

Abstract
Skeletal muscle enables posture, breathing, and locomotion. Skeletal muscle also impacts systemic processes such as metabolism, thermoregulation, and immunity. Skeletal muscle is energetically expensive and is a major consumer of glucose and fatty acids. Metabolism of fatty acids and glucose requires NAD+ function as a hydrogen/electron transfer molecule. Therefore, NAD+ plays a vital role in energy production. In addition, NAD+ also functions as a cosubstrate for post-translational modifications such as deacetylation and ADP-ribosylation. Therefore, NAD+ levels influence a myriad of cellular processes including mitochondrial biogenesis, transcription, and organization of the extracellular matrix. Clearly, NAD+ is a major player in skeletal muscle development, regeneration, aging, and disease. The vast majority of studies indicate that lower NAD+ levels are deleterious for muscle health and higher NAD+ levels augment muscle health. However, the downstream mechanisms of NAD+ function throughout different cellular compartments are not well understood. The purpose of this review is to highlight recent studies investigating NAD+ function in muscle development, homeostasis, disease, and regeneration. Emerging research areas include elucidating roles for NAD+ in muscle lysosome function and calcium mobilization, mechanisms controlling fluctuations in NAD+ levels during muscle development and regeneration, and interactions between targets of NAD+ signaling (especially mitochondria and the extracellular matrix). This knowledge should facilitate identification of more precise pharmacological and activity-based interventions to raise NAD+ levels in skeletal muscle, thereby promoting human health and function in normal and disease states.
Background
The laws of thermodynamics state that energy remains constant within a closed system. Energy is neither created nor destroyed; it just changes form. In biology, the flow of energy through organisms, tissues, and cells is a universal requirement to sustain life. Energy in the form of dietary nutrients is metabolized into adenosine triphosphate (ATP) and other molecules that power necessary cellular processes. Therefore, energy is potentially the most important information that cells interpret. As such, it would logically follow that multiple, highly nuanced cellular mechanisms for nutrient and energy sensing would evolve due to intense selective pressures. This exquisite tailoring of form to function is perhaps best exemplified in the mitochondrial reticulum that facilitates quick energy transfer throughout the muscle cell [1]. Oxidized nicotinamide adenine dinucleotide (NAD+) and its reduced form, reduced nicotinamide adenine dinucleotide (NADH), are critical molecules involved in energy generation because NAD+/NADH participate in oxidation-reduction (redox) reactions in the tricarboxylic acid (TCA) cycle. In this context, NAD+ is not destroyed, it is reversibly reduced. However, NAD+ is also a cosubstrate in multiple post-translational modifications such as deacetylation and ADP-ribosylation. These reactions result in consumption of NAD+, necessitating replenishment of cellular NAD+ stores. Thus, maintaining the balance between NAD+ degradation and biosynthesis is extremely important for cellular homeostasis. In this review, we will discuss evidence and mechanisms of NAD+ function in skeletal muscle development, homeostasis, and disease.
In addition to mediating posture and locomotion, skeletal muscle is a metabolically important organ. Skeletal muscles sense, produce, store, and utilize nutrients and energy transfer molecules that enable muscle to participate in systemic, energy-expensive processes such as temperature regulation and immunity. Skeletal muscle is plastic and readily adapts to change. Examples of skeletal muscle plasticity include hypertrophy in response to weight bearing exercise, atrophy in response to disuse, and fiber type switching due to changes in gene expression, cellular metabolism, or innervation. It seems likely that there is coordination between skeletal muscle’s energy use/sensing and the signaling pathways that regulate its remarkable phenotypic plasticity.
Muscle diseases have a negative effect on health, lifespan, and/or quality of life. Genetic muscle diseases caused by mutations result in a variety of ultrastructural defects in muscle cells and progressive loss of muscle mass and function via multiple different mechanisms. Inflammatory or metabolic diseases (such as diabetes, obesity, autoimmune diseases, cancer, and infections) can result in loss of skeletal muscle as well. Given the integration and interdependence of the nervous and muscular systems, neural disorders or injuries can also impair muscle tissue structure and function. Additionally, skeletal muscle is lost as a natural part of the aging process, and this loss is exacerbated in a condition called sarcopenia. Adequate skeletal muscle mass prior to illness, injury, or aging is predictive of recovery and healthy aging (reviewed in [2]). Skeletal muscle performs a diverse set of crucial functions in organisms and promoting muscle health and exploiting its plasticity could improve locomotion, metabolism, and quality of life in many disorders.
In this review, we will discuss the recent data that document conserved roles for NAD+ in skeletal muscle development, regeneration, aging, and disease as well as interventions targeting skeletal muscle and affecting NAD+ that suggest promising therapeutic benefits. We will also highlight gaps in our knowledge and propose avenues of future investigation to better understand why and how NAD+ regulates skeletal muscle biology.
Introduction to NAD+
A role for NAD+ in cellular ATP production has been known for approximately a century. NAD+ was originally referred to as “cozymase” when first discovered as a hydrogen and electron acceptor and donor in redox reactions (reviewed in [3]). The metabolism of glucose in cells that can occur in either the presence or absence of oxygen (aerobic/oxidative vs. anaerobic/glycolytic, respectively) requires NAD+. NAD+ plays roles in multiple, diverse cellular processes in addition to redox reactions. NAD+ is a cosubstrate in deacetylation reactions as well as in mono- and poly-ADP-ribosylation. While NAD+ can cycle between its oxidized and reduced forms in redox reactions, it is cleaved when it functions as a cosubstrate. Thus, NAD+ must be constantly re-synthesized from dietary precursors in order to maintain cellular pools of NAD+ as well as appropriate NAD+/NADH ratios (Fig. 1). It is not entirely clear whether the critical aspect of NAD+ biology in skeletal muscle is levels of NAD+ or the NAD+/NADH ratio [4]. For simplicity, we will generally just refer to NAD+ levels.
Fig. 1.
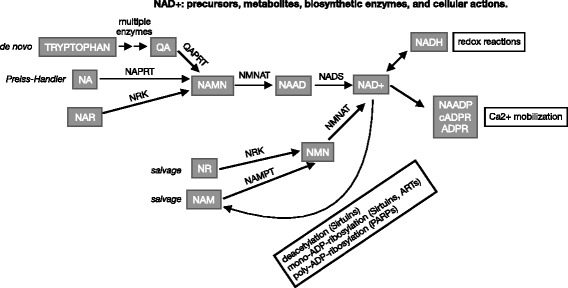
NAD+ biosynthetic pathways. NAD+ can be synthesized via de novo, Preiss-Handler, and multiple salvage pathways. NAD+ precursors and metabolites are represented in gray boxes. The enzymes involved in each conversion are listed above the arrows. The cellular outputs regulated by NAD+ or metabolites are listed in white boxes with black outlines. For explanation of the acronyms, please see the abbreviations list in the main body of the text
Given the large demand for NAD+ as a cosubstrate in many cellular reactions, it has been estimated that grams of NAD+ precursors would need to be ingested daily in order to maintain physiological NAD+ concentrations [5]. As the recommended daily allowance of dietary NAD+ precursors is only in the milligram range, this suggests that there are multiple biosynthetic pathways that generate NAD+ from different dietary precursors and that the nicotinamide byproduct formed upon NAD+ cleavage is efficiently recycled for NAD+ biosynthesis [6].
NAD+ can be formed from dietary ingestion of the amino acid tryptophan or forms of vitamin B3/niacin (nicotinic acid, nicotinamide, and nicotinamide riboside). Quinolinic acid, a metabolite of tryptophan, can also be a starting point for NAD+ biosynthesis (Fig. 1). The de novo pathway of NAD+ biosynthesis from tryptophan involves seven enzymatic steps. In contrast, the salvage pathways of NAD+ biosynthesis from niacin involve only two or three enzymatic steps. Important enzymes involved in NAD+ biosynthesis are nicotinamide phosphoribosyltransferase (NAMPT), which acts on nicotinamide; NRK1 and/or NRK2, which act on nicotinamide riboside; and the NMNAT family of enzymes, which act on multiple substrates in de novo and salvage NAD+ biosynthesis (Fig. 1).
Conserved roles for NAD+ as a mediator of health
NAD+ is linked to healthspan and lifespan in organisms separated by millions of years of evolution. There is a conserved decline in the NAD+/NADH ratio as worms, mice, and humans age [7–9]. Lower levels of NAD+ and lower NAD+/NADH ratios are correlated with functional decline and diseases of aging and mitochondrial dysfunction, such as metabolic and neurodegenerative diseases. Enhancing NAD+ levels is sufficient to increase lifespan in worms [7, 10], and in worm and mouse models of ataxia telangiectasia [11]. NAD+ levels are further implicated in human health in that dietary deficiencies in NAD+ precursors can cause pellagra. Pellagra results in skin, gastrointestinal, psychological, and neurodegenerative symptoms and is fatal without B vitamin or other appropriate NAD+ precursor supplementation. Given the conserved decline of NAD+ with age and the ability of NAD+ to increase lifespan in worms, it has been hypothesized that the loss of skeletal muscle mass and function due to aging or disease could be caused, at least in part, by lower availability of NAD+. We will review roles for NAD+ in muscle development, homeostasis, disease, and aging and highlight interventions that restore NAD+ levels and ameliorate muscle dysfunction.
NAD+ compartmentalization in skeletal muscle
Skeletal muscle is energetically expensive. Skeletal muscle cells take in glucose and store it as glycogen. When there is a need for more energy within muscle fibers, glycogen is converted to glucose that is used to generate ATP. Skeletal muscle cells can contain thousands of mitochondria, which are where the aerobic/oxidative metabolism of glucose occurs. As NAD+ is required as a hydrogen and electron acceptor and donor for aerobic cellular metabolism, NAD+ localization to mitochondria is important for muscle function. Mitochondria are the subcellular compartments that have the highest level of NAD+ in skeletal muscle cells. However, 95% of the NADH in skeletal muscle is also localized to mitochondria. Therefore, the NAD+/NADH ratio is relatively low in skeletal muscle mitochondria despite there being high levels of NAD+ [12–14]. The observation that mitochondria contain the greatest amount of NAD+ in skeletal muscle cells reflects the prioritization of redox and ATP-generating reactions for muscle biology; however, NAD+ is involved in many cellular reactions as discussed below.
NAD+ localization to other organelles facilitates the additional NAD+-consuming reactions that occur in muscle homeostasis (Fig. 2). A cytosolic pool of NAD+ participates in the anaerobic/glycolytic metabolism of glucose into ATP. This type of cellular metabolism is more rapid but less efficient in ATP generation than the aerobic/oxidative metabolism that occurs in mitochondria. Metabolites of cytosolic NAD+, such as nicotinic acid adenine dinucleotide phosphate (NAADP), cyclic ADP-ribose (cADPR), O-acetyl-ADPR (OAADPR), and ADPR, regulate calcium ion channels which can promote calcium influx from the extracellular environment or calcium release from internal stores (reviewed in [6, 15, 16]), (Fig. 2). Like ATP, calcium ions are required for functionality of the contractile apparatus in striated muscle and calcium ions are also important secondary messengers in signal transduction events. Considering that skeletal muscle evolved a subcellular organelle specifically designated for storage and rapid release of calcium ions (the sarcoplasmic reticulum), the role that NAADP, cADPR, OAADPR, and ADPR play in regulating calcium mobilization in skeletal muscle is a burgeoning area of research.
Fig. 2.
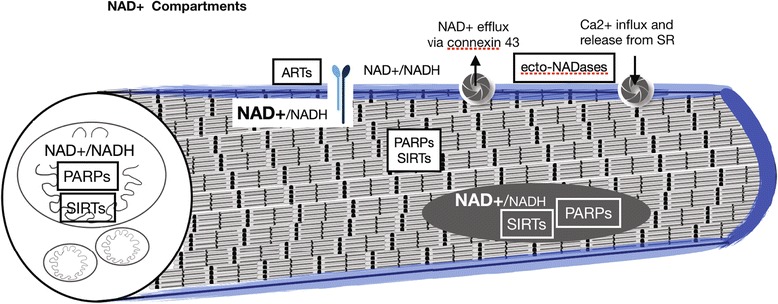
Compartmentalization of NAD+ pools in skeletal muscle. Diagram of a striated skeletal muscle fiber. NAD+ is localized to mitochondrial, nuclear, cytosolic, and membrane proximal pools in muscle cells. Additional NAD+ compartments not diagrammed here include vesicular compartments. The NAD+/NADH ratio is higher in the nuclear and cytosolic compartments compared to the mitochondrial compartment. The ratio is unknown in the membrane proximal compartment in muscle. Enzymes that consume NAD+ and their relative subcellular localizations are found within black or white boxes. Integrin receptors and membrane channels that transport NAD+ and calcium across the sarcolemma can be seen in the diagram
In the nucleus, NAD+ is used as a cosubstrate for sirtuin-mediated deacetylation and mono-ADP-ribosylation reactions as well as poly-ADP-ribosylation reactions carried out by PARP enzymes (Fig. 2). Sirtuins regulate gene expression and protein function via deacetylation of histones or other proteins, respectively. Sirtuin activity is generally beneficial for cellular and organismal health [7, 17]. PARP activity is stimulated by oxidative stress [18, 19]. PARPs are involved in repair of DNA damage. PARP-mediated repair of DNA damage is certainly a beneficial process in times of nutrient excess and oxidative stress; however, PARP activity drastically reduces the pool of nuclear NAD+ and essentially outcompetes sirtuins for their shared cosubstrate. Therefore, the reduction of oxidative stress and the genetic or pharmacological inhibition of PARPs can increase the NAD+ available for sirtuin-mediated deacetylation reactions. This has shown to be a promising therapeutic approach for some muscle conditions [20–23].
Adhesion of muscle cells to their extracellular matrix (ECM) microenvironment is critical for muscle development and homeostasis (reviewed in [24]). Muscle fibers, muscle fiber bundles, and whole muscle are surrounded by ECM. One substructure within the ECM is the basement membrane. Basement membranes are specialized parts of the ECM that serve a myriad of functions, playing both structural and signaling/organizational roles. Laminin, a major component of basement membranes, is necessary for basement membrane assembly [25]. NAD+ is found in the extracellular environment surrounding cells [6, 16], where it is a cosubstrate for ecto-ADP-ribosyltransferases. NAD+ is important for normal structure of the extracellular microenvironment (Fig. 3). There is a membrane tethered, extracellular mono-ADP-ribosyltransferase, ART1, that ADP-ribosylates Integrin alpha7 (ITGA7) at multiple sites in a NAD+ concentration-dependent manner [26–30]. This ADP-ribosylation by ART1 is not readily reversible and leads to an increased laminin-binding affinity of Integrin alpha7beta1 in differentiated myotubes. Thus, this ADP ribosylation event might be a protective mechanism that increases adhesion to laminin when the sarcolemma has been compromised and NAD+ leaks into the extracellular space [27]. Another example of NAD+ function in the plasma membrane proximal compartment of muscle cells involves NRK enzymes. Yeast and human NRK enzymes participate in an alternative salvage pathway that generates NAD+ [31]. NRK2 (aka MIBP2, ITGB1BP3) was found to bind to the cytosolic tail of Integrin beta1 and regulate adhesion of cultured mouse muscle cells to laminin-111 but not fibronectin [32]. We found that zebrafish Nrk2b localizes to the myotendinous junction and is required for proper organization of laminin-111 in the developing myotendinous junction. Exogenous NAD+ rescues laminin integrity at the myotendinous junction in Nrk2b-deficient zebrafish [33], indicating that zebrafish Nrk2b also functions to generate NAD+. Laminin organization at the myotendinous junction is a novel role for NAD+ and the mechanism is still unknown. Taken together, the above data suggest the potential of an additional pool of NAD+, the membrane proximal pool, that mediates skeletal muscle physiology (Fig. 3).
Fig. 3.
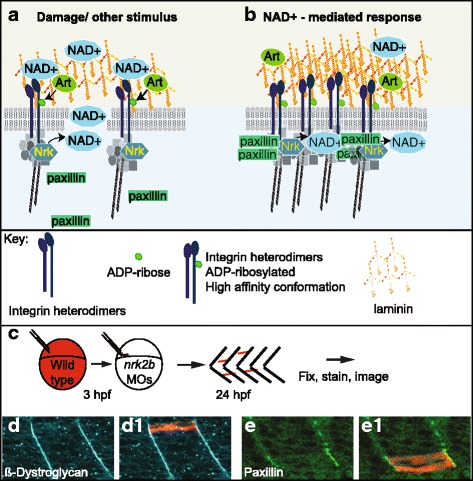
Hypothesized mechanism of membrane proximal NAD+ action in muscle-ECM adhesion. (A–B) Model of membrane proximal NAD+ regulation of subcellular protein localization, post-translational modification, laminin-binding affinity, and ECM organization in muscle. (A) Damage scenario. NRK enzymes localize to cytoplasmic tails of Integrin receptors for laminin and generate an intracellular membrane proximal pool of NAD+. In response to damage, intracellular membrane proximal NAD+ generated by NRK enzymes is actively transported or leaks across the sarcolemma into the extracellular environment. Extracellular membrane proximal NAD+ is consumed by ART enzymes as ADP-ribose moieties are added to Integrin receptors for laminin in mono-ADP-ribosylation reactions. Intracellular localization of Paxillin to cell-matrix adhesion complexes is disrupted. Extracellular organization of laminin is disrupted. (B) NAD+-mediated damage response scenario. Due to the movement of intracellular membrane proximal NAD+ to the extracellular environment, ADP-ribosylation of Integrin receptors for laminin change to a high-affinity binding conformation. Laminin organization is increased, Paxillin localization to cell-matrix adhesion complexes is restored, and Integrin-laminin binding is augmented. (C–E) Reprinted with permission from [33]. c Model of genetic mosaic experiment to determine if wild-type Nrk2b is sufficient in a cell autonomous manner for subcellular localization of beta-Dystroglycan and/or Paxillin to cell-matrix adhesions. Fluorescent dextran labeled wild-type cells were transplanted into a Nrk2b-deficient background and subcellular localization of beta-Dystroglycan and Paxillin were determined by immunohistochemistry. (D–E) Side mount, anterior left, 26 hpf nrk2b morphant hosts with transplanted wild-type cells (red) and beta-Dystroglycan (blue) or Paxillin antibody staining (green). (D-D1) Beta-Dystroglycan robustly localizes to MTJs in nrk2b morphants in the presence and the absence of transplanted wild-type cells. (E-E1) Robust Paxillin localization to cell-matrix adhesion complexes in nrk2b morphants is rescued in the transplanted wild-type cells, but not in the Nrk2b-deficient cells surrounding them. Therefore, Nrk2b is cell autonomously sufficient for subcellular localization of Paxillin, likely through generation of membrane proximal NAD+
NAD+ is also localized to vesicular compartments within cells, such as the Golgi, endoplasmic reticulum, peroxisomes, and lysosomes. It is tempting to speculate that compartmentalization of NAD+ allows for finer spatial and temporal control of NAD+-dependent signaling. To our knowledge, roles for NAD+ in function of these organelles in skeletal muscle have not been elucidated and this represents a major area for future research. We will focus on new data implicating the membrane proximal, mitochondrial, and nuclear/cytoplasmic pools of NAD+ in skeletal muscle development and health.
NAD+ in muscle development and regeneration
Periods of muscle development and/or growth may be coordinated with nutrient availability. As NAD+ provides cells with information about nutrient levels, it is likely that NAD+ is involved in regulating these cellular transitions. A decrease in NAD+ levels is observed in myotubes compared to myoblasts (both in C2C12s and primary muscle cells) [34]. Thus, the current thinking is that decreased NAD+ levels are permissive for muscle differentiation. The regulation of muscle differentiation by NAD+ in cultured mouse myoblasts seems to be mediated by SIRT1 activity. SIRT1 represses muscle differentiation: SIRT1 complexes with myoD and the acetyltransferase p300/CBP-associated factor (PCAF) to inhibit muscle gene transcription in vitro [34]. Fulco and colleagues hypothesized that nutrient sensing of glucose would be integrated with NAD+ levels because this would provide a mechanism where nutrition and myogenesis could be coordinated [4]. They found that energy sensing feeds into NAD+ levels by modulating the levels of NAMPT. NAMPT activity is promoted by AMPK, which is increased with glucose restriction. Thus, glucose restriction activates NAMPT activity, resulting in increased NAD+, increased SIRT1 activity, and repression of muscle differentiation [4] (Fig. 4). There is no evidence that primary (embryonic) myogenesis requires modulation of NAD+ levels in vivo. There is, however, data indicating that secondary (fetal) myogenesis may integrate nutrient sensing with NAD+ levels and SIRT1 activity to mediate muscle growth in vivo. Rats born to calorie-restricted mothers are smaller due to deficits in secondary myogenesis [35]. These data support that muscle differentiation and growth are linked to nutrient availability via NAD+. However, the mechanisms regulating which signal transduction cascades and which protein-protein interactions are invoked downstream of NAD+ inputs are unclear. There are additional unanswered questions regarding roles for NAD+ in modulating SIRT1 activity during muscle development and growth, such as how are NAD+ levels decreased to alleviate SIRT1-mediated repression of differentiation, and how are NAD+ levels restored after differentiation?
Fig. 4.
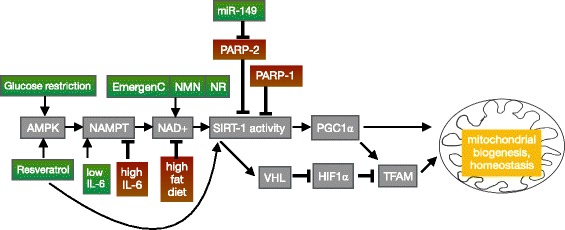
Signaling pathways by which NAD+ regulates mitochondrial biogenesis and homeostasis in muscle. Arrows denote promotion and “T”s signify inhibition of mitochondrial biogenesis or homeostasis. Interventions in green boxes promote NAD+ levels and interventions in red boxes deplete NAD+ levels, thereby regulating mitochondria number and function. For explanation of the acronyms, please see the abbreviations list in the main body of the text
One factor that may contribute to changes in NAD+ levels in the transition from myoblasts to myotubes is PARP-1 (Fig. 4). PARP-1 covalently attaches branched ADP-ribose chains to proteins using NAD+ as a substrate. Thus, PARP-1 is a major NAD+ consumer and NAD+ levels tend to be inversely related to PARP-1 levels in multiple cell types. This phenomenon has been observed in C2C12 myotubes exposed to oxidative stress. H2O2 activated PARP-1 and a subsequent decrease in NAD+ levels were observed [23]. Decreased NAD+ did not reduce SIRT1 protein expression; however, SIRT1 activity, as measured by PGC1-alpha acetylation, was reduced [23]. PARP-1 levels are high in myoblasts and lower in myotubes (C2C12 and a rat skeletal muscle line L6) [22]. Thus, changing levels of PARP-1 likely play a role in SIRT1 activity during muscle differentiation via modulation of NAD+ levels.
An additional effect of PARP-1 levels is sensitivity to oxidative stress. PARP-1 is induced by oxidative stress and plays a role in DNA repair. However, if left unchecked, PARP-1 activity depletes NAD+ levels. This adversely affects electron transport and ATP synthesis and can lead to necrosis. It is also possible that the downregulation of PARP-1 levels in developed myotubes provides a mechanism for increased resistance to oxidative stress [22]. Inhibition of PARP-1 catalytic activity increased protection against oxidative stress in C2C12 myotubes and overexpression of PARP-1 sensitized these cells to oxidant-induced injury. Thus, in the future it will be interesting to determine mechanisms regulating PARP-1 expression during muscle differentiation, to ask whether PARP-1 plays a significant role in differentiation by modulating NAD+ levels, and, if so, whether PARP-1 interacts with nutrient sensing by AMPK.
One question is whether muscle development and muscle repair show similar responses to NAD+ levels during differentiation. Muscle repair is mediated by muscle satellite cells that reside next to muscle fibers under the basement membrane [36, 37]. These self-renewing cells are usually quiescent, but they proliferate and differentiate in response to muscle injury. Recent data suggest SIRT1 plays a role in actively maintaining quiescence in satellite cells by repressing transcription of myogenic genes. Quiescent satellite cells use oxidative-phosphorylation (OXPHOS) to generate energy [38]. Metabolic reprogramming is a hallmark of satellite cells, and Ryall and colleagues asked whether these cells shifted their metabolic state when activated. They observed that satellite cells switch from primarily OXPHOS to increased glycolysis and glutaminolysis when activated and proliferating [38]. One consequence of this metabolic shift is that, similar to muscle development, NAD+ levels decrease, SIRT1 activity decreases, and this change is permissive for myogenic gene transcription [38]. Inactivation of SIRT1 in satellite cells results in initially smaller mice with smaller muscle fibers at postnatal day 9, although the mice recover by postnatal day 14 [38]. In the future, it would be quite interesting to determine the mechanism of size recovery. Taken together, these data indicate that muscle repair and muscle development both rely on changing NAD+ levels and that this process mechanistically links metabolism/energy availability with differentiation in both contexts.
Membrane proximal NAD+ in muscle development
The ECM protein laminin is necessary for primary myotome development. In mice, laminin is thought to act as a barrier that keeps myogenic cells inside the myotome. When laminin polymerization is compromised, myocytes disperse into the sclerotome region [39]. Similarly, in lamb1 or lamc1 mutant zebrafish, some fast-twitch muscle cells extend into adjacent myotomes rather than stopping at myotendinous junctions [40]. Surprisingly, NAD+ biosynthesis plays a role in potentiating laminin organization in the basement membrane at the myotendinous junction. Zebrafish deficient for Nrk2b exhibit defects in laminin organization that are rescued with exogenous NAD+ supplementation. Although the mechanism is not known, NAD+ appears to act via the laminin receptor Integrin alpha6 and promotes subcellular localization of Paxillin to cell adhesion complexes [33] (Fig. 3). This suggests that, at least in zebrafish muscle cells, the NAD+ synthesized in the membrane proximal compartment during muscle development modulates the ECM. Interestingly, there were no observed muscle defects in NRK2 knockout mice [41]. Analysis of NRK1 and NRK2 single and double knock out cells in a primary muscle culture system demonstrated some redundancy between NRK1 and NRK2 in salvage NAD+ biosynthesis from NR [41]. It is possible that the functional redundancy between NRK1 and NRK2 observed in mouse muscle cells does not occur in zebrafish and could explain the discrepancy in muscle development phenotypes between the two species. Regardless of this difference, NAD+ was subsequently implicated in C. elegans muscle development and function [42] and in mouse skeletal muscle health [43–46]. Therefore, a role for NAD+ biosynthesis in muscle development and function appears to be well conserved.
NAD+ and aging muscle
Aging is considered by some to be the tail end of development. If changes that occur throughout the lifespan are thought of as part of a developmental continuum, then it logically follows that developmental robustness and plasticity likely impact the aging process. The loss of skeletal muscle mass and function with aging is called sarcopenia. Sarcopenia decreases quality of life and increases the risk of metabolic diseases. Although there is some controversy regarding whether age decreases mitochondrial content and ATP production (reviewed in [47]), the data in general seem to suggest that mitochondrial dysfunction is a major driver of muscle aging [48–52] (Fig. 5). As described above, mitochondrial homeostasis is strongly influenced by NAD+ via SIRT1. One key target of SIRT1 is PGC-1alpha, which promotes mitochondrial biogenesis [53]. SIRT1 also promotes mitochondrial biogenesis by potentiating mitochondrial transcription of oxidative phosphorylation genes in a PGC-1alpha-independent manner [54]. This pathway involves stabilization of HIF-1alpha, and AMPK appears to mediate the switch between these two SIRT1-dependent pathways of mitochondrial biogenesis [54]. NAD+ levels are reduced in aged muscle in rats, mice, and humans [9, 54, 55], leading to reduced SIRT1 activity, and reduced mitochondrial homeostasis. The mechanisms by which NAD+ levels are modulated are of extreme interest because understanding these mechanisms could lead to approaches to slow or even halt the loss of muscle mass and function during aging. Here, we will discuss recent insights into the mechanisms underlying decreased NAD+ with aging and highlight interventions that increase NAD+ and skeletal muscle function.
Fig. 5.
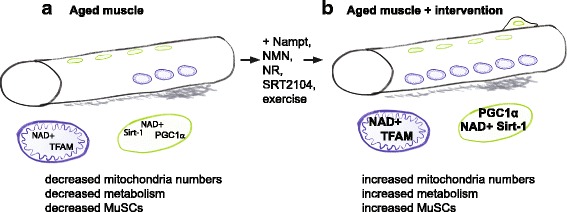
Model of the pathological observations in aged muscle and their reversal by augmentation of nuclear and mitochondrial NAD+ levels. a Fewer mitochondria, fewer muscle stem cells, reduced levels of NAD+ in nuclei and mitochondria, and reduced nuclear SIRT-1 activity have been observed in aged muscle. b Repletion of NAD+ levels in nuclei and mitochondria via various interventions increases mitochondria, increases muscle stem cells, and increases nuclear and mitochondrial NAD+ levels thereby increasing SIRT-1 activity. One mechanism involves nuclear SIRT-1-mediated regulation of TFAM and PGC1alpha gene expression and their effect on mitochondrial metabolism
NAMPT levels decrease during aging, increasing NAMPT improves aged muscle
In aged rats, NAD+ levels have been shown to decrease due to lower levels of the rate limiting enzyme in NAD+ biosynthesis, NAMPT [56]. Knock out of NAMPT in skeletal muscle of mice (mNKO mice) demonstrated that NAMPT is required for muscle function [45]. NAD+ and ATP levels were decreased by 85 and 60%, respectively, in mNKO mice. At 3 months, functional defects were not immediately apparent on a treadmill test: mNKO mice tired at similar rates to controls. However, when extensor digitorum longus (EDL) muscle was repeatedly electrically stimulated ex vivo, force generation was significantly reduced [45]. This deficiency, apparent in response to repeated electrical stimulation, could be because mitochondria have been found to recover more slowly in aged vs young mice, at least in whole lumbrical muscle [49]. mNKO mice were only slightly smaller than wild-type siblings; however, histological changes (smaller fiber diameter, increased Evans blue infiltration, and increased central nuclei) were apparent at 3 months. These phenotypes worsened through time and were classified as “NAD+-deficient myopathy” [45]. Thus, mNKO mice appear to be a good model for investigating the effects of reduced NAD+ on skeletal muscle biology in the context of aging on a developmental continuum.
NAMPT is not only necessary for muscle function, it is sufficient to combat the decline in muscle performance with aging. Overexpression of NAMPT in cardiac and skeletal muscle throughout life increased healthspan [45]. Overexpression of NAMPT in skeletal muscle resulted in improved exercise capacity in aged mice. NAMPT overexpression increased NAD+ levels and, because of this higher baseline, NAD+ levels in aged mice overexpressing NAMPT in skeletal muscle were similar to those of young control mice [45]. This was despite NAMPT overexpressing mice still experiencing a decline in NAD+ levels with aging. Therefore, interventions that bolster NAD+ levels prior to and during aging, such as NAMPT overexpression, maintain the functional capacity and promote the healthy aging of skeletal muscle.
So how can NAMPT levels be increased during aging? Multiple studies in three different mammals indicate that exercise increases NAMPT. Exercise training increased NAMPT levels, NAD+, and SIRT1 activity in aged rats [56]. NAMPT protein was increased 16% in humans in the leg that conducted one-legged knee-extensor exercises compared to the untrained leg [57]. This result is quite compelling, but it suggests that resistance training with the goal of increasing NAMPT expression would need to target all muscle groups. A similar increase in NAMPT expression was observed in the quadriceps of endurance-trained mice (this was the only muscle examined) [57]. Electrostimulation of primary skeletal muscle cells in vitro also increased NAMPT expression [58]. One study found that exercise training increased SIRT3 expression but not NAMPT levels in male rats [59]. The reason for this discrepancy is not clear. However, these studies mostly suggest that exercise training or electrical stimulation could be a tractable approach to increase NAMPT levels until pharmacological interventions are identified/improved.
Experiments elucidating the mechanisms regulating the exercise-mediated increase in NAMPT expression thus far implicate AMPK. Stimulation of AMPK is sufficient to increase NAMPT protein in skeletal muscle: 4 weeks of activating AMPK with the adenosine monophosphate (AMP) analog AICAR (5-aminoimidazole-4-carboxamide ribonucleotide) increased NAMPT expression. AICAR treatment in AMPK knockout mice did not increase NAMPT expression [57]. These experiments showed that AMPK activation is sufficient to increase NAMPT expression. However, a critical experiment showed that AMPK is not necessary for the exercise-induced increase in NAMPT expression in vivo: NAMPT expression was increased in endurance-trained AMPK knockout mice [57]. In the future, it will be interesting to determine the AMPK-independent mechanism of increased NAMPT expression in an effort to maximize NAD+ levels in muscle via exercise and/or pharmacological interventions.
Other approaches to increase NAD+ and SIRT1 activity during aging
A likely benefit of increased NAD+ availability in muscle during aging is potentiation of SIRT1 activity. Many methods of enhancing SIRT1 activity have been discovered and shown to have positive effects on muscle healthspan. Activation of SIRT1 by a small molecule agonist, SRT2104, resulted in increased muscle mitochondrial size and better endurance in aged mice [60]. SRT2104 also reduced loss of muscle mass in the 2-week hindlimb suspension model of muscle atrophy [60]. Another method involves activating NADH-quinone oxidoreductase 1 (NQ01) with beta-lapachone [61]. NQ01 uses NADH as an electron donor and thus depletes NADH and increases the NAD+/NADH ratio [62, 63]. Aged mice fed with beta-lapachone showed increased oxygen consumption, increased heat generation, and improved skeletal muscle mitochondrial biogenesis [61]. Another approach to enhance NAD+ levels is to provide intermediates in NAD+ biosynthesis. Mice fed NMN for 12 months were more active and had improved oxidative metabolism without overt side effects [43]. The particularly interesting aspect of this study is that microarray analysis did not detect a common set of “NMN response genes” between different tissues. This suggests that the response to enhanced NAD+ metabolism is tissue specific. Similar to the above experiments, supplementation with NAM for 5 weeks enhanced SIRT1 activity in young and aged rats [64]. However, combining exercise training with NAM supplementation decreased SIRT1 levels [64]. NAM can inhibit SIRT1 and PARP-1 under stress conditions to preserve NAD+ levels. Thus, NAM supplementation may not be as effective as other methods of enhancing NAD+ levels and SIRT1 activity in aging muscle.
Modulating levels of the major NAD+ consumer PARP-1 have also been shown to be effective in maintaining muscle function during aging. In young mice, isometric exercise increases activity of both SIRT1 and PARP-1, but SIRT1 then binds PARP-1 and inhibits its activity [20]. Thus, young mice are protected from excess activation of PARP-1 and the potential for necrosis that NAD+ depletion can incur. In contrast, aged muscle has higher PARP-1 activity, lower NAD+ content, and lower SIRT1 activity, resulting in decreased performance. The higher PARP-1 activity is partially due to increased acetylation of PARP-1 by GCN5 [20]. NAD+ levels may also indirectly regulate PARP-1 activity. DBC1 (deleted in breast cancer 1) binds and inhibits PARP-1 in a NAD+-dependent manner in vitro [65]. Whether this occurs in skeletal muscle is unknown, but this could be an additional mechanism that regulates PARP-1 activity during aging. Inhibition of PARP-1 by injecting the inhibitor PJ34 in aged mice increased NAD+ content, SIRT1 activity, muscle mitochondrial biogenesis, and performance [20]. PGC-1alpha was not required, which raises the question of whether the increased mitochondrial biogenesis is due to stabilization of HIF-1alpha. Taken together, these results suggest that regulation of NAD+-dependent enzymes, such as decreasing PARP-1 activity, decreasing GCN5 activity, or activating SIRT1, could prevent or slow the age-associated decline in skeletal muscle mass and function.
NR supplementation rejuvenates aged muscle stem cells (MuSCs)
It is easy to imagine that there could be crosstalk between the cellular pathways involved in lifespan extension. The mitochondrial unfolded protein response (UPRmt) is an adaptive pathway that mediates proteostasis in response to stress. Activation of UPRmt correlates with longevity in worms and mice [66]. Thus, it is perhaps not surprising that gene expression analysis of aged MuSCs showed reduced UPRmt, decreased cell cycle, and increased proinflammatory secretome gene expression [46]. NAD+ biosynthesis was found to feed into the UPRmt response. Nicotinamide riboside (NR) treatment increased levels of UPRmt proteins, increased MuSC number in young and aged mice, and quickened muscle regeneration after cardiotoxin-induced injury in both young and old mice [44]. Importantly, infiltration of fibro-adipogenic progenitors (FAPs) was decreased after injury in NR-treated mice. Knockout of SIRT1 in MuSCs abrogated the beneficial effects of NR, indicating that, in this context, NR acts through increased NAD+ and the ensuing increase in SIRT1 activity [46]. The ECM microenvironment provides a niche that regulates MuSC behavior [67]. As mentioned above, NAD+ plays a role in modulating organization of the ECM during muscle development [33]. Thus, one future direction would be to determine if NR supplementation affected MuSC responses by acting on the ECM in the MuSC niche. It would also be interesting to investigate whether NAD+ bioavailability feeds into other pathways that promote longevity.
NAD+ in myopathies and dystrophies
Normal aging and sarcopenia result in decreased muscle performance; however, some individuals lose muscle mass and function due to genetic muscle diseases. Muscular dystrophies and myopathies are stereotypically characterized by muscle wasting, weakness, and impaired locomotion (Fig. 6). Although significant progress has been made with regard to identifying disease causing genes and pathological mechanisms, these conditions are still incurable. One major challenge that remains is to understand if therapeutic crossover exists between the treatments for Duchenne/Becker muscular dystrophy (DMD) and the other types of muscular dystrophies. This is an important question because therapeutic strides are being made for the most common type of muscular dystrophy, DMD, but treatment for most other types of muscle diseases lags behind. Therefore, in the future it is critical to determine whether differences in cellular pathology affect treatment options. Given the critical role of NAD+ in muscle development, homeostasis, and aging, it is tempting to speculate that potentiating NAD+ biosynthesis could be a beneficial adjuvant therapy for a broad spectrum of muscular dystrophies and myopathies.
Fig. 6.
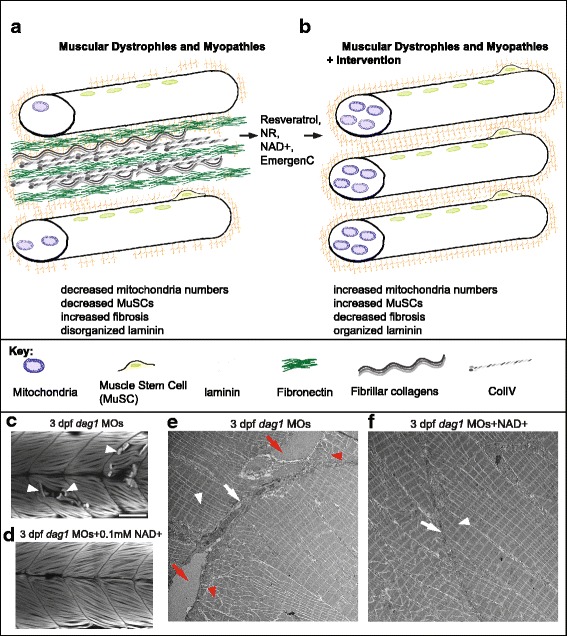
Cellular and molecular mechanisms of amelioration of muscle disease due to NAD+ repletion. (A, B) Model of pathological changes observed in animal models of diseased muscle (A) and the improvement in these disease phenotypes due to provision of dietary NAD+ precursors or NAD+ (B). (C, D) Anterior left, dorsal top, side-mounted, 3 dpf embryos stained with phalloidin to visualize actin. Fiber detachment is readily observed in dag1 morphants (C, white arrowheads), whereas dag1 morphants supplemented with NAD+ display less fiber detachment (D). (E, F) Transmission electron micrographs showing normal muscle ECM (white arrows), disrupted muscle ECM (red arrows), normal sarcomere structure (white arrowheads), and disrupted sarcomere structure (red arrowheads) in a zebrafish model of muscular dystrophy with and without NAD+ supplementation. (E) Dag1-deficient zebrafish. (F) Dag1-deficient zebrafish supplemented with NAD+
Resveratrol is a grape polyphenol that has received attention for its potential to extend lifespan. Resveratrol can directly activate SIRT1 [68] as well as indirectly activate SIRT3 through increasing the mitochondrial NAD+/NADH ratio [69]. Thus, resveratrol may have the potential to improve muscle function in aging and/or disease. Resveratrol was tested for possible efficacy in the mdx mouse model of DMD [70]. Thirty-two weeks of resveratrol supplementation resulted in reductions in both muscle mass lost and fibrosis. The decreased fibrosis presumably resulted from reduced transcription of collagen 1a1, 1a2, and fibronectin [70]. Resveratrol was provided in the diet; thus, it is not known in which tissues/cell types resveratrol functions to reduce fibrosis. Angiotensin-II is known to promote fibrosis via NAD(P)H oxidase-generated reactive oxygen species (ROS) in myoblasts [71]. These results suggest that myoblasts, in addition to fibroblasts and macrophages, can promote fibrosis. Given that CD45+ inflammatory cells were still observed in mdx muscle despite resveratrol treatment [70], it would be interesting to determine whether resveratrol reduced transcription of ECM genes in immune cells, myofibroblasts, or both.
Another method of increasing NAD+ biosynthesis was shown to be beneficial in the mouse model of DMD. NAD+ repletion with NR supplementation improved muscle function in mdx mice [44]. At least some of this improvement is likely due to the NR-mediated increase in number and function of MuSCs in mdx mice [46]. Taken together, these data suggest that NAD+ repletion, via multiple mechanisms, is a worthwhile avenue to explore in humans with DMD.
Could NAD+ repletion benefit other types of muscular dystrophies? As mentioned above, we showed that Nrk2b is necessary for normal muscle ECM development [33]. Laminin in the basement membrane was disorganized in Nrk2b-deficient zebrafish and rescued with exogenous NAD+. We found that muscle atrophy in a zebrafish model of primary dystroglycanopathy due to Dag1 deficiency is preceded by disorganized laminin in the muscle ECM. We asked whether exogenous NAD+ would improve laminin organization in Dag1-deficient zebrafish. We found that either supplementation with NAD+ or EmergenC, which contains vitamin B precursors to NAD+, improved laminin organization, reduced muscle degeneration, and improved swimming in Dag1-deficient zebrafish [72]. We identified the Integrin alpha6 and alpha7 receptors as well as intracellular localization of the focal adhesion protein Paxillin as downstream mechanisms. Using genetic mosaic analysis, we showed that the ECM microenvironment plays a critical role in maintenance of muscle cell-ECM adhesion and fiber viability and thus tissue homeostasis [72]. These results and others suggest that understanding the mechanisms downstream of NAD+-mediated ECM organization could lead to new therapeutic approaches (Fig. 6).
Increasing NAD+ bioavailability also appears to be beneficial for a myopathy. NR supplementation delays the progression of mitochondrial myopathy by stimulating the UPRmt as well as inducing mitochondrial biogenesis [73]. Given the dynamics of mitochondrial remodeling in muscle homeostasis and disease, it seems critical to understand mitochondrial dynamics in live organisms. Generation of tools that allow observation of mitoflashes in living zebrafish provided insight into how mitoflashes change in development and disease [74]. A mitoflash is a burst of ROS that mediates mitochondrial-cytosol/nuclear signaling [75]. It is not currently known whether or how mitoflashes affect the progression of muscle disease. Transgenic zebrafish expressing the biosensor mt-cpYFP [76] showed that mitoflashes are shorter early in muscle development but become longer as development proceeds [74]. Transgenic zebrafish modeling DMD showed the expected decrease in mitochondria numbers, although whether this reduction was due to decreased mitochondrial biogenesis was not investigated. Interestingly, the mitoflashes that were observed in zebrafish with muscle disease were longer than those of wild-type zebrafish [74]. The signaling events downstream of longer bursts of ROS are not known, but do provide an interesting avenue of investigation to pursue in terms of muscle disease pathology and progression.
The above data show that increasing NAD+ levels, whether by supplementing with resveratrol, NR, or EmergenC, is beneficial for multiple types of muscle disease. The mechanisms hypothesized to be responsible for ameliorating disease include increased: mitochondrial biogenesis, UPRmt, MuSC numbers, and laminin organization in the muscle ECM (Fig. 6). Increased NAD+ levels also correlated with reductions in both fibrosis and FAPs. However, none of these studies integrated or looked for connections between all of the different facets of muscle phenotypic improvement. The ECM microenvironment is involved in fibrosis as well as the health of muscle fibers and MuSCs. Thus, it would be interesting in the future to examine whether there is coordination between mitochondrial health and muscle ECM structure or whether these are two entirely separate mechanisms of NAD+ action. It also seems important to investigate whether NAD+ supplementation is beneficial across the spectrum of muscular dystrophies/myopathies to determine if there are muscle diseases that are resistant to NAD+ supplementation. There is one study suggesting that genotype may impact the response to exogenous NAD+. Dystrophic myoblasts (from mdx mice) showed an increase in ERK1/2 phosphorylation in response to exogenous NAD+, whereas control myoblasts did not [77]. The physiological significance of these results is not known, but they do highlight that NAD+ may have varying effects in different genetic environments. Thus, addressing whether activating NAD+ biosynthesis exhibits therapeutic crossover for multiple dystrophies and myopathies should be a priority. Understanding how mechanisms downstream of increased NAD+ cause certain muscle outputs should also be prioritized as this could provide a way to elicit desired muscle responses to NAD+ in different genetic backgrounds.
NAD+ in muscle metabolic diseases
Skeletal muscle mass and function are not only critical for daily life but also whole-body protein metabolism and thus play a critical role in metabolic diseases such as obesity and type 2 diabetes (reviewed in [2]). Metabolic diseases are common and debilitating conditions. Given the roles of skeletal muscle and NAD+ in metabolic regulation, it is possible that NAD+ supplementation could improve metabolic health.
PARP-2 is a major NAD+ consumer and thus indirectly affects SIRT1 activity and metabolic health by modulating NAD+ levels [21]. High fat diet-fed mice have elevated PARP-2 expression in skeletal muscle, which would be predicted to act as a NAD+ sink and lower SIRT1 activity [21]. Therefore, PARP-2 inhibition is a potential strategy to augment NAD+ levels in skeletal muscle in the context of obesity. Mohamed et al. conducted a microRNA (miR) screen to identify potential miRNAs that modulate PARP-2 expression. They identified miR-149 as necessary and sufficient to decrease PARP-2 expression, increase SIRT1 activity, and increase mitochondrial biogenesis in C2C12 cells [21]. Taken together, these results suggest that targeting either PARP-2 expression or enhancing miR-149 expression could be potential approaches to promoting metabolic health via NAD+ repletion in skeletal muscle.
Interestingly, boosting NAD+ levels by 50% in mouse skeletal muscle via tissue-specific overexpression of NAMPT did not induce mitochondrial biogenesis or protect these mice from the negative metabolic consequences of a high fat diet [78]. Therefore, augmenting NAD+ in tissues other than striated muscle is necessary for promoting mitochondrial function and oxidative metabolism [78]. The tissues in which NAD+ repletion is required for whole body metabolic improvement are unknown and are important targets to elucidate for metabolic therapies.
Human trials that analyze the consequences of increasing NAD+ on muscle are limited. However, one recent multicenter trial provided patients with type 2 diabetes either placebo or the NAD+ precursor acipimox for 2 weeks [79]. Muscle biopsies allowed for examination of muscle respiration and gene expression after the treatment. Although mitochondrial DNA content did not differ between the treatments, respiration was higher in acipimox-treated muscle. Upregulation of gene sets that have been previously positively correlated with NAD+ supplementation were observed in this case [79]. While there were side effects to this particular type of NAD+ augmentation in these type 2 diabetics, these data suggest that roles for NAD+ in skeletal muscle and metabolic health are conserved in humans. These data also support the promise of pharmacologically or enzymatically modulating NAD+ biosynthesis and/or utilization as a therapy for many human genetic and metabolic disorders.
Future perspectives
Why is it important to elucidate downstream mechanisms of NAD+ action if, in general, the studies presented here all show benefits to NAD+ supplementation? There is correlative evidence that increased NAD+ can be deleterious in the context of multiple muscle cancers. Higher NAMPT expression levels correlate with increased tumor grade for leiomyosarcomas, as well as embryonal, alveolar, and pleomorphic rhabdomyosarcomas [80]. Thus, it is critically important to learn downstream signaling pathways affected by NAD+ signaling such that the benefits of NAD+ can be leveraged without potentially deleterious side effects. This is especially true because NAMPT expression not only correlates with tumor grade for muscle cancers but also correlates with invasion, metastasis, and chemotherapy resistance for gastric, thyroid, and prostate cancers [81–83]. Although current studies are focused on developing NAMPT inhibitors that are administered along with nicotinic acid to help alleviate toxic side effects of NAMPT inhibitors, it is likely that identifying downstream targets in normal and cancerous cells would lead to improved efficacy with fewer side effects.
Conclusions
It is clear that NAD+ plays a beneficial role in muscle health. The mechanisms underlying promotion of muscle development and homeostasis by NAD+ are best understood in the context of sirtuin regulation in the nucleus, mitochondria, and cytosol. The role of NAD+ in other cellular compartments—particularly the vesicular and membrane proximal compartments—in muscle health is currently understudied. Future research will likely delve into not only the mechanisms of NAD+ action in these different compartments but also the interplay and signaling between NAD+ pools within and between cells.
Acknowledgements
The authors would like to thank the members of the Henry lab for reviewing the manuscript. Thank you to Kell Fremouw for assisting with artistic renderings.
Funding
March of Dimes Foundation Award Number: #1-FY14-284, NIH R15HD088217.
Availability of data and materials
Not applicable.
Abbreviations
- ADP
Adenosine diphosphate
- ADPR
Adenosine diphosphate-ribose
- AICAR
5-aminoimidazole-4-carboxamide ribonucleotide
- AMP
Adenosine monophosphate
- AMPK
Adenosine monophosphate kinase
- ART
ADP-ribosyltransferase
- ATP
Adenosine triphosphate
- C. elegans
Caenorhabditis elegans
- C2C12
Immortalized mouse myoblast cell line
- cADPR
Cyclic ADP-ribose
- CD45
Cluster of differentiation 45
- Dag1
Dystroglycan
- DMD
Duchenne/Becker muscular dystrophy
- DNA
Deoxyribonucleic acid
- ECM
Extracellular matrix
- EDL
Extensor digitorum longus
- ERK1/2
Extracellular signal-regulated protein kinase 1/2
- FAP
Fibro-adipogenic progenitors
- GCN5
Histone acetyltransferase GCN5
- H2O2
Hydrogen peroxide
- HIF-1alpha
Hypoxia inducible factor-1alpha
- IL-6
Interleukin-6
- ITGA7
Integrin alpha7
- ITGB1BP3
Integrin beta1 binding protein3
- L6
Immortalized rat skeletal myoblast cell line
- lamb1
Laminin beta1
- lamc1
Laminin gamma1
- mdx
Mouse DMD model
- MIBP2
Muscle integrin binding protein2
- miR
microRNA
- mNKO
Muscle NAMPT knock out
- MuSC
Muscle stem cell
- NA
Nicotinic acid
- NAAD
Nicotinate adenine dinucleotide
- NAADP
Nicotinic acid adenine dinucleotide phosphate
- NAD+
Oxidized nicotinamide adenine dinucleotide
- NADH
Reduced nicotinamide adenine dinucleotide
- NADPH
Reduced nicotinamide adenine dinucleotide phosphate
- NADS
NAD+ synthase
- NAM
Nicotinamide
- NAMN
Nicotinate mononucleotide
- NAMPT
Nicotinamide phosphoribosyltransferase
- NAPRT
Nicotinate phosphoribosyltransferase
- NAR
Nicotinic acid riboside
- NEFA
Nonesterified fatty acid
- NMN
Nicotinamide mononucleotide
- NMNAT
Nicotinamide nucleotide adenylyltransferase
- NQO1
NADH-quinone oxidoreductase 1
- NR
Nicotinamide riboside
- NRK1/2
Nicotinamide riboside kinase 1 and 2
- Nrk2b
Nicotinamide riboside kinase 2b
- OAADPR
O-acetyl-ADPR
- OXPHOS
Oxidative phosphorylation
- PARP
Poly (ADP-ribose) polymerase
- PCAF
p300/CBP-associated factor
- PGC1-alpha
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PJ34
PARP inhibitor VIII
- QA
Quinolinic acid
- QAPRT
Quinolinate phosphoribosyltransferase
- Redox
Oxidation-reduction
- ROS
Reactive oxygen species
- SIRT
Sirtuin
- SR
Sarcoplasmic reticulum
- SRT2104
Selective activator of SIRT1
- TFAM
Mitochondrial transcription factor A
- UPRmt
Mitochondrial uncoupled protein response
- VHL
Von Hippel-Lindau tumor suppressor
Authors’ contributions
MFG and CAH both wrote, edited, and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Glancy B, Hartnell LM, Malide D, Yu ZX, Combs CA, Connelly PS, et al. Mitochondrial reticulum for cellular energy distribution in muscle. Nature. 2015;523:617–620. doi: 10.1038/nature14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolfe RR. The underappreciated role of muscle in health and disease. Am J Clin Nutr. 2006;84:475–482. doi: 10.1093/ajcn/84.3.475. [DOI] [PubMed] [Google Scholar]
- 3.Cantó C, Menzies KJ, Auwerx J. NAD+ metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, et al. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14:661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiarugi A, Dölle C, Felici R, Ziegler M. The NAD metabolome—a key determinant of cancer cell biology. Nat Rev Cancer. 2012;12:741–752. doi: 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- 6.Nikiforov A, Kulikova V, Ziegler M. The human NAD metabolome: functions, metabolism and compartmentalization. Crit Rev Biochem Mol Biol. 2015;50:284–297. doi: 10.3109/10409238.2015.1028612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Cantó C, et al. The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154:430–441. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu X-H, Lu M, Lee B-Y, Ugurbil K, Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci U S A. 2015;112:2876–2881. doi: 10.1073/pnas.1417921112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, Guillemin GJ. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One. 2012;7:e42357. doi: 10.1371/journal.pone.0042357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hashimoto T, Horikawa M, Nomura T, Sakamoto K. Nicotinamide adenine dinucleotide extends the lifespan of Caenorhabditis elegans mediated by sir-2.1 and daf-16. Biogerontology. 2010;11:31–43. doi: 10.1007/s10522-009-9225-3. [DOI] [PubMed] [Google Scholar]
- 11.Fang EF, Kassahun H, Croteau DL, Scheibye-Knudsen M, Marosi K, Lu H, et al. NAD(+) replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab. 2016;24:566–581. doi: 10.1016/j.cmet.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dash RK, Li Y, Kim J, Beard DA, Saidel GM, Cabrera ME. Metabolic dynamics in skeletal muscle during acute reduction in blood flow and oxygen supply to mitochondria: in-silico studies using a multi-scale, top-down integrated model. PLoS ONE [Internet]. 2008 [cited 2017 Nov 7];3. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2526172/. [DOI] [PMC free article] [PubMed]
- 13.Li Y, Dash RK, Kim J, Saidel GM, Cabrera ME. Role of NADH/NAD+ transport activity and glycogen store on skeletal muscle energy metabolism during exercise: in silico studies. Am J Physiol - Cell Physiol. 2009;296:C25–C46. doi: 10.1152/ajpcell.00094.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White AT, Schenk S. NAD+/NADH and skeletal muscle mitochondrial adaptations to exercise. Am J Physiol - Endocrinol Metab. 2012;303:E308–E321. doi: 10.1152/ajpendo.00054.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fliegert R, Gasser A, Guse AH. Regulation of calcium signalling by adenine-based second messengers. Biochem Soc Trans. 2007;35:109–114. doi: 10.1042/BST0350109. [DOI] [PubMed] [Google Scholar]
- 16.Koch-Nolte F, Fischer S, Haag F, Ziegler M. Compartmentation of NAD+-dependent signalling. FEBS Lett. 2011;585:1651–1656. doi: 10.1016/j.febslet.2011.03.045. [DOI] [PubMed] [Google Scholar]
- 17.Wątroba M, Dudek I, Skoda M, Stangret A, Rzodkiewicz P, Szukiewicz D. Sirtuins, epigenetics and longevity. Ageing Res Rev. 2017;40:11–19. doi: 10.1016/j.arr.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 18.Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012;26:417–432. doi: 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Vos M, Schreiber V, Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem Pharmacol. 2012;84:137–146. doi: 10.1016/j.bcp.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 20.Mohamed JS, Wilson JC, Myers MJ, Sisson KJ, Alway SE. Dysregulation of SIRT-1 in aging mice increases skeletal muscle fatigue by a PARP-1-dependent mechanism. Aging. 2014;6:820–834. doi: 10.18632/aging.100696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohamed JS, Hajira A, Pardo PS, Boriek AM. MicroRNA-149 inhibits PARP-2 and promotes mitochondrial biogenesis via SIRT-1/PGC-1α network in skeletal muscle. Diabetes. 2014;63:1546–1559. doi: 10.2337/db13-1364. [DOI] [PubMed] [Google Scholar]
- 22.Oláh G, Szczesny B, Brunyánszki A, López-García IA, Gerö D, Radák Z, et al. Differentiation-associated downregulation of poly(ADP-ribose) polymerase-1 expression in myoblasts serves to increase their resistance to oxidative stress. PLoS One. 2015;10:e0134227. doi: 10.1371/journal.pone.0134227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bai P, Cantó C, Oudart H, Brunyánszki A, Cen Y, Thomas C, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011;13:461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goody MF, Sher RB, Henry CA. Hanging on for the ride: adhesion to the extracellular matrix mediates cellular responses in skeletal muscle morphogenesis and disease. Dev Biol. 2015;401:75–91. doi: 10.1016/j.ydbio.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hohenester E, Yurchenco PD. Laminins in basement membrane assembly. Cell Adhes Migr. 2013;7:56–63. doi: 10.4161/cam.21831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Z, Gruszczynska-Biegala J, Zolkiewska A. ADP-ribosylation of integrin α7 modulates the binding of integrin α7β1 to laminin. Biochem J. 2005;385:309–317. doi: 10.1042/BJ20040590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zolkiewska A. Ecto-ADP-ribose transferases: cell-surface response to local tissue injury. Physiol Bethesda Md. 2005;20:374–381. doi: 10.1152/physiol.00028.2005. [DOI] [PubMed] [Google Scholar]
- 28.Zolkiewska A, Moss J. Integrin alpha 7 as substrate for a glycosylphosphatidylinositol-anchored ADP-ribosyltransferase on the surface of skeletal muscle cells. J Biol Chem. 1993;268:25273–25276. [PubMed] [Google Scholar]
- 29.Zolkiewska A, Moss J. Processing of ADP-ribosylated integrin alpha 7 in skeletal muscle myotubes. J Biol Chem. 1995;270:9227–9233. doi: 10.1074/jbc.270.16.9227. [DOI] [PubMed] [Google Scholar]
- 30.Zolkiewska A, Moss J. The alpha 7 integrin as a target protein for cell surface mono-ADP-ribosylation in muscle cells. Adv Exp Med Biol. 1997;419:297–303. doi: 10.1007/978-1-4419-8632-0_39. [DOI] [PubMed] [Google Scholar]
- 31.Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell. 2004;117:495–502. doi: 10.1016/S0092-8674(04)00416-7. [DOI] [PubMed] [Google Scholar]
- 32.Li J, Rao H, Burkin D, Kaufman SJ, Wu C. The muscle integrin binding protein (MIBP) interacts with alpha7beta1 integrin and regulates cell adhesion and laminin matrix deposition. Dev Biol. 2003;261:209–219. doi: 10.1016/S0012-1606(03)00304-X. [DOI] [PubMed] [Google Scholar]
- 33.Goody MF, Kelly MW, Lessard KN, Khalil A, Henry CA. Nrk2b-mediated NAD+ production regulates cell adhesion and is required for muscle morphogenesis in vivo: Nrk2b and NAD+ in muscle morphogenesis. Dev Biol. 2010;344:809–826. doi: 10.1016/j.ydbio.2010.05.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, et al. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. 2003;12:51–62. doi: 10.1016/S1097-2765(03)00226-0. [DOI] [PubMed] [Google Scholar]
- 35.Wilson SJ, Ross JJ, Harris AJ. A critical period for formation of secondary myotubes defined by prenatal undernourishment in rats. Dev Camb Engl. 1988;102:815–821. doi: 10.1242/dev.102.4.815. [DOI] [PubMed] [Google Scholar]
- 36.Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev. 2013;93:23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ryall JG, Dell’Orso S, Derfoul A, Juan A, Zare H, Feng X, et al. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell. 2015;16:171–183. doi: 10.1016/j.stem.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bajanca F, Luz M, Raymond K, Martins GG, Sonnenberg A, Tajbakhsh S, et al. Integrin alpha6beta1-laminin interactions regulate early myotome formation in the mouse embryo. Dev Camb Engl. 2006;133:1635–1644. doi: 10.1242/dev.02336. [DOI] [PubMed] [Google Scholar]
- 40.Snow CJ, Goody M, Kelly MW, Oster EC, Jones R, Khalil A, et al. Time-lapse analysis and mathematical characterization elucidate novel mechanisms underlying muscle morphogenesis. PLoS Genet. 2008;4:e1000219. doi: 10.1371/journal.pgen.1000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fletcher RS, Ratajczak J, Doig CL, Oakey LA, Callingham R, Da Silva XG, et al. Nicotinamide riboside kinases display redundancy in mediating nicotinamide mononucleotide and nicotinamide riboside metabolism in skeletal muscle cells. Mol Metab. 2017;6:819–832. doi: 10.1016/j.molmet.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vrablik TL, Wang W, Upadhyay A, Hanna-Rose W. Muscle type-specific responses to NAD+ salvage biosynthesis promote muscle function in Caenorhabditis elegans. Dev Biol. 2011;349:387–394. doi: 10.1016/j.ydbio.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, et al. Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metab. 2016;24:795–806. doi: 10.1016/j.cmet.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryu D, Zhang H, Ropelle ER, Sorrentino V, Mázala DAG, Mouchiroud L, et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci Transl Med. 2016;8:361ra139. doi: 10.1126/scitranslmed.aaf5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frederick DW, Loro E, Liu L, Davila A, Chellappa K, Silverman IM, et al. Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab. 2016;24:269–282. doi: 10.1016/j.cmet.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352:1436–1443. doi: 10.1126/science.aaf2693. [DOI] [PubMed] [Google Scholar]
- 47.Johnson ML, Robinson MM, Nair KS. Skeletal muscle aging and the mitochondrion. Trends Endocrinol Metab TEM. 2013;24:247–256. doi: 10.1016/j.tem.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. J Physiol. 2000;526(Pt 1):203–210. doi: 10.1111/j.1469-7793.2000.t01-1-00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Claflin DR, Jackson MJ, Brooks SV. Age affects the contraction-induced mitochondrial redox response in skeletal muscle. Front Physiol. 2015;6:21. doi: 10.3389/fphys.2015.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lanza IR, Nair KS. Mitochondrial metabolic function assessed in vivo and in vitro. Curr Opin Clin Nutr Metab Care. 2010;13:511–517. doi: 10.1097/MCO.0b013e32833cc93d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lanza IR, Nair KS. Functional assessment of isolated mitochondria in vitro. Methods Enzymol. 2009;457:349–372. doi: 10.1016/S0076-6879(09)05020-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim Y, Triolo M, Hood DA. Impact of aging and exercise on mitochondrial quality control in skeletal muscle. Oxidative Med Cell Longev. 2017;2017:3165396. doi: 10.1155/2017/3165396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Finley LWS, Lee J, Souza A, Desquiret-Dumas V, Bullock K, Rowe GC, et al. Skeletal muscle transcriptional coactivator PGC-1α mediates mitochondrial, but not metabolic, changes during calorie restriction. Proc Natl Acad Sci U S A. 2012;109:2931–2936. doi: 10.1073/pnas.1115813109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gomes AP, Price NL, Ling AJY, Moslehi JJ, Montgomery MK, Rajman L, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One. 2011;6:e19194. doi: 10.1371/journal.pone.0019194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Koltai E, Szabo Z, Atalay M, Boldogh I, Naito H, Goto S, et al. Exercise alters SIRT1, SIRT6, NAD and NAMPT levels in skeletal muscle of aged rats. Mech Ageing Dev. 2010;131:21–28. doi: 10.1016/j.mad.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brandauer J, Vienberg SG, Andersen MA, Ringholm S, Risis S, Larsen PS, et al. AMP-activated protein kinase regulates nicotinamide phosphoribosyl transferase expression in skeletal muscle. J Physiol. 2013;591:5207–5220. doi: 10.1113/jphysiol.2013.259515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim JS, Yoon C-S, Park DR. NAMPT regulates mitochondria biogenesis via NAD metabolism and calcium binding proteins during skeletal muscle contraction. J Exerc Nutr Biochem. 2014;18:259–266. doi: 10.5717/jenb.2014.18.3.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hokari F, Kawasaki E, Sakai A, Koshinaka K, Sakuma K, Kawanaka K. Muscle contractile activity regulates Sirt3 protein expression in rat skeletal muscles. J Appl Physiol Bethesda Md 1985. 2010;109:332–340. doi: 10.1152/japplphysiol.00335.2009. [DOI] [PubMed] [Google Scholar]
- 60.Mercken EM, Mitchell SJ, Martin-Montalvo A, Minor RK, Almeida M, Gomes AP, et al. SRT2104 extends survival of male mice on a standard diet and preserves bone and muscle mass. Aging Cell. 2014;13:787–796. doi: 10.1111/acel.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee J, Park AH, Lee S-H, Lee S-H, Kim J-H, Yang S-J, et al. Beta-lapachone, a modulator of NAD metabolism, prevents health declines in aged mice. PLoS One. 2012;7:e47122. doi: 10.1371/journal.pone.0047122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Siegel D, Gustafson DL, Dehn DL, Han JY, Boonchoong P, Berliner LJ, et al. NAD(P)H: quinone oxidoreductase 1: role as a superoxide scavenger. Mol Pharmacol. 2004;65:1238–1247. doi: 10.1124/mol.65.5.1238. [DOI] [PubMed] [Google Scholar]
- 63.Pink JJ, Planchon SM, Tagliarino C, Varnes ME, Siegel D, Boothman DA. NAD(P)H: quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J Biol Chem. 2000;275:5416–5424. doi: 10.1074/jbc.275.8.5416. [DOI] [PubMed] [Google Scholar]
- 64.Pajk M, Cselko A, Varga C, Posa A, Tokodi M, Boldogh I, et al. Exogenous nicotinamide supplementation and moderate physical exercise can attenuate the aging process in skeletal muscle of rats. Biogerontology. 2017;18:593–600. doi: 10.1007/s10522-017-9705-9. [DOI] [PubMed] [Google Scholar]
- 65.Li J, Bonkowski MS, Moniot S, Zhang D, Hubbard BP, Ling AJY, et al. A conserved NAD+ binding pocket that regulates protein-protein interactions during aging. Science. 2017;355:1312–1317. doi: 10.1126/science.aad8242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lukjanenko L, Jung MJ, Hegde N, Perruisseau-Carrier C, Migliavacca E, Rozo M, et al. Loss of fibronectin from the aged stem cell niche affects the regenerative capacity of skeletal muscle in mice. Nat Med. 2016;22:897–905. doi: 10.1038/nm.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 69.Desquiret-Dumas V, Gueguen N, Leman G, Baron S, Nivet-Antoine V, Chupin S, et al. Resveratrol induces a mitochondrial complex I-dependent increase in NADH oxidation responsible for sirtuin activation in liver cells. J Biol Chem. 2013;288:36662–36675. doi: 10.1074/jbc.M113.466490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hori YS, Kuno A, Hosoda R, Tanno M, Miura T, Shimamoto K, et al. Resveratrol ameliorates muscular pathology in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. J Pharmacol Exp Ther. 2011;338:784–794. doi: 10.1124/jpet.111.183210. [DOI] [PubMed] [Google Scholar]
- 71.Cabello-Verrugio C, Acuña MJ, Morales MG, Becerra A, Simon F, Brandan E. Fibrotic response induced by angiotensin-II requires NAD(P)H oxidase-induced reactive oxygen species (ROS) in skeletal muscle cells. Biochem Biophys Res Commun. 2011;410:665–670. doi: 10.1016/j.bbrc.2011.06.051. [DOI] [PubMed] [Google Scholar]
- 72.Goody MF, Kelly MW, Reynolds CJ, Khalil A, Crawford BD, Henry CA. NAD+ biosynthesis ameliorates a zebrafish model of muscular dystrophy. PLoS Biol. 2012;10:e1001409. doi: 10.1371/journal.pbio.1001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khan NA, Auranen M, Paetau I, Pirinen E, Euro L, Forsström S, et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol Med. 2014;6:721–731. doi: 10.1002/emmm.201403943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang M, Sun T, Jian C, Lei L, Han P, Lv Q, et al. Remodeling of mitochondrial flashes in muscular development and dystrophy in zebrafish. PLoS One. 2015;10:e0132567. doi: 10.1371/journal.pone.0132567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hou T, Wang X, Ma Q, Cheng H. Mitochondrial flashes: new insights into mitochondrial ROS signalling and beyond. J Physiol. 2014;592:3703–3713. doi: 10.1113/jphysiol.2014.275735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, et al. Superoxide flashes in single mitochondria. Cell. 2008;134:279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Young CNJ, Brutkowski W, Lien C-F, Arkle S, Lochmüller H, Zabłocki K, et al. P2X7 purinoceptor alterations in dystrophic mdx mouse muscles: relationship to pathology and potential target for treatment. J Cell Mol Med. 2012;16:1026–1037. doi: 10.1111/j.1582-4934.2011.01397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Frederick DW, Davis JG, Dávila A, Agarwal B, Michan S, Puchowicz MA, et al. Increasing NAD synthesis in muscle via nicotinamide phosphoribosyltransferase is not sufficient to promote oxidative metabolism. J Biol Chem. 2015;290:1546–1558. doi: 10.1074/jbc.M114.579565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van de Weijer T, Phielix E, Bilet L, Williams EG, Ropelle ER, Bierwagen A, et al. Evidence for a direct effect of the NAD+ precursor acipimox on muscle mitochondrial function in humans. Diabetes. 2015;64:1193–1201. doi: 10.2337/db14-0667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vora M, Ansari J, Shanti RM, Veillon D, Cotelingam J, Coppola D, et al. Increased nicotinamide phosphoribosyltransferase in rhabdomyosarcomas and leiomyosarcomas compared to skeletal and smooth muscle tissue. Anticancer Res. 2016;36:503–507. [PMC free article] [PubMed] [Google Scholar]
- 81.Sawicka-Gutaj N, Waligórska-Stachura J, Andrusiewicz M, Biczysko M, Sowiński J, Skrobisz J, et al. Nicotinamide phosphorybosiltransferase overexpression in thyroid malignancies and its correlation with tumor stage and with survivin/survivin DEx3 expression. Tumour Biol J Int Soc Oncodevelopmental Biol Med. 2015;36:7859–7863. doi: 10.1007/s13277-015-3506-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang B, Hasan MK, Alvarado E, Yuan H, Wu H, Chen WY. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene. 2011;30:907–921. doi: 10.1038/onc.2010.468. [DOI] [PubMed] [Google Scholar]
- 83.Bi T-Q, Che X-M, Liao X-H, Zhang D-J, Long H-L, Li H-J, et al. Overexpression of Nampt in gastric cancer and chemopotentiating effects of the Nampt inhibitor FK866 in combination with fluorouracil. Oncol Rep. 2011;26:1251–1257. doi: 10.3892/or.2011.1378. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.