

Falik-Zaccai et al. report a novel human progressive leukoencephalopathy syndrome with spastic quadriparesis, microcephaly, startle response and severe global developmental delay in seven patients, caused by a homozygous sequence variation in PLAA (phospholipase A2-activating protein). The mutation joins other complex phospholipid defects known to affect white matter development and maintenance.
Keywords: phospholipase A2-activating protein (PLAA), progressive leukoencephalopathy, autosomal recessive, startle response, complex phospholipid defects
Abstract
Leukoencephalopathies are a group of white matter disorders related to abnormal formation, maintenance, and turnover of myelin in the central nervous system. These disorders of the brain are categorized according to neuroradiological and pathophysiological criteria. Herein, we have identified a unique form of leukoencephalopathy in seven patients presenting at ages 2 to 4 months with progressive microcephaly, spastic quadriparesis, and global developmental delay. Clinical, metabolic, and imaging characterization of seven patients followed by homozygosity mapping and linkage analysis were performed. Next generation sequencing, bioinformatics, and segregation analyses followed, to determine a loss of function sequence variation in the phospholipase A2-activating protein encoding gene (PLAA). Expression and functional studies of the encoded protein were performed and included measurement of prostaglandin E2 and cytosolic phospholipase A2 activity in membrane fractions of fibroblasts derived from patients and healthy controls. Plaa-null mice were generated and prostaglandin E2 levels were measured in different tissues. The novel phenotype of our patients segregated with a homozygous loss-of-function sequence variant, causing the substitution of leucine at position 752 to phenylalanine, in PLAA, which causes disruption of the protein’s ability to induce prostaglandin E2 and cytosolic phospholipase A2 synthesis in patients’ fibroblasts. Plaa-null mice were perinatal lethal with reduced brain levels of prostaglandin E2. The non-functional phospholipase A2-activating protein and the associated neurological phenotype, reported herein for the first time, join other complex phospholipid defects that cause leukoencephalopathies in humans, emphasizing the importance of this axis in white matter development and maintenance.
Introduction
Leukoencephalopathies are brain white matter disorders categorized by neuroradiological and pathophysiological criteria (van der Knaap, 2001) into:
Hypomyelinating diseases that are primary disturbances in myelin formation; Pelizaeus-Merzbacher disease may be considered the prototype of hypomyelinating disorders. Additionally, Pelizaeus-Merzbacher-like diseases phenotypically resemble Pelizaeus-Merzbacher disease but are inherited as autosomal-recessive disorders. Typically, no gene is identified, but in a small subset of Pelizaeus-Merzbacher-like diseases patients, sequence variations in GJC2 [also known as gap junction protein alpha 12 (GJA12), coding for connexin 46.6 (Uhlenberg et al., 2004; Henneke et al., 2008)], and sequence variations in the gene HSPD1, coding for heat shock 60-kDa protein 1, have been found (Magen et al., 2008). This group also includes syndromes in which hypomyelination is accompanied by other multi-organ involvements such as Cockayne’s and trichothiodystrophy syndromes (Weidenheim et al., 2009), and oculodentodigital dysplasia and 4H syndrome (Atrouni et al., 2003; Timmons et al., 2006).
Dysmyelinating disorders with delayed and disturbed myelination, including most amino-acidopathies and organic acidurias. As the brain (especially glial cells) is very sensitive to accumulation of toxic metabolites, secondary white matter abnormalities can be diagnosed in many metabolic disorders. Dysmyelination is also a main pattern of rare disorders such as SOX10-associated syndromes (the neurological variant of Waardenburg-Shah syndrome) (Pusch et al., 1998). Affected patients present a variable range of neurological symptoms: developmental delay, spasticity, ataxia, nystagmus, and in severe cases, profound neonatal hypotonia and congenital arthrogrypposis due to peripheral hypomyelination. The 18q deletion syndrome, another example for disorders in this category, is characterized by neurological features such as mental retardation, microcephaly, hypotonia, nystagmus and seizures, accompanied by additional multisystem defects, including partial growth hormone deficiency, facial, external ear, cardiac, and skeletal defects. Regions for dysmyelination, congenital aural atresia, and growth hormone insufficiency (18q22.3-q23) were identical and contained five known genes, including the MBP encoding gene (MBP). The dysmyelination region was 100% penetrant (Feenstra et al., 2007).
The third group is disorders with progressive demyelination, including ‘classic’ leukodystrophies: X-linked adrenoleukodystrophy, Alexander’s disease, Metachromatic leukodystrophy, Krabbe’s disease, and disorders with white-matter vacuolization such as Canavan’s disease and vanishing white matter disease.
Several leukoencephalopathies present with cystic degenerations, including megalencephalic leukoencephalopathy with subcortical cysts, first described by van der Knaap et al. in 1995. Early-onset macrocephaly and delayed-onset neurological deterioration, including cerebellar ataxia, spasticity, epilepsy, and mild cognitive decline, are characteristic features. Cystic leukoencephalopathy without megalencephaly has also been described (Henneke et al., 2009).
Disorders secondary to axonal damage include the autosomal-recessive disorder giant axonal neuropathy. Patients present with progressive gait disturbances due to peripheral neuropathy, mental retardation, optic atrophy and spasticity; brain imaging studies show leukoencephalopathy. Pathological hallmarks of giant axonal neuropathy are axonal loss and axonal swellings filled with neurofilaments on nerve biopsy (Tazir et al., 2009). Giant axonal neuropathy is caused by sequence variation in GAN encoding for gigaxonin, located on chromosome 16q24 (Bomont et al., 2000).
Nevertheless, 50% of patients with leukoencephalopathies remain without specific diagnosis.
We report seven individuals from two consanguineous families presenting with a unique phenotype of severe spastic quadriparesis, progressive microcephaly, thin corpus callosum, significant startle response, and severe global developmental delay. Genetic investigation revealed a novel missense variant in the phospholipase A2-activating protein encoding gene (PLAA) and disclosed a new mechanism required for normal development and maintenance of CNS white matter.
Materials and methods
Patients
The Israeli Ministry of Health Ethics Committee for genetic experiments approved the proposed studies. Seven affected and 23 healthy individuals from two consanguineous families were enrolled in the study; they or their legal guardians provided written, informed consent. Clinical investigations included medical procedures, imaging and electrophysiological studies, and muscle biopsies. Skin biopsy was performed as part of the research protocol.
Molecular studies
Genetic linkage analysis
Linkage and haplotype analyses were performed as previously described (Zivony-Elboum et al., 2012).
An analysis of 2050 polymorphic markers, spread across the genome at ∼2 cm intervals was performed for nine family members. Statistical analysis of the logarithm of the odds (LOD) score was performed using the Pedtool-superlink tool. Areas with high LOD score were further examined using Linkage Mapping Set v2.5 HD5 kit and v2.5 MD10 (Applied Biosystems) on 24 family members, according to the manufacturer’s protocol.
Molecular inversion probes and massively parallel sequencing
Molecular inversion probes were designed as described (Teer et al., 2010) to cover the 2 Mb of the candidate region (LC Sciences). A total of 6498 amplimers had an average length of 433 bp (±22 bp). The amplimers covered 97% of the candidate region. DNA capture, library preparation, GAIIx sequencing (Illumina), and data analysis were performed as described (Teer et al., 2010). Potential variants were filtered and visualized with VarSifter (Teer et al., 2012).
Sanger sequencing
For dideoxy sequencing, primers were designed to cover the candidate sequence variations (primer sequences available upon request). Direct sequencing of the polymerase chain reaction (PCR) amplification products was performed using BigDye® 3.1 Terminator chemistry (Applied Biosystems) and separated on an ABI 3130xl genetic analyzer (Applied Biosystems). Data were evaluated using Sequencher v5.0 software (Gene Codes Corporation, Ann Arbor, MI).
Molecular modelling
Molecular modelling of the PLAA protein and assessment of the sequence variation impact was performed using the PyMOL Molecular Graphics System (Schrödinger, New York, NY) (Baugh et al., 2011).
Expression analyses
Reverse transcription of the full-length PLAA transcript
Primary fibroblasts [from healthy controls (nPLAA—for native PLAA) and patients (mPLAA—for mutated PLAA)] were harvested from one near-confluent 25 cm2 flask and RNA extracted using the RNeasy® mini kit (Qiagen). RNA samples were quantified using a Nanodrop Spectrophotometer (Nanodrop Technologies) and qualified by analysis on an RNA NanoChip using the Agilent 2100 Bioanalyzer (Agilent Technologies). Synthesis of cDNA was performed using the TaqMan® Reverse Transcription Reagents Kit (Applied Biosystems). The reaction conditions were as follow: 10 min at 25°C; 30 min at 48°C; and 5 min at 95°C. PCR amplifications of cDNA were performed using FailSafe buffer C (Epicenter Biotechnologies) with PLAA primers 5’CGAGCGGCGCAACCAGGTACC3’ and 5’GCATTCACTTACTTTAGCTGGTTCTG3’ at a final concentration of 1 µM. Thermal conditions for 40 cycles of PCR were as follows: 94°C for 30 s, 60°C for 30 s, and 68°C for 7 min.
Real time quantitative polymerase chain reaction
One microgram of RNA extracted from fibroblasts from healthy controls and patients was subjected to cDNA synthesis followed by real time quantitative polymerase chain reaction (RT-qPCR) using the iTaq™ Universal SYBR® Green mix (Bio-Rad). The final concentration of the PLAA primers (5’GACT TGGGAATCCC AGCTTTTC3’ and 5’TTCCCATACTTGCAGAACCTG3’; Accession # NM_001031689) was 300 nM. RT-qPCR assays were performed with human 18S RNA, glyceraldehyde 3-phosphate dehydrogenase (GADPH), L19 ribosomal protein, and polymerase beta (POLB) as housekeeping protein encoding genes to normalize PLAA transcript levels. Absolute analysis was performed using known amounts of a synthetic transcript of the gene of interest. All RT-qPCR assays were run on the ABI Prism 7500 Sequence Detection System and the conditions were as follow: 50°C for 2 min, 95°C for 10 min, and then 40 cycles of 95°C for 15 s and 60°C for 1 min. The results shown were the averages and standard deviations (SD) from three independent experiments performed in triplicate.
The pro-inflammatory gene expression in fibroblasts with nPLAA or the mPLAA gene was carried out using the appropriate assays-on-demandTM gene expression assay mix consisting of a 20× mix of unlabelled PCR primers and TaqMan® MGB probe, FAMTM dye-labelled (Life Science Technology). Human GADPH, ACTB (β-actin), and 18S RNA encoding genes were used to normalize transcripts for various cytokines. The primer sequences for various cytokine genes are available upon request. The reactions were carried out according to the manufacturer’s instruction using a Bio-Rad Q5 RT-qPCR machine. The results shown were the averages and standard deviations from three independent experiments performed in triplicate.
Western blot analysis
For β-catenin, briefly, 70 µg of protein samples from healthy controls’ and patients’ fibroblasts with or without lipopolysaccharide (LPS; 10 µg/ml) stimulation were electrophoresed on 4–20% Mini-PROTEAN® TGX™ Pre-cast Tris/Glycine gels (Bio-Rad) and then transferred to nitrocellulose membranes. The membranes were probed with non-phospho (active) β-catenin (Cell Signaling Technology) and β-tubulin (Santa Cruz Biotechnology), as a protein loading control, antibodies as described by the manufacturer. An anti-rabbit horseradish peroxidase conjugated secondary antibody (Southern Biotech) was then added, and proteins detected by using enhanced chemiluminescence with SuperSignal® West Femto Maximum Sensitivity substrate (Thermo Fisher Scientific). The membranes were then imaged with GE ImageQuant LAS 4000 (General Electric).
Biochemical studies
Measurement of prostaglandin E2
Primary human fibroblasts from healthy controls and patients were grown in Dulbecco’s modified essential medium with 15% foetal bovine serum at 37°C and 5% CO2. Fibroblasts from patients and control subjects were treated with 10 μg/ml LPS or cholera toxin (CT) for 24 h; and the cell culture supernatants were collected. Prostaglandin E2 (PGE2) levels were determined using enzyme immunoassay kit (Cayman Chemicals). To examine PGE2 levels in mouse tissues, samples were subjected to solid phase extraction on C18 columns (Cayman Chemicals) prior to measurements.
Preparation of membrane fractions from fibroblasts
Membrane fractions from healthy controls and patients unstimulated and LPS-stimulated fibroblasts were isolated using established procedures (Zhang et al., 2008). Protein concentrations in membrane fractions were determined using Bradford Protein Reagent (Bio-Rad).
Measurement of cytosolic phospholipase A2 activity
The cytosolic phospholipase A2 (cPLA2) activity in membrane fractions of fibroblasts from patients and healthy controls was determined using PLA2 activity kit (Cayman Chemicals). The enzymatic activity was normalized to protein concentration for each sample. Bee venom PLA2 was used as a positive control.
Complementation studies
Fibroblasts from patients (with mPLAA) or healthy controls (with nPLAA) were grown and electroporated with the recombinant plasmid or the vector alone using Lonza Nucleofector and Human Dermal Fibroblast kit (Lonza). The mPLAA fibroblasts were electroporated with either CMV promoter-based pIRES2-DsRed2-nPLAA for complementation or pIRES2-DsRed2 vector (Clontech) alone as a control. nPLAA fibroblasts were also electroporated with the vector alone to serve as an additional control.
Mouse model
All animal experiments were performed at the University of Texas Medical Branch (UTMB). Animals were housed in a specific-pathogen free facility at a constant temperature (68–79°F) and humidity (30–70%) on a 12-h light-dark cycle. Autoclaved water and irradiated feed were given to the animals ad libitum. All procedures were performed in accordance with the protocol reviewed and approved by UTMB’s Institutional Animal Care and Use Committee, and in compliance with the institutional policies/guidelines and the Guide for the Care and Use of Laboratory Animals, 8th edition. Euthanasia methods used in the procedures were consistent with the American Veterinary Medical Association Guidelines for the Euthanasia of Animals, 2013 edition. Generation of Plaa-null mice (Plaa gene targeting) and genotyping of the mice are described in the Supplementary material.
Preparation of mouse tissue samples for prostaglandin E2 measurements
In brief, mouse tissues were suspended in homogenization buffer (0.1 M disodium phosphate buffer, pH 7.4, 1 mM EDTA, 10 µM indomethacin) and sonicated. Samples were normalized by measuring protein concentrations using the Bradford Protein Reagent (Bio-Rad). After homogenization, four volumes of ethanol were added and samples centrifuged at 3000g for 10 min at 4°C. Supernatants were collected and ethanol removed by vacuum centrifugation before acidification of the samples with 1 M acetate buffer. The samples were then loaded on prewashed C18 cartridges, washed with H2O, and eluted with ethyl acetate and 1% methanol (99:1 v/v). Ethyl acetate was removed by vacuum centrifugation and samples reconstituted in PGE2 assay buffer for measuring PGE2.
Histopathology
Sections (5 µm) representing mouse skin, lungs, and the brain cerebral cortex from embryonic day (E) 18.5 were fixed in 10% neutral buffered formalin. The tissue sections were mounted on slides and stained with haematoxylin and eosin. For the brain cerebral cortex, sections (10 µm) were also subjected to Luxol® fast blue procedure for staining myelin (Kluver and Barrera, 1953; Hrapchak and Sheehan, 1980). The histopathological evaluation of the tissue sections was performed in a blinded fashion.
Statistical analysis
Where appropriate, at least three independent experiments were performed in triplicate and data analysed using one-way ANOVA with Tukey post hoc correction.
Results
Patients
Seven individuals from two families, all products of consanguineous marriages and uneventful pregnancies, presented with progressive leukoencephalopathy (Fig. 1A and B), defined as dysmyelinating according to the known categories of Van der Knaap and colleagues (van der Knaap, 2001). Affected individuals were normal at birth, with onset of neurological symptoms at age 2–4 months (Table 1). Symptoms included spasticity of lower limbs rapidly progressing to upper extremities, resulting in severe quadriparesis with symptoms of corticospinal tract impairment and posture deformation. Involvement of extrapyramidal system function included dystonic posturing, rigidity/freezing, and hypomimia/amimia. All patients suffered from severe mental and language developmental delay. The motor functions were also prominently impaired (level V, according to Gross Motor Function Classification System; Palisano et al., 1997). Abnormally exaggerated startle reflex to an auditory stimulus was observed in six patients, and seizures developed in three.
Figure 1.

Pedigree of the investigated families. (A) Family I: six affected individuals (filled symbols). (B) Family II: containing another affected individual. A high rate of consanguinity and an autosomal-recessive pattern of inheritance are evident. (C) Photographs of Patient VI5 (Pedigree A) illustrating: coarse facial features (a) pectus carinatum, dystonic posturing, rigidity/freezing and shortening of tendons (b and c), and rocker bottom feet (d). (D) Patients’ brain MRI. [D(a and b)] T1 brain MRI of Patient IV2 (Family II), at 1 year of age, shows white matter atrophy. Corpus callosum is complete but thin. (c) (T2 MRI) and d (T1 MRI). Brain MRI of Patient VI3 (Family I), at 14 years, shows moderate white matter atrophy and severe corpus callosum thinning. (e) T2 MRI and (f) T1 MRI. Brain MRI of Patient V4 (Family I), at age 32 years, shows severe general atrophy. The cortex is usually preserved but very thin, corpus callosum is complete but also very thin. The basal ganglia appear normal.
Table 1.
Clinical characteristics of patients
| Characteristic | Patient | ||||||
|---|---|---|---|---|---|---|---|
| A(VI3) | A(VI4) | A(VI5) | A(V6) | A(VI10) | B(IV2) | A(VI1) | |
| Sex | F | F | M | M | M | M | M |
| Age (y) | 15 | 11 | 16 | 34 | 5 | 3 | 2 |
| AO (m) | 4 | 4 | 3 | 4 | 3 | 2 | 3 |
| FTT | +++ | + | +++ | +++ | ++ | +++ | +++ |
| Progressive microcephaly | +++ | + | +++a | +a | + | +++ | + |
| Pyramidal signs, lower extremities |
|
|
|
|
|
++ | +++ |
| Pyramidal signs, upper extremities | ++ | ++ | ++ | ++ | ++ | ++ | +++ |
| Extra-pyramidal signs | ++ | ++ | +++ | +++ | ++ | +++ | +++ |
| GMFCS (level) | V | V | V | V | V | V | V |
| Cognitive and language development delay | Severe | Severe | Severe | Severe | Severe | Severe | Severe |
| Exaggerated startle response | + | + | + | NA | + | + | + |
| Seizures | - | - | + | + | - | + | - |
| MRI/CT of brain |
|
|
|
Severe general and especially white Matter atrophy and thin corpus callosum) at age 30 y) |
|
|
Delayed myelination (at age 9 m) |
| Kyphosis/pectus carinatum | +/+++ | +/+++ | +/+++ | +++/+++ | +/++ | ++/- | +/+ |
| Hyper-trichosis | + | + | NA | + | + | - | - |
| Small joints hyper-flexibility | + | + | ++ | - | ++ | + | + |
| Large joints contractures |
|
+++ | +++ |
|
+++ | ++ | + |
| Intensive sweating of palms and feet | + | + | - | + | + | + | + |
| Miscellaneous |
|
SSEP: central bilateral disturbance in central conduction above brainstem | Muscle biopsy: mild reduction of cytochrome c oxidase activity. Normal Immunohistochemical staining. Normal muscle cells structure | Occasional horizontal nystagmus. Retinal atrophy with abnormal VEPs and ERG responses | |||
Data were collected regarding medical history, metabolic measurements, imaging, electrophysiological studies and muscle biopsy. Complete physical, neurological, and developmental examinations were performed on seven patients. The disease phenotype in all patients was similarly severe.
AO = age at onset; EM = electron microscopy; ERG = electroretinogram; FTT = failure to thrive; GMFCS = Gross Motor Functional Classification System; m = months; NA = not available; PAS = periodic acid-Schiff; SSEP = somatosensory evoked potentials; VEP = visual evoked potential; y = years.
For all of patients: normal karyotype, level of cholesterol, muscular and lysosomal enzymes.
Until the age of ∼5 years followed by an unexplained gradual increase in head circumference up to 75%.
Head circumferences, normal at birth, decreased to >2 SD below the mean in the ensuing years. In two patients, we observed an unexplained gradual increase in head circumferences up to 75% after the age of ∼5 years. Weight and height, also normal at birth, fell to 3–4 SD below mean in five patients but returned to 50–75% in two of five patients. Progressive chest deformities (kyphosis/pectus carinatum) were observed in all patients. Additional phenotypic characteristics included contractures of large joints, hyperextensibility of small ones, rocker bottom feet, hypertrichosis, and hyperhidrosis of palms and feet [Fig. 1C(a–d)].
Brain MRI demonstrated radiological signs of periventricular and subcortical damage including delayed myelination and atrophy, which worsened with age as a result of enlargement of ventricular system. Thin corpus callosum was a prominent feature in all of the patients. In one case, periventricular lesions were observed (Fig. 1D). Muscle biopsy in Patient VI3 showed normal oxidative phosphorylation and increased aggregation of collagen.
Molecular analyses
Molecular studies
Linkage analysis, performed on 24 individuals from Family I, identified a 1.9 Mb region between markers D9S265 and rs1330920 with a maximal LOD score of 3.24 at D9S1121; the region contained 11 genes (Fig. 2A and B). Seven samples were sequenced (two affected, two obligate carriers, and three unaffected individuals from the same village) with an average coverage of 82 ± 2% (all coding regions were covered). Haplotype analysis (D9S259–D9S169) supported a common ancestral haplotype in Families A and B (Supplementary Table 1).
Figure 2.

Identification of the gene and protein associated with leukoencephalopathy in the studied patients. (A) Haplotypes for each family member were constructed for 11 microsatellite markers spanning the neurodegenerative interval. Markers analysed are given on the left, according to their physical order. Haplotypes are represented by bars, with the disease-associated haplotype shaded in grey. Reduction in the affected linked region to 1.9 Mb was due to healthy individuals VI6 and IV4 who bear fraction of the affected haplotype in a homozygous manner. (B) Physical location of genes and predicted transcripts in the chromosome 9 linked interval. Asterisk denotes genes not approved by the HUGO Gene Nomenclature Committee (HGNC). ‘Strand’ refers to transcription orientation. Bold names indicate gene analysed by direct sequencing. Physical location obtained from UCSC Human Genome Browser Gateway (hg19 assembly). (C) Analysis of the c.2254C > T mutation in exon 14 of PLAA. Sequence analysis is shown for an unaffected individual, an obligatory carrier, and an affected individual. (D) Sequence alignment of human PLAA to orthologues in the mutation area. The leucine at position 752 (boxed) in this protein is highly conserved throughout evolution. (E) Effect of L752F (Leu→Phe) substitution on PLAA structure. Shown is a ribbon diagram of the PUL domain of PLAA (PDB ID 3EBB) which adopts a banana like shaped Armadillo domain. The conserved residues of PLAA (Homo sapiens, Mus musculus, Rattus norvegicus, Xenopus laevis, and S. cerevisiae) are displayed in stick-representation and form the putative binding site of PLAA. Mutation of Leu752 shown in ball-representation disrupts the rigid leucine network that tightly holds together the Armadillo domain.
Next generation sequencing
A total of 4289 variants were identified, but only four of them affected protein sequences (Supplementary Table 2). Out of these four variants, only one was present in a homozygous state in the affected individuals and in a heterozygous state in the obligate carriers. The three others were identified in the samples of unaffected individuals. The missense variant, NM_001031689.2: c.2254C>T [PLAA gene with a mutation corresponding to the amino acid substitution in PLAA protein (p.Leu752Phe)]; was confirmed by Sanger sequencing (Fig. 2C) in all affected individuals and obligate carriers. All seven affected individuals were homozygous for this sequence variation; all their parents were heterozygous.
The above PLAA variant (p.Leu752Phe) was neither observed in the Exome Aggregation Consortium [60 706 unrelated individuals, Exome Aggregation Consortium (ExAC), Cambridge, MA, http://exac.broadinstitute.org/, accessed February 2016] nor in the NHLBI database [6500 unrelated individuals, Exome Variant Server, NHLBI GO Exome Sequencing Project, Seattle, WA (http://evs.gs.washington.edu/EVS/) accessed February 2016]. Population screening of 92 healthy village residents revealed three carriers of this sequence variation (prevalence 3.3%).
The leucine at position 752 in PLAA is highly conserved through Saccharomyces cerevisiae (Fig. 2D), with the exclusion of zebrafish (threonine) and Caenorhabditis elegans (valine). This amino acid substitution is predicted to be deleterious by SIFT (score: 0.04) (http://sift.jcvi.org/www/SIFT_aligned_seqs_submit.html) and probably damaging by PolyPhen-2 (score 0.983) (http://genetics.bwh.harvard.edu/pph2/index.shtml).
Structural effects of p.Leu752Phe substitution in PLAA
The structure of PLAA PUL (PLAP, Ufd3p, and Lub1p) domain, in which Leu752 resides, was recently determined with atomic resolution (Qiu et al., 2010). The PUL domain consists of 15 tightly packed α-helices forming a 6-mer Armadillo domain. This protein fold consists of tightly packed helices in a single rigid structure found in several proteins such as importin-α, β-catenins, and Hsp70 binding protein (Hatzfeld, 1999). The Armadillo domain of PLAA is held together mainly through conserved leucine residues that zip together adjacent α-helices. On an average, leucine is present every three to four residues, corresponding to one turn of the helical wheel. Such Armadillo repeats form banana-shaped domains that generate good binding surfaces, particularly on the inside curvature (Hatzfeld, 1999).
In PLAA, the putative binding site is also paved with a conserved residue (Sievers et al., 2011). Based on these data, the p.Leu752Phe mutation appears to disrupt the tightly packed leucine network of the PLAA PUL domain and to deform the banana-like binding surface (Fig. 2E).
Expression studies
PLAA mRNA expression
At the transcriptional level, fibroblasts from patients were capable of expressing full-length PLAA transcript similar to that of nPLAA fibroblasts (Fig. 3A). Furthermore, based on RT-qPCR, no difference in the levels of PLAA transcript was noted between nPLAA versus mPLAA fibroblasts (Fig. 3B). As amino acid changes can have unexpected effects on protein stability, we sought to confirm production of PLAA protein in fibroblasts with and without the defined mutation in the PLAA gene. Confocal microscopy was performed to localize PLAA in normal and patient fibroblasts. All patient fibroblasts tested showed some localization of PLAA in the nucleus and majority of PLAA in the cytoplasm, which were similar in levels found in the fibroblasts of healthy controls (Fig. 3C and D).
Figure 3.

mRNA levels for PLAA and confocal microscopy of fibroblasts for the presence of PLAA protein. (A) Presence of full-length transcript for PLAA from fibroblasts of affected patients and the control subject based on PCR. (B) RT-qPCR for the detection of PLAA transcript from fibroblasts of a patient versus the healthy control normalized to four house-keeping genes coding for human 18 S RNA, GAPDH, POLB, and L19 ribosomal protein. Arithmetic means ± SD from three biological replicates performed in triplicate are shown. (C) Fibroblasts (nPLAA or mPLAA) were counterstained with DAPI (blue) for the nucleus and with fluorophore conjugated phalloidin (red) for actin. Cells were fixed, subjected to immunofluorescence staining for PLAA (green), and observed by confocal microscopy. (D) Mean fluorescence intensity of regions of interest corresponding to the cytoplasm and nucleus of imaged cells (ImageJ processing software, NIH). Figure represents results from three sets of images and error bars represent SD.
Functional effects of p.Leu752Phe on PLAA
Previous studies showed that PLAA loss causes severe ubiquitin depletion, accumulation of misfolded proteins, and impaired cellular survival, in S. cerevisiae (Mullally et al., 2006; Qiu et al., 2010). However, these effects were neither shown in the growth of S. cerevisiae and its ΔDOA1 (an orthologue of human PLAA in yeast) mutant, nor on ubiquitin depletion in fibroblasts from healthy controls versus patients (Supplementary material). Consequently, we examined other known functions of the PLAA protein. PLAA induces PGE2 production by increasing levels of PLA2 and cyclooxygenase (COX)-2 proteins, two major regulators of prostaglandins (Calignano et al., 1991; Zhang et al., 2008).
Investigating this function, we measured PGE2 levels in our patients’ fibroblasts. Healthy, unstimulated cells expressing nPLAA exhibited ∼2-fold higher levels of PGE2 compared to patients’ fibroblasts (Fig. 4A); this difference became much more prominent after LPS and CT treatment of the cultured nPLAA cells (∼5000-fold and ∼1000-fold, respectively). LPS treatment induced cPLA2 activity in normal fibroblasts, but did not elicit a similar response in patients’ cells (Fig. 4B). These results suggested that p.Leu752Phe mutation in PLAA abrogated its ability to induce prostaglandin biogenesis and properly responded to related stresses. Finally, transfection with a plasmid expressing nPLAA rescued PGE2 levels and cPLA2 activity in both untreated and LPS-stimulated patients’ fibroblasts (Fig. 4C and D).
Figure 4.

PGE2 levels and cPLA2 activity are low in patients' fibroblasts, and could be rescued. (A) Levels of PGE2 in cell culture media after 24 h of stimulation with LPS or cholera toxin (CT). Levels of PGE2 were normalized against protein concentrations in the supernatants. All cells were primary human fibroblasts except RAW 264.7 cells, which are murine macrophage like cells and used as a positive control. (B) Activity of cPLA2 in the membrane fractions of fibroblasts and RAW 264.7 macrophages. Cells were stimulated with or without LPS for 24 h before harvesting and purification of the membrane fractions. The cPLA2 activity was normalized to amount of proteins added to the assay. (C) PGE2 levels in the cell culture media after transfection with CMV promoter-based pIRES2-DsRed2 plasmid containing the native PLAA gene and a fluorescent marker of transfection. Cells were treated as follows: ctl = no transfection; V = transfection with empty vector; PLAA = transfection with plasmid vector containing the wild-type or native PLAA. (D) cPLA2 activity from membrane fractions of fibroblasts after transfection with a plasmid containing the nPLAA in a CMV promoter-based vector system and a fluorescent marker of transfection. (E–G) Fold-changes in transcripts for IL6, IL8, and MIF based on RT-qPCR. Arithmetic means ± SD from three independent experiments performed in triplicate are plotted and the data analysed using one-way ANOVA with Tukey post hoc correction.
We previously observed that PLAA regulated NF-κB-mediated inflammatory responses, and in particular inducible interleukin (IL)-6 (Zhang et al., 2008). Herein, we observed that p.Leu752Phe variation in PLAA abrogated expression of LPS induced IL-6, IL-8, and macrophage migration inhibitory factor (MIF)-encoding genes in patient fibroblasts when compared to fibroblasts from a representative healthy control based on RT-qPCR (Fig. 4E–G).
Cell biology studies
NF-кB recruitment to the nucleus is unaffected by the p.Leu752Phe mutation in PLAA
We investigated the NF-кB signalling pathway which is known to be regulated by PLAA (Zhang et al., 2008) and showed that it was intact in both fibroblasts from patients and healthy controls (Supplementary Fig. 1A and B). For detailed methods, results, and the related figure, see the Supplementary material.
β-catenin Wnt signalling is not affected by the p.Leu752Phe mutation in PLAA
We investigated Wnt signalling by examining levels of non-phospho (active) β-catenin in nPLAA versus mPLAA fibroblasts with and without LPS stimulation. As shown in Supplementary Fig. 1C, the levels of active β-catenin were increased after LPS stimulation to a similar extent in both types of fibroblasts. Detailed results are summarized in the Supplementary material.
Mouse model
Inactivation of the Plaa gene results in perinatal lethality in mice
We generated Plaa-null mice using gene targeting technology (Supplementary Fig. 2). While heterozygous (Plaa+/−) mutants were viable and fertile, the homozygous (Plaa−/−) mutants exhibited perinatal lethality. Initially, we genotyped 66 pups derived from heterozygous intercrosses, typically on postnatal Day 4–9. Twenty-eight pups were wild-type, 38 were Plaa+/−, and there was no Plaa−/− mutants. To determine when the Plaa-null mice died, we set up timed heterozygous intercross mating and examined embryos at different time points (Supplementary Table 3). At embryonic Day 14.5, we recovered live, overtly normal Plaa−/− embryos, which were indistinguishable from Plaa+/− or wild-type littermates. At embryonic Day 18.5, we found mostly live (with a beating heart) but some dead Plaa−/− embryos. Plaa−/− embryos were grossly normal but smaller than Plaa+/− or wild-type littermates. The average body weights of live Plaa−/−, Plaa+/−, and wild-type embryos were 0.83 ± 0.11 g (n = 14), 1.03 ± 0.17 g (n = 32), and 1.34 ± 0.14 (n = 9) at embryonic Day 18.5, respectively. While differences in weights between wild-type and Plaa+/− were not statistically significant, weight differences between wild-type and Plaa−/− (P < 0.0001), and Plaa−/− and Plaa+/− (P < 0.001) were significant by one-way ANOVA with Tukey post hoc correction.
Gross examination revealed that all of the near-term Plaa−/− embryos had abnormal or underdeveloped spleens, which were transparent/pale and smaller. Interestingly, we also found one embryo with exencephaly/microcephaly among a total of 41 Plaa−/− embryos examined. We attempted to resuscitate some of the embryonic Day 18.5 embryos, but Plaa−/− embryos could not be resuscitated. While Plaa+/− and wild-type embryos reacted to the pinch and started gasping for air, Plaa−/− embryos did not make any voluntary or involuntary movement. Subsequently, we also found five Plaa−/− neonates that were naturally delivered, but they were all found dead. Notably, one of the dead Plaa−/− neonates had posterior truncation. The dead Plaa−/− neonates appeared to have no air in their lungs. To date, we have not found any live Plaa−/− neonate. These observations suggested that Plaa-null mice likely died shortly before or after birth.
A tissue-specific prostaglandin E2 reduction and perinatal lethality of Plaa-null mice
To validate the results observed for the human fibroblasts carrying mPLAA, we evaluated the levels of PGE2 in wild-type, Plaa+/−, and Plaa−/− embryos. We isolated lungs, brain, liver, and heart tissues from embryonic Day 18.5 embryos, and determined PGE2 levels for each organ individually (Fig. 5A–D). In the brain, there was a gene copy-dependent reduction of PGE2 with significant reduction in Plaa+/− embryos compared to wild-type (P < 0.001) as well as a significant decreased level of PGE2 in Plaa−/− compared to Plaa+/− (P < 0.05) embryos (Fig. 5B). PGE2 levels were significantly decreased in Plaa−/− lungs, and Plaa+/− and Plaa−/− hearts, but not in the liver (Fig. 5A, C and D). It is unclear why gene copy-dependent reduction of PGE2 was noted in some organs but not in others.
Figure 5.

PGE2 levels in embryonic mouse tissues. Wild-type, Plaa+/−, and Plaa−/− embryos were sacrificed at embryonic Day 18.5 and organs were isolated and prostaglandin levels determined for the lung (A), brain (B), liver (C), and heart (D). Data represented arithmetic means ± SD from tissues representing three wild-type, three Plaa+/−, and four Plaa−/− embryos and obtained from three independent littermates. Significance was determined by one-way ANOVA with Tukey post hoc correction. *P < 0.05 ***P < 0.001.
Histopathological analysis of skin, lungs, and brain cerebral cortex of Plaa-null mice
As shown in Fig. 6A, lungs from wild-type mice embryos exhibited the presence of organized alveolar spaces and thin alveolar walls. However, embryos from Plaa+/− and Plaa−/− mice showed progressively unorganized alveolar spaces and thickening of the alveolar walls, suggesting underdeveloped or immature lungs. Prostaglandins (PGE2) play an important role in the synthesis of lung surfactant, which is crucial in maintaining structural integrity of the alveoli needed for efficient gas exchange during respiration (Akella and Deshpande, 2013). Therefore, reduced PGE2 levels in the lungs of Plaa−/− mouse embryos (Fig. 5A) together with thickened alveolar walls (Fig. 6A) could be partly responsible for the premature deaths of Plaa−/− neonates (Akella and Deshpande, 2013).
Figure 6.

Histopathology of embryonic mouse tissues (5 µm) at embryonic Day 18.5. Lungs (A), brain cerebral cortex (B), and skin (C) were haematoxylin and eosin stained and analysed in a blinded fashion. Tissues representing two wild-type, two Plaa+/−, and four Plaa−/− embryos were analysed. Multiple fields for each tissue were visualized and typical representations are shown with scale bars of 100 µm (magnification ×100; top rows in A–C) and 50 µm (maginification ×200; bottom rows in A and B). Arrows in the haematoxylin and eosin stained slides indicate examples of mature neurons, while the arrowheads indicate dark round cells as examples of immature neurons or oligodendroglia. Plaa−/− mouse embryos showed an increasing number of less matured and undifferentaited neurons.
In the brain cerebral cortex of wild-type embryos, the neurons showed large nuclei and were fully matured, with no indication of degeneration. No signs of apoptotic bodies were noted. There were a few round dark cells that represented either oligodendroglia or granular immature neurons (Fig. 6B). The Plaa+/− embryos had smaller neuronal nuclei and about the same density of the round dark cells. In contrast, Plaa−/− embryos had a vast area of neurons with smaller dark-stained round nuclei that could be described generally as ‘more granular’ in type, an indication of less maturity and differentiation. This was best represented at a lower magnification (100×) which covered a larger tissue section (Fig. 6B). No significant differences were observed in the skin of wild-type versus mutant mouse embryos (Fig. 6C). Typical tissue sections representing multiple fields and from two to four embryos are shown.
Figure 7A–C showed higher magnification (400×) of the brain cerebral cortex of wild-type, Plaa+/−, and Plaa−/− embryos (haematoxylin and eosin stained), respectively, with corresponding myelin staining (Luxol® fast blue) of the sections at the same magnification. The presence of mature neurons and round dark cells, possibly representing oligodendroglia or immature neurons, were seen in the embryos of all mice irrespective of the genotype (haematoxylin and eosin stained) (Fig. 7A–C). However, the neurons appeared less matured and differentiated in the Plaa−/− embryos (Figs 6B and 7C). The role of PGE2 in bradykinin-induced neuroprotection has been reported in microglial and neuronal cells (Hadley et al., 2016). The reduced production of PGE2 in the Plaa−/− embryos brain (Fig. 5B) could thus influence neuronal development and protection (Fig. 6B) and needs further investigation.
Figure 7.
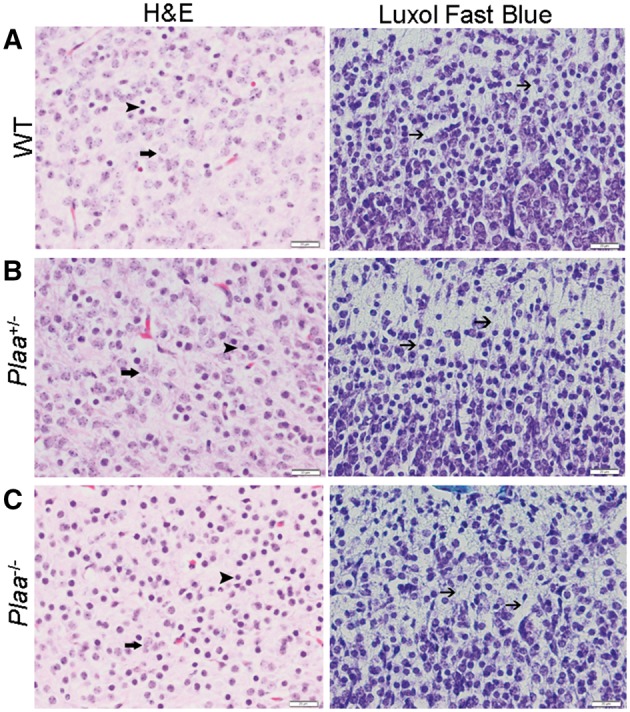
Histopathology and myelin staining of brain cerebral cortex. Sections of the brain cerebral cortex stained either with haematoxylin and eosin (5 µm section) or Luxol® fast blue (10 µm section) for myelin staining. (A) Wild-type mouse embryo; (B) Plaa+/− mouse embryo; and (C) Plaa−/− mouse embryo. Multiple fields for each tissue were visualized and typical representations are shown with a scale bar of 20 µm (magnification ×400). Arrows in the haematoxylin and eosin stained slides indicated examples of mature neurons, while the arrowheads pointed towards dark round cells as examples of immature neurons or oligodendroglia. Plaa−/− mouse embryos showed an increasing number of less matured and undifferentaited neurons. Arrows in all of the Luxol® fast blue stained slides indicated nerve processes with possible minimal early myelin (blue or turquoise colour).
The presence of nerve processes with light blue staining, possibly representing early myelin albeit minimal, was noted in the cerebral cortex sections of all the mouse embryos irrespective of the genotype in the myelin (Luxol® fast blue)-stained sections (Fig. 7A–C).
Discussion
PLAA is a regulatory molecule implicated in modulating production of host cell phospholipases (e.g. PLA2) (Clark et al., 1991; Ribardo et al., 2002). Induction of PLA2 is highly regulated by mitogen-activated protein kinases and NF-κB (Zhang et al., 2008). PLA2 hydrolyses membrane phospholipids to produce arachidonic acid, which is used as a substrate to produce prostaglandins and leukotrienes (eicosanoids) through cyclooxygenase and lipoxygenase pathways, respectively (Ribardo et al., 2002).
In this report, we present seven patients from two families with severe, unique, and progressive leukoencephalopathy. Based on the clinical and radiological findings, these patients could be categorized into the group of primary delay in myelin formation and disturbed myelination (van der Knaap, 2001). Brain biopsies were not performed and histopathological data were not available to confirm or rule out our clinical impression.
All patients were homozygous for a founder sequence variant (p.Leu752Phe) in PLAA that did not lead to the production of unstable transcript or protein.
The PLAA protein is composed of three major domains: N-terminal, multi-protein complex assembly domain contains 7 WD (tryptophan-aspartic acid) 40 repeats; central PFU (PLAA family ubiquitin binding) domain includes a ubiquitin binding region and an SH3 (SRC Homology 3) region; and the C-terminal PUL domain consists of 6 Armadillo repeats and binds to valosin-containing protein, also known as Cdc48 and p97 (Qiu et al., 2010). Our results suggested that the p.Leu752Phe sequence variant of PLAA disrupted the protein’s Armadillo domain, possibly impairing cells’ ability to induce prostaglandin production through a non-NF-кB signalling pathway.
Armadillo folds such as those found in PLAA, importin-α, and β-catenins, play a role in CNS development in Drosophila. Specifically, disruption of cell–cell adhesion function of Armadillo results in construction defects of the axonal scaffold (Loureiro and Peifer, 1998). Interestingly, a recent study suggested that PLA2α regulates the Wnt/β-catenin pathway (Han et al., 2008), which is implicated in neurogenesis, CNS morphogenesis, hirsutism, sweat gland morphology, short tendons, and kyphosis (Toribio et al., 2010; Haara et al., 2011; Joksimovic and Awatramani, 2014; Zhang et al., 2014). In this pathway, β-catenins transduce Wnt signals during embryonic development. Therefore, we hypothesized that PLAA, which activates PLA2, indirectly regulates the Wnt/β-catenin pathway, accounting for the pathology seen in our patients. However, our data indicated that the p.Leu752Phe mutation in PLAA found in our patients did not alter Wnt signalling.
Experimentally, activation of PLAA was recently shown to occur via 1α,25(OH)2D3 binding to a specific membrane-associated receptor, PDIA3, in caveolae, regulating growth zone chondrocytes (Doroudi et al., 2014). These findings might explain non-neurological features of progressive chest deformities (kyphosis/pectus carinatum) present in affected individuals.
Complex phospholipid defects involving CNS have received much attention of late (Lamari et al., 2013), providing insights into late-onset neurodegenerative disease pathophysiology, such as gene PLA2G6 encoding PLA2, underlying autosomal-recessive infantile neuroaxonal dystrophy, neurodegeneration associated with brain iron accumulation, and early-onset dystonia/parkinsonism (Khateeb et al., 2006; Gregory et al., 2008).
Furthermore, PGE2 plays a dual role, both neurotoxic and neuroprotective, in the brain and nervous system, a role modulated by its four receptors (Milatovic et al., 2011). Different binding affinities, varying cellular expression profiles, and attenuation of secondary messengers of these receptors lead to intricate, and sometimes opposing, signal transduction. While in Alzheimer’s disease and amyotrophic lateral sclerosis, it plays a neurotoxic role (Bazan et al., 2002), in excitotoxicity and cerebral ischaemia scenarios, PGE2 is neuroprotective (Gregory et al., 2008). Thus, modulation of PGE2 appears critical for neurological function. When PGE2 levels are reduced by deficiency of synthetic enzymes PLA2 (Gregory et al., 2008), COX-1 and COX-2 (FitzGerald, 2003) or PGE2 receptor (EP1-4), neurological impairment might occur.
Mohri et al. (2006) described prostaglandins as neuroinflammatory molecules that heighten pathological response to demyelination in twitchier mice. Similarly, the arachidonic acid pathway was shown to be modulated during cuprizone neurotoxin induced-demyelination and remyelination processes (Palumbo et al., 2011). Altogether, these data supported a causative relationship between abnormal PLAA activity and the severe leukoencephalopathy seen in our patients.
Of special interest is the prominent feature seen in six patients of exaggerated startle response, previously linked to dysmyelination/hypomyelination disorders, such as multiple sclerosis. In 1978, Mertin and Stackpoole showed that treatment with essential fatty acids, including arachidonic acid, suppressed experimental autoimmune encephalomyelitis in rats, and was abolished by inhibition of prostaglandin biosynthesis (Mertin and Stackpoole, 1978). Additionally, decreased inhibition of startle generator structure was reported to be associated with multiple sclerosis (Ruprecht et al., 2002). The calcium-independent PLA2 inhibitor was also linked to reduced prepulse inhibition of acoustic startle reflex in other studies (Lee et al., 2009).
Multiple studies link PGE2 and arachidonic acid pathways to neurodegenerative disorders, but their exact roles in causing white matter disorders remain unclear. We hypothesize that the prominent startle reflex dysinhibition in our patients may be related to brainstem lesions as part of the diffused axonal and myelin damage.
An additional interesting observation was that p.Leu752Phe substitution in PLAA resulted in an inability of the patient fibroblasts to induce IL-6, IL-8, and MIF in response to the NF-κB activating molecule LPS. While the pathophysiological relevance of this observation is yet to be determined, an association between neurodegenerative disorders and inflammatory cytokine responses has been suggested recently (Schmitz et al., 2015). These cytokines are known for their pleiotropic function and are implicated in activation of microglia, proliferation, migration, and homing of different immune and non-immune cells. In addition, PGE2 has been shown to be important leading to increased production of IL-6 and IL-8 (Cho et al., 2014). Thus, taken together, our observation may suggest that the pathogenesis of the leukoencephalopathy linked to the p.Leu752Phe substitution in PLAA implicates inability to mount appropriate inflammatory responses during pre-/post-neonatal development.
The phenotype of our patients, homozygous for p.Leu752Phe in PLAA and with reduced cPLA2 activity and PGE2 levels, adds new insights into this axis and its role in leukoencephalopathic disorders’ pathogenesis.
Knockout mouse data provide a clue into potential disease mechanisms seen in the patients described in this study. The disturbance in prostaglandin signalling results in a variety of pathological conditions. PLAA-deficient mice showed some phenotypes, specifically perinatal death, immature lungs with reduced PGE2, and reduced body weights similar to that of Ptgs1 (Cox-1)–Ptgs2 (Cox-2) double knockouts (Yu et al., 2006) and Ptgs3 knockouts (Nakatani et al., 2007). The double mutants, as well as Ptgs4 knockouts (Nguyen et al., 1997) died perinatally due to patent ductus arteriosus, Ptgs3 knockouts showed perinatal death, possessed immature lungs, and PGE2 levels were markedly decreased in the organ. The mutants also exhibited decreased body weight and skin morphological and physiological defects. Additionally, prostaglandin signalling is implicated for its roles in a range of physiological processes such as cell fate decision (Nissim et al., 2014), cell differentiation (Li et al., 2000), and ciliogenesis (Jin et al., 2014). The inability of Plaa-null mice to survive, which may stem from impaired neuronal development in the brain, together with our findings that PGE2 levels were significantly reduced in the brain and the lung of Plaa-null mouse embryos, raise the possibility that the pathogenesis of the condition we observed in PLAA-deficient mice and in patients with a non-functional PLAA could be a developmental defect caused at least partly by ineffective prostaglandin signalling.
In addition, with myelin staining of the brain, it was difficult to discern any distinguishing phenotype associated with the wild-type versus Plaa+/− and Plaa−/− embryos (i.e. myelination versus demyelination) These observations are in accordance with previous work that showed myelination occurring in rodents predominantly after birth (Larsen et al., 2006). Studies by Foran and Peterson (1992) also reported that β-galactosidase expressed under the promoter of myelin basic protein gene was first expressed by the oligodendrocytes in the ventral spinal cord only 1 day prior to birth in mice (Foran and Peterson, 1992). Recent studies of Trimarco et al. (2014) indicated that prostaglandin D2 is important in myelination of neuronal, glial, and oligodendrocytes in the CNS and Schwann cells in the peripheral nervous system (PNS). Thus whether PGE2 also plays a similar role in the myelination of CNS and PNS cell types requires further investigation.
Next, we plan to perform histopathological analysis on the brain from homozygous (Plaa−/−), heterozygous (Plaa+/−), and wild-type (Plaa+/+) neonates cut in a sagittal plane across the midline and use one side for histopathology and the contralateral side for optical clearing (OC)/imaging. Such studies will be crucial in further refining the mouse model to study the role of PLAA in this new form of leukoencephalopathy and to better glean the morphology of oligodendrocyte progenitor cells.
Furthermore, the Plaa-null mouse model provides an important tool to study the role of PLAA in CNS development and maintenance. Creating Plaa conditional knockout mice or Plaa knock-in mice carrying the p.Leu752Phe sequence variant would enable us to perform more detailed histopathological studies of the brain and thus to learn what type of leukoencephalopathy is caused by the PLAA sequence variant described here or by PLAA deficiency. Such an animal model would further contribute to the understanding of the significant role of arachidonic acid and PGE2 pathway in the normal development and maintenance of the brain.
In conclusion, we have presented a cohort of patients with progressive microcephaly and leukoencephalopathy, providing the first documentation of a PLAA-related disease. Although the interplay between PLAA, the abnormal production of PGE2, and the resultant hypo-myelination has not been thoroughly delineated, our data clearly indicated an association of this axis with brain development. Supportive evidence from the literature and improved understanding of the new players in this pathway should lead to new therapeutic avenues for intervention in both rare autosomal-recessive disorders and late onset common diseases that involve reduced CNS white matter.
Supplementary Material
Acknowledgements
We thank the families who participated in this study and the physicians and nurses who helped in the care for these patients. Special thanks to Dr Sarah Amit who cared for these patients in Galilee Medical Center’s (GMC) Child Development Unit and referred them to our attention. Tragically, Dr Amit passed away during this study. We would like to thank Dr Raya Rod and Dr Assnat Blum of GMC’s Child Development Unit for performing neurological and clinical work-up on the patients, and Dr Tatiana Freidman who participated in clinical follow-up of patients.
We thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison.
Sequencing of the Molecular Inversion Probe was performed by the NIH Intramural Sequencing Center.
We thank Dr Benjamin B. Gelman, Department of Pathology, UTMB, for examining some of the brain slide pictures. We thank Dr Jian Sha, Department of Microbiology and Immunology, UTMB, for preparing some of the figures. We also thank Molecular Genomics Core, UTMB (specifically Dr Thomas Wood, Director), for his advice and facilities during characterization of the human PLAA.
We thank Ms Tobie Kuritsky for English editing and technical assistance in handling the submission process.
Funding
This study was funded by the Rappaport Institute for Research (to T.F.Z.) by the Rappaport Faculty of Medicine, Technion, Haifa, and by the ‘Izvonot’ foundation of the Israeli Ministry of Justice (to T.F.Z.), Jerusalem, Israel. This research was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland, USA. Funding made available through the endowments of Leon Bromberg Professorship for Excellence in Teaching and Robert E. Shope MD and John S. Dunn Distinguished Chair in Global Health to A.K.C. is greatly acknowledged.
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- LPS
lipopolysaccharide
- PGE
prostaglandin
- RT-qPCR
real time quantitative polymerase chain reaction
References
- Akella A, Deshpande SB. Pulmonary surfactants and their role in pathophysiology of lung disorders. Indian J Exp Biol 2013; 51: 5–22. [PubMed] [Google Scholar]
- Atrouni S, Daraze A, Tamraz J, Cassia A, Caillaud C, Megarbane A. Leukodystrophy associated with oligodontia in a large inbred family: fortuitous association or new entity? Am J Med Genet A 2003; 118A: 76–81. [DOI] [PubMed] [Google Scholar]
- Baugh EH, Lyskov S, Weitzner BD, Gray JJ. Real-time PyMOL visualization for Rosetta and PyRosetta. PLoS One 2011; 6: e21931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan NG, Colangelo V, Lukiw WJ. Prostaglandins and other lipid mediators in Alzheimer's disease. Prostaglandins Other Lipid Mediat 2002; 68–9: 197–210. [DOI] [PubMed] [Google Scholar]
- Bomont P, Cavalier L, Blondeau F, Ben Hamida C, Belal S, Tazir M, et al. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet 2000; 26: 370–4. [DOI] [PubMed] [Google Scholar]
- Calignano A, Piomelli D, Sacktor TC, Schwartz JH. A phospholipase A2-stimulating protein regulated by protein kinase C in Aplysia neurons. Brain Res Mol Brain Res 1991; 9: 347–51. [DOI] [PubMed] [Google Scholar]
- Cho JS, Han IH, Lee HR, Lee HM. Prostaglandin E2 Induces IL-6 and IL-8 Production by the EP receptors/Akt/NF-kappaB pathways in Nasal Polyp-Derived Fibroblasts. Allergy Asthma Immunol Res 2014; 6: 449–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MA, Ozgur LE, Conway TM, Dispoto J, Crooke ST, Bomalaski JS. Cloning of a phospholipase A2-activating protein. Proc Natl Acad Sci USA 1991; 88: 5418–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroudi M, Boyan BD, Schwartz Z. Rapid 1alpha,25(OH)(2)D (3) membrane-mediated activation of Ca(2)(+)/calmodulin-dependent protein kinase II in growth plate chondrocytes requires Pdia3, PLAA and caveolae. Connect Tissue Res 2014; 55 (Suppl 1): 125–8. [DOI] [PubMed] [Google Scholar]
- Feenstra I, Vissers LE, Orsel M, van Kessel AG, Brunner HG, Veltman JA, et al. Genotype-phenotype mapping of chromosome 18q deletions by high-resolution array CGH: an update of the phenotypic map. Am J Med Genet A 2007; 143A: 1858–67. [DOI] [PubMed] [Google Scholar]
- FitzGerald GA. COX-2 and beyond: approaches to prostaglandin inhibition in human disease. Nat Rev Drug Discov 2003; 2: 879–90. [DOI] [PubMed] [Google Scholar]
- Foran DR, Peterson AC. Myelin acquisition in the central nervous system of the mouse revealed by an MBP-Lac Z transgene. J Neurosci 1992; 12: 4890–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory A, Westaway SK, Holm IE, Kotzbauer PT, Hogarth P, Sonek S, et al. Neurodegeneration associated with genetic defects in phospholipase A(2). Neurology 2008; 71: 1402–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haara O, Fujimori S, Schmidt-Ullrich R, Hartmann C, Thesleff I, Mikkola ML. Ectodysplasin and Wnt pathways are required for salivary gland branching morphogenesis. Development 2011; 138: 2681–91. [DOI] [PubMed] [Google Scholar]
- Hadley KB, Ryan AS, Forsyth S, Gautier S, Salem N Jr. The essentiality of arachidonic acid in infant development. Nutrients 2016; 8: 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Lim K, Xu L, Li G, Wu T. Regulation of Wnt/beta-catenin pathway by cPLA2alpha and PPARdelta. J Cell Biochem 2008; 105: 534–45. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hatzfeld M. The armadillo family of structural proteins. Int Rev Cytol 1999; 186: 179–224. [DOI] [PubMed] [Google Scholar]
- Henneke M, Combes P, Diekmann S, Bertini E, Brockmann K, Burlina AP, et al. GJA12 mutations are a rare cause of Pelizaeus-Merzbacher-like disease. Neurology 2008; 70: 748–54. [DOI] [PubMed] [Google Scholar]
- Henneke M, Diekmann S, Ohlenbusch A, Kaiser J, Engelbrecht V, Kohlschutter A, et al. RNASET2-deficient cystic leukoencephalopathy resembles congenital cytomegalovirus brain infection. Nat Genet 2009; 41: 773–5. [DOI] [PubMed] [Google Scholar]
- Hrapchak BB, Sheehan DC. Theory and practice of histotechnology. St. Louis, MO: C. V. Mosby; 1980. [Google Scholar]
- Jin D, Ni TT, Sun J, Wan H, Amack JD, Yu G, et al. Prostaglandin signalling regulates ciliogenesis by modulating intraflagellar transport. Nat Cell Biol 2014; 16: 841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joksimovic M, Awatramani R. Wnt/beta-catenin signaling in midbrain dopaminergic neuron specification and neurogenesis. J Mol Cell Biol 2014; 6: 27–33. [DOI] [PubMed] [Google Scholar]
- Khateeb S, Flusser H, Ofir R, Shelef I, Narkis G, Vardi G, et al. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am J Hum Genet 2006; 79: 942–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluver H, Barrera E. A method for the combined staining of cells and fibers in the nervous system. J Neuropathol Exp Neurol 1953; 12: 400–3. [DOI] [PubMed] [Google Scholar]
- Lamari F, Mochel F, Sedel F, Saudubray JM. Disorders of phospholipids, sphingolipids and fatty acids biosynthesis: toward a new category of inherited metabolic diseases. J Inherit Metab Dis 2013; 36: 411–25. [DOI] [PubMed] [Google Scholar]
- Larsen PH, DaSilva AG, Conant K, Yong VW. Myelin formation during development of the CNS is delayed in matrix metalloproteinase-9 and -12 null mice. J Neurosci 2006; 26: 2207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LY, Farooqui AA, Dawe GS, Burgunder JM, Ong WY. Role of phospholipase A(2) in prepulse inhibition of the auditory startle reflex in rats. Neurosci Lett 2009; 453: 6–8. [DOI] [PubMed] [Google Scholar]
- Li X, Okada Y, Pilbeam CC, Lorenzo JA, Kennedy CR, Breyer RM, Raisz LG. Knockout of the murine prostaglandin EP2 receptor impairs osteoclastogenesis in vitro. Endocrinology 2000; 141: 2054–61. [DOI] [PubMed] [Google Scholar]
- Loureiro J, Peifer M. Roles of Armadillo, a Drosophila catenin, during central nervous system development. Curr Biol 1998; 8: 622–32. [DOI] [PubMed] [Google Scholar]
- Magen D, Georgopoulos C, Bross P, Ang D, Segev Y, Goldsher D, et al. Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am J Hum Genet 2008; 83: 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertin J, Stackpoole A. Suppression by essential fatty acids of experimental allergic encephalomyelitis is abolished by indomethacin. Prostaglandins Med 1978; 1: 283–91. [DOI] [PubMed] [Google Scholar]
- Milatovic D, Montine TJ, Aschner M. Prostanoid signaling: dual role for prostaglandin E2 in neurotoxicity. Neurotoxicology 2011; 32: 312–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohri I, Taniike M, Taniguchi H, Kanekiyo T, Aritake K, Inui T, et al. Prostaglandin D2-mediated microglia/astrocyte interaction enhances astrogliosis and demyelination in twitcher. J Neurosci 2006; 26: 4383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullally JE, Chernova T, Wilkinson KD. Doa1 is a Cdc48 adapter that possesses a novel ubiquitin binding domain. Mol Cell Biol 2006; 26: 822–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani Y, Hokonohara Y, Kakuta S, Sudo K, Iwakura Y, Kudo I. Knockout mice lacking cPGES/p23, a constitutively expressed PGE2 synthetic enzyme, are peri-natally lethal. Biochem Biophys Res Commun 2007; 362: 387–92. [DOI] [PubMed] [Google Scholar]
- Nguyen M, Camenisch T, Snouwaert JN, Hicks E, Coffman TM, Anderson PA, et al. The prostaglandin receptor EP4 triggers remodelling of the cardiovascular system at birth. Nature 1997; 390: 78–81. [DOI] [PubMed] [Google Scholar]
- Nissim S, Sherwood RI, Wucherpfennig J, Saunders D, Harris JM, Esain V, et al. Prostaglandin E2 regulates liver versus pancreas cell-fate decisions and endodermal outgrowth. Dev Cell 2014; 28: 423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palisano R, Rosenbaum P, Walter S, Russell D, Wood E, Galuppi B. Development and reliability of a system to classify gross motor function in children with cerebral palsy. Dev Med Child Neurol 1997; 39: 214–23. [DOI] [PubMed] [Google Scholar]
- Palumbo S, Toscano CD, Parente L, Weigert R, Bosetti F. Time-dependent changes in the brain arachidonic acid cascade during cuprizone-induced demyelination and remyelination. Prostaglandins Leukot Essent Fatty Acids 2011; 85: 29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch C, Hustert E, Pfeifer D, Sudbeck P, Kist R, Roe B, et al. The SOX10/Sox10 gene from human and mouse: sequence, expression, and transactivation by the encoded HMG domain transcription factor. Hum Genet 1998; 103: 115–23. [DOI] [PubMed] [Google Scholar]
- Qiu L, Pashkova N, Walker JR, Winistorfer S, Allali-Hassani A, Akutsu M, et al. Structure and function of the PLAA/Ufd3-p97/Cdc48 complex. J Biol Chem 2010; 285: 365–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribardo DA, Peterson JW, Chopra AK. Phospholipase A2-activating protein–an important regulatory molecule in modulating cyclooxygenase-2 and tumor necrosis factor production during inflammation. Indian J Exp Biol 2002; 40: 129–38. [PubMed] [Google Scholar]
- Ruprecht K, Warmuth-Metz M, Waespe W, Gold R. Symptomatic hyperekplexia in a patient with multiple sclerosis. Neurology 2002; 58: 503–4. [DOI] [PubMed] [Google Scholar]
- Schmitz M, Hermann P, Oikonomou P, Stoeck K, Ebert E, Poliakova T, et al. Cytokine profiles and the role of cellular prion protein in patients with vascular dementia and vascular encephalopathy. Neurobiol Aging 2015; 36: 2597–606. [DOI] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 2011; 7: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazir M, Nouioua S, Magy L, Huehne K, Assami S, Urtizberea A, et al. Phenotypic variability in giant axonal neuropathy. Neuromuscul Disord 2009; 19: 270–4. [DOI] [PubMed] [Google Scholar]
- Teer JK, Bonnycastle LL, Chines PS, Hansen NF, Aoyama N, Swift AJ, et al. Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res 2010; 20: 1420–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teer JK, Green ED, Mullikin JC, Biesecker LG. VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics 2012; 28: 599–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons M, Tsokos M, Asab MA, Seminara SB, Zirzow GC, Kaneski CR, et al. Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia. Neurology 2006; 67: 2066–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toribio RE, Brown HA, Novince CM, Marlow B, Hernon K, Lanigan LG, et al. The midregion, nuclear localization sequence, and C terminus of PTHrP regulate skeletal development, hematopoiesis, and survival in mice. FASEB J 2010; 24: 1947–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimarco A, Forese MG, Alfieri V, Lucente A, Brambilla P, Dina G, et al. Prostaglandin D2 synthase/GPR44: a signaling axis in PNS myelination. Nat Neurosci 2014; 17: 1682–92. [DOI] [PubMed] [Google Scholar]
- Uhlenberg B, Schuelke M, Ruschendorf F, Ruf N, Kaindl AM, Henneke M, et al. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet 2004; 75: 251–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Knaap MS, Barth PG, Stroink H, van Nieuwenhuizen O, Arts WF, Hoogenraad F, et al. Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann Neurol 1995; 37: 324–34. [DOI] [PubMed] [Google Scholar]
- van der Knaap MS. Magnetic resonance in childhood white-matter disorders. Dev Med Child Neurol 2001; 43: 705–12. [DOI] [PubMed] [Google Scholar]
- Weidenheim KM, Dickson DW, Rapin I. Neuropathology of Cockayne syndrome: evidence for impaired development, premature aging, and neurodegeneration. Mech Ageing Dev 2009; 130: 619–36. [DOI] [PubMed] [Google Scholar]
- Yu Y, Fan J, Chen XS, Wang D, Klein-Szanto AJ, Campbell RL, et al. Genetic model of selective COX2 inhibition reveals novel heterodimer signaling. Nat Med 2006; 12: 699–704. [DOI] [PubMed] [Google Scholar]
- Zhang F, Sha J, Wood TG, Galindo CL, Garner HR, Burkart MF, et al. Alteration in the activation state of new inflammation-associated targets by phospholipase A2-activating protein (PLAA). Cell Signal 2008; 20: 844–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Li J, Lea R, Vleminckx K, Amaya E. Fezf2 promotes neuronal differentiation through localised activation of Wnt/beta-catenin signalling during forebrain development. Development 2014; 141: 4794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivony-Elboum Y, Westbroek W, Kfir N, Savitzki D, Shoval Y, Bloom A, et al. A founder mutation in Vps37A causes autosomal recessive complex hereditary spastic paraparesis. J Med Genet 2012; 49: 462–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.