

Abstract
The iron(II)- and 2-(oxo)glutarate-dependent (Fe/2OG) oxygenases catalyze an array of challenging transformations via a common iron(IV)-oxo (ferryl) intermediate, which in most cases abstracts hydrogen (H•) from an aliphatic carbon of the substrate. Although it has been shown that the relative disposition of the Fe–O and C–H bonds can control the rate of H• abstraction and fate of the resultant substrate radical, there remains a paucity of structural information on the actual ferryl states, owing to their high reactivity. We demonstrate here that the stable vanadyl ion [(VIV-oxo)2+] binds along with 2OG or its decarboxylation product, succinate, in the active site of two different Fe/2OG enzymes to faithfully mimic their transient ferryl states. Both ferryl and vanadyl complexes of the Fe/2OG halogenase, SyrB2, remain stably bound to its carrier protein substrate (L-aminoacyl-S-SyrB1), whereas the corresponding complexes harboring transition metals (Fe, Mn) in lower oxidation states dissociate. In the well-studied hydroxylase, taurine:2OG dioxygenase (TauD), the disposition of the substrate C–H bond relative to the vanadyl ion defined by pulse electron paramagnetic resonance methods is consistent with the crystal structure of the reactant complex and computational models of the ferryl state. Vanadyl substitution may thus afford access to structural details of the key ferryl intermediates in this important enzyme class.
Graphical Abstract
The vanadyl ion can be used as a mimic of the key ferryl intermediate in Fe- and 2-oxoglutarate-dependent enzymes. The stability and electronic structure of the vanadyl ion allow structural characterization techniques not permitted by the native intermediate, opening new avenues for understanding catalysis in this important class of enzymes.
Introduction
The iron- and 2-(oxo)glutarate-dependent (Fe/2OG) oxygenases catalyze a variety of chemical transformations at unactivated carbon centers, including hydroxylation, halogenation, desaturation, cyclization, and stereoinversion reactions.1,2 In humans, these reactions play essential roles in connective tissue biosynthesis,3 oxygen and body mass homeostasis,4–6 DNA repair,7–9 epigenetic inheritance, and control of transcription.10–12 In microbes and plants, these reactions are found in both primary metabolism and, notably, in biosynthetic pathways to specialized secondary metabolites that have been widely deployed as drugs.1,2 Remarkably, members of this large enzyme family are believed to initiate these sundry reactions via a common FeIV-oxo (ferryl) intermediate.2,13–18 The strategies used by individual enzymes to direct the ferryl intermediate to different outcomes remain poorly understood. A deeper understanding could facilitate deployment of their broad capabilities in biotechnological applications.2
The mechanistic logic employed by the enzyme class was first elucidated for members of the hydroxylase subclass.13 Dioxygen activation occurs at a mononuclear FeII cofactor, coordinated by a (His)2(Asp)1 “facial triad” of protein ligands,19,20 and is followed by oxidative decarboxylation of 2OG, producing succinate, CO2, and the key ferryl complex.13–15,21 The ferryl intermediate abstracts a hydrogen atom (H•) from a carbon of the substrate,22 producing a substrate radical and an FeIII–OH form of the cofactor. The substrate radical then couples with the hydroxo ligand (often termed “rebound”) to produce the alcohol product and return the cofactor to its FeII oxidation state.23
Fe/2OG halogenases employ a similar strategy but divert the intermediate produced by H• abstraction; the substrate radical instead couples with a halogen (Cl or Br) coordinated cis to the hydroxo.17,24 How these enzymes prevent the facile rebound step to enable cis-halogen transfer has been the subject of much computational and experimental analysis.2 For the halogenase SyrB2, correlations of substrate structure with rate constants for H• abstraction and halogenation/hydroxylation partition ratios suggested that positioning of the target C–H moiety away from the oxo/hydroxo ligand is crucial for selective halogenation.25 Subsequent direct measurements of substrate positioning by pulse electron paramagnetic resonance (EPR) methods on an iron-nitrosyl ({FeNO}7) surrogate for the catalytic intermediates confirmed that the target hydrogen is held farther from the iron center, with an Fe–H vector more nearly perpendicular to the Fe–N vector (which presumably mimics the Fe–O vector of the intermediate states), for a substrate that is primarily halogenated than for a substrate that is primarily hydroxylated.26 Presumably, this configuration poises the substrate radical closer to the halogen ligand to favor its transfer over the normally facile rebound step. Computational analyses suggested that this perpendicular approach engages a π-type frontier molecular orbital (FMO) of the ferryl complex in the H•-abstraction step and promotes halogen transfer in the resultant cis-halo-hydroxo-FeIII/substrate-radical state.27,28 Importantly, these studies suggested that the perpendicular configuration is achieved not by a relocation of the substrate from its usual position above the vacant axial site of the O2-reactive square-pyramidal FeII cofactor form (which is trans to the C-terminal His ligand and has been called the “inline” position; Figure 1A) but rather by relocation of the ferryl oxo group into an adjacent site (“offline”, cis to the C-terminal histidine; Figure 1C).1,27,29 The first crystal structure of an Fe/2OG halogenase with its substrate bound, that of the WelO5•FeII•2OG•12-epi-fischerindole U complex, confirmed substrate binding in the usual (inline) configuration (Figure 1A), lending credence to the hypothesis that the oxo must shift offline in the ferryl state to promote halogenation (Figure 1C).30 It has been suggested that deployment of offline ferryl intermediates could be a general strategy to suppress hydroxylation in other (perhaps all) non-hydroxylation outcomes mediated by Fe/2OG enzymes,31–34 but appropriate experimental methods to test this hypothesis have yet to emerge.
Figure 1.
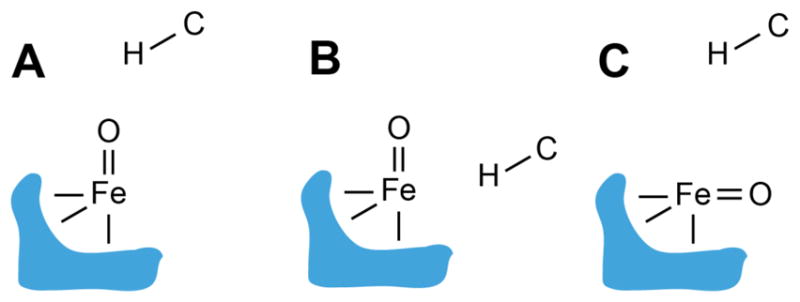
Possible relative dispositions of substrate and ferryl intermediate in Fe/2OG oxygenase reactions. Bonds to the facial triad protein ligands are depicted in cartoon form. (A) Canonical configuration for hydroxylation, with substrate above the inline ferryl oxo group; (B) hypothetical configuration with offline substrate and inline oxo (C) configuration postulated for the halogenases, with inline substrate and offline oxo.
Structural characterization of high-valent iron enzyme intermediates by X-ray crystallography has been impeded by their short lifetimes, which typically do not exceed a few seconds. However, the ferryl complexes of several Fe/2OG enzymes have been trapped by rapid-mixing/freeze-quenching techniques and characterized by spectroscopic methods. Limitations of this approach are that the resultant structural information is limited to the first coordination sphere of the cofactor and interpretation often relies on comparison of measured parameters to those predicted semi-empirically for a set of computationally-derived structural models.
Pulse EPR methods can provide a priori local structural information beyond the first coordination sphere via the distance and orientation dependencies of electron-nuclear hyperfine interactions (e.g., with 1H, 2H, 13C, etc.), but the application of such methods to ferryl complexes in Fe/2OG enzymes has been precluded by the integer-electron-spin (S = 2) ground states of the intermediates.
As introduced above, reaction of the FeII-containing enzyme-substrate complexes with nitric oxide (NO•) has been used to generate EPR-active {FeNO}7 complexes with S = 3/2 ground states, and measurement of hyperfine (HF) couplings between the metal center and deuteria incorporated at strategic positions in the substrates has afforded details of substrate positioning.26,35–37 However, these {FeNO}7 complexes mimic the putative {FeOO}8 adduct which precedes ferryl formation, and it seems unlikely that oxidative addition of NO would accurately reproduce local or global structural changes accompanying ferryl formation, for several reasons. Firstly, NO is a diatomic ligand with greater steric bulk than the oxo group of the ferryl. Secondly, the iron is oxidized by NO formally to the +III oxidation state rather than the +IV state generated by O2 activation. Finally, reaction with NO fails to bring about decarboxylation of 2OG, leaving the co-substrate coordinated in the {FeNO7} complex, whereas the ferryl complex formed by O2 activation has succinate at the corresponding site. These issues cast doubt on the extent to which structural details of the {FeNO}7 complexes can explain or predict the reactivity of the corresponding ferryl intermediates.
Seeking a stable, more accurate mimic of the reactive ferryl state for use in EPR and crystallographic structural studies, we considered the vanadyl ion [(VIVO)2+] as a candidate. Stable for many hours in aqueous solution, vanadyl has previously been used to probe divalent metal (e.g. Zn2+, Mg2+, Ca2+) sites in proteins and would thus be expected to bind in the cofactor sites of Fe/2OG enzymes.38 Structurally, the vanadyl ion has striking similarities to the ferryl moiety. Both have +2 net charges resulting from bonding between the tetravalent metal cation and divalent oxide anion.39 Moreover, the metal-oxo bonds have similar lengths, ranging from 1.58 to 1.63 Å for octahedral vanadyl complexes with oxygen and nitrogen coordination40–45 and from 1.61 to 1.64 Å for the ferryl intermediates in the Fe/2OG hydroxylase, TauD, and halogenases, CytC3 and SyrB2.17,46,47 Importantly, the d1 configuration of VIV gives the vanadyl ion an S = 1/2 ground state, which makes it EPR active and amenable to accurate determination of distances between vanadium and nearby magnetic nuclei by EPR methods.
Results and Discussion
To evaluate the potential of vanadyl to serve as a ferryl mimic, we first tested for its capacity to bind in the cofactor site of TauD, the most extensively studied Fe/2OG oxygenase. A sample prepared by adding vanadyl sulfate to apo TauD, along with succinate and taurine, (Figure 2A) exhibited a strong g ~ 2 X-band continuous-wave (CW) EPR signal with splitting characteristic of hyperfine coupling to the 51V nucleus (~100% natural abundance, I = 7/2). This signal was greatly diminished in intensity in a sample from which the enzyme was omitted, as vanadyl in solution at biological pH exists in a polymeric, EPR-silent form;48 the observed signal therefore arises from a TauD•(VIVO) complex (Figure S1). Global simulation of the X- and Q-band EPR spectra yielded the parameters g = [1.944, 1.979, 1.981] ± 0.001 and AV = [519, 185, 192] ± 3 MHz (Figure S2). The A//component of the vanadium hyperfine tensor can be related to the identity of the ligands in the equatorial plane of the vanadyl (i.e., cis to the -oxo); the observed value of 519 MHz (173 × 10−4 cm−1) is very close to the expected value of 520 MHz (174 × 10−4 cm−1) for three carboxylate oxygen and one histidine nitrogen (with the plane of the histidine ring perpendicular to the V-oxo bond) ligands.38 Although asymmetry and the presence of potentially-bidentate carboxylate ligands may produce deviations from predicted values and thus this agreement is not definitive, the observed value of A//is certainly compatible with the proposed geometry.
Figure 2.
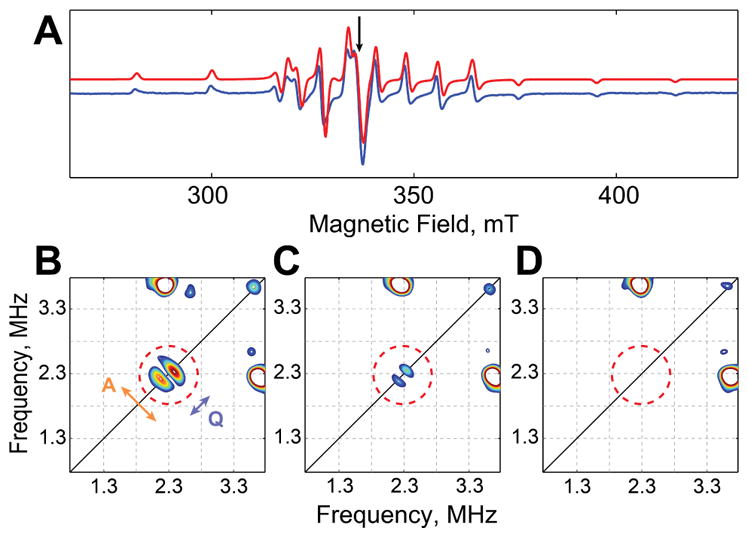
Characterization of the TauD•(VIVO)•taurine•succinate complex by EPR and HYSCORE spectroscopy. (A) Continuous-wave, X-band spectrum (blue) and simulation (red, shifted upwards for clarity). (B–D) HYSCORE spectra collected at the maximum EPR absorbance (indicated by the arrow at 344 mT in A) on samples containing d4-taurine (B), d4-succinate (C), and substrate and co-product of natural isotopic abundance (D). Splittings due to hyperfine coupling (A) and quadrupole coupling (Q) are indicated by arrows in B. The CW spectrum was collected at 80 K, with microwave frequency 9.478 GHz and power 200 μW. HYSCORE spectra were acquired at 35 K with microwave frequency 9.685 GHz.
Further evidence for binding of the vanadyl in the cofactor site was provided by deuterium hyperfine sublevel correlation (2H-HYSCORE) spectra (Figure 2, B–D), which report on interactions between the electron spin and nearby deuterium nuclei. In 2H-HYSCORE spectra, resonances are centered at the Larmor frequency of the deuterium. Hyperfine coupling is manifest by splitting along the anti-diagonal, whereas nuclear quadrupolar interactions cause further splitting along the diagonal (Figure 2B). For samples of the TauD•(VIVO)•succinate•taurine complex prepared with 1,1,2,2-[2H4]-taurine (d4-taurine), a feature centered at ν L = 2.25 MHz was readily observed in the HYSCORE spectrum (Figure 2B) collected at the point of maximum EPR absorption (344 mT). The appearance of such a feature indicates that vanadyl does, as expected, bind in the active site, close to (< 5 Å from) the taurine-binding site. In spectra of samples prepared using 2,2,3,3-[2H4]-succinate, a weaker feature was seen, showing proximity of the vanadyl to the co-product bound in the active site (Figure 2C). No features attributable to deuterium were observed from a sample prepared with unlabeled taurine and succinate (Figure 2D).
To establish that vanadyl substitution can provide a spin probe for structural analysis, we defined the position of taurine relative to the VIVO unit by pulse EPR, as previously done for the {Fe-NO}7 complexes.26,35,36 Field-dependent 2H-HYSCORE spectra were collected for a sample of the TauD•(VIVO)•succinate•d4-taurine complex (Figure 3). The resulting patterns were simulated with a single deuterium hyperfine coupling of [0.26, 0.26, −0.68] ± 0.05 MHz with Euler angles of [0, 35, 15] ± 10° relative to the g tensor (Figure 3); the deuterium nuclear quadrupole interaction was simulated as [−0.06, −0.06, 0.12] ± 0.03 MHz with Euler angles [0, −50, 15] ± 10°. That a single coupling can account for the observed signals strongly suggests that only the most proximal deuteron on C1 (inferred to be the pro-R hydron from crystal structures of TauD)49,50 meaningfully contributes to these spectra. This coupling can be considered in terms of an anisotropic component, T, and an isotropic component, Aiso: A2H = [1, 1, −2]T + Aiso (i.e., an axial tensor of magnitude T = 0.30 MHz and an isotropic component of magnitude Aiso = −0.04 MHz). The anisotropic component can be related to the V-2H distance according to the point-dipole approximation, which yields 3.4 ± 0.2 Å. Moreover, the second Euler angle (35°) corresponds to the O≡V–2H angle, as the unique axis of the (approximately axial) g tensor is collinear with the V≡O bond. Although the observation of isotropic hyperfine coupling is usually indicative of through-bond delocalization of spin density onto the nucleus of interest, through-space isotropic coupling has been observed for atoms in tight van der Waals contact, consistent with the small isotropic component observed here.51–53
Figure 3.
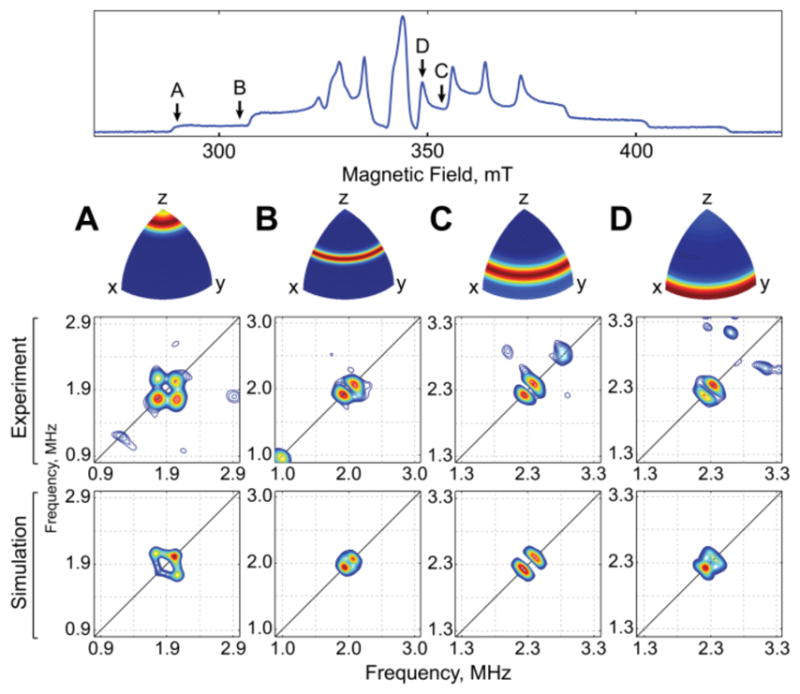
Field-dependent 2H-HYSCORE spectra of the TauD•(VIVO)•taurine•succinate complex in the presence of d4-taurine, recorded at 291.0 mT (A), 304.0 mT (B), 353.0 mT (C), and 348.7 mT (D). Magnetic field positions are indicated by black arrows on the one-dimensional EPR spectrum (top, presented in absorption mode); resulting orientation selectivity patterns are color coded using an “RGB” scheme (red - fully excited, blue - not excited). HYSCORE spectra were collected at 35 K with microwave frequency 9.686 GHz.
Substrate positioning in TauD was previously interrogated for the aforementioned {FeNO}7 complex.36,37 For comparison to the metrics provided by the VIVO probe, we reexamined the iron-nitrosyl complex in the presence of d4-taurine and 2OG by field-dependent 2H-HYSCORE (Figure S3). The data were simulated with a single deuterium hyperfine coupling of [0.26, 0.44, −0.70] ± 0.05 MHz with Euler angles of [0, 30, 93] ± 10° and a deuterium nuclear quadrupole interaction of [−0.04, −0.04, 0.08] ± 0.03 MHz with Euler angles [0, 40, 25] ± 10°. The anisotropic magnitude (T = 0.35 MHz) and second Euler angle (30°) are quite similar to those obtained in the previous study of the {Fe-NO}7 complex and above on the vanadyl complex.36,37
To assess the coordination geometry of the vanadyl ion in TauD and the extent to which it mimics the proposed structure of the ferryl intermediate, we interrogated the complex by x-ray absorption methods. The vanadium K-edge lies at ~ 5480 eV (Figure S4), consistent with the +IV oxidation state.54 The spectrum (Figure S5) also has an intense pre-edge feature with an intensity (99 ± 3 units) that is nearly identical to that of the six-coordinate VO(H2O)52+ ion (106 ± 4 units; Figure S5). This observed intensity is appreciably lower than that seen for formally square pyramidal vanadyl complexes, likely owing to the presence of a weakly coordinating axial ligand in TauD that partially restores centrosymmetry, and hence the observed pre-edge is most consistent with an octahedral geometry distorted by the presence of an oxo ligand.54 Such a geometry could arise from vanadium coordination by the facial triad and bidentate coordination by succinate, a first sphere that would be identical to that favored in previous spectroscopic and computational studies of the ferryl complex.55 Fits to the extended x-ray absorption fine structure (EXAFS) data revealed a coordination sphere composed of two shells: one at 1.60 Å, corresponding to the oxo ligand, and another at 2.05 Å (Figure S6 and Table S1–S3), corresponding to the protein and succinate ligands. The fitting analysis is consistent with previous structural characterization of vanadyl complexes40–45 and notably similar to the first-sphere metrics obtained by EXAFS on the ferryl intermediate in TauD (shells at 1.62 and 2.05 Å).46
From the structural metrics extracted from the HYSCORE and EXAFS analysis of the TauD vanadyl complex, a geometric model for the key atoms in the H• abstraction step effected by the ferryl complex was constructed (Scheme 1). The model has a V≡O–2H angle of (120 ± 10)° and an O–2H distance of (2.3 ± 0.3) Å. Extrapolated to the ferryl complex, this geometry would be consistent with the expectation of efficient σ-channel H• abstraction by the ferryl state in this hydroxylation reaction and is compatible with previous x-ray crystal structures.49,50
Scheme 1.
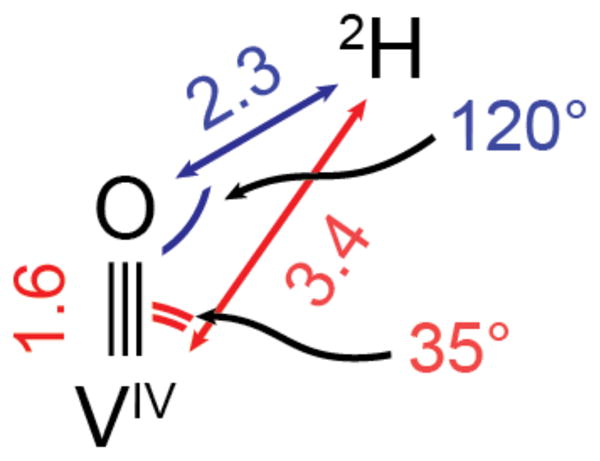
Disposition of the taurine pro-R deuterium nucleus relative to vanadyl in the TauD•(VIVO)•taurine•succinate complex. Experimental measurements are denoted in red, and extrapolated values in blue; distances given are in units of Ångstrom.
The above experiments indicate that the complex of TauD with vanadyl, succinate, and taurine has a structure very similar to that proposed for the ferryl intermediate. Despite this agreement, we sought more compelling evidence of faithful mimicry in a biological context by testing whether binding of vanadyl can replicate a unique feature of the ferryl complex that was found for SyrB2. The substrate of SyrB2 is the carrier protein, SyrB1, with L-threonine appended via thioester linkage to its phosphopantetheine cofactor (L-threoninyl-S-SyrB1); it undergoes chlorination of the L-threonine methyl group. The chloroferryl complex in SyrB2 can be markedly stabilized (t1/2 ~ 110 min) by use of the alternative substrate, L-cyclopropylglycinyl-S-SyrB1, owing to the greater C–H bond strength of the cyclopropyl side chain.17 Neither the apo (metal-free) nor the MnII form (in the presence or absence of 2OG/succinate) of SyrB2 makes a stable complex with L-threoninyl-S-SyrB1; consequently, the two proteins are almost completely resolved by chromatography on an appropriate size-exclusion matrix at 4 °C (Figures 4A, blue and black traces, and S7). By contrast, in the cyclopropyl-stabilized ferryl intermediate state, the two proteins remain associated during chromatography under the same conditions, as evidenced by the shift to shorter elution times (Figure 4A, red trace). The elution time of the new peak is consistent with formation of a 1:1 complex of SyrB1 and SyrB2. In this experiment, chromatography was initiated as soon as possible after formation of the ferryl complex by O2 exposure, and the elution required ~ 30 min, a time scale on which the ferryl complex is stable.17 By contrast, increasing delays between formation of the stabilized ferryl complex and the chromatography step led to decreasing intensity in the ~ 13-min peak of the complex and regain of intensity in the peak of free SyrB2 (Figure 4, orange and gold traces). The time scale for this shift matched the previously published kinetics of decay of the ferryl complex formed in the presence of the non-native substrate. Incomplete formation of the complex (indicated by the presence of the SyrB2 peak at 16 mL) is likely due the combination of decay of the ferryl complex (predicted to be ~20% upon elution from the column) and an inactive protein fraction frequently observed in this enzyme class.18,23 The data indicate that conformational changes coupled to ferryl formation strengthen the protein-protein interactions between SyrB1 and SyrB2 and prevent the release of the substrate during the lifetime of the reactive intermediate, presumably as an adaptation to prevent enzyme auto-oxidation. A similar observation was reported for the class I-a ribonucleotide reductase from E. coli, in which radical translocation from the cofactor (β) subunit into the catalytic (α) subunit was seen to increase their mutual affinity by 25-fold.56
Figure 4.
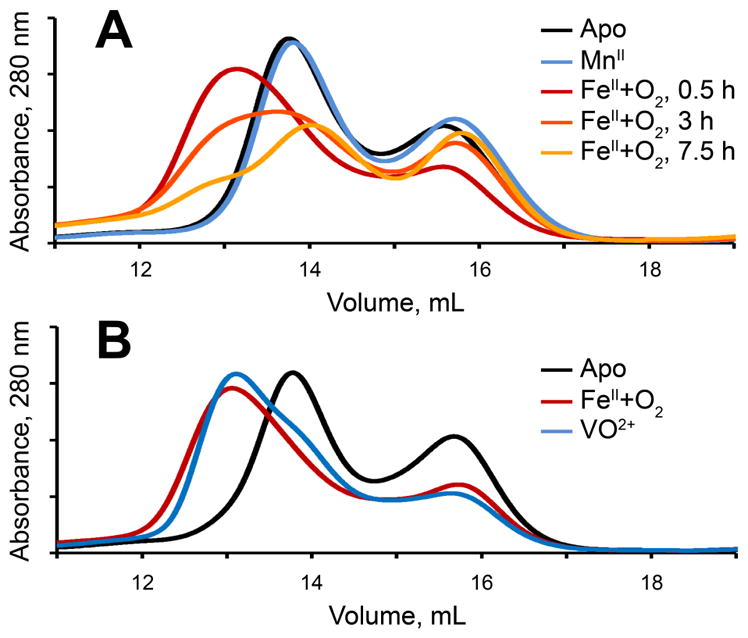
Size-exclusion chromatography of L-cyclopropylglycinyl-S-SyrB1 and SyrB2. (A) SyrB1 and Fe(II)-SyrB2 exposed to oxygen for 0 h (red), 3 h (orange), and 7.5 h (gold) prior to loading. Apo-SyrB2 (black) and Mn(II)-SyrB2 (blue) are depicted for comparison. (B) SyrB1 and SyrB2-VO (blue), Fe(II)-SyrB2 (red), and apo-SyrB2 (black). SyrB2, FeII or VO2+, Cl−, 2OG, and L-cyclopropylglycinyl-S-SyrB1, mixed to final concentrations of 150 μM, 140 μM, 2.7 mM, 0.8 mM, and 150 μM, respectively, were applied to a GE Healthcare Superdex 200 10/300 GL column and eluted at 0.7 mL/min with 50 mM sodium HEPES pH 7.6, 150 mM NaCl.
Remarkably, the presence of vanadyl (along with 2OG) in the active site of SyrB2 reproduces the stability of the enzyme’s complex with its carrier protein substrate (Figure 4B, compare blue and red traces). Even the extent of incomplete formation of the complex is reproduced. Surprisingly, little complex formation is observed in the presence of vanadyl and succinate. The basis for this requirement for the co-substrate rather than co-product is currently unclear and warrants further structural studies. Nevertheless, it is clear that the surrounding protein framework of SyrB2, which has adapted to interact strongly with SyrB1 only in the ferryl state, responds to the presence of vanadyl in the same manner. This profound similarity in a global conformational effect strongly suggests that vanadyl is a faithful mimic of the ferryl state, giving credence to the utility of the vanadyl ion as a structural probe.
The data demonstrate the suitability of the vanadyl ion as an analogue of the ferryl intermediate in Fe/2OG oxygenases and its utility as a structural probe. It binds along with succinate and taurine in the TauD active site, where it adopts a distorted octahedral geometry similar to that previously proposed for the ferryl complex55 and exhibits a metal-oxo bond length (1.60 Å) nearly identical to that determined for the key intermediate state (1.62 Å).46 Remarkably, in addition to accurately reproducing these cofactor-proximal metrics in TauD, the vanadyl ion also reproduces the global conformational effect (manifested by stabilization of its enzyme-substrate complex) that accompanies ferryl formation in SyrB2. Although its utility as an EPR probe to extract precise substrate-positioning information is partly precedented by the prior studies on the {Fe-NO}7 complexes in TauD and other systems,26,35–37 the expectation that it should more accurately mimic the geometries of the ferryl complexes makes it potentially superior for rationalizing the reactivities of the key intermediates. Moreover, the stability of the vanadyl complexes under biological conditions hints at the exciting prospect of obtaining X-ray crystal structures of the ferryl-mimicking states to map local and global protein motions that accompany ferryl formation and control its reactivity.57 Thus, the new structural probe may help unravel incompletely understood mechanisms of reaction control in Fe/2OG oxygenases, thereby enabling their rational reprogramming for biotechnological applications.
Materials and Methods
Materials
Commercially available materials were used without further purification. 1,1,2,2-[2H4]-taurine was purchased from Cambridge Isotope Laboratories. 2,2,3,3-[2H4]-succinic acid was purchased from Sigma.
Preparation of the TauD•(VIVO)•taurine•succinate complex
TauD was purified as previously described.14 Vanadyl sulfate was dissolved in 2.5 mM sulfuric acid at a concentration of 50 mM. TauD, vanadyl, succinate and taurine were mixed to final concentrations of 1.5, 1.0, 5.0, and 5.0 mM, respectively. The solution was then transferred to an EPR tube or x-ray absorption cell and frozen in liquid N2.
Preparation of the TauD•{FeNO}7•taurine•2OG complex
The TauD {Fe-NO}7 complex was prepared in the manner previously described for SyrB2.26 TauD, iron, 2-oxoglutarate and taurine were mixed to concentrations of 1.13, 1.04, 5.67, and 5.67 mM, respectively; the resulting solution was concentrated in a centrifugal filter by approximately four-fold, transferred to an EPR tube, and frozen in liquid N2.
XAS Data Collection and Processing
X-ray absorption spectra were collected on beamline 7-3 at the Stanford Synchrotron Radiation Lightsource under ring conditions of 3 GeV and 500 mA. A Si(220) monochromator (φ=90°) was used for energy selection of the incident beam; harmonic rejection was achieved using a Rh-coated mirror (9 keV) and by detuning the monochromator by 85%. The energy of the incident beam was calibrated by using a vanadium foil upstream of the sample (5465.2 eV). Scans were carried out over the energy range of 5235–6250 eV for a total exposure of 800 seconds. The beam intensity was measured using N2-filled ion chambers before the sample. The sample was placed at 45° relative to the incident beam and the Kα fluorescence was monitored using a 30 element germanium detector. Sample temperature was maintained at 10 K in a liquid helium flow cryostat.
Data processing was performed with the EXAFSPAK software package.58 Three-segment splines (of orders 2, 3, and 3) were removed from the EXAFS using PySpline59 and the EXAFS data were then fit using OPT. Appropriate structural models were adapted from Sinnecker et al.55 (replacing Fe with V), and scattering paths for EXAFS fits were generated using FEFF 9.0 (additional details can be found in the Supporting Information).60 During EXAFS fitting, the distances, Debye-Waller factors, and E0 parameter were all allowed to float while coordination numbers were systematically varied; for all fits the passive electron reduction factor (S02) was fixed at 1. For XANES analysis, the pre-edge region was fit using BlueprintXAS61 and the edge jump obtained from the fits was normalized to 1; reported areas and intensity-weighted average energies are the average of at least 21 physically reasonable fits.
EPR Measurements
X-band continuous wave (CW) measurements were performed on a Bruker ESP 300 spectrometer with an ER 041 MR microwave bridge and an ER 4116DM resonator. All other EPR measurements were performed on a Bruker Elexsys E580 X-band spectrometer equipped with a SuperX-FT microwave bridge. For pulse EPR measurements at X-band, a Bruker ER 4118X-MS5 resonator was used in concert with an Oxford CF935 helium flow cryostat. Microwave pulses generated by the microwave bridge were amplified by a 1 kW traveling wave tube (TWT) amplifier (Applied Systems Engineering, model 117x). Pulse EPR spectra at Q-band frequencies were acquired using a home-built intermediate-frequency extension of the SuperX-FT X-band bridge that has a Millitech 5W pulse power amplifier. All experiments were conducted on a home-built TE011 resonator utilizing the open resonator concept developed by Annino et al.62 and mechanical construction of the probehead similar to that presented by Reijerse et al.63 This setup allows t(π/2) = 12–16 ns at maximum input power with spectrometer dead time (including the resonator ring time) of 100–120 ns. Data acquisition and control of experimental parameters were performed by using Bruker XEPR software.
EPR Analysis
Data processing and spectral simulations were performed using Kazan viewer, a home-written suite of utilities in MATLAB.64 One-dimensional EPR simulations were performed using the “pepper” utility from the EasySpin software package.65 HYSCORE data were analyzed by simultaneous frequency domain simulation of all field-dependent spectra until a satisfactory solution was achieved. The rhombicity of the hyperfine coupling, ε, was defined as
| (1) |
Euler angles (y-convention) are reported with respect to the 51V hyperfine coupling tensor, which is collinear with the g matrix in our simulations.
Geometric information was calculated using a point-dipole model, according to the formula
| (2) |
Deviations from this idealized model could result from significant delocalization of spin density onto the vanadyl oxo ligand. However, available experimental and theoretical results suggest this delocalization is likely to be 5–20%.67,67 At these levels of spin density on oxygen, deviations from the point dipole model are calculated using a geometric model to be within the stated experimental error.
Size Exclusion Chromatography
SyrB1 and SyrB2 were purified and SyrB1 was charged with L-cyclopropylglycine as previously described.17 Oxygen was reomoved from L-cyclopropylglycinyl-S-SyrB1 and SyrB2 as previously described. SyrB2; Fe2+, Mn2+, or VO2+; Cl−; 2OG; and L-cyclopropylglycinyl-S-SyrB1 were mixed in an MBraun (Stratham, NH) anoxic chamber to final concentrations of 0.54 mM, 0.50 mM, 10 mM, 2.6 mM, and 0.56 mM, respectively, with a final volume of 0.150 mL. After removal from the anoxic chamber, samples were mixed with 0.40 mL air-saturated buffer and then centrifuged briefly. Either immediately or after the specified delay, samples were applied to a GE Healthcare Superdex 200 10/300 GL column at 4 °C using a 100 μL sample loop and eluted at 0.7 mL/min with 1.2 column volumes of 50 mM sodium HEPES pH 7.6, 150 mM NaCl.
Supplementary Material
Acknowledgments
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE1255832. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. C.J.P. thanks the NIH for a National Research Service Award (GM113389-01). This work was supported by the National Institutes of Health (GM118812 to J.M.B. and C.K. and GM069657 to C.K. and J.M.B.). Portions of this work were conducted at the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Footnotes
Multifrequency EPR analysis; 2H-HYSCORE analysis of TauD-{FeNO}7; XAS edges, pre-edges, and corresponding fits; EXAFS data and fits; tables of EXAFS fits. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hausinger RP. Fe(II)/α-Ketoglutarate-Dependent Hydroxylases and Related Enzymes. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 2.Bollinger JM, Jr, Chang W-c, Matthews ML, Martinie RJ, Boal AK, Krebs C. 2-Oxoglutarate-Dependent Oxygenases. In: Hausinger RP, Schofield CJ, editors. RSC Metallobiology. Royal Society of Chemistry; 2015. [Google Scholar]
- 3.Kivirikko KI, Myllyharju J. Prolyl 4-hydroxylases and their protein disulfide isomerase subunit. Matrix Biol. 1998;16:357–368. doi: 10.1016/s0945-053x(98)90009-9. [DOI] [PubMed] [Google Scholar]
- 4.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Jr, WGK HIFα Targeted for VHL-Mediated Destruction by Proline Hydroxylation: Implications for O2 Sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 5.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-α to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 6.Gerken T, Girard CA, Tung YCL, Webby CJ, Saudek V, Hewitson KS, Yeo GSH, McDonough MA, Cunliffe S, McNeill LA, Galvanovskis J, Rorsman P, Robins P, Prieur X, Coll AP, Ma M, Jovanovic Z, Farooqi IS, Sedgwick B, Barroso I, Lindahl T, Ponting CP, Ashcroft FM, O’Rahilly S, Schofield CJ. The Obesity-Associated FTO Gene Encodes a 2-Oxoglutarate-Dependent Nucleic Acid Demethylase. Science. 2007;318:1469–1472. doi: 10.1126/science.1151710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature. 2002;419:174–178. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- 8.Falnes PØ, Johansen RF, Seeberg E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature. 2002;419:178–182. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- 9.Yi C, Jia G, Hou G, Dai Q, Zhang W, Zheng G, Jian X, Yang CG, Cui Q, He C. Iron-catalysed oxidation intermediates captured in a DNA repair dioxygenase. Nature. 2010;468:330–333. doi: 10.1038/nature09497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 11.Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- 12.Cloos PAC, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 13.Krebs C, Galonić Fujimori D, Walsh CT, Bollinger JM., Jr Non-Heme Fe(IV)–Oxo Intermediates. Acc Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. The First Direct Characterization of a High-Valent Iron Intermediate in the Reaction of an α-Ketoglutarate-Dependent Dioxygenase: A High-Spin Fe(IV) Complex in Taurine/α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 15.Hoffart LM, Barr EW, Guyer RB, Bollinger JM, Jr, Krebs C. Direct spectroscopic detection of a C-H-cleaving high-spin Fe(IV) complex in a prolyl-4-hydroxylase. Proc Natl Acad Sci U S A. 2006;103:14738–14743. doi: 10.1073/pnas.0604005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galonić DP, Barr EW, Walsh CT, Bollinger JM, Jr, Krebs C. Two interconverting Fe(IV) intermediates in aliphatic chlorination by the halogenase CytC3. Nat Chem Biol. 2007;3:113–116. doi: 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- 17.Matthews ML, Krest CM, Barr EW, Vaillancourt FH, Walsh CT, Green MT, Krebs C, Bollinger JM., Jr Substrate-Triggered Formation and Remarkable Stability of the C-H Bond-Cleaving Chloroferryl Intermediate in the Aliphatic Halogenase SyrB2. Biochemistry. 2009;48:4331–4343. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang W-c, Guo Y, Wang C, Butch SE, Rosenzweig AC, Boal AK, Krebs C, Bollinger JM., Jr Mechanism of the C5 Stereoinversion Reaction in the Biosynthesis of Carbapenem Antibiotics. Science. 2014;343:1140–1144. doi: 10.1126/science.1248000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Que L., Jr One motif - many different reactions. Nat Struct Mol Biol. 2000;7:182–184. doi: 10.1038/73270. [DOI] [PubMed] [Google Scholar]
- 20.Koehntop KD, Emerson JP, Que L., Jr The 2-His-1-carboxylate facial triad: a versatile platform for dioxygen activation by mononuclear non-heme iron(II) enzymes. J Biol Inorg Chem. 2005;10:87–93. doi: 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- 21.Hanauske-Abel HM, Günzler V. A stereochemical concept for the catalytic mechanism of prolylhydroxylase: Applicability to classification and design of inhibitors. J Theor Biol. 1982;94:421–455. doi: 10.1016/0022-5193(82)90320-4. [DOI] [PubMed] [Google Scholar]
- 22.Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM., Jr Evidence for Hydrogen Abstraction from C1 of Taurine by the High-Spin Fe(IV) Intermediate Detected during Oxygen Activation by Taurine:α-Ketoglutarate Dioxygenase (TauD) J Am Chem Soc. 2003;125:13008–13009. doi: 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- 23.Price JC, Barr EW, Hoffart LM, Krebs C, Bollinger JM., Jr Kinetic Dissection of the Catalytic Mechanism of Taurine:α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Biochemistry. 2005;44:8138–8147. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- 24.Blasiak LC, Vaillancourt FH, Walsh CT, Drennan CL. Crystal structure of the non-haem iron halogenase SyrB2 in syringomycin biosynthesis. Nature. 2006;440:368–371. doi: 10.1038/nature04544. [DOI] [PubMed] [Google Scholar]
- 25.Matthews ML, Neumann CS, Miles LA, Grove TL, Booker SJ, Krebs C, Walsh CT, Bollinger JM., Jr Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc Natl Acad Sci U S A. 2009;106:17723–17728. doi: 10.1073/pnas.0909649106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinie RJ, Livada J, Chang W-c, Green MT, Krebs C, Bollinger JM, Jr, Silakov A. Experimental Correlation of Substrate Position with Reaction Outcome in the Aliphatic Halogenase, SyrB2. J Am Chem Soc. 2015;137:6912–6919. doi: 10.1021/jacs.5b03370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong SD, Srnec M, Matthews ML, Liu LV, Kwak Y, Park K, Bell CBI, Alp EE, Zhao J, Yoda Y, Kitao S, Seto M, Krebs C, Bollinger JM, Jr, Solomon EI. Elucidation of the Fe(IV)=O intermediate in the catalytic cycle of the halogenase SyrB2. Nature. 2013;499:320–323. doi: 10.1038/nature12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Srnec M, Wong SD, Matthews ML, Krebs C, Bollinger JM, Jr, Solomon EI. Electronic Structure of the Ferryl Intermediate in the α-Ketoglutarate Dependent Non-Heme Iron Halogenase SyrB2: Contributions to H Atom Abstraction Reactivity. J Am Chem Soc. 2016;138:5110–5122. doi: 10.1021/jacs.6b01151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borowski T, Noack H, Radoń M, Zych K, Siegbahn PEM. Mechanism of Selective Halogenation by SyrB2: A Computational Study. J Am Chem Soc. 2010;132:12887–12898. doi: 10.1021/ja101877a. [DOI] [PubMed] [Google Scholar]
- 30.Mitchell AJ, Zhu Q, Maggiolo AO, Ananth NR, Hillwig ML, Liu X, Boal AK. Structural basis for halogenation by iron- and 2-oxo-glutarate-dependent enzyme WelO5. Nat Chem Biol. 2016;12:636–640. doi: 10.1038/nchembio.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boal AK, Bollinger JM, Jr, Chang W-c. Assembly of the unusual oxacycles in the orthosomycin antibiotics. Proc Natl Acad Sci U S A. 2015;112:11989–11990. doi: 10.1073/pnas.1514689112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCulloch KM, McCranie EK, Smith JA, Sarwar M, Mathieu JL, Gitschlag BL, Du Y, Bachmann BO, Iverson TM. Oxidative cyclizations in orthosomycin biosynthesis expand the known chemistry of an oxygenase superfamily. Proc Natl Acad Sci U S A. 2015;112:11547–11552. doi: 10.1073/pnas.1500964112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan W, Song H, Song F, Guo Y, Wu CH, Sae Her A, Pu Y, Wang S, Naowarojna N, Weitz A, Hendrich MP, Costello CE, Zhang L, Liu P, Zhang YJ. Endoperoxide formation by an α-ketoglutarate-dependent mononuclear non-haem iron enzyme. Nature. 2015;527:539–543. doi: 10.1038/nature15519. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Zhang Z, Smart TJ, Choi H, Hardy F, Lohans CT, Abboud MI, Richardson MSW, Paton RS, McDonough MA, Schofield CJ. Structural and stereoelectronic insights into oxygenase-catalyzed formation of ethylene from 2-oxoglutarate. Proc Natl Acad Sci U S A. 2017;114:4667–4672. doi: 10.1073/pnas.1617760114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang TC, Wolfe MD, Neibergall MB, Mekmouche Y, Lipscomb JD, Hoffman BM. Substrate Binding to NO-Ferro-Naphthalene 1,2-Dioxygenase Studied by High-Resolution Q-Band Pulsed 2H-ENDOR Spectroscopy. J Am Chem Soc. 2003;125:7056–7066. doi: 10.1021/ja0214126. [DOI] [PubMed] [Google Scholar]
- 36.Muthukumaran RB, Grzyska PK, Hausinger RP, McCracken J. Probing the Iron-Substrate Orientation for Taurine/α-Ketoglutarate Dioxygenase Using Deuterium Electron Spin Echo Envelope Modulation Spectroscopy. Biochemistry. 2007;46:5951–5959. doi: 10.1021/bi700562t. [DOI] [PubMed] [Google Scholar]
- 37.Casey TM, Grzyska PK, Hausinger RP, McCracken J. Measuring the Orientation of Taurine in the Active Site of the Non-Heme Fe(II)/α-Ketoglutarate-Dependent Taurine Hydroxylase (TauD) Using Electron Spin Echo Envelope Modulation (ESEEM) Spectroscopy. J Phys Chem B. 2013;117:10384–10394. doi: 10.1021/jp404743d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith TS, II, LoBrutto R, Pecoraro VL. Paramagnetic spectroscopy of vanadyl complexes and its applications to biological systems. Coord Chem Rev. 2002;228:1–18. [Google Scholar]
- 39.Winkler JR, Gray HB. Molecular Electronic Structures of Transition Metal Complexes I. In: Mingos DMP, Day P, Dahl JP, editors. Structure and Bonding. Springer; Berlin Heidelberg: 2011. pp. 17–28. [Google Scholar]
- 40.Ishida T, Mitsubori S, Nogami T, Takeda N, Ishikawa M, Iwamura H. Ferromagnetic Exchange Coupling of Vanadium(IV) dπ Spins across Pyrimidine Rings: Dinuclear Complexes of Oxovanadium(IV) Bis(1,1,1,5,5,5-hexafluoropentane-2,4-dionate) Bridged by Pyrimidine Derivatives. Inorg Chem. 2001;40:7059–7064. doi: 10.1021/ic010730n. [DOI] [PubMed] [Google Scholar]
- 41.Tasiopoulos AJ, Tolis EJ, Tsangaris JM, Evangelou A, Woollins DJ, Slawin AM, Pessoa J, Correia I, Kabanos TA. Model investigations for vanadium-protein interactions: vanadium(III) compounds with dipeptides and their oxovanadium(IV) analogues. J Biol Inorg Chem. 2002;7:363–374. doi: 10.1007/s00775-001-0308-0. [DOI] [PubMed] [Google Scholar]
- 42.Manivannan V, Hoffman JT, Dimayuga VL, Dwight T, Carrano CJ. A comparison of vanadyl acetylacetonate complexes of N2O heteroscorpionate ligands that vary systematically in donor set. Inorg Chim Acta. 2007;360:529–534. [Google Scholar]
- 43.Yuan C, Lu L, Gao X, Wu Y, Guo M, Li Y, Fu X, Zhu M. Ternary oxovanadium(IV) complexes of ONO-donor Schiff base and polypyridyl derivatives as protein tyrosine phosphatase inhibitors: synthesis, characterization, and biological activities. J Biol Inorg Chem. 2009;14:841–851. doi: 10.1007/s00775-009-0496-6. [DOI] [PubMed] [Google Scholar]
- 44.Roy AS, Saha P, Adhikary ND, Ghosh P. o-Iminobenzosemiquinonate and o-Imino-p-methylbenzosemiquinonate Anion Radicals Coupled VO2+ Stabilization. Inorg Chem. 2011;50:2488–2500. doi: 10.1021/ic102296p. [DOI] [PubMed] [Google Scholar]
- 45.Deng Y, Yang Y, Zhang Y, Yan Q, Liu J. Formation of an asymmetrical ligand (H 3 L cyclic) via a metal-induced cyclization of symmetrical thiocarbohydrazone (H 4 L) in synthesizing an oxovanadium(IV) complex VO(HL cyclic)(EtOH)2. J Coord Chem. 2012;65:1409–1416. [Google Scholar]
- 46.Riggs-Gelasco PJ, Price JC, Guyer RB, Brehm JH, Barr EW, Bollinger JM, Jr, Krebs C. EXAFS Spectroscopic Evidence for an FeO Unit in the Fe(IV) Intermediate Observed during Oxygen Activation by Taurine:α-Ketoglutarate Dioxygenase. J Am Chem Soc. 2004;126:8108–8109. doi: 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- 47.Galonić-Fujimori D, Barr EW, Matthews ML, Koch GM, Yonce JR, Walsh CT, Bollinger JM, Jr, Krebs C, Riggs-Gelasco PJ. Spectroscopic Evidence for a High-Spin Br-Fe(IV)-Oxo Intermediate in the α-Ketoglutarate-Dependent Halogenase CytC3 from Streptomyces. J Am Chem Soc. 2007;129:13408–13409. doi: 10.1021/ja076454e. [DOI] [PubMed] [Google Scholar]
- 48.Chasteen ND. In: Biological Magnetic Resonance. Berliner LJ, Reuben J, editors. Springer; US: 1981. pp. 53–119. [Google Scholar]
- 49.Elkins JM, Ryle MJ, Clifton IJ, Dunning Hotopp JC, Lloyd JS, Burzlaff NI, Baldwin JE, Hausinger RP, Roach PL. X-ray Crystal Structure of Escherichia coli Taurine/α-Ketoglutarate Dioxygenase Complexed to Ferrous Iron and Substrates. Biochemistry. 2002;41:5185–5192. doi: 10.1021/bi016014e. [DOI] [PubMed] [Google Scholar]
- 50.O’Brien JR, Schuller DJ, Yang VS, Dillard BD, Lanzilotta WN. Substrate-Induced Conformational Changes in Escherichia coli Taurine/α-Ketoglutarate Dioxygenase and Insight into the Oligomeric Structure. Biochemistry. 2003;42:5547–5554. doi: 10.1021/bi0341096. [DOI] [PubMed] [Google Scholar]
- 51.Horitani M, Byer AS, Shisler KA, Chandra T, Broderick JB, Hoffman BM. Why Nature Uses Radical SAM Enzymes so Widely: Electron Nuclear Double Resonance Studies of Lysine 2,3-Aminomutase Show the 5′-dAdo• “Free Radical” Is Never Free. J Am Chem Soc. 2015;137:7111–7121. doi: 10.1021/jacs.5b00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foner SN, Cochran EL, Bowers VA, Jen CK. Multiple Trapping Sites for Hydrogen Atoms in Rare Gas Matrices. J Chem Phys. 1960;32:963–971. [Google Scholar]
- 53.Morton JR, Preston KF, Strach SJ, Adrian FJ, Jette AN. Anisotropic hyperfine interactions of raregas nuclei near trapped hydrogen atom s. J Chem Phys. 1979;70:2889–2893. [Google Scholar]
- 54.Rees JA, Wandzilak A, Maganas D, Wurster NIC, Hugenbruch S, Kowalska JK, Pollock CJ, Lima FA, Finkelstein KD, DeBeer S. Experimental and theoretical correlations between vanadium K-edge X-ray absorption and Kβ emission spectra. J Biol Inorg Chem. 2016;21:793–805. doi: 10.1007/s00775-016-1358-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sinnecker S, Svensen N, Barr EW, Ye S, Bollinger JM, Jr, Neese F, Krebs C. Spectroscopic and Computational Evaluation of the Structure of the High-Spin Fe(IV)-Oxo Intermediates in Taurine: α-Ketoglutarate Dioxygenase from Escherichia coli and Its His99Ala Ligand Variant. J Am Chem Soc. 2007;129:6168–6179. doi: 10.1021/ja067899q. [DOI] [PubMed] [Google Scholar]
- 56.Minnihan EC, Ando N, Brignole EJ, Olshansky L, Chittuluru J, Asturias FJ, Drennan CL, Nocera DG, Stubbe J. Generation of a stable, aminotyrosyl radical-induced α2β2 complex of Escherichia coli class Ia ribonucleotide reductase. Proc Natl Acad Sci U S A. 2013;110:3835–3840. doi: 10.1073/pnas.1220691110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mitchell AJ, Dunham NP, Martinie RJ, Bergman JA, Pollock CJ, Hu K, Allen BD, Chang W-c, Silakov A, Bollinger JM, Jr, Krebs C, Boal AK. Visualizing the Reaction Cycle in an iron(II)- and 2-(Oxo)-Glutarate-Dependent Hydroxylase. J Am Chem Soc. 2017 doi: 10.1021/jacs.7b07374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.George GN. EXAFSPAK, EXAFSPAK, SSRL, SLAC. Stanford University; Stanford, CA: [Google Scholar]
- 59.Tenderholt A, Hedman B, Hodgson KO. PySpline: A Modern, CrossPlatform Program for the Processing of Raw Averaged XAS Edge and EXAFS Data. AIP Conference Proceedings. 2007;882:105–107. [Google Scholar]
- 60.Rehr JJ, Kas JJ, Vila FD, Prange MP, Jorissen K. Parameter-free calculations of X-ray spectra with FEFF9. Phys Chem Chem Phys. 2010;12:5503–5513. doi: 10.1039/b926434e. [DOI] [PubMed] [Google Scholar]
- 61.Delgado-Jaime MU, Mewis CP, Kennepohl P. Blueprint XAS: a Matlab-based toolbox for the fitting and analysis of XAS spectra. J Synchrotron Radiat. 2010;17:132–137. doi: 10.1107/S0909049509046561. [DOI] [PubMed] [Google Scholar]
- 62.Annino G, Cassettari M, Martinelli M. A New Concept of Open Cavity. IEEE Trans Microw Theory Tech. 2009;57:775–783. [Google Scholar]
- 63.Reijerse E, Lendzian F, Isaacson R, Lubitz W. A tunable general purpose Q-band resonator for CW and pulse EPR/ENDOR experiments with large sample access and optical excitation. J Magn Reson. 2012;214:237–243. doi: 10.1016/j.jmr.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 64. [accessed June 7, 2017];Kazan Viewer - Homepage Alexey Silakov. https://sites.google.com/site/silakovalexey/kazan-viewer.
- 65.Stoll S, Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J Magn Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 66.Baute D, Goldfarb D. The 17O Hyperfine Interaction in V17O(H217O)52+ and Mn(H217O)62+ Determined by High Field ENDOR Aided by DFT Calculations. J Phys Chem A. 2005;109:7865–7871. doi: 10.1021/jp052132q. [DOI] [PubMed] [Google Scholar]
- 67.Matsuoka N, Tsuchimoto M, Yoshioka N. Theoretical Study of Magnetic Properties of Oxovanadium(IV) Complex Self-Assemblies with Tetradentate Schiff Base Ligands. J Phys Chem B. 2011;115:8465–8473. doi: 10.1021/jp111779k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.