

Mutations in the mitochondrial gene SLC25A46 cause optic atrophy spectrum disorder. Wan et al. identify novel mutations in SLC25A46 in individuals with lethal congenital pontocerebellar hypoplasia. Functional studies show that while the latter mutations cause instability or loss of function of SLC25A46, those that are associated with isolated optic atrophy do not.
Keywords: pontocerebellar hypoplasia, SLC25A46, mitochondria, optic atrophy spectrum disorder

Mutations in the mitochondrial gene SLC25A46 cause optic atrophy spectrum disorder. Wan et al. identify novel mutations in SLC25A46 in individuals with lethal congenital pontocerebellar hypoplasia. Functional studies show that while the latter mutations cause instability or loss of function of SLC25A46, those that are associated with isolated optic atrophy do not.
Abstract
Disturbed mitochondrial fusion and fission have been linked to various neurodegenerative disorders. In siblings from two unrelated families who died soon after birth with a profound neurodevelopmental disorder characterized by pontocerebellar hypoplasia and apnoea, we discovered a missense mutation and an exonic deletion in the SLC25A46 gene encoding a mitochondrial protein recently implicated in optic atrophy spectrum disorder. We performed functional studies that confirmed the mitochondrial localization and pro-fission properties of SLC25A46. Knockdown of slc24a46 expression in zebrafish embryos caused brain malformation, spinal motor neuron loss, and poor motility. At the cellular level, we observed abnormally elongated mitochondria, which was rescued by co-injection of the wild-type but not the mutant slc25a46 mRNA. Conversely, overexpression of the wild-type protein led to mitochondrial fragmentation and disruption of the mitochondrial network. In contrast to mutations causing non-lethal optic atrophy, missense mutations causing lethal congenital pontocerebellar hypoplasia markedly destabilize the protein. Indeed, the clinical severity appears inversely correlated with the relative stability of the mutant protein. This genotype–phenotype correlation underscores the importance of SLC25A46 and fine tuning of mitochondrial fission and fusion in pontocerebellar hypoplasia and central neurodevelopment in addition to optic and peripheral neuropathy across the life span.
Introduction
The study of rare congenital neurological disorders has led to the discovery of genes and cellular pathways important in neurodevelopment. Pontocerebellar hypoplasia (PCH) is a phenotypically and genetically heterogeneous group of autosomal recessive congenital disorders characterized by maldevelopment of the cerebellum and brainstem, profound global developmental delay, and often early death (Barth, 1993; Rudnik-Schöneborn et al., 2014). Advances in neuroimaging have facilitated the early detection of combined pontine and cerebellar hypoplasia, while advances in genetic technology have accelerated gene discovery. So far, 10 subtypes of PCH have been designated (omim.org), with causative mutations identified in a growing list of genes in all subtypes except PCH7, for which the genetic locus has not yet been mapped.
In this study, we describe two siblings with PCH and profound weakness with apnoea at birth, in whom we initially considered PCH1, which is distinguished from the other PCH subtypes by its association with spinal muscular atrophy due to spinal motor neuron degeneration (Norman, 1961; Barth, 1993). After no defect in the known PCH1 genes (Renbaum et al., 2009; Wan et al., 2012; Boczonadi et al., 2014) was identified, by whole exome sequencing, we discovered a homozygous missense mutation in the gene SLC25A46 and further examined the clinical features to note optic atrophy and sensorimotor neuropathy. In a similarly affected child in an unrelated family, we identified an allelic homozygous exonic deletion. SLC25A46 encodes a mitochondrial protein that had been unknown to be involved in human disease when we began our investigation, and mutations in which have only recently been shown to cause an optic atrophy spectrum disorder characterized by optic atrophy and axonal neuropathy associated with cerebellar degeneration with broad variability in severity, ranging from early death in two families as the most severe to mild optic atrophy with survival into adulthood as the least severe (Abrams et al., 2015; Nguyen et al., 2016). The prior studies did not address how different mutations in the same gene may lead to such varied severity and manifestations. We performed biochemical and cellular studies to demonstrate that SLC25A46 is a pro-fission mitochondrial outer membrane protein important in the regulation of mitochondrial dynamics, which is consistent with and complementary to the findings by Abrams and colleagues (2015). Importantly, our findings implicate SLC25A46 as a genetic locus for PCH, and we show that the broad clinical spectrum could at least be partially explained by the relative stability of the mutant protein, with loss of protein function correlating with lethal congenital PCH and far milder manifestations correlating with greater abundance of the mutant protein.
Materials and methods
Genetic analysis
The study was approved by the University of California, Los Angeles (UCLA) Institutional Review Board. DNA was extracted from peripheral blood from consenting individuals. We captured the exome using the TruSeq™ Exome Enrichment kit (Illumina FC-121-1048) and sequenced DNA on an Illumina Genome Analyzer 2500. For exome sequencing, each library produced ∼28 million paired-end 100-bp reads. The mean coverage of bases in the target exomes was 38×, with 87% of target bases covered ≥10×. Raw reads passing Illumina’s quality filters were aligned to the reference human genome Build 37 with Novoalign from Novocraft. The GATK UnifiedGenotyper was used to call single-nucleotide variants and indels. The proband had 15 140 non-synonymous and other consequential variants from the GRCh37 reference human genome. Under the hypothesis that the disorder was rare and the causative allele(s) would therefore not be common, and that the variant was likely to be in a long contiguous stretch of homozygosity (LCSH), we filtered out variants with an allele frequency of ≥0.005 in the 1000 Genomes Project or the NHLBI exome dataset, that were predicted to be damaging by PolyPhen or SIFT, and that were located in a LCSH of 5 mB. To compensate for bias in our own analytical system, we filtered out variants that we had identified in 214 exomes from unrelated, unaffected subjects. Under a recessive model, we searched for homozygous variants and compound heterozygous variants (defined as two variants in the same transcript) shared by the two cases. Application of these criteria narrowed the search down to a missense variant in SLC25A46. Sanger sequencing for further validation was performed using standard protocols.
RNA isolation, RT-PCR and cloning procedures
Total RNA was isolated from cultured cells with Isol-RNA Lysis Reagent (5 Prime). To remove residual genomic DNA, the isolated RNA was treated with TURBO DNase (Life Technologies) for 30 min at 37°C and purified by phenol/chloroform extraction. The RNA was reverse transcribed into cDNA using the SuperScript® III First-Strand Synthesis System (Life Technologies) according to the manufacturer’s manual. Reverse transcriptase-polymerase chain reaction (RT-PCR) was carried out with Phusion® DNA Polymerase (New England Biolabs) and primer for GAPDH: forward 5’-ATGGGGAAGGTGAAGGTC-3’, reverse 5’-TTACTCCTTGGAGGCCATG-3’ and SLC25A46 forward 5’-GGCGGATCCGCCACCATGCATCCGCGGCGCCC-3’ and 5’-CCGGTCGACTCA AATGTTATTTTGAAGAAGTGTAGAG-3’. Untagged or N-terminal 2 × HA-tagged SLC25A46 were generated by PCR and cloned into the vectors pSP65 (Promega), pcDNA3 (Life Technologies), or pEGFP-C1 (Clontech). All mutations were introduced by site-directed mutagenesis using Phusion® DNA polymerase and DpnI (New England Biolabs) digestion.
Yeast strains
Standard genetic techniques were used for growth and manipulation of yeast strains (Guthrie, 1991). The strains W303 (wild-type), mim1Δ, and tom70Δ/tom71Δ were gifts from Dr Doron Rapaport, University of Tübingen.
Cell culture, transfection, and imaging
HeLa, HEK293T (ATCC), LAN5 (kindly provided by Dr Joan Valentine, UCLA), Ramos (kindly provided by Dr Michael Teitell, UCLA) and HCT116 (kindly provided by Dr Richard Youle, NIH) were cultured in Dulbecco’s modified Eagle medium (25 mM glucose, sodium pyruvate), RPMI (Life Technologies) and McCoy’s 5A (GE Healthcare), respectively, and grown at 37°C and 5% CO2. Each media was supplemented with 10% foetal bovine serum (FBS) (Atlanta Biological), 100 U/ml penicillin, and 100 μg/ml streptomycin (Life Technologies). Transfections were performed using BioT (Bioland) or Lipofectamine® LTX (Invitrogen) according to manufacturer’s instructions. Forty-eight hours after transfection of HEK293T cells with full-length cDNA of SLC25A46 in the pAcGFP-N1 vector, live cell imaging was performed with an Olympus BX51 Microscope equipped with 40× water immersion objective and GFP filter.
Antibodies
Antibodies used in this study: SLC25A46 (Proteintech), HA (HA.11, Biolegend), β-Tubulin (TU27, Biolegend), MFN2 (D1E9, Cell Signaling), TIMM23 and calnexin (BD Biosciences), LRP130 (H-300, Santa Cruz Biotechnology), TOMM20 (FL-145, Santa Cruz Biotechnology), Mortalin (UC Davis/NIH NeuroMab Facility), PreP, TOMM40, YME1L were generated from recombinant proteins (Pacific Immunology).
Immunoblotting and blue native polyacrylamide gel electrophoresis
Cells were lysed in RIPA buffer (50 mM Tris/HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.1% SDS and 0.5 % sodium deoxycholate, 1 mM EDTA, 10 mM NEM, 10 µM MG132, 1 mM Na3VO4 2.5 mM NaF) for 30 min on ice. After a centrifugation at 21 000g for 10 min at 4°C, the protein concentration was measured using the BCA protein assay kit. Equal amounts of protein extracts were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE). For the analysis of mitochondria by blue native (BN)-PAGE 150–200 µg mitochondria were lysed in BN lysis buffer [20 mM HEPES pH7.4, 50 mM NaCl, 2.5 mM MgCl2, 10% glycerol, 0.1 mM EDTA, 0.5 mM PMSF, 1% digitonin (AG scientific)] at a concentration of 5 mg/ml for 30 min on ice. After a centrifugation at 21 000g for 10 min the mitochondrial extract was separated on a 4–14% BN-PAGE gel. Proteins were transferred to a PVDF membrane and detected by immunoblot using the indicated antibodies.
Immunostaining of cultured cells
LAN5 cells were grown on D-poly-lysine coated coverslips. The cells were fixed in prewarmed 4% paraformaldehyde (PFA) for 15 min at room temperature. After washing with phosphate-buffered saline (PBS), the cells were permeabilized in 0.2% Triton™ X-100 for 10 min and blocked for 30 min in 1% bovine serum albumin (BSA) in PBS. The primary and secondary antibodies (Alexa Fluor® 488 and Alexa Fluor® 568, Life Technologies) were diluted in 1% bovine serum albumin (BSA) in PBS and incubated for 1 h at room temperature. The cells were washed between primary and secondary antibody applications in PBS. To stain nuclear DNA, 4',6-diamidino-2-phenylindole (DAPI, Life Technologies) at a concentration of 300 nM was added to the cells for 2 min. The cells were washed in PBS and mounted in Fluoromount-G® (Southern Biotech). Pictures were taken at a Leica TCS SPE confocal microscope using a 100× objective. Pictures were processed with ImageJ (NIH).
Subcellular fractionation
LAN5 cells were resuspended in homogenization buffer (20 mM HEPES pH 7.4, 220 mM mannitol, 70 mM sucrose, 2.5 mM NaF, 1 mM NaVO4 and 0.5 mM PMSF), and the cells were disrupted with 30 strokes in a glass Teflon homogenizer. The cells were centrifuged at 850 g. The supernatant was saved and the pellet re-extracted in homogenization buffer with another round of 30 strokes in a glass Teflon homogenizer followed by centrifugation at 850 g. The supernatants were combined and centrifuged at 850 g to pellet all remaining nuclei and unbroken cells. The supernatant was centrifuged at 10 000g for 10 min to obtain the 10 000g pellet containing heavy membranes (enriched in mitochondria). The 10 000g pellet was washed once in homogenization buffer and again pelleted at 10 000g. The P10k supernatant was pooled and centrifuged at 100 000g and the pellet (P100k) containing all light membranes including microsomes and the post 100 000g supernatant (S100k) containing soluble proteins were separated. The P100k was washed once in homogenization buffer and subjected to centrifugation again. Protein concentrations of each fraction were determined using the bicinchoninic acid (BCA) protein assay kit (Thermo Fisher) and 50 µg protein of each fraction were resolved by SDS-PAGE.
Isolation of mitochondria from cultured cells and yeast
Isolation of mitochondria from yeast was performed as previously described (Glick, 1991). Mitochondria from cultured cells were isolated as described under subcellular fractionation (heavy membranes).
Alkaline extraction
Fifty micrograms of mitochondria were incubated in isotonic buffer (20 mM HEPES pH 7.4, 220 mM mannitol, 70 mM sucrose), 100 mM sodium carbonate (pH 11.5 or pH 12) or 20 mM HEPES pH 7.4, 1% Triton™ X-100 for 30 min on ice (Fujiki et al., 1982). The samples were centrifuged at 21 000g for 30 min and the resulting supernatants were trichloroacetic acid (TCA)/acetone precipitated. All pellets were resuspended in Thorner buffer (8 M urea, 5% SDS, 40 mM Tris pH 6.8, 0.1 mM EDTA, 5% β-mercaptoethanol) and resolved by SDS-PAGE.
Submitochondrial localization
Fifty micrograms of mitochondria were incubated in isotonic buffer (20 mM HEPES pH 7.4, 200 mM mannitol, 70 mM sucrose), hypotonic buffer (20 mM HEPES pH 7.4) or 20 mM HEPES pH 7.4, 1% Triton™ X-100 with 2.5 µg/ml proteinase K (as indicated) for 30 min on ice. Proteinase K digestion was stopped by the addition of 2 mM PMSF. All reactions were centrifuged at 21 000g. The Triton™ X-100 supernatant was TCA/acetone precipitated. All pellets were resuspended in Thorner buffer and resolved by SDS-PAGE.
In vitro transcription and translation
All precursor proteins were in vitro transcribed and translated from a plasmid using the TnT® SP6/T7 Quick Coupled Transcription/Translation System (Promega). For radioactive labelling TRAN35S-LABEL™, Metabolic Labeling Reagent (MP Biomedicals) was used instead of unlabelled methionine, which was used for cold translation.
Protein import assays into mitochondria
In vitro imports into yeast mitochondria were performed as previously described (Claypool et al., 2011). Twenty micrograms of mitochondria were incubated in import buffer (20 mM HEPES pH 7.4, 220 mM mannitol, 70 mM sucrose, 1 mM ATP, 0.5 mM magnesium acetate, 20 mM sodium succinate, 5 mM NADH) for 5 min at 25°C prior to the addition of the precursor protein. The membrane potential was disrupted by treatment with 50 µM CCCP. After adding the precursor, aliquots were taken at different time points and transferred to new tubes containing cold homogenization buffer (20 mM HEPES pH 7.4, 220 mM mannitol, 70 mM sucrose) and stored on ice until the last time point was taken. Trypsin was then added to a final concentration of 0.5 to 1.25 µg/ml and samples were incubated on ice for an additional 15 min. The trypsin digestion was terminated by adding soybean trypsin inhibitor to a final concentration of 250 µg/ml. Mitochondria were re-isolated by centrifugation at 10 000g for 5 min. Alkaline extraction was performed for membrane proteins in 100 mM Na2CO3 pH 11.5 for 30 min on ice. Membrane sheets containing the imported precursor protein were obtained by centrifugation at 21 000g for 30 min. Samples were resuspended in 2× SDS sample buffer and resolved by SDS-PAGE. For the analysis of imported proteins by BN-PAGE, mitochondria were not treated with trypsin; instead, they were washed once in 20 mM HEPES pH 7.4, 220 mM mannitol, 70 mM sucrose after the import and re-isolated by centrifugation at 10 000g for 10 min. The mitochondria were then lysed as described above. Imported proteins were detected by autoradiography.
Zebrafish morpholino injection
Zebrafish embryos were obtained from the UCLA Zebrafish Core Facility, with procedures approved by the UCLA Animal Research Committee. Morpholino oligonucleotides (Gene Tools) were designed to block translation initiation (GCCGACGAGAAGTCATGCTGTAAAT) or the splice-donor site of exon 3 (CAGTGTCTTATTCTGCATACCTGAC) of zebrafish slc25a46 pre-mRNA. We obtained a standard control from Gene Tools. Embryos at the one-cell or two-cell stage were injected with reagents, and the injection volume ranged from 0.5 to 2.0 nl at a concentration of 4 ng/nl. Embryos were incubated in E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4 and 0.00001% methylene blue; Sigma) at 28.5°C.
Zebrafish neuronal culture and fluorescent imaging of mitochondria
Primary neuronal culture was prepared from zebrafish larva (wnt2-GAL4::UAS-EGFP;UAS-mito-DsRed) at 1 or 2 days post-fertilization (Chen et al., 2013). After 1 or 2 days, the mitochondria signal in neurites was imaged using an epifluorescence microscope (Olympus BX51 equipped with a 40× water immersion objective) and analysed with ImageJ software.
Results
Clinical presentation of siblings in two families with pontocerebellar hypoplasia and apnoea at birth
Among our cohort of patients with profound weakness and neuroimaging findings of pontocerebellar hypoplasia suggestive of PCH1 without a known genetic cause, two sibling pairs presented with particularly severe symptoms at birth. The index patient of Family 1 was born at 38 weeks of gestation to parents who are third cousins, following a pregnancy complicated by polyhydramnios. He was hypotonic at birth, made no respiratory effort, and had to be emergency intubated. Apparently unresponsive, he had little voluntary movement but had myoclonic jerks. His physical examination was otherwise notable for bilateral optic atrophy and flexor contractures at the knees and elbows. Laboratory investigation revealed normal liver function, serum lactate/pyruvate, urine and serum amino acids, ammonia, and glucose. EMG and nerve conduction studies showed absent sensory and motor responses with fibrillations and reduced recruitment of high amplitude polyphasic motor unit action potentials, suggestive of severe axonal sensorimotor neuropathy. An EEG on Day 2 of life revealed only a global encephalopathy without epileptiform discharges. Cerebral MRI on Day 5 of life was notable for a small cerebellum and brainstem (Fig. 1A–C). The patient died at age 4 weeks. An affected sister of the index patient was born at 37.5 week of gestation, following a pregnancy also complicated by polyhydramnios and presented with identical clinical features: hypotonia, severe global developmental delay, and absent respiratory effort. MRI of the brain again demonstrated a small cerebellum and brainstem with diffuse cerebral and cerebellar atrophy (data not shown). She died at age 2 weeks. Respiratory chain enzyme testing on biopsies of the muscle and liver in both newborns was normal.
Figure 1.

Neuroimaging features in two index patients with PCH. (A) Axial, (B) sagittal, and (C) coronal T2-weighted images from the index patient from Family 1 performed on Day 5 of life, demonstrating small cerebellum, pons, and medulla, with prominent ventricles and increased extra-axial spaces, and a thin corpus callosum. (D) Axial, (E) sagittal, and (F) coronal images from the similarly affected index patient from Family 2, performed on Day 6 of life, demonstrating pontocerebellar hypoplasia, prominent ventricles and increased extra-axial spaces, a thin corpus callosum, compared to normal findings from (G) axial, (H) sagittal, and (I) coronal T2-weighted images from a normal subject at 8 days of age.
The index patient of Family 2 was the fifth child of consanguineous parents. The pregnancy was complicated by polyhydramnios, preterm labour, placental haemorrhage, and delivery by caesarean section at 36 weeks of gestation. With a birthweight of 2780 g (<5th percentile), length 48 cm (<25th percentile), and head circumference of 31.5 cm (<5th percentile), she required ventilatory support for respiratory failure. She was hypotonic and areflexic, with occasional myoclonic jerks. She had bilateral optic atrophy. EEG demonstrated generalized slowing with abnormal theta rhythm but no epileptiform changes, while MRI showed pontocerebellar hypoplasia (Fig. 1D–F) compared to an age-matched control individual (Fig. 1G–I). The serum creatine kinase on post-natal Day 2 was normal (99 U/l); her serum lactate was elevated at 8.2 mmol/l. She died at age 6 weeks. An affected sister born 5 years earlier had presented with the same phenotype, with respiratory failure at birth. Her creatine kinase was normal, with serum lactate being slightly increased (2.6 mmol/l) on Day 4. Perinatal EEG showed burst suppression; seizures began at 3 weeks of age. Nerve conduction study at age 18 days showed a mixed sensorimotor neuropathy. Ocular examination at age 6 weeks was notable for optic atrophy. Cerebellar hypoplasia was evident on MRI at age 6 days, with development of pontine and cerebellar atrophy on a subsequent MRI at age 2.5 month. The affected sister died at age 3 months.
Identification of genetic defects in SLC25A46 in patients with pontocerebellar hypoplasia
In Family 1, karyotyping was normal, and SNP microarray analyses showed no genomic rearrangements but revealed LCSH, consistent with the family history of consanguinity. The two patients shared LCSH on chromosomes 5 and 10. To identify the responsible genetic defect, we performed whole exome sequencing of the two patients and their parents. This analysis yielded one candidate variant, fulfilling the requirement of rare biallelic variants in the affected individuals and heterozygous in the parents, in the gene SLC25A46, NC_000005.9:g.5:110097247T>C (ENST00000355943:c.1022T>C; encoding ENSP00000348211:p.Leu341Pro). We did not observe any variants in EXOSC3, VRK1, RARS2, TSEN54 previously reported in PCH1. We confirmed by Sanger sequencing the presence of the genetic variant in the patients and their parents. This variant is rare as it is not found in the public databases dbSNP, EVS, ExA, or the database at the UCLA Clinical Genomics Center.
In the index patient of Family 2, we performed Sanger sequencing of all exons and flanking introns of SLC25A46 to identify no variants in all exons except exon 1, which could not be amplified, raising the possibility of a genomic deletion. We designed multiple primers to ascertain the breakpoints to identify a 1897-nucleotide deletion spanning exon 1 that was homozygous in the index patient and heterozygous in the parents (Supplementary Fig. 1).
The longest transcript among the multiple splice isoforms of SLC25A46 spans 8 exons over 18 895 bases (NM_ 138773) and encodes a 418-amino acid protein that shares structural motifs with members of the mitochondrial solute carrier family (Haitina et al., 2006). The rare genetic variant found in Family 1 c.1022T>C resided in exon 8 of SLC25A46. The deletion in Family 2 eliminated exon 1, which contained the codon ATG for the longest transcript, with the next in-frame ATG in exon 4 encoding Met147.
At the time of our discovery, mutations in SLC25A46 were not known to be associated with human disease. Mutations in SLC25A46 have very recently been reported to cause optic atrophy spectrum disorder, with four missense mutations in exon 8, two frameshift mutations, and a splice site mutation (Abrams et al., 2015) (Fig. 2A).
Figure 2.
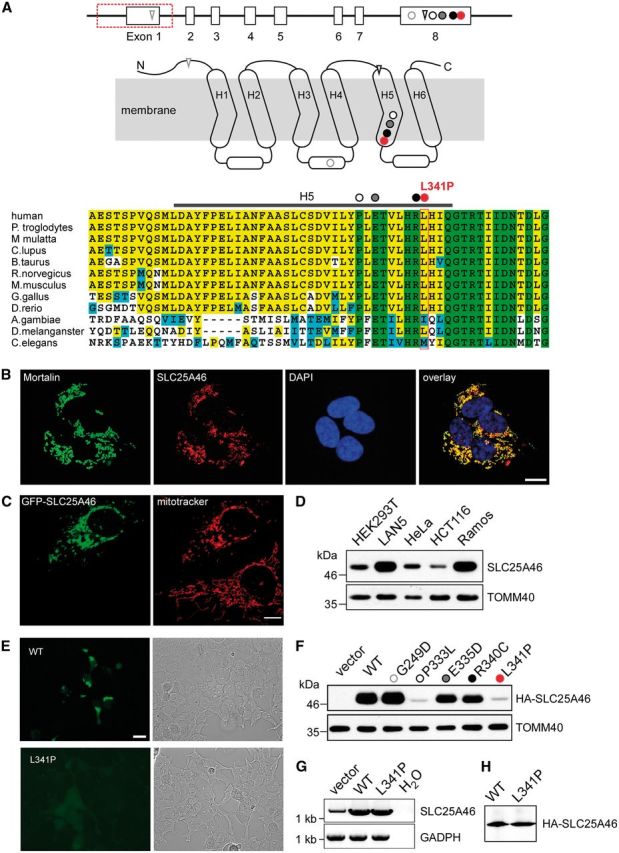
SLC25A46 localizes to the mitochondria with differential mutant protein stability. Genomic structure of SLC25A46, with 8 exons represented by open boxes and the exonic deletion demarcated by a red box along with the mutation c.1022T>C in red in relationship to other recently reported mutations: circle = missense; triangle = frameshift. Predicted membrane-spanning 2D structure of SLC25A46 with three sequence repeats (Repeat 1–3), each with two transmembrane helices linked by an elongated loop with a short alpha-helix. The homozygous nucleotide variant c.1022T>C identified in Family 1 is predicted to alter a highly conserved residue L341 in H5, represented by a red circle in the diagram as well as the alignment of protein sequences encoded by orthologues in human and other eukaryotic organisms. The homozygous 1897-nucleotide deletion identified in Family 2 is predicted to delete exon 1, which contains the start codon ATG, with the next in-frame ATG in exon 4 predicted to encode M147 in the H1-H2 linker. The four previously described missense mutations in exon 8 are predicted to alter highly conserved G249 (grey open circle) in the linker between H3 and H4, and in H5 P333 (black open circle), E335 (grey closed circle), and R340 (black closed circle). The two frameshift mutations in A are predicted to introduce premature stops in the N terminus and H5. (B) Localization to the mitochondria of SLC25A46 by immunohistochemical analysis. LAN5 cells were fixed and incubated with antibodies against the mitochondrial marker mortalin and SLC25A46. Cell nuclei were marked with DAPI (scale bar = 10 µm). (C) Mitochondrial localization of SLC25A46 by mitochondrial labelling with MitoTracker® in a transiently transfected HeLa cell overexpressing GFP-tagged SLC25A46. Note the fragmentation of the mitochondria in the cell overexpressing GFP-SLC25A46, in contrast to the typical extensive long tubular mitochondrial network in the cell below, which was not transfected and not overexpressing SLC25A46. (D) Western blotting analysis of differential SLC25A46 expression in isolated mitochondria from the indicated cell lines. TOMM40 is included as loading control. (E) Visualization under fluorescent microscopy (left) or bright field (right) of transiently transfected HEK293 cells with plasmid constructs for GFP-tagged wild-type (WT) or L341P mutant SLC25A46, demonstrating markedly diminished fluorescent signal intensity of the mutant transfected cells compared to that in cells transfected with the wild-type construct. (F) Western blot analysis of HA-tagged mutant compared to the wild-type SLC25A46 probed with anti-HA antibodies. HEK293T cells were transiently transfected with the pCDNA3 vector containing HA-tagged SLC25A46. TOMM40 is included as a blotting control. (G) Quantitative RT-PCR demonstrated comparable levels of the L341P mutant and the wild-type mRNA. (H) In vitro transcription and translation reaction was performed on the same mRNA as in G.
Leu341Pro causes loss of function of the mitochondrial protein SLC25A46 that promotes fission
The secondary structure of SLC25A46, based on homology with other members of the mitochondrial solute carrier family (Haitina et al., 2006), consists of six putative transmembrane segments H1–H6 with three internal repeats (Fig. 2A). The mutant protein from the deletion identified in Family 2 is predicted to be missing the N-terminus, including at least the H1–H2 linker. By in silico analysis, alternative in-frame ATG in exons 4, 5, and 7 appear to be low probability sites for the initiation of translation. Thus, the elimination of exon 1 would predict the removal of the first 146 amino acid residues, even if the truncated transcript were translated. The rare genetic variant c.1022T>C identified in Family 1 leading to p.Leu341Pro is predicted to be among the top 0.3% of the most damaging substitutions by in silico analysis, with a scaled CADD (combined annotation dependent depletion) score of 25.2 (Kircher et al., 2014). The variant alters a leucine residue in H5 close to the linker of repeat 3 that is conserved in vertebrates (Fig. 2A).
To investigate what functional impact the nucleotide variant c.1022T>C may exert on the mutant gene product, we performed functional studies in mammalian cell lines. Immunohistochemical staining in LAN5 cells, using SLC25A46-specific antibodies, showed co-localization of the endogenous SLC25A46 with mortalin, a mitochondrial protein (Fig. 2B). Similarly, HeLa cells transiently transfected with GFP-tagged SLC25A46 showed co-localization with MitoTracker® labelling of mitochondria, with mitochondrial fragmentation and loss of the mitochondrial network in the transfected cells overexpressing SLC25A46 compared to normal mitochondrial network in untransfected cells (Fig. 2C). We detected differential levels of endogenous SLC25A46 in mitochondria from different cell lines (Fig. 2D). Using TOMM40, a mitochondrial outer membrane protein, as a loading control, SLC25A46 was most abundant in the neuronal cell line LAN5 and the Burkitt’s lymphoma cell line Ramos. In agreement, SLC25A46 mRNA was previously reported to be expressed throughout the CNS in rat, with the highest expression in the hindbrain (Haitina et al., 2006) as well as in the brain and especially in the cerebellum in mouse embryos.
We further compared the expression of the mutant versus wild-type SLC25A46. A GFP fusion with the wild-type protein was readily detected, but the L341P mutant fused to GFP was barely detectable (Fig. 2E). Indeed, by immunoblotting, the expression of the mutant protein Leu341Pro was markedly diminished and could hardly be detected in contrast to the abundant wild-type protein (Fig. 2F). This difference in the abundance of protein was post-translational, as the amount of the mRNA for mutant and wild-type SLC25A46 was comparable when analysed by quantitative RT-PCR (Fig. 2G), and the mutation did not prevent translation of the mRNA as in vitro translation analysis yielded similar levels of mutant and wild-type protein (Fig. 2H).
We also tested the expression of the missense mutants recently reported in patients with optic atrophy spectrum disorder (Abrams et al., 2015). Specifically, G249D correlating with mild optic atrophy was comparable in abundance to the wild-type, while E335D and R340C correlating with optic atrophy with progressive axonal neuropathy and cerebellar atrophy appeared less abundant than the wild-type (Fig. 2F). In contrast, the P333L mutant protein correlating with infantile lethal cerebellar atrophy compatible with PCH demonstrated markedly decreased level comparable to the L341P mutant protein (Fig. 2F).
In summary, SLC25A46 is a mitochondrial protein, and the novel missense mutation c.1022T>C leading to L341P in Family 1 markedly decreases the stability of the protein. Instability of the L341P and P333L mutant proteins and N-terminal truncated mutant protein in Family 2 are correlated with a clinical phenotype of congenital lethal PCH on the extreme end of the optic atrophy spectrum disorder.
SLC25A46 localizes to the mitochondrial outer membrane
We performed additional experiments to probe the submitochondrial localization of SLC25A46. Homology studies suggested that SLC25A46 likely localized to the mitochondrial inner membrane (Haitina et al., 2006); however, a weak homology to yeast protein Ugo1, which resides in the mitochondrial outer membrane, indicated the potential for SLC25A46 to localize to the mitochondrial outer membrane (Sesaki and Jensen, 2001). In addition, other carriers such as MTCH1 and MTCH2 reside in the mitochondrial outer membrane (Lamarca et al., 2007; Zaltsman et al., 2010).
Immunoblot analyses of LAN5 cells that were lysed and fractionated showed that, as expected, SLC25A46 fractionated in the 10 000g pellet (P10k) with the mitochondrial marker TIMM23 and was not recovered in the 100 000g pellet (P100k) that contains microsomes (calnexin as control) or supernatant (S100k) that contains soluble proteins (β-tubulin) (Fig. 3A). We used alkaline extraction to test whether SLC25A46 was an integral membrane protein (Fujiki et al., 1982) (Fig. 3B). SLC25A46 was recovered in the pellet fraction, indicating it is an integral membrane protein like MFN2. In contrast, matrix protein LRP130 is soluble and was recovered in the supernatant fraction.
Figure 3.

Localization of SLC25A46 to the outer mitochondrial membrane. (A) Subcellular fractionation of LAN5 cells by differential centrifugation. SM = starting material, P10k = 10 000g pellet, P100k = 100 000g pellet, S100k = 100 000g post-supernatant. Selected antibodies detected SLC25A46, TIMM23 (mitochondria), calnexin (ER, endoplasmic reticulum), and β-tubulin (cytosol). (B) Alkaline extraction in 0.1 M Na2CO3 solution at pH11 and 12 was performed on mitochondria isolated from LAN5 cells. The samples were separated into pellet (P) and supernatant (S) fractions. 1% Triton™ X-100 (TX-100) treatment and the starting material incubated in isotonic buffer were included as controls. Selected antibodies detected SLC25A46, MFN2 (outer membrane), and LRP130 (matrix). (C) Sub-mitochondrial localization was tested by osmotic shock (OS). After lysis of mitochondria in hypotonic buffer, the pellet fraction was collected and subject to proteinase K treatment. As a control, intact mitochondria were treated with protease and the pellet from the osmotic shock fraction was treated with 1% Triton™ X-100. Selected antibodies detected SLC25A46, MFN2 (OM, outer membrane), YME1L (IM, inner membrane), and LRP130 (matrix). (D) Import of radiolabelled SLC25A46, yeast Oac1 (IM), and yeast Tom40 (OM) into HEK293T mitochondria followed by subsequent trypsin treatment as indicated and alkaline extraction. The membrane potential was disrupted by CCCP (−ΔΨ). (E) As in C, isolated mitochondria from HEK293T cells stably expressing 2 × HA-SLC245A46 wild-type (WT) or L341P were treated with proteinase K in isotonic or hypotonic buffer to determine the sub-mitochondrial localization of wild-type and mutant SLC25A46. Controls include TOMM20 (OM), YME1L (IM) and PreP (matrix). (F) As in B, alkaline extraction (pH 12) on isolated mitochondria from stable HEK239T cells expressing 2 × HA-SLC245A46 wild-type (WT) or L341P. (G) Import of radiolabelled wild-type SLC25A46 or L341P mutant into HEK293T mitochondria followed by carbonate extraction and trypsin treatment. CCCP was used to disrupt the membrane potential (−ΔΨ).
Osmotic shock analysis on isolated LAN5 mitochondria indicated that SLC25A46 localized to the mitochondrial outer membrane, like control MFN2 (Fig. 3C). Specifically, treatment of mitochondria with proteinase K led to the digestion of SLC25A46 and outer membrane protein MFN2, but left intact YME1L (inner membrane facing the intermembrane space) and LRP130 (matrix). When the outer membrane was disrupted with hypotonic buffer, SLC25A46, MFN2, and YME1L were degraded, and finally LRP130 was also degraded when samples were treated with detergent to lyse the inner membrane.
To confirm an outer membrane localization, radiolabelled SLC25A46, inner membrane carrier protein yeast Oac1 and yeast Tom40 were imported into isolated HEK293T mitochondria in a time course assay followed by carbonate extraction (Fig. 2D). Samples were then treated with trypsin to remove outer membrane proteins. Again, SLC25A46 and yeast Tom40 imported to the outer membrane but yeast Oac1 translocated into the inner membrane. Thus, SLC25A46, despite homology to other mitochondrial solute carriers in the inner membrane, localizes to the mitochondrial outer membrane, similar to MTCH2 and Ugo1.
The amino acid change at residue 341 in SLC25A46 does not impair localization to the mitochondrial outer membrane
The submitochondrial localization of mutant SLC25A46 L341P was investigated to confirm that the mutant protein assembled as expected. HEK293T cell lines that stably expressed HA-tagged SLC25A46 wild-type or L341P mutant proteins were generated and mitochondria were purified. As in Fig. 3C, osmotic shock experiments were performed in the presence of proteinase K (Fig. 3E). Using an anti-HA antibody, SLC25A46 wild-type and mutant as well as control TOMM20 localized to the mitochondrial outer membrane because the proteins were sensitive to protease when mitochondria were incubated in isotonic buffer. Only when the outer membrane was disrupted with hypotonic buffer was YME1L degraded. In contrast PreP, localized to the matrix, was protected from protease because the inner membrane remained intact. As shown in Fig. 3B, alkaline extraction was performed (Fig. 3F). SLC25A46 wild-type and mutant localized to the pellet fraction, whereas PreP was soluble in the matrix. Finally, we analysed the import of radiolabelled SLC25A46 wild-type and mutant proteins into isolated HEK293T mitochondria in a time course assay followed by carbonate extraction to confirm insertion into the outer mitochondrial membrane (Fig. 3G). After import, the samples were treated with trypsin as indicated, again to confirm insertion into the outer membrane. Both the mutant and wild-type SLC25A46 were imported and assembled into the outer membrane. In summary, SLC25A46 is a mitochondrial outer membrane protein, and the L341P mutation does not preclude the protein from being imported and assembled into the outer membrane.
As SLC25A46 resides in the outer membrane, we investigated the import pathway to determine if the SLC25A46 might follow a pathway similar to Ugo1. Using blue-native gels, we examined assembly of SLC25A46 into complexes in mitochondria from the HEK293T cells stably expressing HA-SLC25A46 (Supplementary Fig. 2A). Mitochondria were solubilized in digitonin, and the lysate was separated on blue native gels. SLC25A46 migrated primarily in a complex of ∼100 kDa and a smaller amount localized to a larger complex of ∼400 kDa. Similarly, with in vitro import assays that were separated on blue native gels, SLC25A46 wild-type migrated primarily in a complex of ∼100 kDa (Supplementary Fig. 3B). Moreover, the mutant L341P assembled in a complex of identical size, albeit in lower abundance. Ugo1 relies on Mim1 and outer membrane receptors Tom70 and Tom71 for import (Papic et al., 2011). Because protein import is highly conserved in yeast and mammalian mitochondria (Bauer et al., 2002), we switched to yeast mitochondria to investigate the outer membrane components required for SLC25A46 import. As in Fig. 3D with HEK293T mitochondria, SLC25A46, yeast Oac1, and yeast Tom40 showed similar import into yeast mitochondria (Supplementary Fig. 2C). The outer membrane receptor requirements for SLC25A46 and Ugo1 import were compared in import assays into mitochondria from yeast strains deleted for MIM1 (Δmim1) and TOM70/TOM71 (Δtom70Δtom71) followed by blue native gel analysis (Supplementary Fig. 2D), SLC25A46 was imported into a ∼100 kDa complex similar to that observed of endogenous SLC25A46 in HEK293T mitochondria (Supplementary Fig. 2A and D). Moreover, import of SLC25A46 and Ugo1 was markedly decreased in mitochondria lacking Mim1 or Tom70/Tom71 (Supplementary Fig. 2D). In summary, SLC25A46 import requirements are similar to Ugo1, supporting SLC25A46 assembly in the outer membrane.
Knockdown of slc25a46 expression in zebrafish embryos disrupts neurodevelopment and mitochondrial fission
To examine the functional consequences of the mutation c.1022T>C, we knocked down endogenous slc25a46 expression in zebrafish embryos (Supplementary Fig. 3) by slc25a46-specific antisense morpholino injection (Fig. 4A). Embryos injected with antisense morpholinos directed against the start codon (AUG) or the splice-donor site of exon 3 (SPL) led to a dose-dependent phenotype of brain maldevelopment in the midbrain as well as the hindbrain (Fig. 4A). Furthermore, there was loss of spinal motor neurons in the more than half of the embryos injected with the AUG (Fig. 4B) but not the SPL morpholinos.
Figure 4.
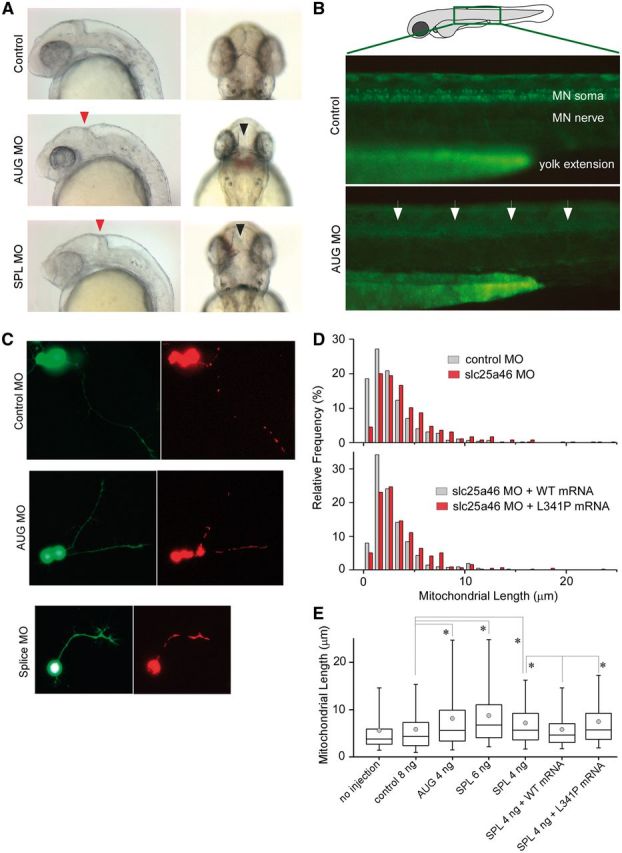
Knockdown of slc25A46 in zebrafish causes brain maldevelopment, loss of spinal motor neurons, and abnormal increase in mitochondrial length in neurites. (A) Zebrafish embryos injected with slc25A46-specific antisense morpholinos AUG (targeted against the start codon) or SPL (targeted against the splice-donor site for exon 3) compared to those injected with non-specific control demonstrate abnormal indentation at the midbrain-hindbrain junction on lateral view (red arrowhead) as well as an abnormal gap between the optic tectum on dorsal view (black arrowhead). (B) Zebrafish embryos from the ET2 line (in which GFP is specifically expressed in the caudal primary motor neurons) injected with AUG morpholino demonstrate under epifluorescent microscopy decreased abundance of spinal motor neurons (arrows) on lateral view compared to embryos injected with control morpholinos. (C) Visualization of dsRed-labelled mitochondria in GFP-labelled neurons isolated from morpholino-injected embryos. Cells were dissociated from the brain dissected from 1–2 days post-fertilization zebrafish embryos injected with different morpholinos and maintained in culture for 1–2 days. (D) Distribution of the length of mitochondria in neurites. AUG morpholino injection led to an increase in length with a rightward shift in distribution compared to the results from control morpholino injection. Co-injection of the wild-type but not the mutant slc25a46 mRNA reverses mitochondrial length. (E) Summary of mitochondrial length measurements in cultured neurons isolated from zebrafish embryos injected with different constructs. Over 100 neurons were used in each condition, with 400–800 mitochondria being measured. Both AUG and SPL morpholino injection led to an increase in the length of mitochondria, which was reversed by the coinjection of the wild-type but not the mutant slc25a46 mRNA. Box = 25–75th percentile; whisker = 5–95th percentile; line = median; circle = mean; *P < 0.0001, two-tailed Mann-Whitney U-test. MO = morpholino.
Although the human SLC25A46 does not share strong homology to the yeast Ugo1 at the protein level, SLC25A46 is similar to Ugo1 in that it has the characteristics of a mitochondrial solute carrier but resides in the outer membrane, which suggested to us that SLC25A46, like Ugo1, may be important in the regulation of mitochondrial dynamics. Given the preferential expression of slc25a46 in the brain and the spinal cord during zebrafish embryonic development (Supplementary Fig. 3), we examined mitochondrial dynamics in dissociated neurons from zebrafish embryos injected with antisense slc25a46-specific AUG or SPL morpholinos, control morpholinos, and/or co-injection with the wild-type or mutant slc25a46 mRNA. We found that mitochondria in cultured neurons from antisense slc25a46-specific morpholino-injected embryos are longer compared to those from neurons isolated from control morpholino-injected embryos (Fig. 4C and D). The increase in the mitochondrial length of the morpholino-injected neurons is reversed by co-injection with the wild-type but not the mutant slc25a46 mRNA (Fig. 4D). Mitochondrial length measurements of neurons from uninjected zebrafish embryos and those injected with morpholinos with or without mRNA are summarized in Fig. 4E. Shown in Fig. 5 are serial live images taken every 2 min of mitochondria in representative neurites in cultured neurons isolated from zebrafish embryos injected with either the control or AUG morpholino, demonstrating unusually large and immobile mitochondria in the AUG morpholino-injected embryos, in contrast to mitochondria of average size that move along the neurites and undergo fusion and fission observed in cultured neurons from control morpholino-injected embryos.
Figure 5.

Knockdown of slc25A46 causes an increase in mitochondrial length in neurites of cultured neurons. Serial live imaging, taken every 2 min, shown from left to right, of dsRed-labelled mitochondria in cultured GFP-labelled neurons isolated from the brain of zebrafish embryos injected with control or AUG morpholino as in Fig. 4C.
Discussion
We report the identification of a homozygous missense mutation c.1022T>C and a homozygous genomic deletion involving exon 1 in SLC25A46 encoding a mitochondrial protein leading to lethal pontocerebellar hypoplasia with apnoea and profound weakness suggestive of PCH1. The extended clinical phenotype of this condition also includes optic atrophy and severe axonal sensorimotor neuropathy distinctive from PCH1, which is characterized by spinal muscular atrophy with intact sensory function. There are other rare subtypes of PCH. PCH3 with optic atrophy and seizures (Rajab et al., 2003) is caused by mutations in the PCLO gene thought to be important in synaptic function (Ahmed et al., 2015). PCH6 with mitochondrial respiratory chain defects is caused by mutations in the mitochondrial arginyl-transfer RNA synthetase gene RARS2 (Edvardson et al., 2007). Our patients did not harbour mutations in the PCH3 and PCH6 causative genes. We were therefore surprised that our investigation converged on the same gene SLC25A46 in which recessive mutations were recently identified in a cohort of patients with optic atrophy and peripheral neuropathy. The SLC25A46-associated syndrome was named optic atrophy spectrum disorder and has since been designated HMSNVIB (hereditary motor and sensory neuropathy type VIB, OMIM #616505), which is characterized by a broad clinical spectrum with early-onset optic atrophy along with peripheral neuropathy and cerebellar degeneration of highly variable age of onset and severity (Abrams et al., 2015).
The homozygous missense mutation c.1022T>C in Family 1 predicts an amino acid alteration p.Leu341Pro that markedly destabilizes the gene product, raising consideration of a loss of function as a disease mechanism in our patients. We were intrigued that a mutation leading to p.Arg340Cys was found in an individual with optic atrophy spectrum disorder who was in his 50 s (Abrams et al., 2015), while our patients with a change in an adjacent amino acid residue p.Leu341Pro died soon after birth. Meanwhile, a mutation leading to p.Pro333Leu was found in an individual who had been tested for mutations causing PCH with profound cerebellar atrophy and early death soon after birth (Abrams et al., 2015). What could be the basis for such a contrast in the phenotypes resulting from changes in neighbouring amino acid residues? In vitro, an obvious difference among mutant proteins appears to be their relative stability: we observed marked instability of mutant proteins p.Leu341Pro and p.Pro333Leu correlating with lethal congenital PCH, in contrast to stable expression of the p.Gly249Asp mutant protein correlating with the least severe manifestations of optic atrophy spectrum disorder, with the other mutant proteins of intermediary stability correlating with intermediary severity. That is, the more stable and abundant the mutant protein, the less severe the clinical manifestations.
The identification of a homozygous 1897-nucleotide genomic deletion spanning exon 1 of SLC25A46 in the index patient in Family 2, with practically identical clinical features as Family 1, strengthens the correlation of PCH with loss of function of the mutant gene product. Furthermore, a novel homozygous splice site mutation in SLC25A46 recently described in optic atrophy spectrum disorder was identified in a newborn with polyhydramnios, respiratory insufficiency, optic atrophy, diffuse cerebral atrophy with severe cerebellar atrophy, and death within days after birth (Nguyen et al., 2016), with identical presentation as our patients. The early lethality in the setting of PCH with optic atrophy and axonal neuropathy likely reflects the critical importance of SLC25A46 that is essential for survival and in both central and peripheral neurodevelopment.
What is the function of SLC25A46? Little was known at the time when we began our investigation, which led us to similar findings and conclusions as Abrams et al. (2015). Knockdown of slc25a46 expression in zebrafish embryos caused brain malformation, spinal motor neuron loss, and poor motility. At the cellular level, we observed abnormally elongated mitochondria in neurites of cultured neurons isolated from the brain of zebrafish embryos injected with slc25a46-specific morpholinos. This phenotype was rescued by co-injection of the wild-type, but not the mutant slc25a46 mRNA. Conversely, overexpression of the wild-type protein led to mitochondrial fragmentation and disruption of the mitochondrial network, suggesting a pro-fission function of SLC25A46.
Although slc25a46-specific morpholino knockdown in zebrafish embryos was an approach used by both our group and Abrams et al. (2015), the two teams examined different cells; Abrams and colleagues were focused on optic atrophy and peripheral neuropathy, whereas we were curious about brain development. Other than the abnormal gap between the optic tectum of the morphant zebrafish embryos noted by both groups, our observations were distinct from those by Abrams et al. (2015). Yet, cellular data from both groups led to the same conclusion that SLC25A46 appeared to be a pro-fission mitochondrial protein, as the most prominent defect in the knockdown experiments appeared to be impaired fission.
In a separate report, we describe the identification of interaction partners of SLC25A46 (Steffen et al., submitted for publication) including mitofilin as reported by Abrams et al. (2015) and the mechanism of turnover of the mutant protein L341P. Efforts are underway to further investigate how specific mutations may disrupt protein function to cause variable clinical phenotypes throughout the life span. Our findings of defects in pro-fission SLC25A46 causing lethal PCH and the observation that defects in fusion-mediating MFN2 could drastically disrupt the development and maturation of cerebellar Purkinje neurons (Chen et al., 2007) emphasize how a fine balance between fission and fusion may be particularly important in cerebellar development and degeneration.
In summary, the investigation of the cause of a lethal congenital pontocerebellar hypoplasia in our patients has led to the identification of SLC25A46 as a new locus for PCH on one extreme of the optic atrophy spectrum disorder/HMSNVIB. We present data for SLC25A46 as a pro-fission component in the regulation of mitochondrial dynamics of critical importance in neurodevelopment that is conserved in human and zebrafish. We propose that the broad clinical severity and manifestations of optic atrophy spectrum disorder may be dictated by the relative stability of SLC25A46; specifically, lethal congenital PCH syndrome appears to correlate with the marked instability and loss of function of the mutant protein. Pleiotropy is a common phenomenon, yet the recognition that defects in SLC25A46 may cause PCH has clinical relevance to a neurologist taking care of a newborn with cerebral malformations and central hypoventilation as well as scientific relevance in the efforts to understand the molecular components and functional roles of mitochondrial fusion and fission in neurodevelopment and neurodegeneration.
Supplementary Material
Acknowledgements
The authors are grateful for the generosity of the families who participated in this study. The sequencing and analytical work were supported by the Bioinformatics and Genomics Core of the UCLA Muscular Dystrophy Core Center within the Center for Duchenne Muscular Dystrophy. We thank Drs Diane M. Papazian and Robert H. Baloh for their critical reading of the manuscript.
Funding
This research was supported by Deutsche Forschungsgemeinschaft (DFG) STE 2045/1-1 to J.S.; Australian National Health and Medical Research Council (NHMRC) Centre for Research Excellence Grant ID 1031893 to M.M.R.; NHMRC Career Development Fellowship GNT1032364 to P.J.L. and Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS; NIH R01 GM61721 and CIRM RT307678 to C.M.K.; NIH R01 NS064183, National Center for Advancing Translational Sciences UCLA CTSI Grant UL1TR000124 and UCLA Children’s Discovery and Innovation Institute Award, and the Gochman Fund to J.C.J.
Supplementary material
Supplementary material is available here.
Glossary
Abbreviation
- PCH
pontocerebellar hypoplasia
References
- Abrams AJ, Hufnagel RB, Rebelo A, Zanna C, Patel N, Gonzalez MA, et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat Genet 2015; 47: 926–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed MY, Chioza BA, Rajab A, Schmitz-Abe K, Al-Khayat A, Al-Turki S, et al. Loss of PCLO function underlies pontocerebellar hypoplasia type III. Neurology 2015; 84: 1745–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth PG. Pontocerebellar hypoplasias: an overview of a group of inherited neurodegenerative disorders with fetal onset. Brain Dev 1993; 15: 411–22. [DOI] [PubMed] [Google Scholar]
- Bauer MF, Hofmann S, Neupert W. Import of mitochondrial proteins. Int Rev Neurobiol 2002;53: 57–90. [DOI] [PubMed] [Google Scholar]
- Boczonadi V, Muller JS, Pyle A, Munkley J, Dor T, Quartararo J, et al. EXOSC8 mutations alter mRNA metabolism and cause hypomyelination with spinal muscular atrophy and cerebellar hypoplasia. Nat Commun 2014;5:4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007; 130: 548–62. [DOI] [PubMed] [Google Scholar]
- Chen Z, Lee H, Henle SJ, Cheever TR, Ekker SC, Henley JR. Primary neuron culture for nerve growth and axon guidance studies in zebrafish (Danio rerio). PLoS One 2013; 8: e57539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claypool SM, Whited K, Srijumnong S, Han X, Koehler CM. Barth syndrome mutations that cause tafazzin complex lability. J Cell Biol 2011; 192: 447–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Shaag A, Kolesnikova O, Gomori JM, Tarassov I, Einbinder T, et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet 2007; 81: 857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki Y, Hubbard AL, Fowler S, Lazarow PB. Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J Cell Biol 1982; 93: 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick BS. Protein import into isolated yeast mitochondria. Methods Cell Biol 1991;34: 389–99. [DOI] [PubMed] [Google Scholar]
- Guthrie C. Messenger RNA splicing in yeast: clues to why the spliceosome is a ribonucleoprotein. Science 1991; 253: 157–63. [DOI] [PubMed] [Google Scholar]
- Haitina T, Lindblom J, Renstrom T, Fredriksson R. Fourteen novel human members of mitochondrial solute carrier family 25 (SLC25) widely expressed in the central nervous system. Genomics 2006; 88: 779–90. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014; 46: 310–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarca V, Sanz-Clemente A, Perez-Pe R, Martinez-Lorenzo MJ, Halaihel N, Muniesa P, et al. Two isoforms of PSAP/MTCH1 share two proapoptotic domains and multiple internal signals for import into the mitochondrial outer membrane. Am J Physiol Cell Physiol 2007; 293: C1347–61. [DOI] [PubMed] [Google Scholar]
- Nguyen M, Boesten I, Hellebrekers D, Mulder-den Hartog NM, de Coo I, Smeets H, et al. Novel pathogenic SLC25A46 splice-site mutation causes an optic atrophy spectrum disorder. Clin Genet 2016. PMID:26951855 [DOI] [PubMed] [Google Scholar]
- Norman RM. Cerebellar hypoplasia in Werdnig-Hoffmann disease. Arch Dis Child 1961; 36: 96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papic D, Krumpe K, Dukanovic J, Dimmer KS, Rapaport D. Multispan mitochondrial outer membrane protein Ugo1 follows a unique Mim1-dependent import pathway. J Cell Biol 2011; 194: 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajab A, Mochida GH, Hill A, Ganesh V, Bodell A, Riaz A, et al. A novel form of pontocerebellar hypoplasia maps to chromosome 7q11-21. Neurology 2003; 60: 1664–7. [DOI] [PubMed] [Google Scholar]
- Renbaum P, Kellerman E, Jaron R, Geiger D, Segel R, Lee M, et al. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet 2009; 85: 281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnik-Schöneborn S, Barth PG, Zerres K. Pontocerebellar hypoplasia. Am J Med Genet C Semin Med Genet 2014; 166C: 173–83. [DOI] [PubMed] [Google Scholar]
- Sesaki H, Jensen RE. UGO1 encodes an outer membrane protein required for mitochondrial fusion. J Cell Biol 2001; 152: 1123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen J, Vashisht A, Wan J, Jen JC, Claypool SM, Wohlschlegel JA, et al. Rapid degradation of mutant SLC25A46 by the ubiquitin-proteasome system results in MFN1/2 mediated hyperfusion of mitochondria. Submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Yourshaw M, Mamsa H, Rudnik-Schöneborn S, Menezes MP, Hong JE, et al. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat Genet 2012; 44: 704–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaltsman Y, Shachnai L, Yivgi-Ohana N, Schwarz M, Maryanovich M, Houtkooper RH, et al. MTCH2/MIMP is a major facilitator of tBID recruitment to mitochondria. Nat Cell Biol 2010; 12: 553–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.