in compliance with University Laboratory Animal Resources regulations. This work does not involve any applicable consent to participate.
Consent for publication
Not applicable
Competing interests
Mark J. Fisher has received support from Boehringer-Ingelheim and Otsuka Pharmaceutical Company (research grants).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Rachita K. Sumbria, Email: rsumbria@kgi.edu
Mher Mahoney Grigoryan, Email: mgrigory@uci.edu.
Vitaly Vasilevko, Email: vvasilev@uci.edu.
Annlia Paganini-Hill, Email: apaganin@uci.edu.
Kelley Kilday, Email: kkilday@uci.edu.
Ronald Kim, Email: rckim@uci.edu.
David H. Cribbs, Email: cribbs@uci.edu
Mark J. Fisher, Phone: 714-456-6856, Email: mfisher@uci.edu
References
- 1.Greenberg SM, Vernooij MW, Cordonnier C, Viswanathan A, Al-Shahi Salman R, Warach S, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 2009;8(2):165–174. doi: 10.1016/S1474-4422(09)70013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sumbria RK, Grigoryan MM, Vasilevko V, Krasieva TB, Scadeng M, Dvornikova AK, et al. A murine model of inflammation-induced cerebral microbleeds. J Neuroinflammation. 2016;13(1):218. doi: 10.1186/s12974-016-0693-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ungvari Z, Tarantini S, Kirkpatrick AC, Csiszar A, Prodan CI. Cerebral microhemorrhages: mechanisms, consequences, and prevention. Am J Physiol Heart Circ Physiol. 2017;312(6):H1128–H1H43. doi: 10.1152/ajpheart.00780.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li L, Fisher M, Lau WL, Moradi H, Cheung A, Thai G, et al. Cerebral microbleeds and cognitive decline in a hemodialysis patient: case report and review of literature. Hemodial Int. 2015;19(3):E1–E7. doi: 10.1111/hdi.12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee JS, Ko K, Oh JH, Park JH, Lee HK, Floriolli D, et al. Cerebral microbleeds, hypertension, and intracerebral hemorrhage in cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Front Neurol. 2017;8:203. doi: 10.3389/fneur.2017.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cordonnier C, van der Flier WM. Brain microbleeds and Alzheimer’s disease: innocent observation or key player? Brain. 2011;134(Pt 2):335–344. doi: 10.1093/brain/awq321. [DOI] [PubMed] [Google Scholar]
- 7.Vernooij MW, van der Lugt A, Ikram MA, Wielopolski PA, Niessen WJ, Hofman A, et al. Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology. 2008;70(14):1208–14. doi: 10.1212/01.wnl.0000307750.41970.d9. [DOI] [PubMed] [Google Scholar]
- 8.Miwa K, Tanaka M, Okazaki S, Furukado S, Sakaguchi M, Kitagawa K. Relations of blood inflammatory marker levels with cerebral microbleeds. Stroke. 2011;42(11):3202–3206. doi: 10.1161/STROKEAHA.111.621193. [DOI] [PubMed] [Google Scholar]
- 9.Shoamanesh A, Preis SR, Beiser AS, Vasan RS, Benjamin EJ, Kase CS, et al. Inflammatory biomarkers, cerebral microbleeds, and small vessel disease: Framingham Heart Study. Neurology. 2015;84(8):825–832. doi: 10.1212/WNL.0000000000001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schreiber S, Bueche CZ, Garz C, Braun H. Blood brain barrier breakdown as the starting point of cerebral small vessel disease?—new insights from a rat model. Exp Transl Stroke Med. 2013;5(1):4. doi: 10.1186/2040-7378-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Nieuwenhuizen KM, Hendrikse J, Klijn CJM. New microbleed after blood-brain barrier leakage in intracerebral haemorrhage. BMJ Case Rep. 2017. 10.1136/bcr-2016-218794 [DOI] [PMC free article] [PubMed]
- 12.Dziedzic T. Systemic inflammation as a therapeutic target in acute ischemic stroke. Expert Rev Neurother. 2015;15(5):523–531. doi: 10.1586/14737175.2015.1035712. [DOI] [PubMed] [Google Scholar]
- 13.Rouhl RP, Damoiseaux JG, Lodder J, Theunissen RO, Knottnerus IL, Staals J, et al. Vascular inflammation in cerebral small vessel disease. Neurobiol Aging. 2012;33(8):1800–1806. doi: 10.1016/j.neurobiolaging.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 14.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4–S9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 15.Pauletto P, Rattazzi M. Inflammation and hypertension: the search for a link. Nephrol Dial Transplant. 2006;21(4):850–853. doi: 10.1093/ndt/gfl019. [DOI] [PubMed] [Google Scholar]
- 16.Sumbria RK, Vasilevko V, Grigoryan MM, Paganini-Hill A, Kim R, Cribbs DH, et al. Effects of phosphodiesterase 3A modulation on murine cerebral microhemorrhages. J Neuroinflammation. 2017;14(1):114. doi: 10.1186/s12974-017-0885-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cacciottolo M, Morgan TE, Finch CE. Rust on the brain from microbleeds and its relevance to Alzheimer studies: invited commentary on Cacciottolo Neurobiology of Aging, 2016. J Alzheimers Dis Parkinsonism. 2016;6(6):287. [DOI] [PMC free article] [PubMed]
- 18.Romero JR, Preis SR, Beiser AS, DeCarli C, Lee DY, Viswanathan A, et al. Lipoprotein phospholipase A2 and cerebral microbleeds in the Framingham Heart Study. Stroke. 2012;43(11):3091–3094. doi: 10.1161/STROKEAHA.112.656744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charidimou A, Werring DJ. A raging fire in acute lacunar stroke: inflammation, blood-brain barrier dysfunction and the origin of cerebral microbleeds. J Neurol Sci. 2014;340(1–2):1–2. doi: 10.1016/j.jns.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Banks WA, Gray AM, Erickson MA, Salameh TS, Damodarasamy M, Sheibani N, et al. Lipopolysaccharide-induced blood-brain barrier disruption: roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J Neuroinflammation. 2015;12:223. doi: 10.1186/s12974-015-0434-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosh A, Birngruber T, Sattler W, Kroath T, Ratzer M, Sinner F, et al. Assessment of blood-brain barrier function and the neuroinflammatory response in the rat brain by using cerebral open flow microperfusion (cOFM) PLoS One. 2014;9(5):e98143. doi: 10.1371/journal.pone.0098143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagyoszi P, Wilhelm I, Farkas AE, Fazakas C, Dung NT, Hasko J, et al. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem Int. 2010;57(5):556–564. doi: 10.1016/j.neuint.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Fang H, Wang PF, Zhou Y, Wang YC, Yang QW. Toll-like receptor 4 signaling in intracerebral hemorrhage-induced inflammation and injury. J Neuroinflammation. 2013;10:27. doi: 10.1186/1742-2094-10-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang AT, Choi JP, Kotzin JJ, Yang Y, Hong CC, Hobson N, et al. Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature. 2017;545(7654):305–310. doi: 10.1038/nature22075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung DW, Yoo KY, Hwang IK, Kim DW, Chung JY, Lee CH, et al. Systemic administration of lipopolysaccharide induces cyclooxygenase-2 immunoreactivity in endothelium and increases microglia in the mouse hippocampus. Cell Mol Neurobiol. 2010;30(4):531–541. doi: 10.1007/s10571-009-9477-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosidi NL, Zhou J, Pattanaik S, Wang P, Jin W, Brophy M, et al. Cortical microhemorrhages cause local inflammation but do not trigger widespread dendrite degeneration. PLoS One. 2011;6(10):e26612. doi: 10.1371/journal.pone.0026612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Min H, Jang YH, Cho IH, Yu SW, Lee SJ. Alternatively activated brain-infiltrating macrophages facilitate recovery from collagenase-induced intracerebral hemorrhage. Mol Brain. 2016;9:42. doi: 10.1186/s13041-016-0225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29(43):13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ni W, Mao S, Xi G, Keep RF, Hua Y. Role of erythrocyte CD47 in intracerebral hematoma clearance. Stroke. 2016;47(2):505–511. doi: 10.1161/STROKEAHA.115.010920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeerakathil T, Wolf PA, Beiser A, Hald JK, Au R, Kase CS, et al. Cerebral microbleeds: prevalence and associations with cardiovascular risk factors in the Framingham Study. Stroke. 2004;35(8):1831–1835. doi: 10.1161/01.STR.0000131809.35202.1b. [DOI] [PubMed] [Google Scholar]
- 31.Cai KC, van Mil S, Murray E, Mallet JF, Matar C, Ismail N. Age and sex differences in immune response following LPS treatment in mice. Brain Behav Immun. 2016;58:327–337. doi: 10.1016/j.bbi.2016.08.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Community-established norms of data sharing are fully respected.