

There are as yet no disease-modifying treatments for Machado-Joseph disease (spinocerebellar ataxia type 3). Costa et al. report that the atypical antipsychotic aripiprazole reduces levels of the mutant protein ATXN3 in animal models of this disease. Aripiprazole may have therapeutic potential for Machado-Joseph disease and possibly other neurodegenerative proteinopathies.
Keywords: spinocerebellar ataxia type 3, Machado-Joseph disease, therapeutics, drug screen, neurodegeneration
There are as yet no disease-modifying treatments for Machado-Joseph disease (spinocerebellar ataxia type 3). Costa et al. report that the atypical antipsychotic aripiprazole reduces levels of the mutant protein ATXN3 in animal models of this disease. Aripiprazole may have therapeutic potential for Machado-Joseph disease and possibly other neurodegenerative proteinopathies.
Abstract
No disease-modifying treatment exists for the fatal neurodegenerative polyglutamine disease known both as Machado-Joseph disease and spinocerebellar ataxia type 3. As a potential route to therapy, we identified small molecules that reduce levels of the mutant disease protein, ATXN3. Screens of a small molecule collection, including 1250 Food and Drug Administration-approved drugs, in a novel cell-based assay, followed by secondary screens in brain slice cultures from transgenic mice expressing the human disease gene, identified the atypical antipsychotic aripiprazole as one of the hits. Aripiprazole increased longevity in a Drosophila model of Machado-Joseph disease and effectively reduced aggregated ATXN3 species in flies and in brains of transgenic mice treated for 10 days. The aripiprazole-mediated decrease in ATXN3 abundance may reflect a complex response culminating in the modulation of specific components of cellular protein homeostasis. Aripiprazole represents a potentially promising therapeutic drug for Machado-Joseph disease and possibly other neurological proteinopathies.
Introduction
Machado-Joseph disease (MJD), also known as spinocerebellar ataxia type 3 (SCA3), is a fatal neurodegenerative disease that currently lacks disease-modifying treatment. The most prevalent dominant hereditary ataxia (Ruano et al., 2014), MJD is characterized by progressive ataxia, ophthalmoplegia and pyramidal signs often accompanied by extrapyramidal signs (Coutinho and Andrade, 1978; Coutinho and Sequeiros, 1981; Paulson, 2007). These clinical features reflect neuronal degeneration and pathological changes in the cerebellum, brainstem, substantia nigra, thalamus, basal ganglia, and spinal cord (Coutinho and Andrade, 1978; Coutinho and Sequeiros, 1981; Schols et al., 2004; Rub et al., 2008).
MJD is one of nine known polyglutamine (polyQ) diseases caused by expanded CAG repeats that encode abnormally long polyQ tracts in the disease proteins. In this disease, the polyQ expansion occurs near the carboxyl-terminus of ATXN3, a deubiquitinase encoded by the ATXN3 gene (Kawaguchi et al., 1994). While normal ATXN3 alleles contain 12–44 CAG repeats, disease alleles are expanded to ∼60–87 triplets (Maciel et al., 2001; Lima et al., 2005). The polyQ expansion in ATXN3 increases its propensity to aggregate leading to the formation of intracellular aggregates (Ellisdon et al., 2006; Costa Mdo and Paulson, 2012). These aggregates are found primarily in the nuclei of neurons (Paulson et al., 1997) but also occur in the cytoplasm and neurites (Hayashi et al., 2003; Yamada et al., 2004, 2008). Though the full biological function of ATXN3 remains unknown, it is known to regulate the stability of proteins involved in diverse pathways (Todi and Paulson, 2011; Costa Mdo and Paulson, 2012). ATXN3 carrying an expanded polyQ tract becomes neurotoxic and is known to trigger several pathogenic cascades, but the primary event leading to MJD pathogenesis is yet unknown (Paulson, 2007; Matos et al., 2011; Costa Mdo and Paulson, 2012). While many therapeutic strategies targeting pathogenic pathways or mutant ATXN3 expression/stability have been proposed, none has yet advanced to human clinical trials (Di Prospero and Fischbeck, 2005; Shao and Diamond, 2007; Chen et al., 2008; Chou et al., 2010; Menzies et al., 2010; Nascimento-Ferreira et al., 2011; Shakkottai et al., 2011; Silva-Fernandes et al., 2014).
Here, we sought to identify small molecules that reduce levels of mutant ATXN3 protein as potential therapeutic agents for MJD. Reducing the abundance of mutant ATXN3 or its oligomers is a compelling therapeutic approach because these species represent upstream targets in the pathogenic cascade. The success of such a strategy does not require a detailed understanding of disease mechanisms. Moreover, the fact that mice lacking ATXN3 appear normal (Schmitt et al., 2007) suggests that strategies to reduce total levels of ATXN3 (i.e. normal and mutant) should not result in adverse consequences due to loss of ATXN3 function. Although we and others have developed gene-silencing strategies that effectively reduce levels of mutant ATXN3 in MJD mouse models (do Carmo Costa et al., 2013; Nobrega et al., 2013; Rodriguez-Lebron et al., 2013), achieving broad brain delivery of silencing agents for optimal efficacy in patients remains a challenge (do Carmo Costa et al., 2013). Accordingly, we generated a cell-based assay to screen small molecules that reduce ATXN3 levels to find broadly deliverable oral agents that might have a disease-modifying effect. Using this assay to screen a library of small molecules including 1250 Food and Drug Administration (FDA)-approved drugs followed by subsequent testing in several MJD models, we found that the atypical antipsychotic aripiprazole suppresses soluble ATXN3 levels in Drosophila and mouse models of disease. As a suppressor of aggregation-prone protein abundance, aripiprazole may hold promise as a repurposed drug for MJD and possibly other neurodegenerative proteinopathies.
Materials and methods
Experimental design
The goal of this study was to identify a novel therapeutic small molecule for MJD. We carried out an unbiased screen to identify compounds that can reduce levels of ATXN3 in mammalian cells. Following secondary screens, the best hit was tested in transgenic mice that model this disease (Cemal et al., 2002). Groups of mice were assembled randomly and drug and vehicle were administered blindly. Building on documented ATXN3 levels assessed by immunoblotting, sample size was calculated assuming a power of 0.99 and P < 0.05 (two-tailed) to achieve 50% of ATXN3 reduction (n = 9).
Plasmids and cell culture
Full-length ATXN3 (accession number ABS29269) carrying either a normal or an expanded polyQ tract (Q26 or Q81) was subcloned in the vector pcDNA3.1 FLAG.firefly Luciferase. The mammalian expression plasmids pcDNA.FLAG.ATXN3Q26.Luc and pcDNA.FLAG.ATXN3Q81.Luc were confirmed by sequencing and shown to express N-terminal FLAG-tagged ATXN3Q26 or ATXN3Q81 fused with firefly Luciferase (Luc), respectively. These neomycin-resistant plasmids were transfected into human embryonic kidney (HEK) 293 cells using Lipofectamine® LTX (Invitrogen) for 4 h, after which medium was replaced by growth medium [Dulbecco’s modified Eagle medium (DMEM)/10% foetal bovine serum (FBS)/ 1% penicillin/streptomycin]. On the following day, medium was replaced for selection medium consisting of growth medium with 1000 mg/ml geneticin, which was shown to kill all the parent HEK 293 cells in a killing curve carried out just before the transfection experiments. After 1 month of passage in selection medium, stably transfected clones for each cell line (293.ATXN3Q26Luc and 293.ATXN3Q81Luc) were pooled and frozen in liquid nitrogen. Stably transfected cell lines were maintained in selection medium except during treatment with specific compounds that were diluted in growth medium.
Small molecule screen
Primary screen
The screen was carried out at the Center for Chemical Genomics (CCG) of the University of Michigan. We screened 2880 small molecules including the Microsource Spectrum Collection of 2000 compounds comprising 1000 drugs (800 of which are FDA-approved), the NCC 450 FDA-approved drugs, and another collection of 430 natural products with known biological activity available through the CCG. Stably transfected 293.ATXN3Q81Luc cells were plated in columns 1 to 22 of 384-well plates (2.5 × 103 cells/well) in a final volume of 40 μl of DMEM/10%FBS/1% penicillin/streptomycin. Parental HEK293 cells were plated in columns 23 and 24, and a total of nine plates were incubated at 37°C/5% CO2. After 24 h, columns 3 to 22 were spotted with 0.2 μl of library compounds in dimethyl sulphoxide (DMSO; final concentration of 8 μM) using a Biomek FX laboratory automation workstation with high-density replication (pin tool). Columns 1–2, and 23–24 were spotted with 0.2 μl of DMSO, corresponding to negative and positive controls, respectively. After 48 h of incubation at 37°C/5% CO2, media was aspirated and 10 μl of Steady-Glo® Luciferase Assay System (Promega) was added to each well, and after a 10 min incubation at room temperature the activity of firefly luciferase was measured in a PHERAstar® plate reader (BMG Labtech).
Actives and triage criteria
Actives were defined using standard deviation (SD) values computed by the MScreen Database (Jacob et al., 2012) for the negative control on a plate by plate basis. Samples with a SD by plate ≥3 were defined as actives. This criterion produced 162 active samples. Additional triage criteria excluded 28 molecules that were active in three or more additional luciferase-based assays carried out at the CCG, 11 molecules that represented general promiscuity [(% assays ≥ 3 SD for negative control) >30.0%], and three compounds showing black structure alert. A total of 120 small molecules were selected for further dose response titration.
Concentration response screen
The 120 small molecules selected for dose response were screened in duplicate by direct spotting (60–0.47 μM). Two sets of six plates were prepared in parallel following the same protocol described above. Forty-eight hours after compound addition, one set of plates was assayed for firefly luciferase activity as described above. The other set of plates was assayed for cellular viability by adding 5 μl of alamarBlue® (Invitrogen) to each well, incubating 90 min at 37°C/5% CO2, and measuring fluorescence (excitation 560 nm, emission 600 nm, cut-off 590 nm) in a SpectraMax® M5 microplate reader (Molecular Devices).
Fresh powder compounds
Nine of the 10 selected compounds from the dose response screen were available for purchase from vendors and were acquired as fresh powders: artemether, CAS 71963-77-4 (A29361, Sigma Aldrich); aripiprazole, CAS 129722-12-9 (565444, AK Scientific); AM251, CAS 183232-66-8 (1117, Tocris Bioscience); cefamandole sodium, CAS 30034-03-8 (C7145, Sigma Aldrich); clotrimazole, CAS 23593-75-1 (C6019, Sigma Aldrich); mifepristone, CAS 84371-65-3 (M8046, Sigma Aldrich); monensin sodium, CAS 22373-78-0 (M5273, Sigma Aldrich); salinomycin sodium, CAS 55721-31-8 (anhydrous) (46729, Sigma Aldrich); tranilast, CAS 53902-12-8 (T0318, Sigma Aldrich).
Mouse procedures and treatment of YACMJD84.2 mice with aripiprazole
All mouse procedures were approved by the University of Michigan Committee on the Use and Care of Animals (Protocol PRO00005029). A colony of YACMJD84.2 transgenic mice has been maintained by us in the same animal husbandry since 2007. Mice were housed in cages with a maximum number of five animals and maintained in a standard 12-h light/dark cycle with food and water ad libitum. Genotyping was performed using DNA isolated from tail biopsy at the time of weaning, as previously described (Cemal et al., 2002) and genotypes of all studied mice were confirmed using DNA extracted from tails collected post-mortem. For mouse treatments, aripiprazole was dissolved in DMSO/Tween-80 in a 1:1 ratio and its pharmacokinetic parameters were determined for intraperitoneal injections: 12-week-old wild-type littermates from our YACMJD84.2 colony were intraperitoneally injected with aripiprazole (15 mg/kg at 20 ml/kg in 96% saline/2% DMSO/2% Tween-80 as vehicle), and plasma and brain were collected at four time points after injection (0.5, 4, 8 and 24 h; two females per group). For the subchronic treatment, a group of 12–14 week old hemizygous YACMJD84.2 (Q84) mice were injected (intraperitoneally) daily with aripiprazole 15 mg/kg, 20 ml/kg or vehicle for 10 days (n = 9 mice per group; five females and four males). Five hours after the final injection, mice were anaesthetized with ketamine/xylazine and perfused transcardially with phosphate-buffered saline (PBS; for RNA and protein studies) and brains were immediately placed on dry ice and stored at −80°C. Another group of 12–14 week old Q84 mice were treated with aripiprazole or vehicle and sacrificed in the same way [n aripiprazole = 10 (four females and six males); n vehicle = 7 (two females and five males)] but the right brain hemisphere was stored at −80°C and the left hemisphere was fixed in 4% paraformaldehyde for at least 3 days, and then impregnated in 30% sucrose (for immunofluorescence).
Organotypic mouse brain slice cultures
Sagittal brain slices (300 -μm thick) from Q84 mice (8–9 weeks old) were prepared as previously reported (Shakkottai et al., 2011). Nine slices per brain were used in each set of cultures. After a quick wash in the corresponding medium, each slice was placed on a cell culture insert (0.4 μm pore size, 30 mm diameter; Millipore), which was previously placed on a well (6-well plate) containing 1.2 ml of culture medium (50% minimal essential medium with Earle’s salts, 25% horse serum, 25% Hank’s balanced salts solution, 25 mM HEPES, 2 mM L-glutamine, 6.5 mg/ml glucose) and the tested compound or its vehicle. After 48 h of incubation at 37°C/5% CO2, brain slices were assessed for ATXN3 levels by immunoblotting or immunofluorescence and for cell viability. For immunoblotting, each slice was macrodissected into separate cerebellum and brainstem that were immediately frozen at −80°C in 80 μl and 150 μl, respectively, of cold RIPA buffer containing protease inhibitors (Complete, Roche Diagnostics). Cell viability was assessed on treated slices by incubation in propidium iodide 1 μg/ml in culture medium for 1 h. Slices were then mounted in ProLong® Gold Antifade Reagent (Invitrogen) and imaged using a FV500 Olympus confocal microscope.
Western blotting
Protein lysates from cell cultures, mouse brain slices or mouse brains were obtained by lysis in RIPA buffer containing protease inhibitors (Complete, Roche Diagnostics), followed by sonication and centrifugation. The supernatants (soluble protein fractions) were collected, total protein concentration was determined using the BCA method (Pierce) and then stored at −80°C. Total proteins (50 μg from cell and slice cultures or 75 μg from brain regions of mice) were resolved in 10% sodium dodecyl sulphate-polyacrylamide electrophoresis (SDS-PAGE) gels, and corresponding polyvinyl difluoride membranes were incubated overnight at 4°C with primary antibodies: mouse anti-ATXN3 (1H9) (1:2000; MAB5360, Millipore), goat anti-Luciferase (1:500; G7451, Promega), mouse anti-FLAG (M5) (1:500; IB13091, Sigma), rabbit anti-LC3 (1:500; PM036, MBL International Corporation), mouse anti-HSP90β (1:1000; ADI-SPA842, Enzo Life Sciences), rabbit anti-HSP90α (1:1000; ab2928, Abcam), mouse anti-HSP70 (1:500; SPA810, Enzo Life Sciences), rabbit anti-HSP40 (1:1000; #4868, Cell Signaling), rabbit anti-HSP25 (1:1000; ADI-SPA801, Enzo Life Sciences), rabbit anti-HSF1 (1:1000, ADI-SPA-901, Enzo Life Sciences), rabbit anti-ubiquitin (1:1000, Z 0458, Dako), rabbit anti-RAD23A (1:5000; TA307264, Origene), rabbit anti-RAD23B (1:2000; A302-306 A, Bethyl Labs), rabbit anti-α-Tubulin (11H10) (1:10000; #2125, Cell Signaling), and mouse anti-GAPDH (1:1000; MAB374, Millipore). Pellets corresponding to insoluble protein fractions were resuspended in 100 μl of Laemmli buffer 2× and boiled for 10 min. Insoluble proteins were then quantified and 100 μg were loaded in a filter trap assay apparatus and transferred to a nitrocellulose membrane (0.20 μm pore) that was incubated overnight with rabbit anti-MJD antibody (1:5000) at 4°C. Bound primary antibodies were visualized by incubation with a peroxidase-conjugated anti-mouse or anti-rabbit secondary antibody (1:10000; Jackson Immuno Research Laboratories) followed by treatment with the ECL-plus reagent (Western Lightning®, PerkinElmer) and exposure to autoradiography films. Band intensity was quantified by densitometry using ImageJ.
Quantitative reverse transcriptase polymerase chain reaction
Total RNA from brainstem fractions of mice treated with aripiprazole or vehicle was obtained by an initial extraction using TRIzol® Reagent (Invitrogen) followed by purification using the RNeasy® mini kit (Qiagen) following the manufacturer’s instructions. Reverse transcription of 1.5 μg of total RNA per sample was performed using the iScript™ cDNA synthesis kit (Bio-RAD). Human ATXN3 and mouse Atxn3, Drd2, 5HT1A, 5HT2A and Gapdh (housekeeping) transcript levels were accessed by quantitative real-time polymerase chain reaction as previously reported (do Carmo Costa et al., 2013), using primers described in Supplementary Table 1. Relative gene expression was determined using the ΔΔCT method, normalizing for Gapdh mRNA levels.
Immunofluorescence, quantification of nuclear ATXN3 and counting of ATXN3-positive puncta
Brains from mice perfused with 4% paraformaldehyde were post-fixed overnight at 4°C in the same fixative, immersed in 30% sucrose/PBS, and sectioned on a sledge microtome (SM200R Leica Biosystems). Free-floating 40 μm sagittal sections were collected and stored in cryoprotectant solution at −20°C. Brain sections from treated mice or brain slices from organotypic cultures processed for immunofluorescence were initially subjected to antigen retrieval and incubated using the Vector® M.O.M.™ immunodetection kit (Vector Laboratories). For double or single immunofluorescence, sections were incubated with mouse anti-ATXN3 (1H9) (1:1000; MAB5360 Millipore) and rabbit anti-NeuN (1:1000; ABN78 Millipore), and then incubated with the corresponding secondary Alexa Fluor® 488 and/or 568 antibodies (1:1000; Invitrogen). All sections were then stained with DAPI (Sigma), mounted with ProLong® Gold Antifade Reagent (Invitrogen), and imaged using a FV500 Olympus confocal microscope. Single-plan images of ventral pons from mice treated with aripiprazole or vehicle were acquired using a 60× water objective. Neuronal nuclear ATXN3 was quantified using ImageJ by quantifying ATXN3 fluorescence in DAPI and NeuN double-positive nuclei. CTCF was obtained by measuring ATXN3 fluorescence in 15 nuclei/image field/mouse and normalizing for nuclei area using equation CTCF = fluorescence intensity − (nucleus area × mean fluorescence of background readings). ATXN3 positive puncta were quantified using ImageJ by assessing particle fluorescence (applying the triangle threshold) in the same image fields described above.
Treatment of MJD flies with aripiprazole
Drosophila stocks were reared on standard cornmeal media at 25°C in diurnal environments with ∼60% humidity. New fly lines were generated using the site-specific pHiC31 integration system (Keravala and Calos, 2008) into site attP2 of the third chromosome of the fruit fly. Full-length human ATXN3 cDNA with 77 CAG was cloned into pWalium10-moe (Perrimon Lab, Harvard Medical School). We generated flies that carry ATXN3 in site attP2 or the empty vector control into the same site as an isogenic control line. Injections were done by the Duke University Model Organism core into y[1], w[*]; +; attP2 (landing site). For aripiprazole treatments, instant media (Nutri-Fly Instant, Genesee Scientific) was prepared with vehicle (1:1 DMSO–Tween-80, Sigma Aldrich) or vehicle with compound (aripiprazole, AK Scientific) at 50 µM. Adult flies of the following genotypes, w[*]; sqh-Gal4/+; UAS-ataxin-3Q77/+ or w[*], sqh-Gal4/+; Empty-Vector-Control/+ were switched from standard media to instant media as soon as they eclosed from their pupal case (Day 0), and thereafter maintained on instant media with vehicle compound. Media was switched every 3 days, and death was noted daily.
Recombinant protein expression and purification
pGEX-6P1 plasmids encoding GST-ATXN3Q26 and GST-ATXN3Q55 were transformed in Rosetta (DE3) BL21 cells. Colonies grown overnight on LB/ampicillin/chloramphenicol plates at 37°C were resuspended in 150 ml of the same selection medium and incubated at 37°C, 230 rpm for 2 h. Bacterial cultures of 1 l of medium were prepared by inoculating 50 ml of the pre-culture and incubating at 37°C, 230 rpm until reaching an OD600 of 0.6–0.8. Expression of fusion proteins was then induced by adding 1 mM isopropyl-1-thio-D-galactopyranoside for 3 h at 37°C. Cells were collected by centrifugation and stored at −20°C. Cell pellets were resuspended in 20 ml of lysis buffer [150 mM NaCl, 50 mM Tris (pH7.5), 0.5% NP-40, protease inhibitors (COMPLETE, Roche Diagnostics), phenylmethane sulphonyl fluoride (PMSF), lysozyme], incubated on ice for 30 min, additionally lysed by sonication, and finally centrifuged at 15000 rpm, 20 min, at 4°C. The supernatants were collected and incubated with 1 ml of glutathione Sepharose® beads (GE Healthcare) for 3 h at 4°C, with rotation. Beads were then washed in cold PBS, resuspended in 5 ml of PBS containing 40 μl of PreScission Protease (2000 units/ml, GE Healthcare) and incubated at room temperature for 15 min. Cleaved ATXN3 was collected in the supernatant after centrifugation at 1200 rpm for 5 min. Additional ATXN3 was recovered from beads after three subsequent steps of resuspension in PBS, incubation at room temperature and centrifugation. ATXN3Q26 and ATXN3Q55 solutions were concentrated in Ultra-15 centrifugal filter units (Amicon) and proteins were purified by fast protein liquid chromatography (FPLC) using a Superdex™ 200 column (GE healthcare) and 50 mM Na2PO4, 100 mM NaCl, 1 mM NaN3 (pH 7.4) buffer. Chromatography fractions were analysed by SDS-PAGE and the ones containing pure proteins were concentrated and protein concentration was determined in the Nanodrop (Thermo Scientific) by absorption at 280 nm.
ATXN3 fibril formation
Thioflavin T assay
Solutions of ATXN3Q26 or ATXN3Q55 at a final concentration of 10 μM were prepared in the presence of aripiprazole 400 μM or vehicle in 25 mM Na2PO4, 200 mM NaCl, thioflavin T 10 μM, pH7.4. Samples and blank control (only buffer) (110 μl) were dispensed in each well of a black/clear flat bottom 96-well plate (Corning), which was sealed and incubated at 37°C with agitation in a FLUOstar Omega (BMG Labtech Inc) plate reader. Fluorescence of three replicates of each sample was measured every 10 min for up to 5 days. The emission and excitation wavelengths of the filter were 440 nm and 490 nm, respectively, and readings were taken using 90% gain adjustment. Values for protein solutions were normalized to readings of blank buffer.
Native gel electrophoresis
ATXN3Q26 and ATXN3Q55 protein solutions (10 μl of each sample) were monitored before and after the fibrillation assay in the presence of aripiprazole or vehicle using a NativePAGE™ Novex® Bis-Tris gel system (Life technologies) following the manufacturer’s protocol.
Transmission electron microscopy
Negatively stained specimens for transmission electron microscopy (TEM) were prepared by applying 5 μl of sample on hydrophilic 400 mesh carbon-coated support films mounted on copper grids (Ted Pella, Inc.). The samples were allowed to adhere for 4 min, rinsed twice with distilled water, and stained for 60–90 s with 1% uranyl acetate (Ted Pella, Inc.). Samples were imaged at an accelerating voltage of 60 kV in a Philips CM-100 microscope.
Statistical analysis
Levels of proteins and transcripts, and fluorescence intensity were compared using Student’s t-test (comparison of two groups) whenever distributions were normal and homogeneous. In the other cases, comparisons were assessed using the non-parametric Mann-Whitney U-test. Fly survival was analysed using Kaplan-Meier curves and the Log-rank Mantel-Cox test was used to compare survival curves. A P < 0.05 was considered statistically significant in all analyses. Data were analysed using IBM SPSS Statistics 22 software.
Results
Identification of small molecules that reduce levels of mutant ATXN3 in a cell-based assay
We developed cell-based assays to identify small molecules that reduce levels of ATXN3. We generated HEK 293 cell lines that stably express FLAG-tagged full-length human ATXN3 with a normal or expanded polyQ tract (Q26 or Q81) fused to firefly luciferase (Luc)—termed ATXN3Q26.Luc and ATXN3Q81.Luc, respectively (Fig. 1A and B). In this cell assay, levels of ATXN3/Luciferase fusion proteins are measured by chemiluminescence. We used the ATXN3Q81.Luc cells in a 384-well format to screen 2880 small molecules, including 1250 FDA-approved drugs, through the CCG of the University of Michigan. These molecules, comprising 2402 unique chemical structures, were screened at 8 μM for 48 h of treatment (average plate Z factor 0.81) (Fig. 1C and D). Among 162 actives with standard deviation by plate of ≥3, we selected 120 compounds for dose response screen (Fig. 1E) (see ‘Materials and methods’ section for triage criteria). Luminescence and viability dose response screen were run in parallel using duplicates of eight concentrations for each molecule (range 0.47–60 μM). Ten structurally diverse compounds met criteria for follow-up screens (IC50 < 100 μM, viability > 70%, and luminescence inhibition ≥20%), nine of which were available for purchase from vendors (Fig. 1E and Supplementary Fig. 1).
Figure 1.
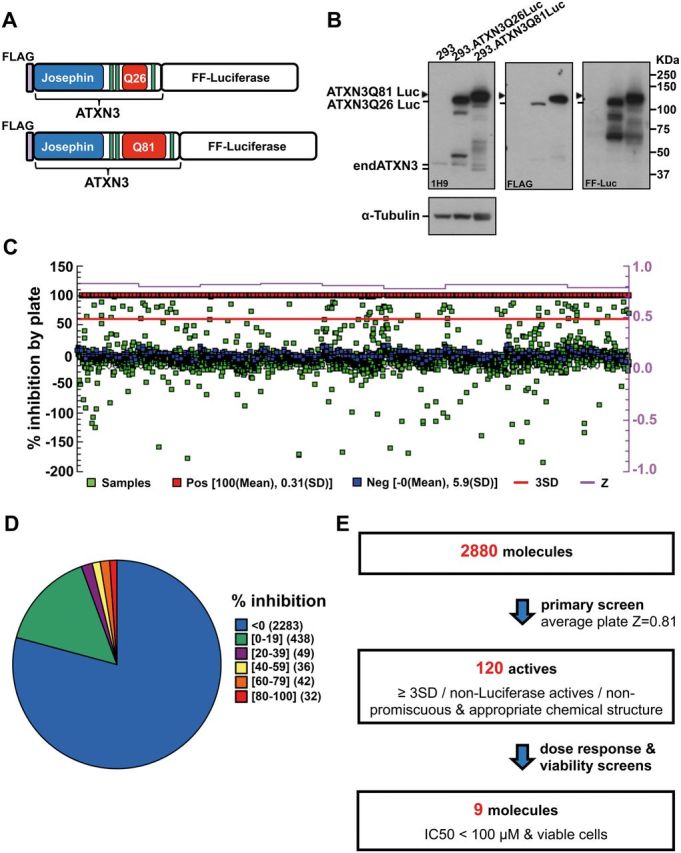
Cell-based screen identifies small molecules that reduce levels of mutant ATXN3. (A) Schematic of ATXN3 fusion proteins expressed in stably transfected HEK 293 cell lines. (B) Immunoblots using anti-ATXN3 (1H9), anti-Luciferase (Luc) and anti-FLAG antibodies show the expression of ATXN3 fusion proteins in 293.ATXN3Q26Luc and 293.ATXN3Q81Luc cell lines. (C) Schematic view of the primary high throughput screen of 2880 small molecules using 293.ATXN3Q81Luc cells. Percentage of luminescence inhibition by plate (nine plates) is shown relative to the positive control (red squares) corresponding to parental HEK 293 cells treated with vehicle (DMSO). 293.ATXN3Q81Luc cells treated with small molecules at 8 μM (samples) are represented as green squares, and cells treated with DMSO (negative control) as blue squares. Z-factor is also represented for each plate in purple (average plate Z = 0.81). (D) Distribution of compounds in primary screen sorted by percentage of luminescence inhibition (n = 2880). (E) Stepwise strategy followed to select nine small molecules that reduce levels of mutant ATXN3 for follow-up studies. FF = firefly.
These nine compounds, showing an IC50 that ranged from 0.2 to 50.1 µM (Supplementary Fig. 1), were tested in independent dose-response experiments in both 293.ATXN3Q26Luc and 293.ATXN3Q81Luc cell lines, with the efficacy of each molecule assessed by measuring ATXN3 levels by immunoblotting. Five of the nine tested compounds (salinomycin sodium, AM251, aripiprazole, clotrimazole and mifepristone) were confirmed to decrease levels of ATXN3Q81.Luc fusion protein (Fig. 2A and B). These molecules reduce ATXN3 levels in a polyQ-length independent manner, as they also reduce the amount of non-expanded ATXN3Q26Luc (data not shown) and of endogenous ATXN3 (Fig. 2A and C), further indicating that they can act on ATXN3 expressed at physiological levels.
Figure 2.

Five small molecules decrease levels of expanded ATXN3 in confirmation screens using 293.ATXN3Q81.Luc cells. (A) Representative anti-ATXN3 immunoblots show the efficacy of sodium salinomycin (Na Salinomycin), AM251, aripiprazole, clotrimazole and mifepristone to reduce the amount of mutant ATXN3 fusion protein after 48 h treatment of 293.ATXN3Q81Luc cells. Compounds were dissolved in DMSO except aripiprazole, which was dissolved in 1:1 DMSO/Tween-80. Quantification of bands corresponding to ATXN3Q81Luc and endogenous ATXN3 (endATXN3) is shown in B and C, respectively. Bars represent the mean percentage of each protein relative to vehicle-treated cells and normalized to α-tubulin (±SEM) in three independent experiments. Comparisons between cells treated with a specific compound concentration and cells treated with vehicle were performed using Student’s t-test with statistical significance, as indicated: *P < 0.05 and **P < 0.01.
Aripiprazole, AM251 and salinomycin sodium reduce human mutant ATXN3 in brain slice cultures from YACMJD84.2 transgenic mice
To assess the efficacy of these five compounds to reduce ATXN3 levels in the mammalian brain we performed secondary screens in organotypic brain slice cultures derived from hemizygous YACMJD84.2 (Q84) transgenic mice. These mice harbour the full-length human ATXN3 disease gene with an expanded repeat of 84 CAGs (Cemal et al., 2002) and therefore express all human pathogenic ATXN3 isoforms, the precise target in patients with MJD.
In sagittal brain slices treated for 48 h, aripiprazole, AM251 or salinomycin sodium reduced levels of human mutant ATXN3Q84 and mouse ATXN3 in the cerebellum and brainstem, the two most affected brain regions in patients with MJD (Fig. 3A–C). In contrast, clotrimazole and mifepristone did not alter ATXN3 levels (Supplementary Fig. 2). Treatments did not affect slice viability assessed by propidium iodide uptake (Supplementary Fig. 3) except for salinomycin sodium at 10 μM. Aripiprazole, AM251 and salinomycin sodium are structurally unrelated and have different pharmacological properties: aripiprazole (PubChem CID 60795) is an atypical antipsychotic agent; AM251 (PubChem CID 2125) is a cannabinoid receptor 1 (CB1) antagonist; and salinomycin sodium (PubChem CID 5748657) is an antibacterial and coccidiostat compound with selective toxicity against cancer stem cells. Because the goal of this small molecule screen is to identify a potential therapeutic agent for MJD we selected the sole FDA-approved drug among these three, aripiprazole, for further in vivo testing in fly and mouse models of this disorder.
Figure 3.

Sodium Salinomycin, AM251, and aripiprazole reduce human mutant ATXN3 levels in organotypic brain slice cultures from YACMJD84.2 transgenic mice (Q84). (A) Anti-ATXN3 immunoblots show that sodium salinomycin (Na Salinomycin), AM251 and aripiprazole decrease human mutant ATXN3 levels in brainstem and cerebellar fractions of brain slices from hemizygous YACMJD84.2 mice (Q84). Sagittal brain slices (300 μM) from the same mouse were cultured with specific concentrations of the compound or its vehicle at 37°C/5% CO2. After 48 h of treatment, brainstem and cerebellar regions from each slice were processed separately. Quantification of bands corresponding to human mutant ATXN3 and mouse ATXN3 shown in B and C, respectively. Bars represent the mean percentage of protein relative to levels in vehicle-treated slices and normalized to α-tubulin (±SEM) for three independent experiments using different mice. Comparison between slices treated with a specific compound/concentration and slices treated with vehicle was performed using Student’s t-test with statistical significance, as indicated: *P < 0.05 and **P < 0.01.
Aripiprazole delays onset of mutant ATXN3-mediated toxicity in MJD flies
To test the efficacy of aripiprazole in vivo, we generated novel transgenic Drosophila lines that express full-length human ATXN3 with a pathogenic polyQ tract of 77 glutamines (MJD) through the Gal4-UAS system of targeted expression. When the sqh-Gal4 driver expresses mutant UAS-ATXN3 throughout Drosophila, MJD flies have a markedly shortened lifespan (mean survival 11.5 days ± 0.376) compared to flies containing the empty vector control (mean survival 50.5 ± 1.041 days) (Fig. 4A) inserted at the same chromosomal site as ATXN3 (see ‘Materials and methods’ section).
Figure 4.
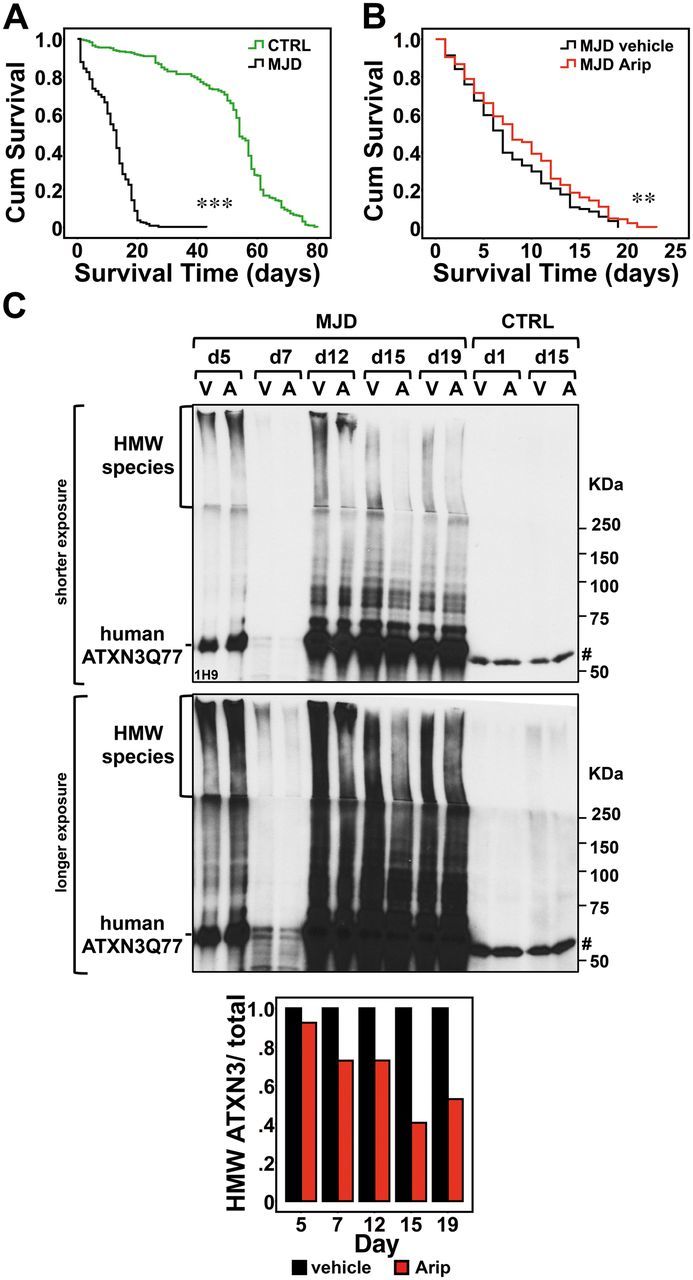
Aripiprazole modestly enhances the longevity of flies expressing mutant ATXN3 and reduces HMW ATXN3 species. (A) We generated new transgenic flies that express full-length human ATXN3 with a pathogenic expansion of 77 repeats (MJD). Kaplan-Meier survival curves show that, when ATXN3 expression is driven throughout the fly by the sqh-Gal4 driver, MJD flies (n = 318) have a markedly shortened lifespan compared to flies containing empty vector control (CTRL) (n = 284). Genotypes: w*; sqh-Gal4/+; Empty-Vector-Ctrl/+, w*; sqh-Gal4/+; UAS-ATXN3Q77(MJD)/+. (B) Upon eclosion, MJD flies were placed in instant formula food containing either the vehicle (1:1 DMSO:Tween-80) or aripiprazole (50 μM). Aripiprazole-treated flies (Arip) (n = 242) show a modest but significant increase in mean survival compared to vehicle-treated flies. Genotype: w*; sqh-Gal4/+; UAS-ATXN3Q77(MJD)/+. (C) ATXN3 immunoblotting of lysates from whole flies reveals decreased HMW ATXN3 species in flies treated with aripiprazole (A) compared to flies treated with vehicle (V) (n = 10 per group, except n = 3 for Day 7). Histogram shows the relative amount of HMW ATXN3 species to total ATXN3 quantified by band intensity shown in the immunoblot. Hash symbol represents a non-specific ATXN3 band. Kaplan-Meier survival curves were compared using the Log-Rank Mantel-Cox test. **P < 0.01 and ***P < 0.001.
To mirror the treatment in patients with MJD, which would start in adult life, we started to treat MJD flies upon eclosion from the pupal case (Day 0 in Fig. 4A and B) by placing them in quick formula food containing either vehicle or aripiprazole (50 μM, the effective dosage in mouse brain slice cultures). At least 200 flies in groups of 9–17 flies per treatment vial were monitored. Aripiprazole-treated MJD flies showed increased mean survival of 1.3 days (9.0 ± 0.367 days) compared with vehicle-treated MJD flies (7.7 ± 0.343 days) (Fig. 4B). ATXN3 immunoblotting of protein lysates from whole flies (n = 10 per group, except n = 3 for Day 7) revealed that aripiprazole decreases high molecular weight (HMW) ATXN3 species to 0.73, 0.41 and 0.53 of levels in vehicle-treated flies at Days 12, 15 and 19, respectively (Fig. 4C), concomitant with the increased longevity (Fig. 4B).
MJD transgenic mice treated with aripiprazole show decreased levels of some aggregated forms of mutant ATXN3 in brain
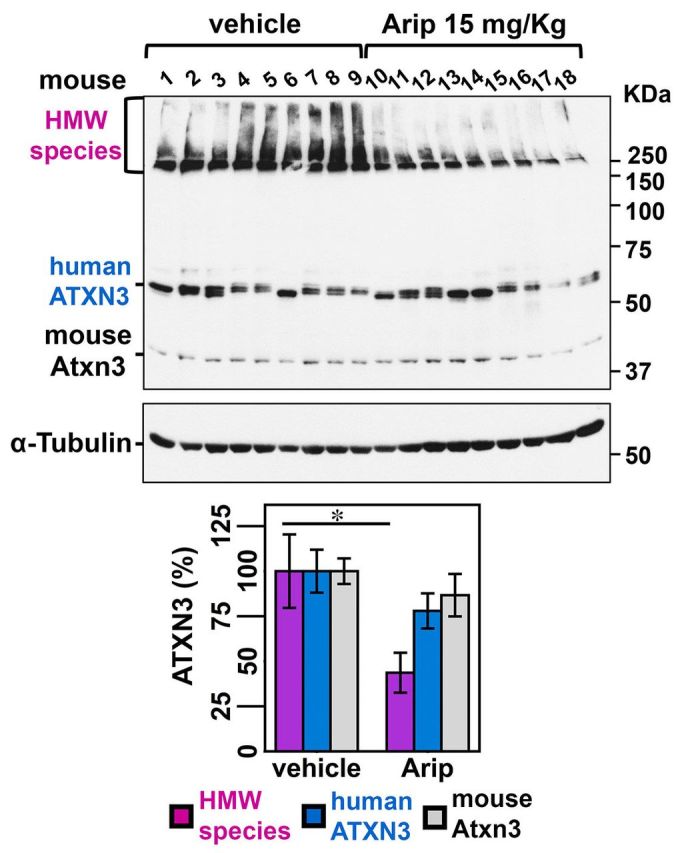
We next assessed whether aripiprazole could decrease levels of pathogenic ATXN3 in vivo in brains of Q84 mice. We treated 12-week-old Q84 mice (nine mice per group, comprising five females and four males) for 10 days with daily intraperitoneal injections of vehicle or aripiprazole 15 mg/kg, which, to our knowledge, is the maximum tolerable dose reported in chronically treated mice (Madhavan et al., 2007). This concentration of aripiprazole is rapidly absorbed, showing maximal concentration in the brain 30 min post-injection (Supplementary Fig. 4). Five hours after the final injection on Day 10, mice were sacrificed and brains collected for total protein and RNA extraction from different regions: brainstem, cerebellum, cervical spinal cord and forebrain.
Immunoblot analysis of lysates revealed a reduction in HMW ATXN3 species in the brainstem from aripiprazole-treated mice, whether male or female, to 44% of levels in vehicle-treated mice (Fig. 5A and B). A similar reduction of HMW ATXN3 was observed in the cerebellum (Supplementary Fig. 5A and B), spinal cord and forebrain (data not shown). A trend toward decreased levels of monomeric human ATXN3 and endogenous mouse ATXN3 was also observed (Fig. 5A and B), suggesting that longer treatments with aripiprazole might decrease all forms of ATXN3.
Figure 5.

Subchronic treatment of Q84 mice with aripiprazole reduces soluble HMW species of ATXN3 in the brainstem/midbrain. Twelve-week-old Q84 mice were injected daily with aripiprazole 15 mg/kg (10–18) or vehicle (1–9) for 10 days (males 1–4 and 10–13; females 5–9 and 14–18). (A) Anti-ATXN3 immunoblotting of solubilized protein extracts from brainstem reveals decreased HMW ATXN3 species in aripiprazole-treated mice. (B) Quantification of ATXN3 species (in A) shows that aripiprazole reduced HMW ATXN3 species to 44% of levels found in vehicle-treated mice. Bars represent the average percentage of protein species relative to vehicle-treated mice, corrected for α-tubulin (±SEM). Comparison between groups was made using Student’s t-test and statistical significance is indicated as *P < 0.05. (C) Aripiprazole and vehicle-treated mice show similar levels of human ATXN3 and mouse Atxn3 transcripts in brainstem. Values were normalized for Gapdh expression and referenced to the average of vehicle-treated mice of the correspondent gender. Bars represent the average of transcript fold change per mouse group (n = 9) ± SEM. (D) Filter trap assay using anti-MJD antibody shows insoluble ATXN3 in the brainstem/midbrain of aripiprazole and vehicle-treated mice. (E) Quantification of bands in D, normalized for total protein levels revealed by Ponceau staining (Supplementary Fig. 6), shows no differences of insoluble ATXN3 between the two groups of mice. Bars represent the average of insoluble ATXN3 relative to vehicle-treated mice (±SEM). (F) Confocal single plan images of pontine neurons from aripiprazole or vehicle-treated mice labelled for ATXN3 (green), NeuN (red) and nuclei with DAPI (blue). No major differences of ATXN3 staining between the two groups of mice are noted except for a slight reduction of ATXN3 in the cytoplasm in mice treated with aripiprazole. Scale bar = 20 μm. (G) Both groups of mice display similar number of ATXN3-positive puncta in ventral pontine nuclei. Bars represent the average of puncta (±SEM). (H) Quantification of nuclear ATXN3 fluorescence in pontine neurons reveals no differences between mice treated with aripiprazole (n = 10) or vehicle (n = 7). Bars correspond to the average corrected total cell fluorescence (CTCF) of ATXN3 (±SEM).
A potential effect of aripiprazole at the ATXN3 transcription level was ruled out, as human ATXN3 and mouse Atxn3 transcript levels proved to be similar in brainstems from aripiprazole and vehicle-treated mice (Fig. 5C).
As aripiprazole preferentially decreased HMW ATXN3 levels from solubilized protein fractions, we asked whether this drug similarly affected levels of highly aggregated ATXN3 that may fail to be solubilized by standard lysis. Filter trap analysis of insoluble fractions from the same brainstem and cerebellar samples described above revealed some variability among samples but no differences in insoluble ATXN3 between the two treatments in these brain areas (brainstem: Fig. 5D, E and Supplementary Fig. 6; cerebellum: Supplementary Fig. 5C–E). Furthermore, anti-ATXN3 immunofluorescence of ventral pons of Q84 mice treated with aripiprazole showed similar abundance of ATXN3-positive puncta as in vehicle-treated mice (Fig. 5F and G). Because mutant ATXN3 accumulates and tends to form insoluble aggregates in the nucleus (Paulson et al., 1997; Chai et al., 2002), we assessed ATXN3 levels in neuronal nuclei of ventral pons by immunofluorescence (Fig. 5F). Fluorescence quantification revealed no differences in ATXN3 nuclear levels between the two treatments (Fig. 5H). Compared to control mice, pons from aripiprazole-treated mice showed a non-significant trend towards a decrease of total ATXN3 fluorescence (Supplementary Fig. 7), which could correspond to the apparent slight reduction of cytoplasmic ATXN3 fluorescence in these mice (Fig. 5F). In summary, aripiprazole effectively decreases solubilized ATXN3, in particular the HMW species observed by immunoblotting, but not more highly insoluble ATXN3 species detected by filter-trap assay or immunofluorescence (puncta).
By binding to membrane receptors, aripiprazole is known to alter intracellular Ca2+ levels (Stark et al., 2007). Therefore, we assessed whether aripiprazole treatment altered levels of the ATXN3 cleavage fragment generated by the Ca2+ sensitive protease calpain, the production of which is correlated with ATXN3 aggregation (Haacke et al., 2007; Koch et al., 2011; Simoes et al., 2012). Aripiprazole treatment did not alter levels of the ATXN3 fragment (∼30–34 kDa) in brainstem and cerebellum of Q84 mice compared to control mice (Supplementary Fig. 8).
The pharmacological action of aripiprazole is complex and not fully understood. Aripiprazole exerts its efficacy as an atypical antipsychotic by partial agonism at dopamine D2 receptors (D2Rs) and serotonin 5-HT1A receptors (encoded by Htr1a) together with antagonism at serotonin 5-HT2A receptors (encoded by Htr2a) (de Bartolomeis et al., 2015). As aripiprazole has been shown to increase D2 receptor mRNA levels in the ventral tegmental area of rats (Han et al., 2009), we assessed transcript levels of Drd2 and the other main target receptors, Htr1a and Htr2a, in aripiprazole-treated mice. Indeed, aripiprazole was able to engage its targets by increasing Drd2 and Htr2a transcripts in the brainstem of Q84 mice (Supplementary Fig. 9). This effect is mediated by aripiprazole rather than the ATXN3 transgene because Drd2, Htr1a and Htr2a transcripts are similarly abundant in brainstems of 12-week-old Q84 mice and littermate wild-type controls (Supplementary Fig. 10).
Aripiprazole does not interfere with fibrillation of ATXN3 in vitro
Because aripiprazole is effective in reducing certain HMW forms of ATXN3 in vivo, we asked whether aripiprazole directly modulates ATXN3 fibril formation. We incubated recombinant ATXN3 carrying a modestly expanded glutamine tract (ATXN3Q55) with aripiprazole or vehicle in the presence of thioflavin T and monitored the change of fluorescence that occurs upon incorporation into amyloid-like fibrils (Fig. 6A). The kinetics of ATXN3Q55 fibril formation was identical in the presence or absence of aripiprazole. Similarly, aripiprazole did not interfere with fibril formation by non-pathogenic ATXN3Q26 (data not shown). Corroborating this result, native PAGE analysis of samples at the end of the thioflavin T assay revealed no differences in HMW or other species of ATXN3Q55 in the presence of aripiprazole (Fig. 6B). Furthermore, imaging by electron microscopy revealed ATXN3 spheroidal particles and short chains (‘beads on strings’) (Gales et al., 2005; Masino et al., 2011) in the presence or absence of aripiprazole (Fig. 6C).
Figure 6.
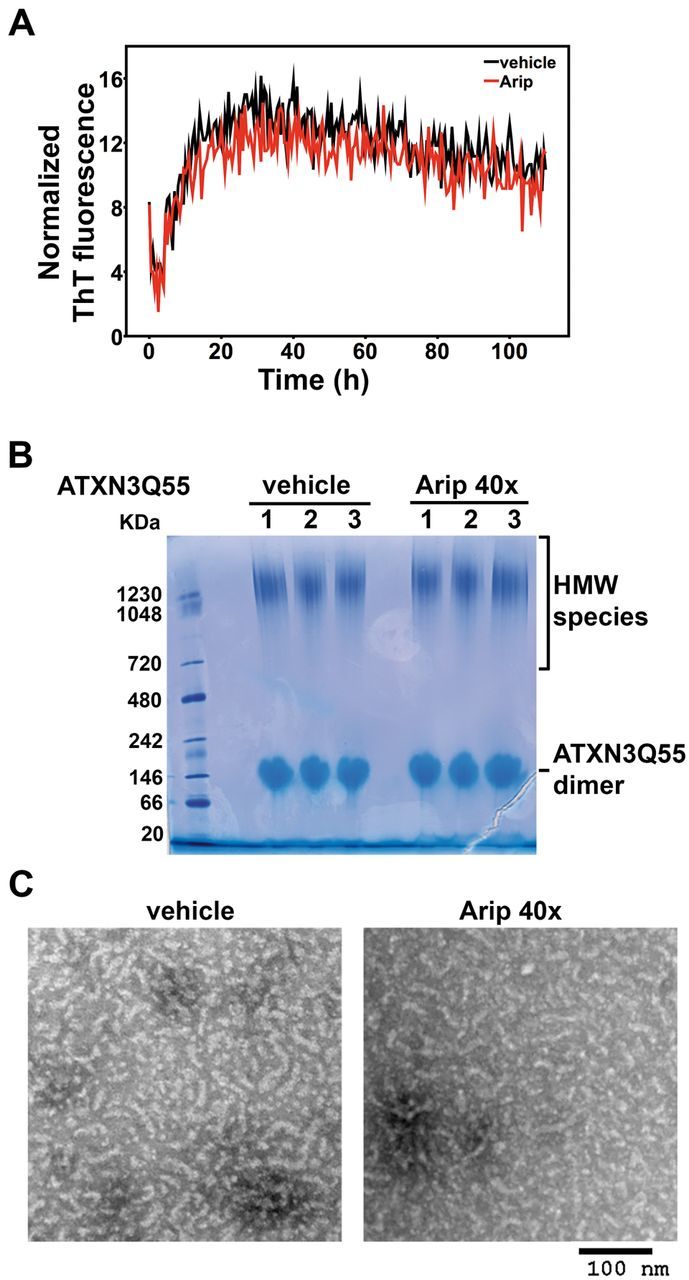
Aripiprazole does not interfere with ATXN3 fibril formation in vitro. (A) Thioflavin T fluorescence assay shows that ATXN3Q55 fibril formation is not affected by aripiprazole. Curves of recombinant ATXN3Q55 (10 μM) incubated with 40 times molar excess of aripiprazole (400 μM) in (red) or vehicle (black) were normalized by fluorescence values for the blank control (buffer). Each point on thioflavin T fluorescence assays corresponds to the average of three replicates in two independent experiments. (B) Blue native PAGE gel analysis of samples at the end of Thioflavin T assay shows similar ATXN3 species in samples of ATXN3Q55 incubated with aripiprazole or vehicle. Two major species are present equally in both samples: HMW species and ATXN3Q55 dimers. (C) Transmission electron microscopy images of the same samples used in B show spheroidal and short fibrillar ATXN3 complexes formed in the presence of aripiprazole or vehicle. Scale bar = 100 nm.
Aripiprazole alters components of protein homeostasis machinery in MJD transgenic mouse brains
While aripiprazole does not directly modulate ATXN3 fibril formation in vitro, we hypothesized that it decreases soluble aggregates of ATXN3 by regulating key components of cellular protein homeostasis in vivo. First we assessed levels of such components in Q84 mice in the absence of aripiprazole. Brainstem lysates from 12-week-old Q84 mice showed dysregulated levels of several components of chaperone machinery compared to wild-type littermate controls (Fig. 7A–D): Hsp40 was decreased (36% of control), wherever Hsp90β and Hsf1 were increased (122% and 152% of control, respectively). Reduction of Hsp40 levels appears to be a consistent marker of mutant ATXN3 pathogenesis in MJD mouse brains as it was previously reported in the cerebellum of an unrelated MJD mouse model (Chou et al., 2008). On the other hand, increased levels of Hsp90β and Hsf1 in Q84 brainstems are also consistent with the fact that Hsp90 inhibition and activation of Hsf1 pathway led to improved phenotypes of MJD animal models (Teixeira-Castro et al., 2011; Silva-Fernandes et al., 2014).
Figure 7.
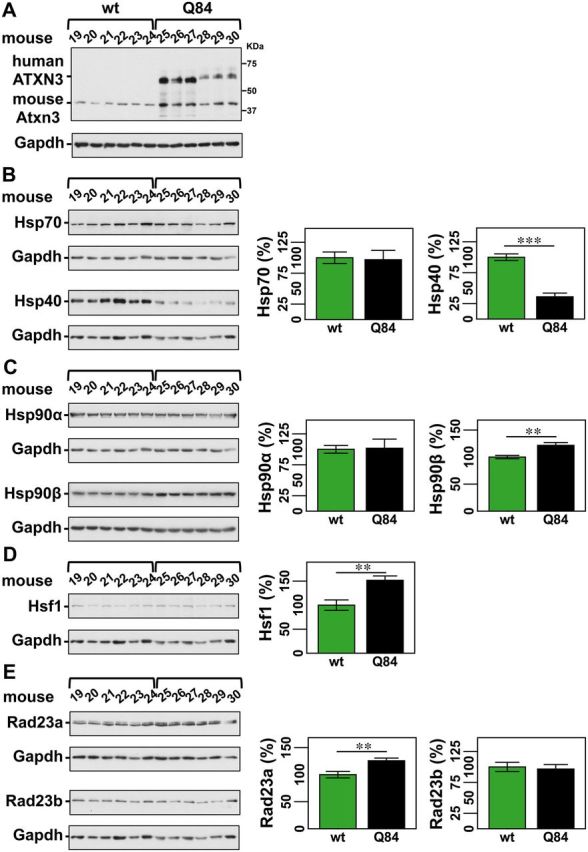
Q84 mice show dysregulation of key proteostasis components. Western blot analysis of total soluble protein lysates from brainstems of 12-week-old Q84 mice (n = 6; mouse number 25–30) and wild-type littermate controls (wt) (n = 6; mouse number 19–24) (A) show decreased Hsp40 (B), increased Hsp90β (C), increased HSF1 (D), and increased RAD23A (E) in Q84 mice. (A) Immunoblot showing ATXN3 expression in the brainstems of tested mice. (B–E) Left panels show immunoblots the indicated proteins in soluble fractions of brainstem extracts from Q84 or wild-type littermate mice. Right panels display quantification of band intensity, with values normalized to GAPDH. Bars represent the average percentage of protein relative to vehicle-treated mice (±SEM). Comparison between groups was made using Student’s t-test and statistical significance is indicated as **P < 0.01, and ***P < 0.001.
Aripiprazole is known to increase levels of HSP90α in PC12 neural cells (Ishima et al., 2012). Therefore, we anticipated that treatment of Q84 mice with aripiprazole could affect the abundance of molecular chaperones in the brains, thereby decreasing misfolded, aggregated ATXN3 species. Analysis of soluble fractions from the brainstems of aripiprazole-treated Q84 mice (Fig. 5A and B) revealed decreased levels of Hsp70 to 62% (Fig. 8A), Hsp90α to 79% and Hsp90β to 22% (Fig. 8B) of levels in vehicle-treated mice. No differences were observed in levels of Hsp40 (Fig. 8A). We observed, however, that heat shock transcription factor, Hsf1, was increased in samples from aripiprazole-treated mice (Fig. 8C), which may be related to the observed marked reduction of cytosolic forms of Hsp90, in particular Hsp90β.
Figure 8.

Aripiprazole affects components of the protein quality control machinery in brains of treated Q84 mice. In mice treated with aripiprazole evaluation of chaperone machinery components shows decreased levels of Hsp70 (A), Hsp90α and Hsp90β (B), increased levels of HSF1 (C), and a trend for decreased amount of RAD23A and RAD23B (D). (A–D) Left panels show immunoblots detecting the indicated proteins in soluble fractions of brainstem extracts from aripiprazole and vehicle-treated mice (same animals as in Fig. 5). Right panels display the quantification of band intensity, with values normalized for α-tubulin or GAPDH. Bars represent the average percentage of protein relative to vehicle-treated mice (±SEM). Comparison between groups was made using Student’s t-test and statistical significance is indicated as *P < 0.05, **P < 0.01, and ***P < 0.001.
Macroautophagy and the proteasome have also been implicated in degrading mutant ATXN3 (Matsumoto et al., 2004; Jana et al., 2005; Menzies et al., 2010; Nascimento-Ferreira et al., 2011). While we did not observe treatment differences in LC3I, LC3II or LC3II/LC3I ratio (Supplementary Fig. 11), we noted in aripiprazole-treated mice a trend toward decreased levels of Rad23a and Rad23b, ATXN3 interactors that prevent its proteasomal degradation (Blount et al., 2014) (Fig. 8). As Rad23a levels are increased in Q84 mice (Fig. 7E), aripiprazole treatment appears to restore Rad23a abundance to normal (lower) levels, which should promote ATXN3 proteasomal degradation.
No differences in transcript levels of Hsf1, Hspa1b (Hsp70), Hsp90ab1 (Hsp90β), Hsp90aa1 (Hsp90α), Rad23a, and Rad23b were found between aripiprazole and vehicle-treated Q84 mice suggesting that the observed effect of aripiprazole on the encoded proteins is downstream of transcription (data not shown).
In summary, subchronic treatment of Q84 mice with aripiprazole may affect proteostasis by altering levels of molecular chaperones and ATXN3 interactors implicated in its stability/degradation.
Discussion
No preventive therapy exists for MJD or any other neurodegenerative disease. Molecular chaperones, the proteasome and macroautophagy are components of cellular protein quality control known to regulate mutant ATXN3 and/or promote its degradation (Warrick et al., 1999; Tsai et al., 2003; Matsumoto et al., 2004; Jana et al., 2005; Berger et al., 2006; Boeddrich et al., 2006; Mishra et al., 2008; Ying et al., 2009; Durcan et al., 2010; Sugiura et al., 2010; Teixeira-Castro et al., 2011; Blount et al., 2014). While several pharmacological approaches have targeted specific protein quality control pathways to ameliorate disease phenotypes in animal models of this disease, none of the tested agents has yet advanced to clinical trials (Menzies et al., 2010; Nascimento-Ferreira et al., 2011; Wang et al., 2013; Silva-Fernandes et al., 2014). Here, we sought to identify a potential disease-modifying therapy for MJD that would act upstream in the cascade of disease pathogenesis. We took an unbiased approach to screen small molecules, including 1250 FDA-approved drugs, to identify agents that promote reduction of mutant ATXN3 levels.
The cell-based assays generated here display excellent signal to noise properties (average plate Z-factor = 0.81) and high reproducibility. Efficacy of selected small molecules identified by this assay was first confirmed ex vivo using brain slice cultures from MJD transgenic mice (Q84) expressing the full-length human disease ATXN3 gene (Cemal et al., 2002), and subsequently in vivo in MJD flies and in the same MJD mice. The ATXN3Q81.Luc cell line proved to be a robust assay for this initial, as well as for subsequent, high-throughput screens to identify modulators of ATXN3 abundance.
Five of nine compounds selected from dose response screens were confirmed to be effective in independent testing using fresh compound powders and immunoblotting for ATXN3 as the readout. Among these five compounds, salinomycin sodium, AM251 and aripiprazole reduced levels of human mutant ATXN3 in brainstem and cerebellar areas of brain slices from Q84 mice, which are the precise target regions in MJD patients. Despite their different chemical properties, all three molecules interfere with intracellular Ca2+ levels either by binding to membrane receptors [AM251 (Szabo et al., 2014) and aripiprazole (Stark et al., 2007)], or by acting as an ionophore in cell membranes [salinomycin (Boehmerle et al., 2014)]. Calcium homeostasis is indeed dysregulated in MJD models (Chen et al., 2008; Pellistri et al., 2013). At the cellular level, it is known that regulation of Ca2+ levels to decrease ATXN3 proteolysis correlates directly with a reduction on ATXN3 aggregation (Haacke et al., 2007; Koch et al., 2011; Simoes et al., 2012). The efficacy of aripiprazole in reducing ATXN3 abundance, however, is likely independent of this particular mechanism, as this drug does not appear to alter ATXN3 proteolysis in Q84 mice. Future studies will be needed to determine whether salinomycin, AM251 and aripiprazole modulate intracellular Ca2+ balance to alter the stability/clearance of mutant ATXN3.
Because aripiprazole is a FDA-approved, atypical antipsychotic commonly used in the clinic, we carried out further in vivo testing of this drug. Aripiprazole modestly decreased mutant ATXN3-mediated toxicity in MJD flies by increasing survival, which correlated with the observed reduction of HMW ATXN3 species in these flies. In Q84 mice, a 10-day course of treatment with aripiprazole led to a reduction of soluble ATXN3, in particular HMW (aggregated) species but not of more highly insoluble ATXN3 aggregates in the brain. It is perhaps to be expected that heavily aggregated, insoluble material would not be cleared in a short treatment trial whereas soluble, oligomeric forms of ATXN3 would be. While the relative toxicity of soluble versus insoluble polyQ protein aggregates is still debated, soluble oligomeric intermediates reside more upstream in the pathogenic cascade and are thought to be a critical toxic species in various neurodegenerative diseases (Williams and Paulson, 2008).
While preclinical trials of aripiprazole now become a priority to assess whether this drug can be repurposed to treat MJD, knowledge of its mechanism of action towards ATXN3 abundance could shed light on cellular pathways that could be targeted to develop improved therapeutics for MJD. Aripiprazole effects on signalling and intracellular pathways are complex but its efficacy as an antipsychotic mainly reflects combined action on dopaminergic and serotonergic signalling (de Bartolomeis et al., 2015).
Antipsychotics such as aripiprazole should be used cautiously in any patient (Muench and Hamer, 2010), including those with MJD in who decreased dopamine transporter binding, extrapyramidal signs, and nigrostriatal cell loss may predispose to anti-psychotic-induced parkinsonism (Woods and Schaumburg, 1972; Coutinho and Andrade, 1978; Yen et al., 2000). As a second generation antipsychotic, however, aripiprazole shows fewer adverse effects than typical antipsychotics that act mainly as dopamine D2 receptor antagonists (Muench and Hamer, 2010). Depending on endogenous dopamine levels, aripiprazole can act as a full antagonist, a moderate antagonist or a partial agonist at D2 receptors (de Bartolomeis et al., 2015). Presently, the potential involvement of dopaminergic signalling in the regulation of ATXN3 abundance is unexplored. Whether dopaminergic signalling contributes to the action of aripiprazole to reduce ATXN3 requires further investigation. Because Drd2 transcripts are increased in the brainstem of Q84 mice treated with aripiprazole, agonism of these receptors may contribute to its observed efficacy. While it is possible that aripiprazole acts as a dopamine D2 receptor antagonist in MJD patients, because these patients often show deficits in the dopaminergic system, it is conceivable that this atypical antipsychotic could act as a dopamine receptor partial agonist and further improve dopamine-mediated signalling in these patients.
Modulation of serotonergic signalling by citalopram, a selective serotonin reuptake inhibitor, was recently reported to suppress ATXN3 aggregation and neurotoxicity in models of MJD (Teixeira-Castro et al., 2015). While the specific 5-hydroxytryptamine (5-HT) receptors necessary for citalopram efficacy in transgenic mice were not identified, studies in a C. elegans model implicated 5HT1A and 5HT2A in its mechanism (Teixeira-Castro et al., 2015). Similarly, agonism of 5HT1A and antagonism of 5HT2A by aripiprazole could underlie a disease-modifying pathway in MJD. Whereas citalopram and aripiprazole may share pathways to alter ATXN3 abundance, it is also possible that the manner in which these two drugs work differs. The neuropathological and molecular outcomes measured differed in the two studies: for example, citalopram was shown to decrease intraneuronal inclusions of ATXN3 but not levels of ATXN3 monomers in a cDNA transgenic mouse model (Teixeira-Castro et al., 2015) whereas in the current study, aripiprazole reduced oligomeric and monomeric species of ATXN3 with no effect on large insoluble aggregates (inclusions) in a transgenic mouse model expressing the full human ATXN3 disease gene (Q84). Future studies will be needed to compare the effects and mechanisms of action of aripiprazole versus citalopram using the same MJD mouse model.
The fact that aripiprazole does not alter in vitro ATXN3 fibrillation suggests that its protective effect is not due to direct interaction with ATXN3 but rather occurs indirectly through one or more cellular pathways. Which cellular pathways are triggered by aripiprazole to reduce ATXN3 abundance? Though more work is required to define the precise mechanism of action, we observed that aripiprazole affects the levels of select protein quality control proteins in a manner that should favour degradation of mutant ATXN3.
RAD23A and RAD23B are known to interact with ATXN3 and prevent its degradation by the proteasome (Blount et al., 2014). As we observed increased levels of RAD23A in the brainstem of Q84 mice, proteasomal degradation of ATXN3 could be reduced in Q84 mouse brains. Observations in mice treated with aripiprazole are consistent with the drug enhancing mutant ATXN3 clearance: (i) the decreased levels of RAD23A and RAD23B would be expected to increase ATXN3 accessibility to the proteasome; and (ii) decreased levels of Hsp70 paradoxically could increase the targeting of mutant ATXN3 to the proteasome, as has been observed for inhibitors of Hsp70 ATPase activity in other neurodegenerative proteinopathies (Jinwal et al., 2009).
Molecular chaperones are key protein quality control components that ensure proper protein folding and help target misfolded proteins for degradation. Indeed, Q84 transgenic mice showed altered levels of important components of the molecular chaperone machinery in the brainstem: reduced Hsp40 and increased Hsp90β and Hsf1. Treatment of Q84 mice with aripiprazole decreased levels of Hsp90α and Hsp90β, which may explain the observed further increase in Hsf1 abundance. These results concur with previous reports showing neuroprotection by activating the Hsf1 pathway in a C. elegans model of MJD (Teixeira-Castro et al., 2011) or by inhibiting Hsp90 in an unrelated transgenic mouse model of this disease (Silva-Fernandes et al., 2014).
In conclusion, using a combination of in vitro, ex vivo and in vivo assays in human cell, mouse and fly models we identified aripiprazole as a potential therapeutic agent for MJD. Because aripiprazole reduced levels of presumed oligomeric forms of mutant ATXN3, this drug conceivably could act similarly towards other aggregate-prone proteins.
Supplementary Material
Acknowledgements
The authors thank: Dr Masayoshi Tada and Dr Stuart Decker for generating the ATXN3-firefly Luciferase plasmids; Dr Paul Kirchhoff for assessing compound triage; Dr Vikram Shakkottai for helping with mouse brain slicing in the vibratome; and members of Paulson lab for fruitful discussions.
Funding
M.C.C. was a recipient of fellowships from Fundação para a Ciência e a Tecnologia (FCT) Portugal (SFRH/BPD/28560/2006) and National Ataxia Foundation (NAF Research Fellowship Award 2011). This work was funded by NIH R01NS038712 and R01NS086778, the Mateus Ataxia Research Fund, and the National Ataxia Foundation (NAF Pioneer SCA Translational Award 2012).
Supplementary material
Supplementary material is available here.
Glossary
Abbreviations
- CTCF
corrected total cell fluorescence
- DMSO
dimethyl sulphoxide
- FDA
Food and Drug Administration
- HMW
high molecular weight
- MJD
Machado-Joseph disease
- polyQ
polyglutamine
- SCA3
spinocerebellar ataxia type 3
References
- Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet 2006; 15: 433–42. [DOI] [PubMed] [Google Scholar]
- Blount JR, Tsou WL, Ristic G, Burr AA, Ouyang M, Galante H, et al. Ubiquitin-binding site 2 of ataxin-3 prevents its proteasomal degradation by interacting with Rad23. Nat Commun 2014; 5: 4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeddrich A, Gaumer S, Haacke A, Tzvetkov N, Albrecht M, Evert BO, et al. An arginine/lysine-rich motif is crucial for VCP/p97-mediated modulation of ataxin-3 fibrillogenesis. EMBO J 2006; 25: 1547–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehmerle W, Muenzfeld H, Springer A, Huehnchen P, Endres M. Specific targeting of neurotoxic side effects and pharmacological profile of the novel cancer stem cell drug salinomycin in mice. J Mol Med 2014; 92: 889–900. [DOI] [PubMed] [Google Scholar]
- Cemal CK, Carroll CJ, Lawrence L, Lowrie MB, Ruddle P, Al-Mahdawi S, et al. YAC transgenic mice carrying pathological alleles of the MJD1 locus exhibit a mild and slowly progressive cerebellar deficit. Hum Mol Genet 2002; 11: 1075–94. [DOI] [PubMed] [Google Scholar]
- Chai Y, Shao J, Miller VM, Williams A, Paulson HL. Live-cell imaging reveals divergent intracellular dynamics of polyglutamine disease proteins and supports a sequestration model of pathogenesis. Proc Natl Acad Sci USA 2002; 99: 9310–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Tang TS, Tu H, Nelson O, Pook M, Hammer R, et al. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J Neurosci 2008; 28: 12713–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou AH, Chen SY, Yeh TH, Weng YH, Wang HL. HDAC inhibitor sodium butyrate reverses transcriptional downregulation and ameliorates ataxic symptoms in a transgenic mouse model of SCA3. Neurobiol Dis 2010; 41: 481–8. [DOI] [PubMed] [Google Scholar]
- Chou AH, Yeh TH, Ouyang P, Chen YL, Chen SY, Wang HL. Polyglutamine-expanded ataxin-3 causes cerebellar dysfunction of SCA3 transgenic mice by inducing transcriptional dysregulation. Neurobiol Dis 2008; 31: 89–101. [DOI] [PubMed] [Google Scholar]
- Costa Mdo C, Paulson HL. Toward understanding Machado-Joseph disease. Prog Neurobiol 2012; 97: 239–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho P, Andrade C. Autosomal dominant system degeneration in Portuguese families of the Azores Islands. A new genetic disorder involving cerebellar, pyramidal, extrapyramidal and spinal cord motor functions. Neurology 1978; 28: 703–9. [DOI] [PubMed] [Google Scholar]
- Coutinho P, Sequeiros J. Clinical, genetic and pathological aspects of Machado-Joseph disease [in French]. J Genet Hum 1981; 29: 203–9. [PubMed] [Google Scholar]
- de Bartolomeis A, Tomasetti C, Iasevoli F. Update on the mechanism of action of aripiprazole: translational insights into antipsychotic strategies beyond Dopamine Receptor Antagonism. CNS Drugs 2015; 29: 773–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Prospero NA, Fischbeck KH. Therapeutics development for triplet repeat expansion diseases. Nat Rev Genet 2005; 6: 756–65. [DOI] [PubMed] [Google Scholar]
- do Carmo Costa M, Luna-Cancalon K, Fischer S, Ashraf NS, Ouyang M, Dharia RM, et al. Toward RNAi therapy for the Polyglutamine Disease Machado-Joseph Disease. Mol Ther 2013; 21: 1898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcan TM, Kontogiannea M, Thorarinsdottir T, Fallon L, Williams AJ, Djarmati A, et al. The Machado-Joseph disease-associated mutant form of ataxin-3 regulates parkin ubiquitination and stability. Hum Mol Genet 2010; 20: 141–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellisdon AM, Thomas B, Bottomley SP. The two-stage pathway of ataxin-3 fibrillogenesis involves a polyglutamine-independent step. J Biol Chem 2006; 281: 16888–96. [DOI] [PubMed] [Google Scholar]
- Gales L, Cortes L, Almeida C, Melo CV, Costa MC, Maciel P, et al. Towards a structural understanding of the fibrillization pathway in Machado-Joseph's disease: trapping early oligomers of non-expanded ataxin-3. J Mol Biol 2005; 353: 642–54. [DOI] [PubMed] [Google Scholar]
- Haacke A, Hartl FU, Breuer P. Calpain inhibition is sufficient to suppress aggregation of polyglutamine-expanded Ataxin-3. J Biol Chem 2007; 282: 18851–6. [DOI] [PubMed] [Google Scholar]
- Han M, Huang XF, Deng C. Aripiprazole differentially affects mesolimbic and nigrostriatal dopaminergic transmission: implications for long-term drug efficacy and low extrapyramidal side-effects. Int J Neuropsychopharmacol 2009; 12: 941–52. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Kobayashi K, Furuta H. Immunohistochemical study of neuronal intranuclear and cytoplasmic inclusions in Machado-Joseph disease. Psychiatry Clin Neurosci 2003; 57: 205–13. [DOI] [PubMed] [Google Scholar]
- Ishima T, Iyo M, Hashimoto K. Neurite outgrowth mediated by the heat shock protein Hsp90alpha: a novel target for the antipsychotic drug aripiprazole. Transl Psychiatry 2012; 2: e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob RT, Larsen MJ, Larsen SD, Kirchhoff PD, Sherman DH, Neubig RR. MScreen: an integrated compound management and high-throughput screening data storage and analysis system. J Biomol Screen 2012; 17: 1080–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana NR, Dikshit P, Goswami A, Kotliarova S, Murata S, Tanaka K, et al. Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J Biol Chem 2005; 280: 11635–40. [DOI] [PubMed] [Google Scholar]
- Jinwal UK, Miyata Y, Koren J, 3rd, Jones JR, Trotter JH, Chang L, et al. Chemical manipulation of hsp70 ATPase activity regulates tau stability. J Neurosci 2009; 29: 12079–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 1994; 8: 221–8. [DOI] [PubMed] [Google Scholar]
- Keravala A, Calos MP. Site-specific chromosomal integration mediated by phiC31 integrase. Methods Mol Biol 2008; 435: 165–73. [DOI] [PubMed] [Google Scholar]
- Koch P, Breuer P, Peitz M, Jungverdorben J, Kesavan J, Poppe D, et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 2011; 480: 543–6. [DOI] [PubMed] [Google Scholar]
- Lima M, Costa MC, Montiel R, Ferro A, Santos C, Silva C, et al. Population genetics of wild-type CAG repeats in the Machado-Joseph disease gene in Portugal. Hum Hered 2005; 60: 156–63. [DOI] [PubMed] [Google Scholar]
- Maciel P, Costa MC, Ferro A, Rousseau M, Santos CS, Gaspar C, et al. Improvement in the molecular diagnosis of Machado-Joseph disease. Arch Neurol 2001; 58: 1821–7. [DOI] [PubMed] [Google Scholar]
- Madhavan A, Argilli E, Bonci A, Whistler JL. Loss of D2 dopamine receptor function modulates cocaine-induced glutamatergic synaptic potentiation in the ventral tegmental area. J Neurosci 2007; 33: 12329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino L, Nicastro G, De Simone A, Calder L, Molloy J, Pastore A. The Josephin domain determines the morphological and mechanical properties of ataxin-3 fibrils. Biophys J 2011; 100: 2033–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos CA, de Macedo-Ribeiro S, Carvalho AL. Polyglutamine diseases: the special case of ataxin-3 and Machado-Joseph disease. Prog Neurobiol 2011; 95: 26–48. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Yada M, Hatakeyama S, Ishimoto H, Tanimura T, Tsuji S, et al. Molecular clearance of ataxin-3 is regulated by a mammalian E4. EMBO J 2004; 23: 659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies FM, Huebener J, Renna M, Bonin M, Riess O, Rubinsztein DC. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 2010; 133 (Pt 1): 93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, Dikshit P, Purkayastha S, Sharma J, Nukina N, Jana NR. E6-AP promotes misfolded polyglutamine proteins for proteasomal degradation and suppresses polyglutamine protein aggregation and toxicity. J Biol Chem 2008; 283: 7648–56. [DOI] [PubMed] [Google Scholar]
- Muench J, Hamer AM. Adverse effects of antipsychotic medications. Am Fam Physician 2010; 81: 617–22. [PubMed] [Google Scholar]
- Nascimento-Ferreira I, Santos-Ferreira T, Sousa-Ferreira L, Auregan G, Onofre I, Alves S, et al. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado-Joseph disease. Brain 2011; 134 (Pt 5): 1400–15. [DOI] [PubMed] [Google Scholar]
- Nobrega C, Nascimento-Ferreira I, Onofre I, Albuquerque D, Hirai H, Deglon N, et al. Silencing mutant ataxin-3 rescues motor deficits and neuropathology in machado-joseph disease transgenic mice. PLoS One 2013; 8: e52396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson HL. Dominantly inherited ataxias: lessons learned from Machado-Joseph disease/spinocerebellar ataxia type 3. Semin Neurol 2007; 27: 133–42. [DOI] [PubMed] [Google Scholar]
- Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, et al. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 1997; 19: 333–44. [DOI] [PubMed] [Google Scholar]
- Pellistri F, Bucciantini M, Invernizzi G, Gatta E, Penco A, Frana AM, et al. Different ataxin-3 amyloid aggregates induce intracellular Ca(2+) deregulation by different mechanisms in cerebellar granule cells. Biochim Biophys Acta 2013; 1833: 3155–65. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Lebron E, Costa MD, Luna-Cancalon K, Peron TM, Fischer S, Boudreau RL, et al. Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice. Mol Ther 2013; 21: 1909–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 2014; 42: 174–83. [DOI] [PubMed] [Google Scholar]
- Rub U, Brunt ER, Deller T. New insights into the pathoanatomy of spinocerebellar ataxia type 3 (Machado-Joseph disease). Curr Opin Neurol 2008; 21: 111–16. [DOI] [PubMed] [Google Scholar]
- Schmitt I, Linden M, Khazneh H, Evert BO, Breuer P, Klockgether T, et al. Inactivation of the mouse Atxn3 (ataxin-3) gene increases protein ubiquitination. Biochem Biophys Res Commun 2007; 362: 734–9. [DOI] [PubMed] [Google Scholar]
- Schols L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol 2004; 3: 291–304. [DOI] [PubMed] [Google Scholar]
- Shakkottai VG, do Carmo Costa M, Dell'orco JM, Sankaranarayanan A, Wulff H, Paulson HL. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci 2011; 31: 13002–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao J, Diamond MI. Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum Mol Genet 2007; 16 Spec No. 2: R115–23. [DOI] [PubMed] [Google Scholar]
- Silva-Fernandes A, Duarte-Silva S, Neves-Carvalho A, Amorim M, Soares-Cunha C, Oliveira P, et al. Chronic treatment with 17-DMAG improves balance and coordination in a new mouse model of Machado-Joseph disease. Neurotherapeutics 2014; 11: 433–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes AT, Goncalves N, Koeppen A, Deglon N, Kugler S, Duarte CB, et al. Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado-Joseph disease. Brain 2012; 135 (Pt 8): 2428–39. [DOI] [PubMed] [Google Scholar]
- Stark AD, Jordan S, Allers KA, Bertekap RL, Chen R, Mistry Kannan T, et al. Interaction of the novel antipsychotic aripiprazole with 5-HT1A and 5-HT 2A receptors: functional receptor-binding and in vivo electrophysiological studies. Psychopharmacology 2007; 190: 373–82. [DOI] [PubMed] [Google Scholar]
- Sugiura A, Yonashiro R, Fukuda T, Matsushita N, Nagashima S, Inatome R, et al. A mitochondrial ubiquitin ligase MITOL controls cell toxicity of polyglutamine-expanded protein. Mitochondrion 2010; 11: 139–46. [DOI] [PubMed] [Google Scholar]
- Szabo GG, Lenkey N, Holderith N, Andrasi T, Nusser Z, Hajos N. Presynaptic calcium channel inhibition underlies CB(1) cannabinoid receptor-mediated suppression of GABA release. J Neurosci 2014; 34: 7958–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira-Castro A, Ailion M, Jalles A, Brignull HR, Vilaca JL, Dias N, et al. Neuron-specific proteotoxicity of mutant ataxin-3 in C. elegans: rescue by the DAF-16 and HSF-1 pathways. Hum Mol Genet 2011; 20: 2996–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira-Castro A, Jalles A, Esteves S, Kang S, da Silva Santos L, Silva-Fernandes A, et al. Serotonergic signalling suppresses ataxin 3 aggregation and neurotoxicity in animal models of Machado-Joseph disease. Brain 2015; 138 (Pt 11): 3221–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todi SV, Paulson HL. Balancing act: deubiquitinating enzymes in the nervous system. Trends Neurosci 2011; 34: 370–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YC, Fishman PS, Thakor NV, Oyler GA. Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J Biol Chem 2003; 278: 22044–55. [DOI] [PubMed] [Google Scholar]
- Wang HL, Hu SH, Chou AH, Wang SS, Weng YH, Yeh TH. H1152 promotes the degradation of polyglutamine-expanded ataxin-3 or ataxin-7 independently of its ROCK-inhibiting effect and ameliorates mutant ataxin-3-induced neurodegeneration in the SCA3 transgenic mouse. Neuropharmacology 2013; 70: 1–11. [DOI] [PubMed] [Google Scholar]
- Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet 1999; 23: 425–8. [DOI] [PubMed] [Google Scholar]
- Williams AJ, Paulson HL. Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci 2008; 31: 521–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods BT, Schaumburg HH. Nigro-spino-dentatal degeneration with nuclear ophthalmoplegia. A unique and partially treatable clinico-pathological entity. J Neurol Sci 1972; 17: 149–66. [DOI] [PubMed] [Google Scholar]
- Yamada M, Sato T, Tsuji S, Takahashi H. CAG repeat disorder models and human neuropathology: similarities and differences. Acta Neuropathol 2008; 115: 71–86. [DOI] [PubMed] [Google Scholar]
- Yamada M, Tan CF, Inenaga C, Tsuji S, Takahashi H. Sharing of polyglutamine localization by the neuronal nucleus and cytoplasm in CAG-repeat diseases. Neuropathol Appl Neurobiol 2004; 30: 665–75. [DOI] [PubMed] [Google Scholar]
- Yen TC, Lu CS, Tzen KY, Wey SP, Chou YH, Weng YH, et al. Decreased dopamine transporter binding in Machado-Joseph disease. J Nucl Med 2000; 41: 994–8. [PubMed] [Google Scholar]
- Ying Z, Wang H, Fan H, Zhu X, Zhou J, Fei E, et al. Gp78, an ER associated E3, promotes SOD1 and ataxin-3 degradation. Hum Mol Genet 2009; 18: 4268–81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.