

Abstract
The aim of the present study was to investigate the cytotoxic effect and multi-drug resistance (MDR) of Clostridium difficile toxin A (TcdA) on K562/A02 cells, and understand its underlying molecular pathways. K562/A02 cells were treated with TcdA at different concentrations for 24, 48 and 72 h, and the inhibition effect and drug resistance of TcdA on K562/A02 cell proliferation was assessed by methyl thiazolyl tetrazolium colorimetric assay. Furthermore, cell cycle-apoptosis was analyzed by flow cytometry, P-glycoprotein (P-gp) expression was determined by western blot analysis and caspase-3 activity was measured using a caspase-3 activity kit. TcdA inhibited K562/A02 cell proliferation in a time- and dose-dependent manner. The inhibition rate of K562/A02 cells reached 8.76±0.76, 28.55±0.43, 47.89±0.27, 58.08±0.06 and 57.70±0.79% following treatment with 50, 100, 200, 400 and 800 ng/ml TcdA, respectively, for 24 h. K562/A02 cells in the G0/G1 phase increased and cells in the S phase decreased following treatment with TcdA (P<0.05), and the apoptotic rates in the 200 and 400 ng/ml concentration groups were 14.05 and 22.89%, respectively. In addition, TcdA (50 ng/ml) significantly inhibited the proliferation of K562/A02 cells and reduced the half maximal inhibitory concentration of these drugs in combination with chemotherapy drugs. The reversal folds were 3.09, 2.89 and 2.79, respectively. Furthermore, TcdA treatment was associated with the upregulation of P-gp in K562/A02 cells, and caspase-3 activity was observed to increase in K562/A02 cells following TcdA treatment, when compared with untreated controls (P<0.05). These findings suggested that TcdA may be able to inhibit K562/A02 cell growth, induce cell apoptosis by decreasing P-gp levels and caspase-3 activation, and partially reverse MDR. Further studies are required to evaluate the potential of TcdA as a candidate for the chemotherapy of hematologic malignancies.
Keywords: leukemia, multi-drug resistance, K562/A02 cell, apoptosis, Clostridium difficile toxin A
Introduction
Multi-drug resistance (MDR) is a phenomenon of simultaneous resistance to several structurally and functionally distinct drugs, and is the major cause of treatment failure in cancer chemotherapy. Many mechanisms involved in MDR, including reduced accumulation of intracellular drugs (decreased drug uptake and/or increased drug efflux), enhanced ability of DNA repair, redistributed drug accumulation, added detoxification, the modified expression of genes, and proteins related to cell apoptosis have been recognized for several decades (1,2). Multidrug resistance-associated proteins (MRPs) are considered as the main mechanism of MDR. These transmembrane proteins lead to MDR either by decreasing the intracellular retention of drugs or altering intracellular drug distribution (3). In addition, the ATP-binding cassette (ABC) super family of membrane transporters requires ATP hydrolysis to transport substrates across membranes such as P-gp (4). P-glycoprotein (P-gp) serves as an ATP-dependent efflux pump for a variety of chemicals, including many antineoplastic agents (5). Increasing studies (6,7) have shown that P-gp expression correlates with poor response to chemotherapy and unfavorable prognosis. Therefore, such agents that inactivate P-gp in tumor cells or affect its function may overcome MDR. The first (Verapamil, Cyclosporin A) and second (VX-710, PSC833) generation of P-gp inhibitors have been applied to clinical practice. However, these are restricted for adverse reactions. Recently, third generation P-gp inhibitors including S9788 and LY335979 (Zosuquidar) have been tested in clinical trials. However, their therapeutic effects remain unclear. Therefore, determining how to overcome the MDR of tumor cells to increase the success rate of chemotherapy remains an urgent problem in the present treatment of leukemia.
Clostridium difficile (C. difficile) is a common inhabitant of the colon flora in human infants and sometimes in adults, which produces toxins [Clostridium difficile toxin A (TcdA) and Clostridium difficile toxin B (TcdB)] that could cause pseudomembranous enterocolitis in some patients receiving antibiotic therapy. TcdA has been intensively studied since its initial recognition as a major C. difficile virulence factor (8,9). It causes necrosis, mucosal inflammation, hemorrhagic colitis and increased intestinal permeability, and inhibits protein synthesis (10,11). Mahida et al (12) provided the first experimental evidence that epithelial cells (T84 cells) exposed to TcdA undergoes apoptosis. Furthermore, some studies (6,13,14) have shown the same effect caused by TcdA due to the inactivation of Rho, the activation of caspases 3, 6, 8 and 9 and Bid, and mitochondrial damage followed by cytochrome C release. To date, apoptosis induced by TcdA has been confirmed in several nonmalignant and malignant human cell lines including A549 (lung carcinoma), 009P (breast carcinoma), PC3 (prostate carcinoma), SW480 (colonic carcinoma), SMMC7721 (hepatocellular carcinoma), Raji (lymphoma), breast epithelium (006FA), HCS (diploid fibroblast), etc. Our previous studies indicated that TcdA inhibits K562 (erythroleukemia cell line) cell growth by inducing apoptosis, and the mechanism may be associated with the upregulation of Bcl-2 Assaciated X (Bax) Protein, the activation of caspase-3, the decrease in mitochondrial membrane potential, and the release of cytochrome C from the mitochondria matrix. The present study aimed to explore the effects of C. difficile TcdA on the proliferation, apoptosis and MDR of human chronic myeloid leukemia cell line K562/A02.
Materials and methods
Preparation of C. difficile Toxin A
TcdA was purified, as previously (15) described. C. difficile VPI 10463 bacterial brain heart infusion was centrifuged at 4°C, precipitated with 50% (NH4)2SO4 and acetic acid precipitation at pH 5.5, followed by ion-exchange chromatography on a DEAE-Toyopearl 650 M column. Coomassie brilliant blue (Dishen Biotechnology, Shanghai, China) G-250-based colorimetric assay was performed with Bovine Serum Albumin (BSA) as the standard to draw the standard curve, the concentration of the protein purified toxin was measured, and stored at −20°C.
Cell culture and reagents
K562 and K562/A02 cells were obtained from the Central Laboratory of the First Hospital of Lanzhou University. Cells were maintained in a nutrient medium (RPMI 1640; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml of penicillin and 100 U/ml of streptomycin at 37°C in a humidified incubator atmosphere with 5% CO2. Cells were passage every 2–3 days. In the training process of K562/A02 cells, 1 g/ml of adriamycin (ADM; Zhejiang Hisun Pharmaceutical Co., Ltd., Taizhou, China) was added to maintain its resistance at two weeks before the experiment was stopped. Living cells were strained by trypan blue staining in more than 95% for the experiment.
RPMI 1640 was purchased from Gibco; Thermo Fisher Scientific, Inc. Serum was purchased from Hangzhou Sijiqing Biological Engineering Materials Co., Ltd., (Hangzhou, Zhejiang, China). Methyl thiazolyl tetrazolium (MTT) colorimetric assay was purchased from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). The Annexin V-FITC/PI apoptosis detection kit was purchased from Thermo Fisher Scientific, Inc. The activity test kit of caspase-3 was purchased from Beyotime Institute of Biotechnology (Beijing, China). The antibody against P-gp was purchased from Santa Cruz Biotechnology, Inc., Dallas, TX, USA. Adriamycin (ADM) and Daunorubicin (DNR) were purchased from Zhejiang Hisun Pharmaceutical Co., Ltd. Cytosine Arabinoside (AraC) was purchased from the Shanghai New Hualian Pharmaceutical Co., Ltd., (Shanghai, China).
Cell growth inhibition assay
K562 and K562/A02 cells were seeded in 96-well plates (Corning Incorporated, Corning, NY, USA) with 200 µl of medium. Approximately 2×105 K562/A02 cells were seeded to each well, treated with various concentrations of TcdA (50, 100, 200, 400 and 800 ng/ml). The control group was left untreated, while the blank control group was fixed with RPMI 1640. After 24, 48 and 72 h, the inhibitory concentration of 50% (IC50) value was calculated using the MTT method, and 20 µl of medium containing 5 mg/ml of MTT was added to each well at four hours before the end of the test. Then, 50 µl of triad lysis fluid [10% dodecyl sulfate, sodium salt (SDS), 5% isobutanol and 0.012 mol/l of HCL] was used to dissolve the sample at 37°C overnight. The absorbance of the mixture (A value) at 570 nm was measured using a microplate enzyme-linked immunosorbent assay reader. The inhibition rate of cell growth (%) was calculated as [A570 (mean negative control group)-A570 (mean TcdA treatment group)/(A570 (mean negative control group)-A570 (mean blank control group)] ×100%.
The growth curve was plotted with the action time as the cross axis and the corresponding absorbance (A value) as the vertical axis. The IC50 was calculated according to the linear regression equation.
Cell-cycle analysis
Propidium iodide (PI; Sigma-Aldrich; Merck KGaA) staining and flow cytometry (FCM; Beckman Coulter, Inc., Brea, CA, USA) were used to determine the stage of the cell cycle. Cells were treated with or without 200 and 400 ng/ml of TcdA for 24 h at 37°C. Then, cells were collected and washed twice with phosphate-buffered saline (PBS), suspended in 2 ml of 70% ice-cold ethanol, and stored overnight at 4°C. On the following day, samples were centrifuged and incubated with 1 mg/ml of RNaseA (Fermentas; Thermo Fisher Scientific, Inc.) in the dark at 4°C. Next, cells were stained with 100 µg/ml of PI for 30 min at 4°C. The samples were analyzed by FCM using cell analysis software to determine the population of cells in each cycle phase.
Cell apoptosis was detected with the Annexin V-FITC/PI Apoptosis Detection kit according to the manufacturer's instructions. Data acquisition was performed using FCM and analyzed with CellQuest software.
Multidrug resistance reversal assay
The reversal effect of TcdA on K562/A02 cells was calculated by MTT assay. Briefly, K562/A02 cells were seeded in a 96-well plate at a density of 1×105/ml. ADM (0.1, 1, 10 and 100 µg/ml), DNR (0.01, 0.1, 1 and 10 µg/ml) and Ara-C (0.1, 1, 10 and 100 µg/ml) were respectively added, and treated with or without TcdA (50 ng/ml). MTT assay was performed for measurements, as previously described. Furthermore, the inhibition rate of cell growth (%) and IC50 of TcdA were calculated. The resistance folds and reversal folds were calculated as follows: Resistance Folds=IC50 of resistance cell/IC50 of sensitive cell; Reversal Folds=IC50 of no reversal agent/IC50 of plus reversal agent.
Detection of P-gp expression
Western blot assay was performed to examine the effect of TcdA on the expression of P-gp in K562 and K562/A02 cells. Cells collected by centrifugation were treated with or without 50 ng/ml TcdA for 24 and 48 h, washed twice with PBS, added with lysis buffer and protease inhibitor phenylmethylsulfonyl fluoride (PMSF) (lysis buffer: PMSF=l ml: 10 µl), placed in an ice bath for 30 min, centrifuged at 12,000 rpm for 10 min at 4°C, and stored at −20°C. Then, the sample was mixed with 20 µl of the sample buffer, separated by 6% dodecyl sulfate, sodium salt (SDS)-Polyacrylamide gel electrophoresis (SDS-PAGE) [40% Acr/Bic, gel buffer solution (pH 8.8), Concentrate buffer (pH 6.8), 10% SDS, 10% AP, ultrapure water and TEMED], and transferred onto a polyvidyline fluoride (PVDF) membrane (Shanghai Bioscience and Biotechnology, Shanghai, China). The transfer buffer comprised of 5.89 g of Tris, 2.99 g of glycine, 0.37 g of SDS and 200 ml methanol, plus 1,000 ml of ultrapure water at 30 V. After completion of the transfer, the sample was transferred on a decolorization shaking bed with 1× Li Chunhong dyeing liquid (2 g of 1xLi Chunhong dyeing liquid S, 30 g of trichloroacetic acid and 30 g of sulfonyl salicylic acid, plus 100 ml of distilled water to) dye for five min, and rinse with water until the albumen was clear on the membrane. Then, these were closed by nonfat dry milk and added with antibody diluted in Tris-Buffered Saline and Tween 20 (TBS-T) buffer [100 mmol/l Tris-HCL (pH 7.5), 150 mmol/l NaCl] contain 0.05% Tween 20 at 4°C overnight. Next, the membranes were washed by TBS-T twice for 10 min each time, added with horseradish peroxidase (HRP)-labeled secondary antibody, incubated for one hour at room temperature, and washed again with TBS-T. In addition, the A and B reagents of the enzyme-catalyzed chemiluminescent (ECL) kit (NEN Life Science Inc., Boston, MA, USA) were mixed in the plastic film, the membrane protein was placed faced down to allow full contact with the mixture for one minute, and the film was transferred onto another plastic wrap to remove the residue. Then, these were exposed with x-ray films in the darkroom for two min, the x-ray films were removed and these were immersed in 1× developer for one minute, and washed with running water for one minute. Next, these were immersed in fixative for one minute, washed with running water again, and dried at room temperature. Finally, a gel imaging system was used to determine the gray value of the protein bands and quantitatively analyze the protein expression with a ratio of β-actin values.
Flow cytometric analysis of caspase activity
Cells collected by centrifugation were treated with or without different concentrations of TcdA (50, 100, 200, 400 and 800 ng/ml) for 24, 48 and 72 h, and washed with PBS. Then, these were suspended in cell lysate (10 µl of lysate/2×l06 cells), ice bath cracking for approximately 15 min, and centrifugation at 16,000 g for 15 min at 4°C. The blank control contained 90 µl of Buffer solution, while the experimental group was 80 µl of Buffer solution and 10 µl of Sample. Next, caspase-3 substrate (2 mM of Ac-DEVD-pNA) was added, mixed thoroughly, and incubated for one hour at 37°C. Absorbance at 405 nm of the mixture (A value) was measured when the color change was most obvious. The A405 value of the sample was deducted from the A405 value of the blank control, which was the absorbance produced by the pNA produced by caspase-3. This was used to indicate the activity of caspase-3.
Statistical analysis
All experiments were performed in triplicate. Data were presented as the mean ± standard deviation (x±s) of results obtained from three independent experiments with similar patterns. All data were analyzed using SPSS 17.0 to perform the correlation analysis. Multiple groups were compared by single factor analysis of variance and LSD-t test. The average of these two groups was compared by t-test. P<0.05 was considered to indicate a statistically significant difference.
Results
Effect of TcdA on K562/A02 cell growth
The viability of K562/A02 cells was inhibited by TcdA treatment in a time and dose-dependent manner (Fig. 1). The inhibition rate of cell growth (%) was 8.76±0.76, 28.55±0.43, 47.89±0.27, 58.08±0.06 and 57.70±0.79%, respectively, after treatment with TcdA at different concentrations (50, 100, 200, 400 and 800 ng/ml) for 24 h. Furthermore, the IC50 values were 367.8, 297.1 and 241.5 ng/ml after 24, 48 and 72 h, respectively. The inhibition rate of cell growth (%) in the group treated with 50 ng/ml of TcdA was <10%, when compared with the control group (Table I). However, this change was not statistically significant. Therefore, this concentration was used to treat K562/A02 cells for the remaining experiments in the present study.
Figure 1.
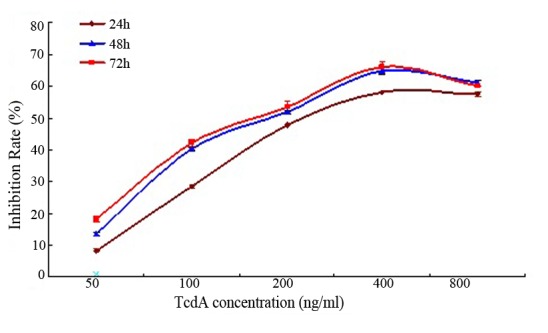
Effect of TcdA on K562/A02 cell growth. The viability of K562/A02 cells was inhibited by TcdA treatment in a time-and dose-dependent manner. TcdA, Clostridium difficile toxin A.
Table I.
Differences in A570 value following treatment with different concentrations of Clostridium difficile toxin A on K562/A02 cells.
| Group | 24 h | 48 h | 72 h |
|---|---|---|---|
| Control | 0.583±0.008 | 0.473±0.010 | 0.405±0.012 |
| TcdA (ng/ml) | |||
| 50 | 0.543±0.008a | 0.422±0.013a–c | 0.348±0017a–c |
| 100 | 0.444±0.011a,b | 0.323±0.006a–c | 0.274±0.009a–c |
| 200 | 0.350±0.002a,b | 0.279±0.006a–c | 0.239±0011a–c |
| 400 | 0.301±0.001a,b | 0.231±0.011a–c | 0.200±0.008a,b |
| 800 | 0.303±0.004a | 0.245±0.004a | 0.219±0.011a |
Data are presented as the mean ± standard deviation.
P<0.05 vs. control group
P<0.05 vs. the previous concentration group at the same time point
P<0.05 vs. the previous time point at the same concentration. TcdA, Clostridium difficile toxin A.
Effect of TcdA on the cell cycle of K562/A02 cells
Fig. 2A presents the changes in cell-cycle distribution in TcdA-treated K562/A02 cells. Mean cells in the G0/G1 phase increased and a decreased in the percentage of cells was observed in the S phase after treatment with TcdA (P<0.05). In addition, there are obvious apoptosis peaks. This suggests that G0/G1 phase arrest occurred after treatment with 200 and 400 ng/ml of TcdA.
Figure 2.
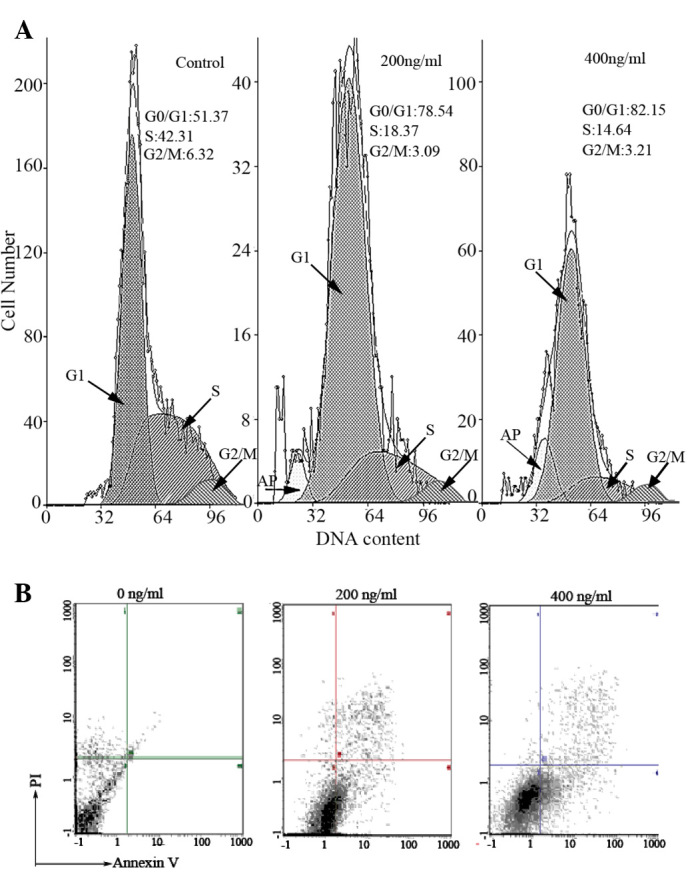
(A) Effect of TcdA on apoptosis and cell cycle distribution in K562/A02 cells in the Control, TcdA (200 ng/ml) and TcdA (400 ng/ml) treatment groups. Arrows indicate apoptosis peaks. Mean cells in the G0/G1 phase increased and then decreased in the S phase following treatment with TcdA, and marked apoptosis peaks were observed. (B) TcdA induced apoptosis in K562/A02 cells. The results revealed a significant accumulation of Annexin V-fluorescein isothiocyanate positive cells and the percentage of apoptotic cells was 14.05 and 22.89%, following treatment with 200 and 400 ng/ml of TcdA, respectively. TcdA, Clostridium difficile toxin A; PI, propidium iodide.
TcdA induced apoptosis in K562/A02 cells
The results showed the significant accumulation of Annexin V-FITC positive cells and indicated that TcdA could induce K562/A02 cells apoptosis, and the percentage of apoptotic cells was 14.05 and 22.89%, respectively, after treatment with 200 and 400 ng/ml of TcdA (Fig. 2B).
The reversal effect of TcdA on K562/A02 cells
Table II shows that the Resistance Folds of DNR and Ara-C on K562/A02 cells were 21.91 and 7.46, respectively; while ADM reached up to 87.06. There was a significant inhibitory effect on the proliferation of K562/A02 cells after the combination of 50 ng/ml of TcdA with drugs (Fig. 3). TcdA (50 ng/ml) could significantly reduce the IC50 of these drugs, and the reversal times were 3.09, 2.89 and 2.79, respectively (Table II).
Table II.
Half maximal inhibitory concentration of chemotherapy drugs used alone or combined with Clostridium difficile toxin A on K562 and K562/A02 cells for 24 h.
| Chemotherapy drugs alone | Chemothe-rapy drugs plus TcdA | ||||
|---|---|---|---|---|---|
| Group | K562 | K562/A02 | K562/A02 | Resistant folds | Reversal folds |
| IC50 (µg/ml) | |||||
| ADM | 0.64±0.014 | 55.72±5.140 | 8.03±3.472 | 87.06 | 3.09 |
| DNR | 0.11±0.001 | 2.41±0.800 | 1.20±0.049 | 21.91 | 2.89 |
| Ara-C | 2.59±0.650 | 19.31±2.160 | 6.93±1.803 | 7.46 | 2.79 |
Data are presented as the mean ± standard devation (n=3). IC50, half maximal inhibitory concentration; TcdA, Clostridium difficile toxin A; ADM, Adriamycin; DNR, Daunorubicin; AraC, Cytosine Arabinoside.
Figure 3.

Effect of chemotherapy drugs alone or combined with 50 ng/ml TcdA on K562/A02 cells proliferation. There was a significant inhibitory effect on the proliferation of K562/A02 cells following the combined treatment of 50 ng/ml TcdA and chemotherapy drugs. *P<0.05 vs. chemotherapy drugs alone. TcdA, Clostridium difficile toxin A; ADM, Adriamycin; DNR, Daunorubicin; AraC, Cytosine Arabinoside.
Effect of TcdA on the expression of P-gp in K562/A02 cells
As shown in Fig. 4, the effect of TcdA on the expression of P-gp in K562 and K562/A02 cells by western blot. The relative expression level of P-gp in K562 and K562/A02 cells were 0.23 and 0.49, respectively. The level decreased to 0.47 and 0.29, respectively, after TcdA (50 ng/ml) treatment for 24 and 48 h on K562/A02 cells.
Figure 4.

Effect of TcdA on P-gp expression in K562/A02 cells. Lane 1, K562 cells; lane 2, K562/A02 cells treated with TcdA for 48 h; lane 3, K562/A02 cells treated with TcdA for 24 h; lane 4, K562/A02 cells. The relative expression levels of P-gp in K562 and K562/A02 cells were 0.23 and 0.49, respectively. The level decreased to 0.47 and 0.29 following TcdA (50 ng/ml) treatment for 24 and 48 h, respectively. TcdA, Clostridium difficile toxin A; P-gp, P-glycoprotein.
Effect of TcdA on the activition of caspase-3 in K562/A02 cells
As shown in Fig. 5, the caspase-3 activity of K562/A02 cells was very low, and the absorbance values were 0.19±0.014, 0.20±0.017 and 0.19±0.012 after 24, 48 of 72 h, respectively. TcdA could significantly enhance the activity of caspase-3 in the experimental group, and the absorbance values were statistically significant (P<0.05), compared with the control group. Furthermore, enzyme activity gradually increased with the increase in concentration.
Figure 5.
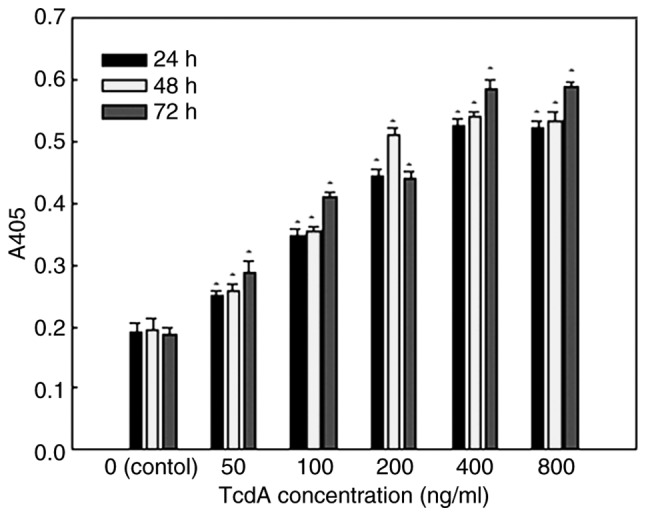
Caspase-3 activity of K562/A02 cells treated with different concentrations of TcdA for 24, 48 and 72 h, respectively. TcdA significantly enhanced the activity of caspase-3 in the experimental group following 24, 48 and 72 h, and the absorbance values were statistically significant when compared with the control group. Furthermore, enzyme activity gradually increased with the increase in concentration. *P<0.05 vs. controls. TcdA, Clostridium difficile toxin A.
Discussion
The MDR of tumor cells was defined as a phenomenon, in which resistance occurs in structurally and mechanically diverse anticancer drugs. K562/A02 cells with MDR property and hindered apoptosis has been the human leukemia cell line with the stable MDR phenotype induced in medium containing ADM for a long time (16). TcdA is a kind of soluble protein that combines with the cell membrane via receptors that induce cytopathic effects after being phagocytized by immune cells. Its susceptibility differs in many mammalian cells (17). The present study revealed that cancer and normal cells have a much different sensitivity to the cytotoxicity of toxins, which was particularly pronounced at small doses (18). The present study revealed that TcdA could inhibit K562/A02 cell growth in a time and dose-dependent manner, and the inhibition rate was statistically significant, compared with the control group (P<0.05). This is in accordance with previous study (13). Meanwhile, FCM revealed that the percentage of apoptotic cells was 14.05 and 22.89%, respectively, after 200 and 400 ng/ml of TcdA treatment, suggesting that TcdA could inhibit cell growth by inducing apoptosis.
Cell-cycle control is the major regulatory mechanism of cell growth. Many cytotoxic agents arrest the cell cycle at the G1, S, or G2/M phase before inducing apoptotic cell death (19,20). Disturbance in the cancer-cell cycle is one therapeutic target for the development of new anticancer drugs (21). It has been found that cell-cycle arrest may result in apoptosis due to the existence of cell-cycle checkpoints and feedback control (22). In the present study, the results of the FCM analysis suggests that a significant accumulation occurred at the G0/G1 phase in K562/A02 cells after TcdA treatment, which was associated with a significant decrease in cells at the S phases. This suggests that G0/G1 phase arrest occurred after treatment with TcdA.
Studies (6) have confirmed that P-gp is the main mechanism of MDR of tumor cells. P-gp, is the product of MDR1 gene, can insert in the cell membrane and discharge a variety of antitumor drugs through its own pump, which decrease intracellular drug concentration and result in drug resistance. MDR1and P-gp is the main target for the treatment of refractory/relapsed leukemia. The results showed that P-gp was highly expressed in multidrug resistant leukemic cell lines, this consistent with Peng et al (5) study. The K562/A02 cell lines are highly resistant to traditional chemotherapy drugs such as ADM, DNR and Ara-C, especially for ADM. The IC50 of ADM, DNR and Ara-C decreased significantly, after treatment with non-cytotoxic doses of (50 ng/ml) TcdA for 24 h. This suggested that TcdA could reverse MDR of cells in addition to inhibiting cell growth. The P-gp expression level of K562/A02 cells decreased significantly compared with the control group after treatment with TcdA (P<0.05), suggesting that TcdA could inhibit cell growth and reverse MDR of cells by reducing P-gp expression. Zu et al (23) found that expression of P-gp inhibits apoptosis in tumor cells, P-gp inhibitor leads to cell death and directly induces apoptosis. In addition, they pointed that inhibition of P-gp function is an effective way to induce apoptosis and overcome MDR. Those are consistent with our studies. Therefore, TcdA could act as a drug resistance reversal agent to improve the antitumor effect of other anti-tumor drugs and reduce the dosage, in order to achieve the purpose of treatment. This greatly increased the possible application of TcdA in leukemia treatment.
In addition, P-gp can inhibit apoptosis by inhibiting the activity of cysteine proteinase, and the downregulation of P-gp expression can activate caspase and promote apoptosis, conversely (24). Caspase-3 is considered to be the most important effectors due to the sequence of the cascade downstream in apoptosis, and its activation marks the irreversible phase of apoptosis. Caspase-3 are down regulated in many malignant tumors, and even lack expression (25), suggesting that the expression level of caspase plays an important role in the development and progression of malignant tumors. Cell apoptosis can be completely blocked by the caspase-3 inhibitor. In addition, increase of caspase-3 expression can inhibit cell proliferation and accelerate its apoptosis (26). This study revealed that the expression level of P-gp protein in K562/A02 cells was high, while the activity of caspase was significantly inhibited. It is suggested that the expression defects of caspase enzyme may be related to the MDR of cells. After TcdA treatment, the expression of P-gp was down regulated, and the activity of caspase subsequently increased in a dose-dependent manner, suggesting that TcdA could promote apoptosis by inhibiting the expression of P-gp and activating caspase. This may be one of the mechanisms by which TcdA induce cell apoptosis and reverse drug resistance. In accordance with our finding, Brito et al (13). also have found that TcdA activates caspase-3, −6, −8, and −9 activity, caspase-3 activity increased more quickly than the other caspases, and demonstrated the involvement of caspase-activating cascades in toxin A-induced apoptosis. In addition, Zu et al (23) also found that P-gp related apoptosis was caspase-mediated.
In conclusion, TcdA could induce apoptosis, obviously enhance the inhibitory effect of other antitumor drugs with non-cytotoxic doses, and partly reverse the drug resistance of K562/A02 cells. This mechanism may be related to the inhibition of P-gp expression and the activation of caspase. All of which indicate the potential of TcdA in the treatment of leukemia, especially refractory leukemia. However, determining how to combine this with some carrier to increase the specificity of tumor cell toxicity is worth further research, due to the low cell selectivity and intestinal toxicity of TcdA.
Acknowledgements
The authors acknowledge the financial support provided by the Lanzhou Science and Technology Plan Project under project number 2014-1-37. The authors would also like to thank Professor Fu Siwu from the Northwest University for Nationalities for providing the Clostridium difficile VPI 10463 strain, and we also acknowledge the University.
References
- 1.Aberuyi N, Rahgozar S, Moafi A. The role of ATP-binding cassette transporter A2 in childhood acute lymphoblastic leukemia multidrug resistance. Iran J Ped Hematol Oncol. 2014;4:118–126. [PMC free article] [PubMed] [Google Scholar]
- 2.Gillet JP, Gottesman MM. Mechanisms of multidrug resistance in cancer. Methods Mol Biol. 2010;596:47–76. doi: 10.1007/978-1-60761-416-6_4. [DOI] [PubMed] [Google Scholar]
- 3.Valera ET, Scrideli CA, Queiroz RG, Mori BM, Tone LG. Multiple drug resistance protein (MDR-1), multidrug resistance-related protein (MRP) and lung resistance protein (LRP) gene expression in childhood acute lymphoblastic leukemia. Sao Paulo Med J. 2004;122:166–171. doi: 10.1590/S1516-31802004000400007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toyoda Y, Hagiya Y, Adachi T, Hoshijima K, Kuo MT, Ishikawa T. MRP class of human ATP binding cassette (ABC) transporters: Historical background and new research directions. Xenobiotica. 2008;38:833–862. doi: 10.1080/00498250701883514. [DOI] [PubMed] [Google Scholar]
- 5.Peng Z, Xiao Z, Wang Y, Liu P, Cai Y, Lu S, Feng W, Han ZC. Reversal of P-glycoprotein-mediated multidrug resistance with small interference RNA (siRNA) in leukemia cells. Cancer Gene Ther. 2004;11:707–712. doi: 10.1038/sj.cgt.7700738. [DOI] [PubMed] [Google Scholar]
- 6.Plasschaert SL, de Bont ES, Boezen M, vander Kolk DM, Daenen SM, Faber KN, Kamps WA, de Vries EG, Vellenga E. Expression of multidrug resistance-associated proteins predicts prognosis in childhood and adult acute lymphoblastic leukemia. Clin Cancer Res. 2005;11:8661–8668. doi: 10.1158/1078-0432.CCR-05-1096. [DOI] [PubMed] [Google Scholar]
- 7.Swerts K, De Moerloose B, Dhooge C, Laureys G, Benoit Y, Philippe J. Prognostic significance of multidrug resistance-related proteins in childhood acute lymphoblastic leukaemia. Eur J Cancer. 2006;42:295–309. doi: 10.1016/j.ejca.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 8.Lyerly DM, Lockwood DE, Richardson SH, Wilkins TD. Biological activities of toxins A and B of Clostridium difficile. Infect Immun. 1982;35:1147–1150. doi: 10.1128/iai.35.3.1147-1150.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamiya S, Reed PJ, Borriello SP. Purification and characterisation of Clostridium difficile toxin A by bovine thyroglobulin affinity chromatography and dissociation in denaturing conditions with or without reduction. J Med Microbiol. 1989;30:69–77. doi: 10.1099/00222615-30-1-69. [DOI] [PubMed] [Google Scholar]
- 10.Brito GA, Carneiro-Filho B, Orià RB, Destura RV, Lima AA, Guerrant RL. Clostridium difficile toxin A induces intestinal epithelial cell apoptosis and damage: Role of Gln and Ala-Gln in toxin A effects. Dig Dis Sci. 2005;50:1271–1278. doi: 10.1007/s10620-005-2771-x. [DOI] [PubMed] [Google Scholar]
- 11.Nottrott S, Schoentaube J, Genth H, Just I, Gerhard R. Clostridium difficile toxin A-induced apoptosis is p53-independent but depends on glucosylation of Rho GTPases. Apoptosis. 2007;12:1443–1453. doi: 10.1007/s10495-007-0074-8. [DOI] [PubMed] [Google Scholar]
- 12.Mahida YR, Makh S, Hyde S, Gray T, Borriello SP. Effect of Clostridium difficile toxin A on human intestinal epithelial cells: Induction of interleukin 8 production and apoptosis after cell detachment. Gut. 1996;38:337–347. doi: 10.1136/gut.38.3.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brito GA, Fujji J, Carneiro-Filho BA, Lima AA, Obrig T, Guerrant RL. Mechanism of Clostridium difficile toxin A-induced apoptosis in T84 cells. J Infect Dis. 2002;186:1438–1447. doi: 10.1086/344729. [DOI] [PubMed] [Google Scholar]
- 14.Carneiro BA, Fujii J, Brito GA, Alcantara C, Orià RB, Lima AA, Obrig T, Guerrant RL. Caspase and bid involvement in Clostridium difficile toxin A-induced apoptosis and modulation of toxin A effects by glutamine and alanyl-glutamine in vivo and in vitro. Infect Immun. 2006;74:81–87. doi: 10.1128/IAI.74.1.81-87.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meng XQ, Kamiya S, Yamakawa K, Ogura H, Nakamura S. Purification and characterisation of intracellular toxin A of Clostridium difficile. J Med Microbiol. 1993;38:69–73. doi: 10.1099/00222615-38-1-69. [DOI] [PubMed] [Google Scholar]
- 16.Meng QQ, Chen BA, Wu W, Shao ZY, Gao F, Zhao HH. Antitumor effects of low-frequency ultrasound combined with adriamycin on human leukemia multidrug resistance cell line K562/A02. Ai Zheng. 2008;27:1182–1185. (In Chinese) [PubMed] [Google Scholar]
- 17.D'Auria KM, Kolling GL, Donato GM, Warren CA, Gray MC, Hewlett EL, Papin JA. In vivo physiological and transcriptional profiling reveals host responses to Clostridium difficile toxin A and toxin B. Infect Immun. 2013;81:3814–3824. doi: 10.1128/IAI.00869-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu D, Joyee AG, Nandagopal S, Lopez M, Ma X, Berry J, Lin F. Effects of Clostridium difficile toxin A and B on human T lymphocyte migration. Toxins. 2013;5:926–938. doi: 10.3390/toxins5050926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torres K, Horwitz SB. Mechanisms of Taxol-induced cell death are concentration dependent. Cancer Res. 1998;58:3620–3626. [PubMed] [Google Scholar]
- 20.Murray AW. Recycling the cell cycle: Cyclins revisited. Cell. 2004;116:221–234. doi: 10.1016/S0092-8674(03)01080-8. [DOI] [PubMed] [Google Scholar]
- 21.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 22.Pietenpol JA, Stewart ZA. Cell cycle checkpoint signaling: Cell cycle arrest versus apoptosis. Toxicology. 2002;182:475–481. doi: 10.1016/S0300-483X(02)00460-2. [DOI] [PubMed] [Google Scholar]
- 23.Zu Y, Yang Z, Tang S, Han Y, Ma J. Effects of P-glycoprotein and its inhibitors on apoptosis in K562 cells. Molecules. 2014;19:13061–13075. doi: 10.3390/molecules190913061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gibalova L, Seres M, Rusnak A, Ditte P, Labudova M, Uhrik B, Pastorek J, Sedlak J, Breier A, Sulova Z. P-glycoprotein depresses cisplatin sensitivity in L1210 cells by inhibiting cisplatin-induced caspase-3 activation. Toxicol In Vitro. 2012;26:435–444. doi: 10.1016/j.tiv.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 25.Hoshi T, Sasano H, Kato K, Yabuki N, Ohara S, Konno R, Asaki S, Toyota T, Tateno H, Nagura H. Immunohistochemistry of Caspase3/CPP32 in human stomach and its correlation with cell proliferation and apoptosis. Anticancer Res. 1998;18:4347–4353. [PubMed] [Google Scholar]
- 26.Qa'Dan M, Ramsey M, Daniel J, Spyres LM, Safiejko-Mroczka B, Ortiz-Leduc W, Ballard JD. Clostridium difficile toxin B activates dual caspase-dependent and caspase-independent apoptosis in intoxicated cells. Cell Microbiol. 2002;4:425–434. doi: 10.1046/j.1462-5822.2002.00201.x. [DOI] [PubMed] [Google Scholar]