

Abstract
Objective
To investigate the relationship of in vivo microglial activation to clinical and MRI parameters in MS.
Methods
Patients with secondary progressive MS (n = 10) or relapsing-remitting MS (n = 10) and age-matched healthy controls (n = 17) were studied. Microglial activation was measured using PET and radioligand [11C](R)-PK11195. Clinical assessment and structural and quantitative MRI including diffusion tensor imaging (DTI) were performed for comparison.
Results
[11C](R)-PK11195 binding was significantly higher in the normal-appearing white matter (NAWM) of patients with secondary progressive vs relapsing MS and healthy controls, in the thalami of patients with secondary progressive MS vs controls, and in the perilesional area among the progressive compared with relapsing patients. Higher binding in the NAWM was associated with higher clinical disability and reduced white matter (WM) structural integrity, as shown by lower fractional anisotropy, higher mean diffusivity, and increased WM lesion load. Increasing age contributed to higher microglial activation in the NAWM among patients with MS but not in healthy controls.
Conclusions
PET can be used to quantitate microglial activation, which associates with MS progression. This study demonstrates that increased microglial activity in the NAWM correlates closely with impaired WM structural integrity and thus offers one rational pathologic correlate to diffusion tensor imaging (DTI) parameters.
In most cases, MS initially presents as the relapsing-remitting form (RRMS) but, with time, most patients advance into the secondary progressive phase (SPMS) with cumulative disability.1 While the armamentarium of potent immunomodulatory therapies for RRMS has expanded over the past years, an efficient treatment of SPMS is still scarce owing to the lack of thorough understanding of the pathology in progressive MS. One common hypothesis holds that the progression of MS results from chronic inflammation associated with diffuse demyelination, axonal injury, mitochondrial dysfunction, and neurodegeneration.2
Despite being an established method in the clinical setting, the alterations observed in conventional MRI during the evolution of MS are somewhat nonspecific in terms of neuropathology. Thus, MRI alone is insufficient for detailed evaluation of the cellular changes associated with progressive MS.2–4 By contrast, PET allows in vivo quantification of specific molecular structures, such as the 18-kDa translocator protein (TSPO). TSPO is mainly expressed on the outer mitochondrial membrane of activated, but not resting microglia and macrophages, and on some astrocytes.5,6 It can be detected in vivo using TSPO-specific radioligands and hence is used for the evaluation of neuroinflammation in various neurodegenerative conditions, including MS.5
Neuropathology studies have produced evidence that there is increased microglial activation in the SPMS brain tissue both in the normal-appearing white matter (NAWM) and at the rim of T1-hypointense lesions.2–4 We considered that detecting this in vivo would be of significant interest in assessing progressive patients with MS and, thus, the primary objective of this study was to evaluate whether microglial activation in the NAWM, measured crosssectionally using in vivo PET, correlates with the progressive MS type and with various MRI parameters.
Methods
Standard protocol approvals, registrations, and patient consents
The study was conducted as a cross-sectional, investigator-initiated academic study in the Turku PET Centre (TPC) and Turku University Hospital, Finland, and the Wolfson Molecular Imaging Centre (WMIC), University of Manchester, the United Kingdom. The study protocol was approved by the Ethics Committee of the Hospital District of Southwest Finland, the Greater Manchester East Research Ethics Committee, and the Administration of Radioactive Substances Advisory Committee in the United Kingdom. Written informed consent was obtained from all participants before participation in the study, and the study was conducted according to the principles of the Declaration of Helsinki.
Study participants
Seventeen healthy controls, 10 patients with SPMS, and 10 patients with RRMS/clinically isolated syndrome (CIS) were included in the final evaluation. All patients with MS were imaged in the TPC. In addition, 8 controls age-matched to the SPMS group were imaged in the TPC. Nine controls, age-matched to the RRMS group, were imaged in the WMIC as part of previous PET studies.7,8 The inclusion and exclusion criteria of the study participants have been described in our earlier study9 and in appendix e-1 (links.lww.com/NXI/A37). None of the patients with SPMS had been treated with immunomodulatory treatment within 3 months before imaging (median 4.2 years from the cessation of disease-modifying treatment), and none of the patients with CIS/RRMS had yet initiated a disease-modifying therapy.
In vivo brain MRI and [11C](R)-PK11195 PET imaging
Brain MRI was performed with a Philips Gyroscan Intera 1.5 T Nova Dual scanner (Philips, Best, the Netherlands) in the TPC, and similarly, with a Philips Achieva 1.5 T scanner (Philips, Best, Netherlands) in the WMIC. In both centers, PET imaging was performed using an ECAT high resolution research tomograph scanner (CTI/Siemens, Knoxville, TN)10 that has an intrinsic spatial resolution of approximately 2.5 mm3.11 The protocols for the brain MRI and volumetric analyses and for the [11C](R)-PK11195 radioligand production and brain PET imaging with [11C](R)-PK11195 are explained in detail in appendix e-1 (links.lww.com/NXI/A37). The analyses of the diffusion tensor imaging (DTI) data with evaluation of global mean fractional anisotropy (FA) and mean diffusivity (MD) in the NAWM were performed according to the methodology reported earlier.12
The coregistration of magnetic resonance to the PET sum images was performed using statistical parametric mapping (SPM8, version 8; Wellcome Trust Center for Neuroimaging) run on MATLAB 2011 (The MathWorks, Natick, MA). The procedures for the coregistration, performing partial volume correction, and the region of interest (ROI) definition are described in detail in appendix e-1 (links.lww.com/NXI/A37). The ROIs evaluated for the specific radioligand binding were as follows: T1-hypointense lesions as a single lesion mask (gadolinium-enhancing and gadolinium-negative lesion masks evaluated separately), perilesional areas with 0–3 and 3–6 mm radiuses from the T1-negative lesion masks (figure 1), NAWM (cerebral white matter [WM] segment − [T1 lesions + perilesional masks]), neocortex, thalamus, striatum, and cerebellum.
Figure 1. Illustration of white matter– and lesion-associated regions of interest (ROIs).
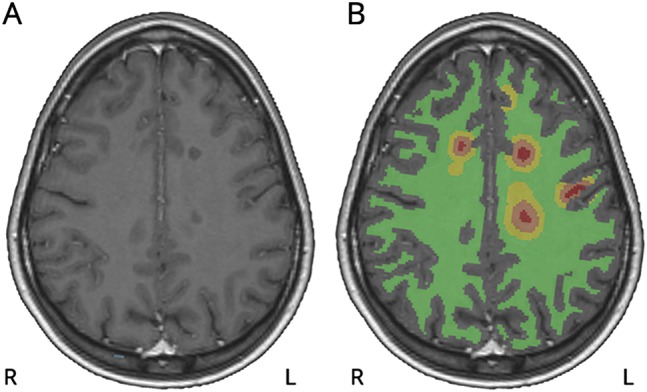
The images are from a 25-year-old female patient with relapsing-remitting MS and an Expanded Disability Status Scale score of 1.0. (A) Axial view of gadolinium-enhanced 3DT1 MRI showing T1-hypointense white matter lesions. (B) Respective MRI with overlaid ROIs of T1-hypointense lesions (red), perilesional areas with 0-3 mm (brown) and 3-6 mm (yellow) radiuses, and normal-appearing white matter (green). Note that some of the perilesional ROIs visible in this slice are not associated with T1-hypointense lesions, but are due to larger perilesional ROIs associated with T1-hypointense lesion cores in adjacent sections. The gray matter areas have been masked out from all the lesion-associated ROIs for all analyses.
The regional specific binding of [11C](R)-PK11195 was evaluated as distribution volume ratios (DVRs) using the Logan method within a time interval of 20–60 minutes. For the reference region input in DVR estimation, we used clustered gray matter (GM) derived with supervised cluster algorithm approach using 4 predefined kinetic tissue classes (SuperPK software, SCA4)13,14 as described in our previous study.9 Further parameters regarding the GM clustering are described in appendix e-1 (links.lww.com/NXI/A37).
Statistical methods
The statistical analyses of the clinical, MRI, and PET measurements were performed with SAS System for Windows (SAS, version 9.4, SAS Institute Inc.). On the basis of previous brain PET studies with [11C](R)-PK11195,13,15–17 we estimated that ten participants per study group would be sufficient to reveal a 15% difference in [11C](R)-PK11195 binding between the 2 groups (controls vs patients with MS), with 90% power at p < 0.05 based on estimated regional DVR values ranging from 1.2 (SD 0.08) to 1.4 (SD 0.10). The methodology for performing the statistical analyses is provided in detail in appendix e-1 (links.lww.com/NXI/A37). In short, the groupwise comparisons of the MRI and DTI parameters between the MS patient groups and age-matched controls were performed using analysis of variance (ANOVA) and between the RRMS and SPMS groups using analysis of covariance (ANCOVA, age as a covariate), with the Tukey-Kramer post hoc test for multiple comparisons. For the groupwise comparison of [11C](R)-PK11195 binding within ROIs, the controls were pooled into 1 group (n = 17) after finding no statistically significant differences in radioligand binding between younger and older controls. The statistical testing was performed using ANCOVA (age as a covariate), with the Tukey-Kramer post hoc test for multiple comparisons.
The interaction between regional DVR and clinical or MRI variables was examined within the pooled data of all patients with MS (n = 20) using repeated-measures ANCOVA (rm ANCOVA) adjusted for age and regional DVRs or volumes considered as repeated measures using unstructured covariance structure. The effect of age on DVR values was compared between pooled healthy controls and patients with MS using ANCOVA, where the interaction between groups and age was examined yielding an age coefficient for each group. For all analyses, p < 0.05 was considered statistically significant.
Results
Association of structural and quantitative MRI findings with the level of clinical disability in MS
The patients with SPMS had advanced disease associated with higher age, longer disease duration, and higher Expanded Disability Status Scale (EDSS) and MS severity scale (MSSS) scores compared with the patients recently diagnosed with RRMS/CIS (table 1). In the age-adjusted analyses, no differences were observed in the normalized brain volumes measured as WM and GM parenchymal fractions and total brain parenchymal fraction (BPF) between patients with RRMS and patients with SPMS, although the patients with SPMS had higher T2, total T1, and Gd− T1 lesion loads than patients with RRMS. No difference was noted in the Gd+ T1 lesion loads between the MS subgroups. WM, GM, and total BPFs were not different in patients compared with the respective control groups (table 1). Clinical and MRI characteristics of controls and patients with MS are shown in detail in table 1.
Table 1.
Clinical and MRI parameters of healthy controls and patients with MS
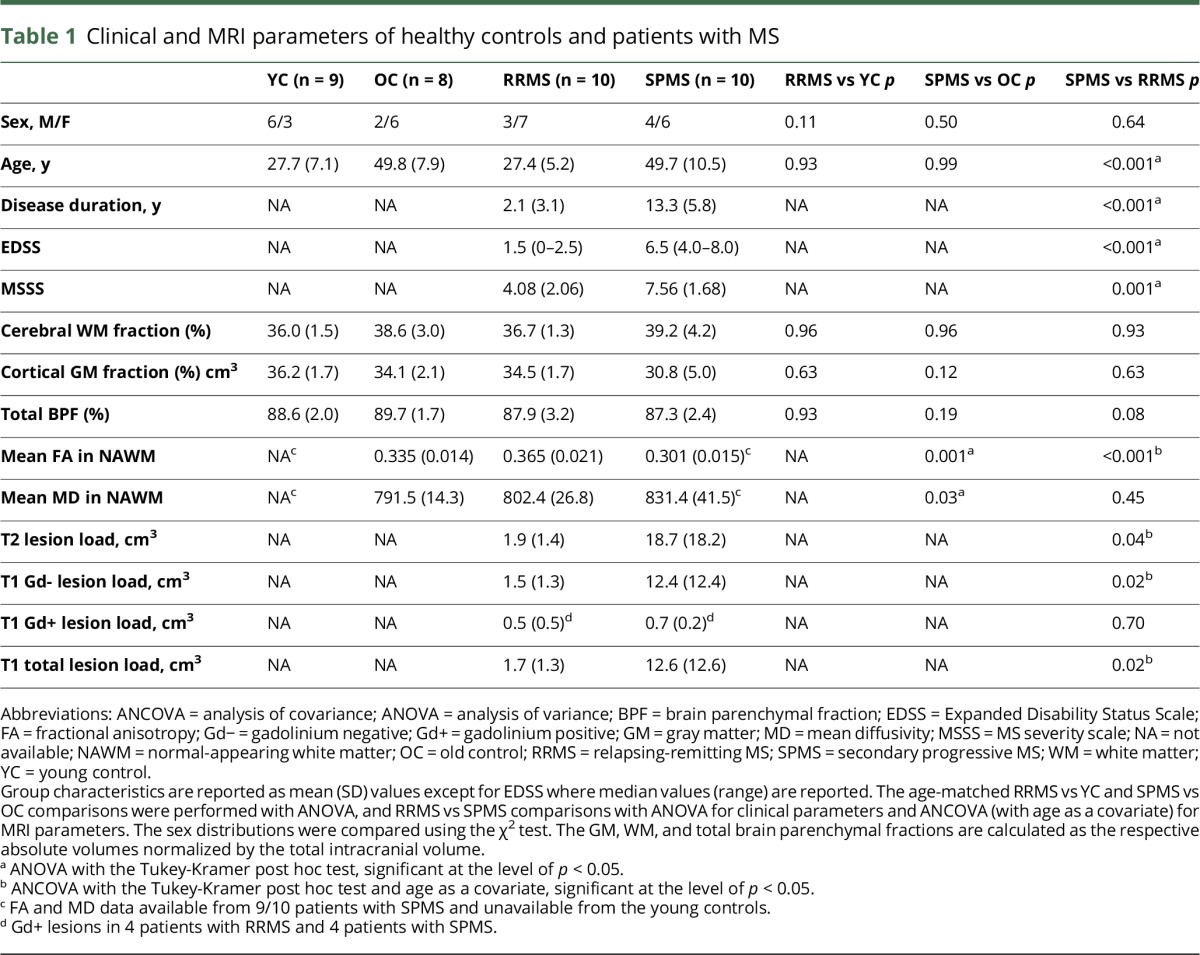
Quantitative MRI revealed differences within the NAWM DTI measures of patients with SPMS vs healthy controls and patients with RRMS (table 1). The reduced mean FA was observed in the NAWM tracts as a sign of impaired structural integrity in the SPMS group compared with patients with RRMS and controls. MD was increased in patients with SPMS compared with controls (table 1).
DTI data were unavailable for the younger controls. However, when compared with the older control group and adjusted for age, the mean FA was higher among patients with RRMS (ANCOVA, p = 0.029). In the mean MD, no differences were observed between patients with RRMS and older controls (ANCOVA p = 0.82).
Both structural and quantitative MRI determinants had a strong interaction with the clinical status of the patients with MS (table e-1, links.lww.com/NXI/A37). Higher total T1 lesion load associated positively with higher EDSS and MSSS scores, whereas higher T2 lesion load was associated with higher EDSS scores. In addition, reduced FA correlated with higher EDSS and MSSS scores. The WM, GM, and total BPFs did not correlate with the clinical parameters (table e-1). Microglial activation was increased in the NAWM and in the thalami of patients with SPMS compared with patients with RRMS and healthy controls.
Binding of [11C](R)-PK11195, measured as DVR, was higher in the NAWM of patients with SPMS compared with healthy controls (ANCOVA, p = 0.003). The binding in the NAWM was similarly higher in the SPMS group vs the RRMS group (ANCOVA, p = 0.018) (figure 2A).
Figure 2. Region of interest (ROI)-specific [11C](R)-PK11195 binding in patients with relapsing-remitting MS (RRMS), patients with secondary progressive MS (SPMS), and controls.
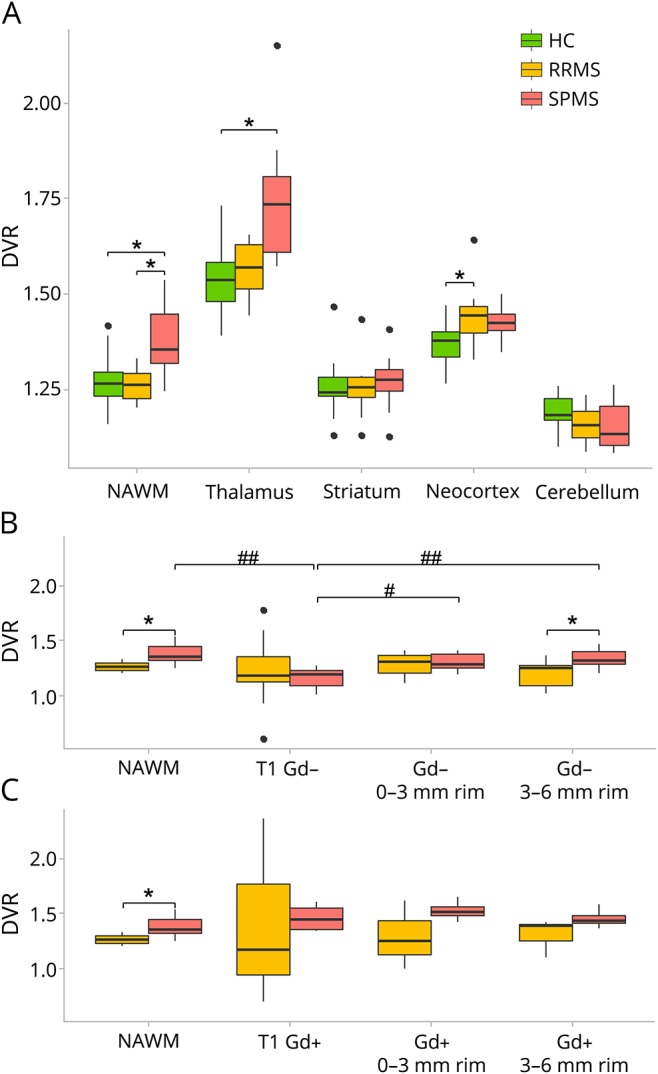
Radioligand binding by group among healthy controls (HC, n = 17), patients with RRMS (n = 10), and patients with SPMS (n = 10) in non–lesion-associated ROIs (A), gadolinium-negative lesion-associated ROIs (B), and gadolinium-positive lesion-associated ROIs (C). Each boxplot represents the group median, 1st and 3rd quartiles, minimum and maximum, and outliers (black dots) of distribution volume ratio (DVR) values in each ROI. NAWM = normal-appearing white matter; T1 Gd− = T1-hypointense, gadolinium-negative lesion ROI (including all T1 Gd− lesions); Gd− 0–3 mm rim = perilesional circular ROI 0–3 mm from the edge of gadolinium-negative lesions; Gd− 3–6 mm rim = perilesional circular ROI 3–6 mm from the edge of gadolinium-negative lesions; T1 Gd+ = T1-hypointense, gadolinium-positive lesion ROI (including all T1 Gd+ lesions); Gd+ 0–3 mm rim = perilesional circular ROI 0–3 mm from the edge of gadolinium-positive lesions; Gd+ 3–6 mm rim = perilesional circular ROI 3–6 mm from the edge of gadolinium-positive lesions. *Analysis of , age as a covariate, p < 0.05. #Repeated-measures analysis of variance (ANOVA), p < 0.05. ##Repeated-measures ANOVA, p < 0.001.
In the analyses of GM structures, higher radioligand binding was observed in the thalami of patients with SPMS compared with controls (ANCOVA, p = 0.022). For the other GM ROIs investigated, no differences between SPMS vs RRMS or SPMS vs control groups were found (figure 2A). However, the DVR values in the cortical GM were higher among the patients with RRMS compared with controls (ANCOVA, p = 0.003). The absolute groupwise DVR values are reported in detail in table e-2 (links.lww.com/NXI/A37).
Microglial activation is increased in the perilesional area of patients with SPMS compared with patients with RRMS
The radioligand binding was increased in the outer perilesional 3–6 mm circular ROI surrounding the Gd− T1-hypointense chronic lesions in patients with SPMS when compared with respective ROI in patients with RRMS (age-adjusted ANCOVA, p = 0.043), but not in the inner 0–3 mm perilesional ROI (p = 0.40) (figure 2B). There were no group differences in radioligand binding in the core area of the Gd− T1-hypointense lesions. In these lesions, the binding was always low. In the evaluation of Gd+ lesion–associated ROIs, the mean perilesional binding in patients with SPMS appeared to be higher compared with patients with RRMS, but the difference did not reach statistical significance because of the low number of patients (n = 4 + 4) and high variance (figure 2C).
Within-group evaluation of the patients with SPMS showed that TSPO binding inside Gd− T1-hypointense lesions was lower when compared with the NAWM in the same patients (rm ANOVA, p < 0.001), whereas in the RRMS group, no such difference was observed. Similarly, in patients with SPMS, the perilesional binding was higher than that in the center of the lesions (rm ANOVA; perilesional 0–3 mm zone vs core of Gd− T1 lesions, p = 0.003 and perilesional 3–6 mm zone vs Gd− T1 lesions, p < 0.001). In patients with RRMS, such differences were not observed (figure 2B).
In the Gd+ T1-hypointense lesions and in the perilesional areas surrounding them, no differences within groups were found in the DVR values. The absolute groupwise DVR values are reported in detail in table e-2 (links.lww.com/NXI/A37).
Increased [11C](R)-PK11195 binding in the NAWM is associated with more clinical disability and higher age in patients with MS
There were notable interactions between radioligand binding and the clinical characteristics of the patients with MS in a pooled analysis of the 2 MS subgroups (table e-1, links.lww.com/NXI/A37). The increased DVR of [11C](R)-PK11195 in the NAWM was associated with higher EDSS and higher MSSS scores, illustrated in figure 3, A and B. In addition, higher binding in the perilesional 3–6 mm region was associated with higher EDSS scores. In the other ROIs investigated, no significant associations between radioligand binding and clinical disability measures were found.
Figure 3. Associations of [11C](R)-PK11195 binding to disability and diffusion tensor imaging (DTI) changes in patients with MS.
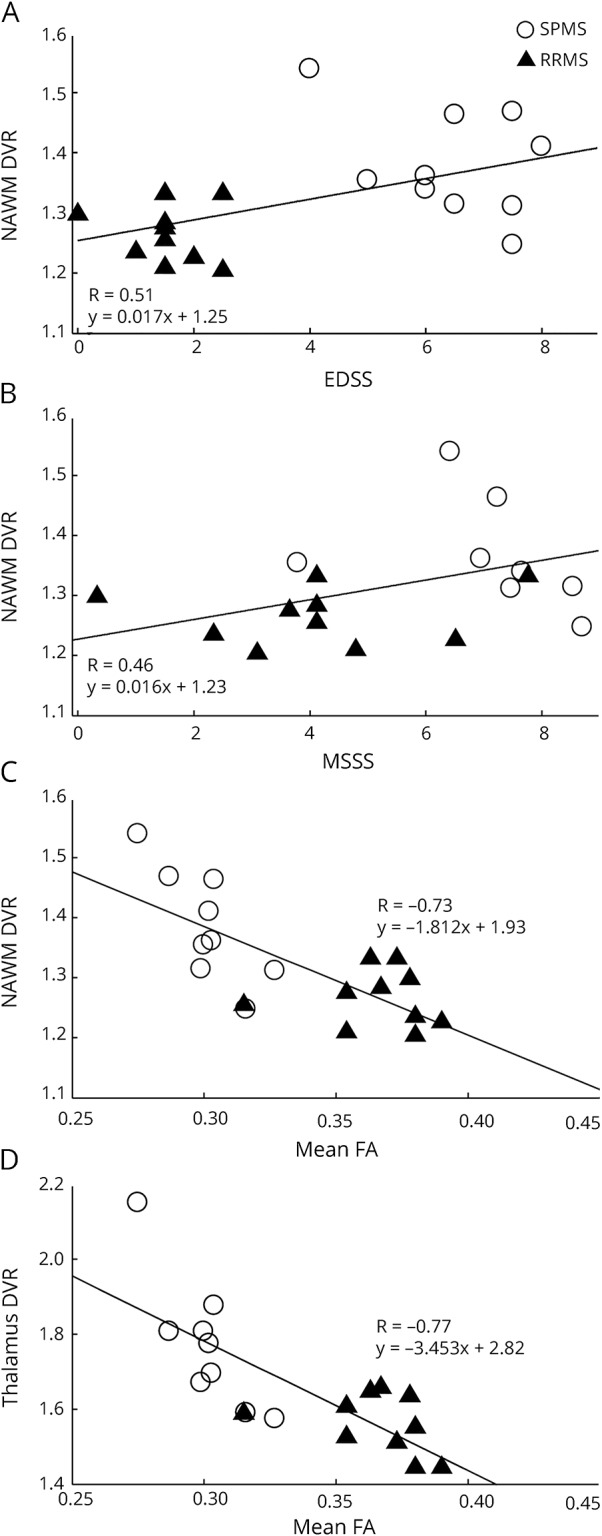
Region of interest–specific binding of [11C](R)-PK11195 measured as the distribution volume ratio (DVR) in the normal-appearing white matter (NAWM) plotted with (A) Expanded Disability Status Scale (EDSS), (B) MS severity score (MSSS), (C) mean fractional anisotropy (FA) in the NAWM, and (D) the DVR in the thalami plotted with mean FA in the NAWM in the pooled MS patient group. The correlations are visualized with linear regression lines corresponding to the results from repeated-measures analysis of covariance (rm ANCOVA) reported in tables e-1 and e-3 (links.lww.com/NXI/A37). SPMS = secondary progressive MS; RRMS = relapsing-remitting MS.
The effect of age on microglial activation in different ROIs within the pooled MS and control groups was further evaluated. In the ANCOVA analysis, increasing age correlated with higher [11C](R)-PK11195 DVR in the NAWM (age coefficient 0.003, standard error [SE] 0.001, p = 0.02) and thalami (age coefficient 0.005, SE 0.002, p = 0.03) among the patients with MS, but not in healthy controls (age coefficient 0.001, SE 0.001, p = 0.58 and age coefficient 0.004, SE 0.002, p = 0.13, respectively; figure 4). Conversely, healthy controls had higher cortical GM binding with increasing age (age coefficient 0.003, SE 0.001, p = 0.012), whereas a similar effect was absent in the MS group (age coefficient −0.001, SE 0.001, p = 0.60). No age dependence was observed in any of the other ROIs investigated.
Figure 4. Correlation of age with [11C](R)-PK11195 binding in the normal-appearing white matter (NAWM) and thalami.
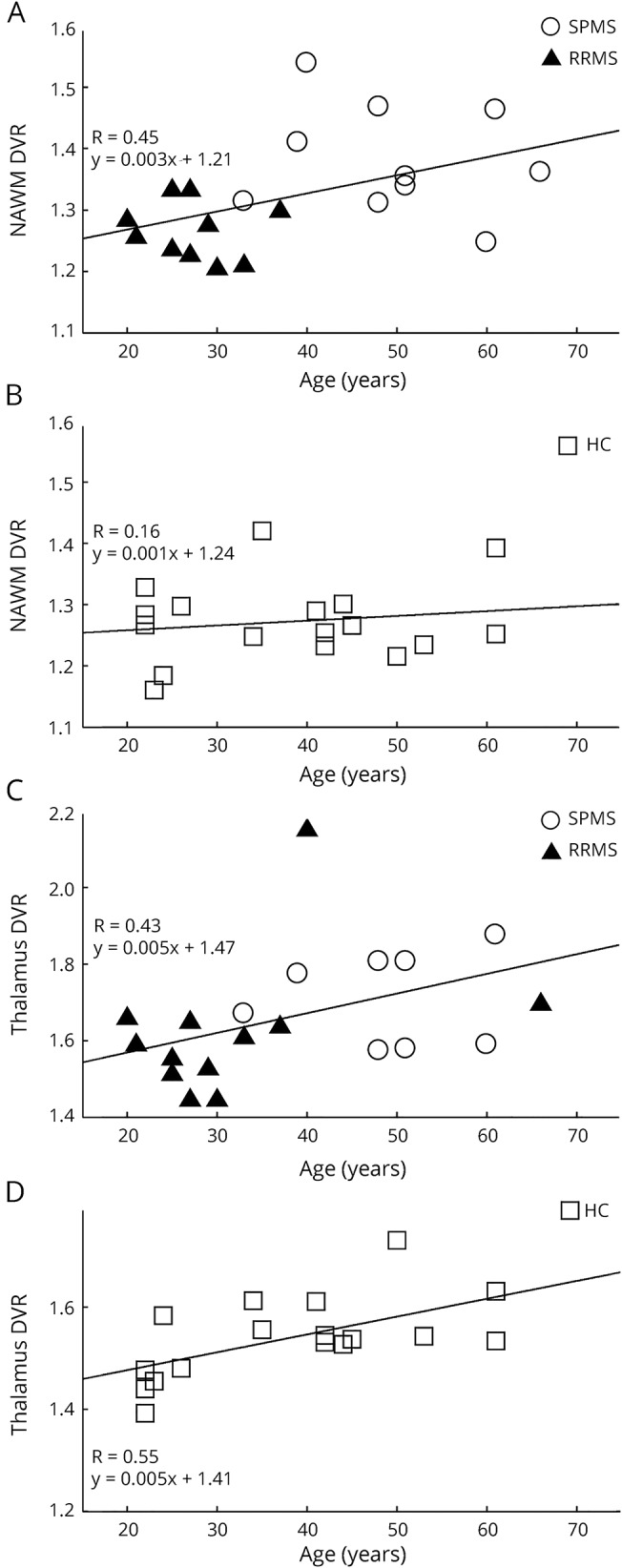
(A and C) Patients with MS, n = 20. (B and D) Controls, n = 17. The correlations are visualized with linear regression lines corresponding to the results from repeated-measures analysis of covariance (rm ANCOVA). SPMS = secondary progressive MS; RRMS = relapsing-remitting MS; HC = healthy control.
Increased [11C](R)-PK11195 binding in the NAWM, thalami, and the perilesional area correlate with MRI-detectable WM and GM pathology
We noted several associations between the [11C](R)-PK11195 binding and both structural and quantitative MRI parameters (tables e-3 and e-4, links.lww.com/NXI/A37). It is important that increased [11C](R)-PK11195 binding in the NAWM and thalami was associated with reduced WM tract integrity shown as lower mean global FA (figure 3, C and D) and higher MD in the NAWM (table e-3). The higher binding in the NAWM and thalami, as well as in the striatum, was also positively associated with higher T1 and T2 lesion loads in the WM. The same was true for the perilesional radioligand binding in the 3–6 mm ring around the lesions. Increased binding in the thalami was also associated with reduced cortical GM fraction (table e-4).
Discussion
We demonstrate that increased microglial activation in the NAWM of patients with MS correlates with advanced clinical disability, higher age, and reduced WM tract integrity in the NAWM, measured by DTI and expressed as a reduction in the mean FA. Microglial activation is increased in patients with SPMS compared with patients with RRMS and healthy controls. The finding is most pronounced in the NAWM, thalami, and in areas surrounding T1-hypointense chronic lesions. Based on these findings and previous work in the field, it is evident that microglial activation is relevant to the process of MS pathogenesis.9,17–19 Hence, in vivo TSPO PET can complement other imaging methodologies to improve our understanding of the kinetics and the cellular pathology leading to MS progression and help measure the overall inflammatory burden in progressive MS.
DTI has the ability to detect the diffuse pathologic changes in the WM20 by analyzing the movement of water molecules in the tissue. FA quantifies the preferential directionality of water diffusion along WM tracts and thereby provides in vivo information about the pathologic processes that modify brain microstructure.21 The seminal early studies already demonstrated the in vivo measurable decrease in the NAWM FA in patients with MS compared with controls.22,23 Reduced FA correlates with clinical disability,24 cognitive dysfunction,25 and increased lesion quantity in connected brain regions.26,27 The latter implies that Wallerian degeneration of axons transected by remote, but connected focal lesions, is an important pathogenic mechanism of NAWM damage in MS. Because activated microglial cells can contribute to axonal damage by producing proinflammatory cytokines and reactive oxygen and nitrogen species,28 it is likely that lesion-independent diffuse axonopathy also results in NAWM damage measurable by DTI.29
Our work demonstrates that in SPMS, the mean FA is reduced in the NAWM compared with patients with RRMS and healthy controls, which is in agreement with the neuropathologic and molecular imaging findings.18,22,23 The somewhat higher mean FA values in the NAWM among the RRMS cohort compared with the older cohort of controls could possibly be explained by the age difference because even normal aging induces alterations in the WM, which is reflected by a reduction in FA and increase in microglial activation.30 This study is the first direct comparative analysis of DTI measures and microglial activation in MS in vivo. Our results demonstrate the coupling of increased microglial activation in the NAWM and thalami to the diffuse DTI pathology in the NAWM, and to decreased cortical GM volume, and support the theory of the thalamus being a key MS-related disconnection node.31
Future studies will show whether the microglial activation–correlating global mean FA measurements of the NAWM can be validated for use as an independent indicator or a predictor of potentially reversible SPMS-associated diffuse pathology. This would be a much more widely available method to measure diffuse WM pathology than TSPO PET imaging. Improved technologies for assessing FA values might also be of use as demonstrated in a multicenter study of FA measurement in amyotrophic lateral sclerosis.32
Aging generally brings widespread, subtle microglial activation in healthy individuals and in patients with neurodegenerative disease.14,30,33 There may be age-dependent accumulation of dystrophic microglia that are incapable of conventional response to injury and may present a more proinflammatory, disease-promoting phenotype.34 This study shows that microglial activation is increased in the thalami and NAWM, with increasing age more steeply among patients with MS than in age-matched healthy individuals, which supports the notion that subtle microglial activation may initially be an age-related phenomenon but is accentuated following neuroinflammation.
Our results of the increased TSPO binding in the NAWM and thalami in MS are in agreement with earlier in vivo PET studies.15,17,19,35 In our study, the patients either had relatively early RRMS disease or late SPMS disease, which was probably helpful for demonstrating groupwise differences in the pathology despite the limited number of patients. Importantly, none of the patients were on immunomodulatory treatment, thus offering a view into the pathology related to the natural course of the disease. In contrast to 2 earlier studies by other research groups,17,19 we observed increased TSPO binding in the cortical GM only in the patients with RRMS, but not in the patients with SPMS when compared with age-matched controls. This might be due to the differences in the methods for reference region extraction, partial volume correction and in the accuracy of the segmentation of cortical structures effected also by the different resolutions of MRI scanners. Our data sets implicated increasing cortical TSPO binding in the GM with age among healthy controls, but not within patients with MS. We suspect that this could be attributed to image reconstruction–related issues concerning attenuation and scatter correction because the younger and older controls were scanned and reconstructed in different facilities, with cortical and basal structures being more prone to such bias than the WM and deep GM.
TSPO PET imaging can be used to assess the total inflammatory burden in MS, and our results support the close association between neuroinflammation and neurodegeneration that is suspected in progressive MS. The study demonstrates, in vivo, a concomitant presence of structural disintegrity of the NAWM and microglial activity both in the perilesional WM and in the NAWM. Importantly, these were associated with increasing clinical disability scores. Based on our results, and on earlier TSPO PET and neuropathology studies, it is likely that the increased TSPO binding, i.e., microglial activation, is associated with myelin degradation, gliosis and inflammation in the perilesional area and diffuse inflammation in the NAWM, and correlates with neurodegeneration. PET imaging can thus contribute to the multiparametric evaluation of progressive MS pathology and may therefore provide a complementary tool for clinical treatment studies of progressive MS.
Acknowledgment
The authors gratefully acknowledge Prof. Federico Turkheimer (Imperial College London) for providing SuperPK software for academic use and Prof. Ronald Boellaard (VU University Medical Center Groningen and University of Groningen) for his kind assistance in the optimization of the supervised cluster algorithm. They thank Dr. David Smith for critical reading of the manuscript and the expert staff of Turku PET Centre for their work in radiochemical production and imaging procedures. They also acknowledge Ms. Salla Nuutinen and Ms. Marjo Nylund (Turku University Hospital) for their technical assistance in finalizing the manuscript.
Glossary
- ANCOVA
analysis of covariance
- ANOVA
analysis of variance
- BPF
brain parenchymal fraction
- CIS
clinically isolated syndrome
- DTI
diffusion tensor imaging
- DVR
distribution volume ratio
- EDSS
Expanded Disability Status Scale
- FA
fractional anisotropy
- GM
gray matter
- MD
mean diffusivity
- MSSS
MS severity scale
- NAWM
normal-appearing white matter
- rm ANCOVA
repeated-measures analysis of covariance
- ROI
region of interest
- RRMS
relapsing-remitting MS
- SPMS
secondary progressive MS
- TPC
Turku PET Centre
- TSPO
translocator protein
- WM
white matter
- WMIC
Wolfson Molecular Imaging Centre
Author contributions
Eero Rissanen: study concept and design, acquisition, analysis, or interpretation of data, and drafting of the manuscript. Jouni Tuisku: acquisition, analysis, or interpretation of data and drafting of the manuscript. Tero Vahlberg, Marcus Sucksdorff, Teemu Paavilainen, Riitta Parkkola, Johanna Rokka, Alexander Gerhard, Rainer Hinz, and Peter S. Talbot: acquisition, analysis, or interpretation of data. Juha O. Rinne and Laura Airas: study concept and design and drafting of the manuscript.
Study funding
The study was funded by the Finnish Academy, the Sigrid Juselius Foundation, Merck Serono S.A. Geneva, Switzerland, the Finnish MS Foundation, and the European Union's Seventh Framework Programme (FP7/2007-2013) under grant agreement no. HEALTH-F2-2011-278850 (INMiND).
Disclosure
E. Rissanen received speaker honoraria from Teva, Biogen, and Roche; consulted for Merck; and received research support from Turku University Hospital and the Finnish MS Foundation. J. Tuisku, T. Vahlberg, M. Sucksdorff, and T. Paavilainen report no disclosures. R. Parkkola received research support from the Sigrid Juselius Foundation, the Maud Kuistilla Memorial Foundation, the Emil Aaltonen Foundation, and the Tor, Joe, and Pentti Borg Foundation. J. Rokka served on the editorial board of Neuropathology and Applied Neurobiology and received research support from GE Healthcare, MRC, European Commission INMiND, NIHR, PSP Association, and the Michael J. Fox Foundation. A. Gerhard reports no disclosures. R. Hinz served on the editorial board of the Journal of Neuroimaging and received research support from the European Union's Seventh Framework Programme. P.S. Talbot served on the editorial board of the Journal of Psychopharmacology. J.O. Rinne served as a consultant neurologist for Clinical Research Services Turku (which includes contract research with pharmaceutical companies); received speaker honoraria from Biogen, Novartis, and Orion Pharma; is an associate editor of the Journal of Movement Disorders; and received research support from the Academy of Finland and the Sigrid Juselius Foundation. L. Airas served on the scientific advisory board of Teva, Roche, Biogen, Genzyme, and Novartis; received travel funding and/or speaker honoraria from Teva, Genzyme, Biogen, Sanofi-Aventis, and Merck; and received research support from Merck, Novartis, Biogen, and Genzyme. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NN.
References
- 1.Compston A, Coles A. Multiple sclerosis. Lancet 2008;372:1502–1517. [DOI] [PubMed] [Google Scholar]
- 2.Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 2012;8:647–656. [DOI] [PubMed] [Google Scholar]
- 3.Filippi M, Rocca MA, Barkhof F, et al. . Association between pathological and MRI findings in multiple sclerosis. Lancet Neurol 2012;11:349–360. [DOI] [PubMed] [Google Scholar]
- 4.Frischer JM, Bramow S, Dal-Bianco A, et al. . The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009;132:1175–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ching AS, Kuhnast B, Damont A, Roeda D, Tavitian B, Dolle F. Current paradigm of the 18-kDa translocator protein (TSPO) as a molecular target for PET imaging in neuroinflammation and neurodegenerative diseases. Insights Imaging 2012;3:111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lavisse S, Guillermier M, Herard AS, et al. . Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci 2012;32:10809–10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su Z, Herholz K, Gerhard A, et al. . [11C]-(R)PK11195 tracer kinetics in the brain of glioma patients and a comparison of two referencing approaches. Eur J Nucl Med Mol Imaging 2013;40:1406–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hunter HJ, Hinz R, Gerhard A, et al. . Brain inflammation and psoriasis: a [11 C]-(R)-PK11195 positron emission tomography study. Br J Dermatol 2016;175:1082–1084. [DOI] [PubMed] [Google Scholar]
- 9.Rissanen E, Tuisku J, Rokka J, et al. . In vivo detection of diffuse inflammation in secondary progressive multiple sclerosis using positron emission tomography imaging and radioligand [11C]PK11195. J Nucl Med 2014;55:939–944. [DOI] [PubMed] [Google Scholar]
- 10.Heiss WD, Habedank B, Klein JC, et al. . Metabolic rates in small brain nuclei determined by high-resolution PET. J Nucl Med 2004;45:1811–1815. [PubMed] [Google Scholar]
- 11.de Jong HW, van Velden FH, Kloet RW, Buijs FL, Boellaard R, Lammertsma AA. Performance evaluation of the ECAT HRRT: an LSO-LYSO double layer high resolution, high sensitivity scanner. Phys Med Biol 2007;52:1505–1526. [DOI] [PubMed] [Google Scholar]
- 12.Rissanen E, Virta JR, Paavilainen T, et al. . Adenosine A2A receptors in secondary progressive multiple sclerosis: a [11C]TMSX brain PET study. J Cereb Blood Flow Metab 2013;33:1394–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turkheimer FE, Edison P, Pavese N, et al. . Reference and target region modeling of [11C]-(R)-PK11195 brain studies. J Nucl Med 2007;48:158–167. [PubMed] [Google Scholar]
- 14.Yaqub M, van Berckel BN, Schuitemaker A, et al. . Optimization of supervised cluster analysis for extracting reference tissue input curves in [11C]-(R)-PK11195 brain PET studies. J Cereb Blood Flow Metab 2012;32:1600–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banati RB, Newcombe J, Gunn RN, et al. . The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain 2000;123:2321–2337. [DOI] [PubMed] [Google Scholar]
- 16.Okello A, Edison P, Archer HA, et al. . Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology 2009;72:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Politis M, Giannetti P, Su P, et al. . Increased PK11195 PET binding in the cortex of patients with MS correlates with disability. Neurology 2012;79:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moll NM, Rietsch AM, Thomas S, et al. . Multiple sclerosis normal-appearing white matter: pathology-imaging correlations. Ann Neurol 2011;70:764–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herranz E, Gianni C, Louapre C, et al. . Neuroinflammatory component of gray matter pathology in multiple sclerosis. Ann Neurol 2016;80:776–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmierer K, Wheeler-Kingshott CA, Boulby PA, et al. . Diffusion tensor imaging of post mortem multiple sclerosis brain. Neuroimage 2007;35:467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basser PJ, Pierpaoli C. Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J Magn Reson B 1996;111:209–219. [DOI] [PubMed] [Google Scholar]
- 22.Werring DJ, Clark CA, Barker GJ, Thompson AJ, Miller DH. Diffusion tensor imaging of lesions and normal-appearing white matter in multiple sclerosis. Neurology 1999;52:1626–1632. [DOI] [PubMed] [Google Scholar]
- 23.Ciccarelli O, Werring DJ, Wheeler-Kingshott CA, et al. . Investigation of MS normal-appearing brain using diffusion tensor MRI with clinical correlations. Neurology 2001;56:926–933. [DOI] [PubMed] [Google Scholar]
- 24.Harrison DM, Shiee N, Bazin PL, et al. . Tract-specific quantitative MRI better correlates with disability than conventional MRI in multiple sclerosis. J Neurol 2013;260:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welton T, Kent D, Constantinescu CS, Auer DP, Dineen RA. Functionally relevant white matter degradation in multiple sclerosis: a tract-based spatial meta-analysis. Radiology 2015;275:89–96. [DOI] [PubMed] [Google Scholar]
- 26.Werring DJ, Brassat D, Droogan AG, et al. . The pathogenesis of lesions and normal-appearing white matter changes in multiple sclerosis: a serial diffusion MRI study. Brain 2000;123:1667–1676. [DOI] [PubMed] [Google Scholar]
- 27.Ciccarelli O, Werring DJ, Barker GJ, et al. . A study of the mechanisms of normal-appearing white matter damage in multiple sclerosis using diffusion tensor imaging—evidence of Wallerian degeneration. J Neurol 2003;250:287–292. [DOI] [PubMed] [Google Scholar]
- 28.Witte ME, Mahad DJ, Lassmann H, van Horssen J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol Med 2014;20:179–187. [DOI] [PubMed] [Google Scholar]
- 29.Friese MA, Schattling B, Fugger L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat Rev Neurol 2014;10:225–238. [DOI] [PubMed] [Google Scholar]
- 30.Cornejo F, von Bernhardi R. Age-dependent changes in the activation and regulation of microglia. Adv Exp Med Biol 2016;949:205–226. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Duan Y, Huang J, et al. . Multimodal quantitative MR imaging of the thalamus in multiple sclerosis and neuromyelitis optica. Radiology 2015;277:784–792. [DOI] [PubMed] [Google Scholar]
- 32.Muller HP, Turner MR, Grosskreutz J, et al. . A large-scale multicentre cerebral diffusion tensor imaging study in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2016;87:570–579. [DOI] [PubMed] [Google Scholar]
- 33.Kumar A, Muzik O, Shandal V, Chugani D, Chakraborty P, Chugani HT. Evaluation of age-related changes in translocator protein (TSPO) in human brain using [11C]-(R)-PK11195 PET. J Neuroinflammation 2012;9:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michell-Robinson MA, Touil H, Healy LM, et al. . Roles of microglia in brain development, tissue maintenance and repair. Brain 2015;138:1138–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Debruyne JC, Versijpt J, Van Laere KJ, et al. . PET visualization of microglia in multiple sclerosis patients using [11C]PK11195. Eur J Neurol 2003;10:257–264. [DOI] [PubMed] [Google Scholar]