

Abstract
Hydrogels are formed from hydrophilic polymer chains surrounded by a water-rich environment. They have widespread applications in various fields such as biomedicine, soft electronics, sensors, and actuators. Conventional hydrogels usually possess limited mechanical strength and are prone to permanent breakage. Further, the lack of dynamic cues and structural complexity within the hydrogels has limited their functions. Recent developments include engineering hydrogels that possess improved physicochemical properties, ranging from designs of innovative chemistries and compositions to integration of dynamic modulation and sophisticated architectures. We review major advances in designing and engineering hydrogels and strategies targeting precise manipulation of their properties across multiple scales.
Engineering functional hydrogels with enhanced physicochemical properties
Advances have been made to improve the mechanical properties of hydrogels as well as to make them shear-thinning, self-healing, and responsive. In addition, technologies have been developed to manipulate the shape, structure, and architecture of hydrogels with enhanced control and spatial precision.
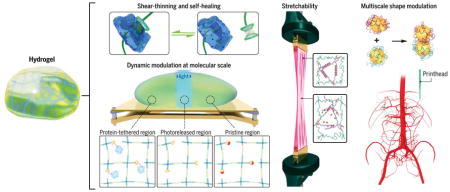
Hydrogels, a class of three-dimensional (3D) networks formed by hydrophilic polymer chains embedded in a water-rich environment, possess broadly tunable physical and chemical properties (1–3). A variety of naturally derived and synthetic polymers can be processed into hydrogels, from those formed through physical entanglement to ones stabilized via covalent cross-linking. Hydrogels may be further tuned toward the integration of chemically and biologically active recognition moieties such as stimuli-responsive molecules and growth factors that enhance their functionality. The versatility of the hydrogel system has endowed it with widespread applications in various fields, including biomedicine (1–3), soft electronics (4, 5), sensors (6–8), and actuators (9–14). As an example, when a hydrogel is created with proper stiffness and bioactive moieties, it modulates the behavior of the embedded cells (15, 16). In addition, chemically active moieties and light-guiding properties allow hydrogels to sense substances of interest and perform on-demand actuation (7, 17).
Advances in chemical methods—such as click chemistry, combination of gelation mechanisms, and doping with nanomaterials—have produced hydrogels with more controlled physicochemical properties. Furthermore, the static and uniform microenvironments characteristic of traditional hydrogel matrices do not necessarily replicate the hierarchical complexity of the biological tissues. Innovative fabrication strategies have been developed not only to achieve dynamic modulation of the hydrogels that can evolve their shapes along predefined paths, but also to control the spatial heterogeneity that will determine localized cellular behaviors, tissue integration, and device functions.
Hydrogel formation
Hydrogels are formed by cross-linking polymer chains dispersed in an aqueous medium through a myriad of mechanisms, including physical entanglement, ionic interactions, and chemical cross-linking (Fig. 1). Majority of the physical gelation methods depend on the intrinsic properties of the polymers. This dependence limits the ability to fine-tune the attributes of hydrogels, but gelation is easy to achieve without the need for modifying the polymer chains and is usually easy to reverse when necessary. Conversely, chemical approaches can be used to allow for more controllable, precise management of the cross-linking procedure, potentially in a spatially and dynamically defined manner.
Fig. 1. Cross-linking of hydrogels.

(A to D) Physical cross-linking. (A) Thermally induced entanglement of polymer chains. (B) Molecular self-assembly. (C) Ionic gelation. (D) Electrostatic interaction. (E) Chemical cross-linking.
Many natural polymers such asseaweed-derived polysaccharides and proteins from animal origin form thermally driven hydrogels. During the gelation process, physical entanglement of the polymer chains occurs in response to a temperature change. This change is typically caused by an alteration in their solubility and the formation of packed polymer backbones that are physically rigid (Fig. 1A) (18, 19). An increase or decrease in temperature may result in thermal gelation, in which the transition temperatures are defined as lower critical solution temperature (LCST) and upper critical solution temperature (UCST), respectively (20, 21). The gelation mechanism, however, may vary with specific types of polymers. Macromolecules exhibiting UCST include natural polymerssuch as gelatin as well as synthetic polymers such as poly-acrylic acid (PAA) that gel as the temperature drops to below their respective UCSTs. In contrast, some other macromolecules show LCST behavior, such as synthetic polymer poly(N-isopropylacrylamide) (PNIPAAM), that gels above its LCST. The LCST/UCST of the thermo-responsive polymers may be tuned by their molecular weight, ratio of the copolymers, and/or the balance of the hydrophobic/hydrophilic segments (22, 23).
Noncovalent molecular self-assembly is adopted as a common strategy, especially for protein-based hydrogels (24). Weak noncovalent bonding mechanisms—including hydrogen bonds, van der Waals forces, and hydrophobic interactions—cause macromolecules to fold into scaffolds possessing well-defined structures and functionality. A notable example is the hierarchical self-assembly of collagen, the most abundant protein in the human body (Fig. 1B). The assembly procedure relies on the regular arrangement of the amino acids in collagen molecules that are rich in proline or hydroxyproline (25). These molecules facilitate the formation of the triple helix termed tropocollagen. Subsequent stabilization upon further packing of the tropocollagen subunits into fibrils/fibers eventually forms a collagen hydrogel (24–26). Inspired by this mechanism, biomimetic supramolecular formulations have been designed that can follow similar hierarchical self-assembly processes, such as collagen-mimetic peptides (26) and those based on peptide-amphiphiles and hydrogelators (27, 28).
Spontaneous physical gelation may alternatively depend on chelation or electrostatic interactions. Alginate, a polysaccharide composed of α-L-guluronic acid (G) and β-D-mannuronic acid (M) residues derived mainly from brown algae, is a prominent example of hydrogel formation based on chelation (29). The G-blocks in alginate rapidly gel in presence of certain species of divalent cations such as Ca2+ or Ba2+ in an “egg-box” form, in which pairs of helical chains pack and surround ions that are locked in between (Fig. 1C) (30). The sophisticated structures of natural macromolecules often render them varying degrees of electrostatic charges along the backbone. Although many natural polymers are negatively charged at neutral pH because of the presence of carboxyl groups (such as hyaluronic acid and alginate), some may also present positive charges when amine groups dominate (such as gelatin and chitosan). In contrast, synthetic polyelectrolytes offer much better control over the electrostatic properties. A common example is the poly(L-lysine) (PLL)/PAA pair (31, 32). When the solutions containing polyelectrolytes of opposite charges are mixed, the polymer chains entangle to form complexes that become insoluble because of mutual shielding (Fig. 1D) (33).
Chemical cross-linking is better at stabilizing a hydrogel matrix because it allows substantially improved flexibility and spatiotemporal precision during the gelation process as compared with physical methods. Chemically active moieties pendant to the backbones or side chains of macromolecules in an aqueous solution can form covalent bonds under proper circumstances to obtain hydrogels (Fig. 1E). Conventional mechanisms include condensation reactions (for example, carbodiimide chemistry between hydroxyl groups/amines and carboxylic acids), radical polymerization, aldehyde complementation, high-energy irradiation, and enzyme-enabled biochemistry, among others (34). Over the past decade, click chemistry—proposed to be fast, orthogonal, high-yield, and typically amenable to cells and bioactive agents—has undergone tremendous development. This class of precise chemical reactions aims to address the challenges associated with traditional chemical reactions, such as low yield, prolonged reaction times, and extreme reaction conditions (35–37). A rich variety of bio-compatible click reactions have been devised for use in direct bioconjugation and hydrogel formation in the presence of cells within the matrices, such as thiol-vinyl sulfone and thiol-maleimide Michael addition reactions, azide-alkyne and azide-alkyne cycloaddition reactions, and thiolene photo-coupling reaction (37).
Although each of these gelation methods has its advantages and limitations, the field has been marching toward combining multiple components and/or mechanisms to achieve improved hydrogel formulations (38–42). These hydrogels typically exhibit excellent physicochemical properties, such as substantially enhanced mechanics, injectability, self-healing, and possibility to undergo dynamic modulation, as further discussed below.
Tuning the strength
Conventional hydrogels typically possess a low to intermediate stretchability within a few times their original length, and fracture energies of <100 J m−2. Applications such as load-bearing bio-materials (39, 43), soft robotics (44), and wearable devices (4–6) have spurred interest in developing tough and highly stretchable hydrogels as a class of soft and hydrated substrates.
One strategy lies in the use of hydrogels derived from natural proteins that may present exceptional elasticity (45). For example, elastin is a widely distributed structural protein required to maintain tissue integrity and confer elasticity and is present in tissues such as blood vessels, heart, bladder, and skin. To this end, recombinant tropoelastin, and its enzymatically cross-linked form termed elastin-like polypeptides (ELPs), have been engineered (46, 47). These engineered elastin-based hydrogels typically possess strong elasticity by using a pentapeptide repeat, VPGXG, where X is any amino acid except proline (46). They can achieve stretchability of up to ~400% their original lengths (47), which is much higher than most existing naturally derived hydrogels. Nevertheless, the high cost of these protein-based materials limits their use for applications requiring large hydrogel amounts.
Alternatively, hydrogels formed through hybridization with nanomaterials (39, 48), via crystallite cross-linking (49), or by mixing multiple components (38, 41, 50), may possess substantially improved mechanical properties (51). For all these mechanisms, the key lies in combination of carefully selected components that formulate the hydrogels. Nanomaterials (such as inorganic nanoparticles, carbon nanotubes, and graphene), because of their highly tunable surface properties and usually strong mechanics, contribute to enhanced mechanical performances of bulk hydrogels through interacting with polymer chains forming the hydrogels (39, 42, 48). The use of nanomaterials as cross-linkers would reduce the restrictions exerted by dense cross-links that are otherwise present in pure polymer networks. For example, by use of inorganic clay nanoplatelets as anchors for grafting the polymer chains upon cross-linking, the resulting PNIPAAM hydrogel network could be stretched to up to 1400% its original length (48). The stable grafting points of these clay-PNIPAAM networks was attributed to the tunable surface properties of the clay nano-platelets (48). The filler materials may provide additional functionality, such as enhancement of electrical conductivity (39), or promotion of tissue regeneration (52).
Multiple cross-linking mechanisms, often involving physical entanglement and chemical bonding of two types of polymer chains, have been combined to achieve hydrogels with superior toughness and high fracture energy (41, 53–56). A highly stretchable and tough hydrogel was synthesized by mixing alginate, ionically cross-linked by Ca2+, and covalently cross-linked long-chain polyacrylamide (PAAm). The two components, alginate and PAAm, were further intertwined through covalent bonding between their respective carboxyl and amine groups (Fig. 2A) (41). During the stretching process, the PAAm network remains intact while the alginate network unzips progressively to efficiently dissipate energy, which can reform upon removal of the stress, thus exhibiting excellent hysteresis and negligible gross deformation. As such, this class of hybrid hydrogels could achieve >20 times the elongation of their original lengths with fracture energies of up to 9000 J m−2. Alginate molecules of different chain lengths may further be coembedded in the alginate-PAAm system in order to synthesize hydrogels containing improved density of chelation, thus achieving extraordinarily high fracture energies of up to 16,000 J m−1 without compromising the toughness (38). Introduction of crystallization within an entangled polymer network represents another approach to achieve tough and stretchable hydrogels (49, 57). As an example, rather than the alginate component, polyvinyl alcohol (PVA) molecules could be incorporated, which upon annealing formed crystallites within the intertwined network of PVA and PAAm (49, 57). The dried network was then rehydrated to form the tough and stretchable hydrogel, in which the crystallites of PVA functioned as the zippable, energy-dissipating segments.
Fig. 2. Tuning the mechanics of hydrogels.

(A) Stretchable hydrogel made from long-chain polymers and reversible physically cross-linked polymers. (Right) The hydrogel could be stretched to 21 times its initial length, where the stretch λ is the final length of the unclamped region divided by the original length; stress-stretch curves of the alginate, PAAm, and alginate-PAAm hydrogels, each stretched to rupture, where the nominal stress s is defined as the force applied on the deformed hydrogel, divided by the cross-sectional area of the undeformed hydrogel. [Adapted with permission from (41), copyright 2012 Nature Publishing Group] (B) Stretchable hydrogel based on a sliding ring mechanism. (Right) Photo of an elongated NIPAAM-AAcNa-HPR-C hydrogel, and stress-strain curves of different hydrogels: (i) NIPAAM-AAcNa-BIS [0.65 weight % (wt %)], (ii) NIPAAM-AAcNa-BIS (0.065 wt %), (iii) NIPAAM-AAcNa-HPR-C (2.00 wt %), (iv) NIPAAM-AAcNa-HPR-C (1.21 wt %), and (v) NIPAAM-AAcNa-HPR-C (0.65 wt %). The percentages denote those of the cross-linkers. [Adapted with permission from (50), copyright 2014 Nature Publishing Group] (C) Tough bonding of hydrogels with smooth surfaces. (Right) The peeling process of a tough hydrogel chemically anchored on a glass substrate, and curves of the peeling force per width of hydrogel sheet versus displacement for various types of hydrogel-solid bonding. [Adapted with permission from (59), copyright 2016 Nature Publishing Group]
In another strategy, molecular sliding was devised to synthesize extremely stretchable hydrogels with good toughness (50). Hydrogels were derived from a network of structures composed of a copolymer, of N-isopropylacrylamide (NIPAAM) and sodium acrylic acid (AAcNa), and α-cyclodextrin conjugated with poly(ethylene glycol) (PEG) (Fig. 2B). The free movement and sliding of the α-cyclodextrin rings along the polymer chains, enabled by the inclusion of the ionic monomer AAcNa to modulate ionization, rendered the hydrogels highly stretchable to up to 400 to 800% their original lengths. The degree of stretching, however, was also dependent on the type of cross-linker used, either hydroxypropylated polyrotaxane cross-linker (HPR-C) or N,N′-methylenebisacrylamide (BIS).
Strong bonding between hydrogels and solid materials as diverse as metals, glass, ceramics, and silicone is required for a wide range of applications in areas spanning from biomedicine to soft electronics (5, 58). Nevertheless, rational design of a robust interaction between these two mechanically distinctive classes of materials has remained historically challenging. Following an approach used for making a tough composite hydrogel from intertwined polymer networks, the long-chain polymer molecules were further covalently attached to the surface of a solid through anchored chemical bonds (Fig. 2C) (40, 59). The resistance to scission of the anchored polymer chains during peeling generates the adhesion, and the energy is dissipated by the reversible physical chelation of the ionically cross-linked alginate. This type of bonding may reach interfacial toughness values of over 1000 J m−2, which is comparable with the toughest bonding found between a tendon and a bone in humans (800 J m−2). In addition to potential applications in biomedicine, optimization of hydrogel bonding with other types of materials, such as elastomers, may further expedite the development of the field of soft electronics. For example, stretchable soft electronics have been designed by coating an electrolyte-containing hydrogel layer onto a substrate of elastomeric tape without strong bonding between the two surfaces (4).
Break and heal
Hydrogels capable of regaining their initial mechanical properties and undergoing autonomous healing upon damage can be useful in various applications such as in biomedicine, external coatings, and flexible electronics. Such properties typically rely on reestablishing molecular interactions in the water-rich microenvironment after the hydrogel is subjected to external forces or damage. This ability is achieved by the incorporation of moieties that present reversible but typically strong physical interactions in the hydrogel’s polymer network. These mechanisms can range from electrostatic interactions (55, 60, 61) and hydrogen bonding (62, 63) to hydrophobic interactions (64, 65) and caged guest-host interactions (66, 67). The resulting hydrogels may further be stimuli-dependent that are modulated by, for example, pH (62) or oxidative state (66).
Besides functioning as chemical cross-linkers, clay nanoplatelets may also be physically mixed with polymer chains to form shear-thinning hydrogels. Synthetic clay nanoplatelets that have an anisotropic charge distribution, positive along the edges and negative on the two surfaces, may result in a net negative charge in an aqueous medium (60). Therefore, when complexed with positively charged supramolecules such as gelatin type A at neutral pH, a shear-thinning hydrogel is formed because of the strong electrostatic interactions between the clay nanoplatelets and the polymer chains (Fig. 3A). The resulting hydrogel temporarily reduces its viscosity upon application of shear stress to endow injectability through a plug flow process, while returning to its original viscosity at low shear owing to electrostatic interactions (68). The system is simple and may be extended to many charged species of nanoparticles and polymer chains to produce injectable formulations (61). These shear-thinning hydrogels have found utility in hemostatic dressing (60), endovascular embolization (68), tissue engineering (69), and drug and gene delivery (61).
Fig. 3. Shear-thinning and self-healing hydrogels.

(A) Shear-thinning hydrogel through nanocompositing. (Bottom) Recovery of the nanocomposites was observed by subjecting the hydrogel to alternating high and low strain conditions (100% strain and 1% strain) while monitoring the moduli of the composite. [Adapted with permission from (60), copyright 2014 American Chemical Society] (B to D) Self-healing hydrogels based on ionic interactions, hydrogen bonds, and host-guest coupling. (B) Ionic interactions. (Bottom left) Recovery of the sample for different waiting times. (Bottom right) Self-healing between either two freshly cut surfaces (red and blue) or a fresh and an aged surface (white) of samples; [Adapted with permission from (55), copyright 2013 Nature Publishing Group] (C) Hydrogen bonds. (Bottom) The healed hydrogels at low pH separate after exposure to a high-pH solution (with pH > 9), and the separated hydrogels could reheal upon exposure to acidic solution (pH < 3). [Adapted with permission from (62), copyright 2012 the National Academy of Sciences] (D) Host-guest coupling. (Bottom) The cut hydrogel spread with NaClO aqueous solution did not heal after 24 hours, but readhesion was observed 24 hours after spreading reuced glutathione aqueous solution onto the oxidized cut surface. [Adapted with permission from (66), copyright 2011 Nature Publishing Group]
Systems with electrostatic interactions between polymer chains containing oppositely charged side groups have also been developed (Fig. 3B) (55). Polyampholytes, synthesized through random copolymerization of oppositely charged monomers, were induced to spontaneously form physical hydrogels containing electrostatic bonds with a wide distribution of strengths. In this scenario, the strong ionic bonds serve as semipermanent cross-links to maintain the shape of the hydrogel, whereas the weak bonds dissipate energy through reversible bond breakages and formations, enabling improved toughness as well as shear-thinning properties. The physical cross-linking imparted by the electrostatic interactions, however, further enable these types of hydrogels to self-heal even upon complete damage between two freshly cut or aged surfaces (55).
Self-healing through hydrogen bonding can be attained by incorporating side groups such as amine or carboxyl into the polymer backbone of the hydrogel network (Fig. 3C) (62). It further allows a pH-dependent healing capacity of the damaged hydrogels. In the case of carboxyl pendant groups, at pH lower than the pKa values of the molecules (where Ka is the acid dissociation constant), the functional groups become protonated and can form hydrogen bonds, whereas in their deprotonated state at pH values above pKa, the charged groups exhibit strong electrostatic repulsion. A host-guest system forms another basis of generating self-healing hydrogels. The classical molecule cyclodextrin, when conjugated to the polymer backbone, functions as the host to engulf hydrophobic moieties dangling as side chains of another polymer backbone to achieve self-healing. If an environment-responsive moiety—such as PAAm-ferrocene (PAAm-Fc), which undergoes a reversible hydrophobic-charge transition at reduced/oxidized states—is included in a hydrogel, self-healing could further be selectively promoted upon introduction of favorable stimuli (Fig. 3D) (66). Whereas current strategies in synthesizing self-healing hydrogels heavily rely on physical and chemical methods, we envision the emergence of bioinspired approaches in which bioactive species may as well facilitate the healing process of the hydrogels upon damage. In nature, self-healing of biological tissues occurs frequently through an orchestrated cascade of cellular events, in which cells residing in the local microenvironment actively respond to the wound by selectively clearing the debris, secreting bioactive cues, and depositing new extracellular matrix (ECM) molecules to achieve healing. Therefore, rational combination of the physicochemical methods and biologically active species (such as enzymes, microorganisms, or even mammalian cells) may represent a new self-healing strategy.
Dynamic modulation
No single existing system, whether it is biological or synthetic, is purely static. Rather, dynamic evolutions are universally present, in which the capability to modulate hydrogel behavior in a temporally dependent manner becomes particularly attractive. When engineering biologically relevant systems, it is strongly desirable that the hydrogel micro-environment evolves with time through intrinsically embedded moieties that respond to external stimuli, either in an explicitly user-defined fashion or imposed by coexisting cells. Various approaches have been devised in the past decade toward incorporation of dynamic physiological signals within hydrogel platforms to precisely modulate cell-matrix interactions (1, 70). Dynamic modulation of hydrogels further opens up opportunities for construction of actuators and robotics (9–12).
Photopatterning is a long-established approach for introducing heterogeneity in a hydrogel volume by means of controlled spatial immobilization of bioactive molecules (71–73). The addition of specific biochemical cues into the matrix through photopatterning allows localized presentation of signals for cells to respond. These signals may include growth factors, proteins, hormones, cell-adhesion peptides, and cell-repelling moiety (73). Notably, the reverse process—on-demand release of certain moieties from a hydrogel matrix—is also sometimes desirable to trigger temporal changes within the hydrogel. In realizing this capability, photodegradation (as opposed to photocross linking), has been developed on the basis of the inclusion of photolabile moieties that undergo photolysis upon two-photon illumination (Fig. 4A) (74). These moieties can be designed to be biocompatible, enabling photodegradation of the hydrogels in the presence of bioactive molecules or cells, as well as time-dependent degradation. Photopatterning and photodegradation may be combined to achieve full control over the dynamics of the spatial patterns in a hydrogel system (36, 75). For example, a photo-reversible patterning strategy can be used to first define spatial distribution of biomolecules through photomediated ligation, followed by subsequent removal upon further exposure to a different light (Fig. 4B) (75). More recently, attention has been focused on dynamic modulation of biophysical cues inside the hydrogel such as matrix mechanics, which can be used to tune the behavior of encapsulated cells (76, 77).
Fig. 4. Dynamic modulation of hydrogel microenvironment.

(A) Photo-degradation through photolabile moieties. (Bottom left) Hydrogels demonstrated surface erosion upon irradiation. (Bottom right) Hydrogel was eroded spatially through masked flood irradiation, where feature dimensions were quantified with profilometry after different periods of irradiation. Scale bars, 100 μm. [Adapted with permission from (74), copyright 2009 American Association for the Advancement of Science] (B) Dynamic photopatterning and photorelease. (Bottom) Patterned primary antibodies are visualized with a fluorescent secondary antibody, which were subsequently photoreleased to form a secondary pattern. Scale bar, 3 mm. [Adapted with permission from (75), copyright 2015 Nature Publishing Group] (C) Cell-responsive cleavage and anticleavage negative feedback system. (Bottom) recombinant TIMP-3 (rTIMP-3) activity was measured by its ability to inhibit a recombinant MMP-2 (rMMP-2) solution, in which (left) bound rTIMP-3 did not substantially reduce rMMP-2. (Bottom) Hydrogels with (solid symbols) and without (open symbols) encapsulated rTIMP-3 were incubated with (squares) or without (triangles) rMMP-2, in which encapsulated rTIMP-3 attenuated rMMP-2-mediated hydrogel degradation. [Adapted with permission from (80), copyright 2014 Nature Publishing Group] (D) Thermo-responsive shape morphing hydrogel. (Bottom) Serial images of a closed gripper with poly(propylene fumarate) (PPF) segments on the outside opening as the temperature was decreased to below 36°C and then folding back on itself to become a closed gripper, but with the PPF segments on the inside. Scale bar, 2 mm. [Adapted with permission from (85), copyright 2015 American Chemical Society]
Cells may be another source of external stimuli to trigger a dynamic alteration of the hydrogel matrix in which they reside (78–80). In the human body, a critical characteristic of the ECM lies in its susceptibility to proteolysis triggered by the residing cells to facilitate their mobility and subsequent remodeling of the microenvironment. To achieve this functionality within a hydrogel, a specially designed linker moiety typically sensitive to an enzyme can be copolymerized into the hydrogel so as to allow for localized protease-mediated degradation of the polymeric network (79). In combination with a growth factor-sequestering component, these cell-instructive hydrogel matrices are able to reversibly immobilize growth factors on demand (81). The concept of protease-mediated degradation has been recently taken a step forward by implementing a negative feedback loop through a combinatory effect of an enzyme-labile moiety and an enzyme inhibitor coembedded in the hydrogel matrix (80). In thisscenario, an enzyme-sensitive peptide [for example, to matrix metalloproteinase (MMP)] is used as the linker of the hydrogel network, whereas the enzyme-inhibitor [for example, tissue inhibitor of MMPs (TIMP)] is simultaneously liberated by the same cleaving event. Concurrent inclusion of the enzyme-inhibitor pair induces balanced local activation and inhibition of the enzyme activity, therefore modulating the hydrogel degradation (Fig. 4C). Compared with conventional unconfined stimuli-responsiveness, the introduction of the feedback system clearly demonstrates its advantage in providing more precise manipulation of the matrix properties. This mechanism, based on cell-responsive MMPs, has also been adapted for other types of hydrogels [such as PEG-heparin (82)]. Alternatively, instead of introducing stimuli-responsive moieties, selective modification of the macromolecules before formation of hydrogels may allow for manipulation of hydrogel properties. For example, when heparin was selectively desulfated, the resulting hydrogels demonstrated altered release profiles of affinity-immobilized growth factors compared with the release in the case of pristine heparin (83). This strategy can be extended to chemical modifications so as to further enable dynamic control of biomolecule sequestration and release.
Morphogenesis is an important feature of living organisms. As an organism develops, it alters morphologies along a certain path, to produce the final shape and architecture working in a coordinated manner that renders the entire organism functional. Through meticulous selection of polymers that make up the hydrogels, they can also be processed to ensue bioinspired shape-morphing capability upon desired stimulation (12) such as humidity (10, 84), temperature (85), pH (86, 87), ionic strength (87, 88), magnetism (89, 90), or light (11), (among other stimulants. A heterogeneous or multilayer configuration containing materials with varying degree of responsiveness is typically required to enable a dynamic shape change. For example, a dual-layer hydrogel formed by two polymers with distinctive swelling capacities (for example, PEG and alginate) presents directional bending upon absorption of water (84). Another example is the combination of thermo-sensitive (such as PNI-PAAM) and thermo-inert hydrogel materials that modulate bidirectional bending of fabricated structure, at temperatures above or below which the thermo-sensitive hydrogel component is shaped (Fig. 4D) (85). Also, the inclusion of pH-sensitive moieties into a hydrogel leads to pH-mediated swelling or deswelling (86). Although these models rely on the intrinsic properties of the polymers that constitute hydrogels, functional materials such as magnetic nanoparticles (90) and carbon nanomaterials (11) may be added to the hydrogel matrices in order to achieve shape actuation upon external stimuli of magnetic field and light-induced local heating, respectively.
Cell-mediated traction force can also contribute to shape-morphing of hydrogels (91). Carefully designed microstructures seeded with cells are able to self-fold into predefined architectures through origami (92). Alternatively, cells that spontaneously contract enable mechanical movement of the substrate on which they reside, leading to generation of soft bioactuators (93, 94). Whereas earlier bioactuators were mainly based on elastomeric materials, hydrogel materials have recently attracted increasing attention because of their better biocompatibility. To this end, a combination of myocytes (skeletal muscle cells or cardiomyocytes) and hydrogels that enable communications between these cells is usually adopted. For example, carbon nanotube–composited gelatin methacryloyl hydrogels have been processed into flexible substrates as large as centimeter scales, and cardiomyocytes spontaneously and synchronously beating on the surfaces allow these bioactuators to exhibit rhythmic contraction or extension and swimming behaviors(95, 96). Engineered skeletal muscles, when bound to bioprinted hydrogel frameworks, could also actuate and move the devices in defined patterns (13, 14). These bioactuators relying on cell traction forces may be remotely controlled by a variety of external stimuli such as electrical signals (95–97) and light (14, 94). To be suited for building robust bioactuators, hydrogels must be both bio-compatible and mechanically stable.
Shaping the hydrogels
Biological tissues are exceedingly complex and may possess a hierarchically assembled architecture featuring a variety of nano- or microscale bioactive molecules in conjunction with multiple cell populations functioning in synergy. For example, almost all organs in the human body contain repeating building units that assemble into distinctive compartments and interfaces [for example, the myocardium, endocardium, and pericardium in the heart (98, 99) and the lobules constituting the liver (99, 100)]. Most organs are embedded with perfusable vasculature of sophisticated tortuosity that supplies oxygen and nutrients and transports bioactive molecules (such as growth factors and hormones). Such complexity further extends to the architecture of the blood vessels, themselves defined by layered structures of tunica intima, tunica media, and tunica externa (99). Consequently, engineering a biomimetic organ requires the capacity to reconstitute its native characteristics and physiology through rational design that ensures compositional, architectural, mechanical, and functional accuracies. Soft actuators and electronic devices made from hydrogels may similarly demand spatiotemporal control to render heterogeneity required for functionality.
Microengineering provides a versatile approach to engineer well-defined hydrogels from miniaturized building blocks through programmed spontaneous assembly. Multiple types of hydrogel units, individually fabricated to contain desired cell types and physicochemical cues, can be used to mimic the microtissue units in the human organs. Earlier versions of packing strategies rely on shape complementarity in which these different blocks can be processed by using, for example, lithographic or photopatterning techniques. These engineered building blocks of microgels with complementary geometrical configurations subsequently undergo directed assembly into the bulk form under a combinatory influence of external energy, surface tension, and microgel dimensions at liquid-liquid interfaces (Fig. 5A) (101). Assembled microgels may be further cross-linked to stabilize the bulk architecture. Using this strategy, arrays of ring-shaped microgels were assembled into a 3D tubular construct with interconnected lumens in a biphasic system via stimulation applied by fluid shear (102). Microgels could also be assembled into large-scale structures on a surface patterned with hydrophilic and hydrophobic regions at high fidelity (103). A class of hydrophobic hydrogels has been proposed by stabilizing a layer of hydrophobic microparticles on the surface of conventional hydrogels to allow them to float on aqueous media (104). These hydrophobic hydrogels with complementary shapes could self-assemble because of hydrophobic interactions directly on the surface of water when brought into proximity with each other. Although this type of self-assembly is convenient and scalable, it does not confer strong control over the assembly process because it is limited by the sole reliance on the miniaturization of interfacial surface free energy (101).
Fig. 5. Shaping macroscale hydrogels.

(A) Self-assembly through shape complementarity. (Bottom) (Top row) Hydrogels assembled by capillary force. Scale bar, 200 μm. (Bottom row) Hydrophobic hydrogels assembled through hydrophobic interactions on the surface of water. Scale bars, 5 cm. [Adapted with permission from (101), copyright 2008 the National Academy of Sciences, and (104), copyright 2016 American Chemical Society] (B) Self-assembly using sequence-complementing DNA glues. (Bottom) (Top row) DNA-assisted hydrogel self-assembly across multiple length scales. Scale bars, 1 mm. (Bottom row) Directed formation regular dimers and T-junction based on their surface DNA glue patterns. Scale bars, 1 mm. [Adapted with permission from (111), copyright 2013 Nature Publishing Group] (C) Embedded nozzle-based bioprinting. (Bottom left) A continuous hollow knot written with fluorescent microspheres in a granular hydrogel. Scale bar, 3 mm. (Bottom right) A freely floating hydrogel jellyfish model retrieved after printing and dissolution of the granular hydrogel. (Inset) The printed structure before the removal of the supporting hydrogel matrix. Scale bars, 5 and (inset) 10 mm. [Adapted with permission from (125), copyright 2015 American Association for the Advancement of Science] (D) 4D biomimetic printing through a printed dual-layer structure with unbalanced swelling. (Bottom) Time-lapse photographs showing bioprinted simple flower-like structures undergoing shape-morphing during the swelling process, for the double layers of different orientations. Scale bars, 5 mm. [Adapted with permission from (140), copyright 2016 Nature Publishing Group]
Alternatively, the self-assembly properties of nucleic acids provide a powerful approach for the fabrication of sophisticated patterns, structures, and devices. Sequence complementarity in DNA/RNA strands can be encoded to induce self-organization into a predefined structure, when the complementary segments pair up under proper physical conditions (105). On the basis of this principle, a plethora of molecular assemblies from synthetic nucleic acids have been generated such as tubes (106–108), ribbons (108), lattices (107, 109), and other complex shapes (106, 110). This concept has been adapted to assemble microgel units, similar to the abovementioned approach but in a much more precisely controlled manner. In this sense, the “giant DNA glue” is decorated onto the prescribed surfaces of a nonspherical hydrogel unit to produce asymmetric glue patterns on heterogeneous surfaces. These microscale building units, through combination of the molecular programmability of the DNA glue and the shape controllability of the microgels, can achieve multiplexed assembly of complex structures of hydrogels across multiple scales (Fig. 5B) (111). The assembly of diverse hydrogel structures—including dimers, linear chains, and large-scale networks—could potentially be used to build hierarchical tissue architectures and biomedical devices. Nucleic acid–directed self-assembly may further be adapted for programmed synthesis of 3D biological tissues (112). Instead of hydrogels, dissociated single cells may be chemically functionalized with oligonucleotides, which are complementary to DNA sequences coated on desired surfaces to allow for cellular assembly into the third dimension via a layer-by-layer approach. The oligonucleotides were designed to be degradable, thus achieving on-demand reversible binding once the micro-tissues formed (112).
Three-dimensional printing represents another facile technique for constructing volumetric objects (113, 114). Three-dimensional printing empowers extremely high precision through the robotic manipulation of the prepolymer (the ink), thus allowing for reproducible manufacturing of objects with minimal aberration in their shape, architecture, and functionality. The 3D printing technology was initially introduced in the form of stereolithography, in which defined objects are manufactured through a pull-out procedure from a liquid bath of prepolymer via layer-by-layer photopatterned cross-linking (115). Precise control of locally available oxygen concentrations during the stepwise radical polymerization further enables continuous manufacture of large-sized 3D shapes at substantially improved speed (116). Using such a manufacturing principle, hydrogel objects with complex 3D architecture and multiplexed materials may be generated with high spatial resolution (100, 117).
Complementary to stereolithography, nozzle-based 3D printing strategies typically provide higher degrees of flexibility in the production of hydrogel objects with defined architecture. The ink, deposited through positioned ejection from an orthogonally movable printhead, builds 3D structures in a layer-by-layer manner. Whereas stereolithography solely relies on photopolymerization, a rich variety of cross-linking mechanisms may be used for nozzle-based printing through meticulous selection of the inks combined with rational design of the printhead. For example, physical cross-linking of alginate by Ca2+ can be achieved with a core-sheath nozzle design to co-deliver the two components during the extrusion procedure, whereas photocrosslinking may still be applied to the methacryloyl-modified hydrogels after printing (118, 119). Alternatively, shear-thinning inks may be directly printed, followed by subsequent chemical cross-linking (120). Also, they may function as sacrificial templates in certain 3D printing applications so as to obtain hollow hydrogel structures after selective removal (121–123). Tough and highly stretchable hydrogels can also be printed into complex architecture (124). The capacity of nozzle-based 3D printing has been substantially boosted with recent development of the technique based on embedded printing (125–127). Conventionally, extruded hydrogel materials would not stand their own weights when complex structures containing large cavities, thin-walled tubes, and suspended elements were created. The ability to resist gravitational force has thus been proposed by the use of a supporting hydrogel bath, which is both shear-thinning, allowing for convenient deposition of the ink, and self-healing, ensuring shape-maintenance of the printed fine structures within the volume (Fig. 5C).
Often, the ability to combine multiple types of hydrogels is desired over a single material to fabricate compositionally complex patterns. Printers carrying several printheads have thus been developed to deposit selected hydrogel inks, or inks with polymer scaffolding in a prescribed manner (120, 128, 129). The emergence of microfluidic printheads capable of codelivering dual material types further enhanced the speed of multimaterial printing via improved switching between the different materials (118, 119, 130, 131). Instead, by using a microscale fluid mixing device inside the printhead, the two materials may be homogenized to achieve direct printing of gradient structures through precise control over the ratio between the inks infused into the printhead (132).
Outlook
Hydrogels represent an important class of materials possessing a watery environment and broadly tunable physicochemical properties. Efforts devoted to engineering hydrogels with enhanced properties in the past decade have expanded their opportunities in numerous applications, including biomedicine, soft electronics, sensors, and actuators. For instance, the mechanics of hydrogels have been engineered to become stiff and tough while maintaining a high water content. Self-healing mechanisms and dynamic modulation have been incorporated within hydrogel systems to achieve control over their behaviors over time. Advanced biofabrication techniques have further improved our capability to construct sophisticated hydrogel architectures that feature hierarchically assembled structures across multiple length scales.
However, several key challenges persist. Clinical translation of hydrogels requires rigorous testing (133), and their approval by the U.S. Food and Drug Administration for clinical applications has been limited to a few types of hydrogel materials (134). Maintaining or improving the mechanics of hydrogels in a wide range of media has remained another challenge. Swelling is a common phenomenon of hydrogels owing to water uptake caused by unequal osmotic pressure when immersed in an aqueous medium of lower osmolarity. Swelling often causes the weakening of the hydrogel networks upon long exposures in aqueous environments. In tackling the mechanical instability caused by swelling of the hydrogel network, an “anti-swelling” (or “nonswellable”) hydrogel consisting of a network integrating hydrophilic and thermo-responsive polymer units was proposed (135). It resisted swelling at elevated temperature when the thermo-responsive polymer chains condensed, thus preserving the mechanics of the pristine hydrogel. Although this strategy seems universal, continuation in the innovation of new chemistries and compositions to further optimize the processes becomes critical.
The potential to integrate hydrogel formulations with advanced biofabrication techniques is exciting as well, despite that strict optimization processes should be carried out to meet proper fabrication requirements. For example, when highly stretchable hydrogels are combined with bio-printing, elastic substrates of any arbitrary shape, pattern, and architecture may be obtained (124). As an alternative strategy to direct hydrogel printing, the mechanical properties of hydrogels can be reinforced by integrating them with printed micro-fibrous scaffolds (136). When encapsulation of cells is desired during the fabrication processes, the need for hydrogels with suitable mechanical properties and biocompatibility becomes critical (114). In addition, printing should be carried out under physiologically relevant conditions in order to ensure cell viability and phenotype maintenance (137).
Furthermore, printed hydrogel structures may be dynamically modulated. Through material designs, it is feasible to add an extra dimension—time—into the 3D architecture. In 4D printing (138–143), compositionally heterogeneous inks are deposited to form the initial layout, which is capable of shape transformation in a preprogrammed manner over time under stimuli. This technique allows for performance-driven functionality to be designed into the printed materials. For example, a hydrophilic polymer printed in between rigid, nonswelling segments swells after encountering water and expands into hydrogel structures, forcing the rigid materials to bend until they stop when hitting neighboring elements (138, 139). The folding pattern of printed structures can be accurately predicted through mathematical modeling, which serves as a guideline for the design of the printing path to achieve prescribed temporal transformation (140).
We envision that with increasing adoption of interdisciplinary approaches, it is possible to rationally combine multiple strategies, leading to creation of hydrogels that possess enhanced properties. The design of materials in which multiple constituents or phases are assembled across multiple length scales may harness synergistic effects and facilitate novel functionalities. The combinations of properties can emerge as a result of the particular arrangement or interactions between the multiscale constituents, providing opportunities for further innovations in hydrogels. Ultimately, the advancements in engineering hydrogels should be coupled with their end applications in a feedback loop to achieve optimal designs through iterative optimization cycles.
Acknowledgments
The authors acknowledge funding from the National Institutes of Health (grants AR057837, DE021468, D005865, AR068258, AR066193, EB022403, and EB021148) and the Office of Naval Research Presidential Early Career Award for Scientists and Engineers (PECASE). Y.S.Z. acknowledges the National Cancer Institute of the National Institutes of Health Pathway to Independence Award (K99CA201603). We greatly appreciate S. Masoud Moosavi-Basri for assistance with figure illustrations. We also thank S. Hassan, J. Seo, and W. Wang for their valuable discussions.
REFERENCES AND NOTES
- 1.Burdick JA, Murphy WL. Moving from static to dynamic complexity in hydrogel design. Nat Commun. 2012;3:1269. doi: 10.1038/ncomms2271. [DOI] [PubMed] [Google Scholar]
- 2.Seliktar D. Designing cell-compatible hydrogels for biomedical applications. Science. 2012;336:1124–1128. doi: 10.1126/science.1214804. [DOI] [PubMed] [Google Scholar]
- 3.Hoffman AS. Hydrogels for biomedical applications. Adv Drug Deliv Rev. 2012;64:18–23. doi: 10.1016/j.addr.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 4.Keplinger C, et al. Stretchable, transparent, ionic conductors. Science. 2013;341:984–987. doi: 10.1126/science.1240228. [DOI] [PubMed] [Google Scholar]
- 5.Lin S, et al. Stretchable hydrogel electronics and devices. Adv Mater. 2016;28:4497–4505. doi: 10.1002/adma.201504152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larson C, et al. Highly stretchable electroluminescent skin for optical signaling and tactile sensing. Science. 2016;351:1071–1074. doi: 10.1126/science.aac5082. [DOI] [PubMed] [Google Scholar]
- 7.Chan KWY, et al. MRI-detectable pH nanosensors incorporated into hydrogels for in vivo sensing of transplanted-cell viability. Nat Mater. 2013;12:268–275. doi: 10.1038/nmat3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heo YJ, Shibata H, Okitsu T, Kawanishi T, Takeuchi S. Long-term in vivo glucose monitoring using fluorescent hydrogel fibers. Proc Natl Acad Sci USA. 2011;108:13399–13403. doi: 10.1073/pnas.1104954108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J, Hanna JA, Byun M, Santangelo CD, Hayward RC. Designing responsive buckled surfaces by halftone gel lithography. Science. 2012;335:1201–1205. doi: 10.1126/science.1215309. [DOI] [PubMed] [Google Scholar]
- 10.Jeong K-U, et al. Three-dimensional actuators transformed from the programmed two-dimensional structures via bending, twisting and folding mechanisms. J Mater Chem. 2011;21:6824–6830. doi: 10.1039/c0jm03631e. [DOI] [Google Scholar]
- 11.Wang E, Desai MS, Lee S-W. Light-controlled graphene-elastin composite hydrogel actuators. Nano Lett. 2013;13:2826–2830. doi: 10.1021/nl401088b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ionov L. Hydrogel-based actuators: Possibilities and limitations. Mater Today. 2014;17:494–503. doi: 10.1016/j.mattod.2014.07.002. [DOI] [Google Scholar]
- 13.Cvetkovic C, et al. Three-dimensionally printed biological machines powered by skeletal muscle. Proc Natl Acad Sci USA. 2014;111:10125–10130. doi: 10.1073/pnas.1401577111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raman R, et al. Optogenetic skeletal muscle-powered adaptive biological machines. Proc Natl Acad Sci USA. 2016;113:3497–3502. doi: 10.1073/pnas.1516139113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 16.Huebsch N, et al. Harnessing traction-mediated manipulation of the cell/matrix interface to control stem-cell fate. Nat Mater. 2010;9:518–526. doi: 10.1038/nmat2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi M, et al. Light-guiding hydrogels for cell-based sensing and optogenetic synthesis in vivo. Nat Photonics. 2013;7:987–994. doi: 10.1038/nphoton.2013.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Djabourov M, Leblond J, Papon P. Gelation of aqueous gelatin solutions. I. Structural investigation. J Phys. 1988;49:319–332. doi: 10.1051/jphys:01988004902031900. [DOI] [Google Scholar]
- 19.Djabourov M, Leblond J, Papon P. Gelation of aqueous gelatin solutions. II. Rheology of the sol-gel transition. J Phys. 1988;49:333–343. doi: 10.1051/jphys:01988004902033300. [DOI] [Google Scholar]
- 20.Gasperini L, Mano JF, Reis RL. Natural polymers for the microencapsulation of cells. J R Soc Interface. 2014;11:20140817. doi: 10.1098/rsif.2014.0817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ward MA, Georgiou TK. Thermoresponsive polymers for biomedical applications. Polymers (Basel) 2011;3:1215–1242. doi: 10.3390/polym3031215. [DOI] [Google Scholar]
- 22.Lutz J-F, Akdemir O, Hoth A. Point by point comparison of two thermosensitive polymers exhibiting a similar LCST: Is the age of poly(NIPAM) over? J Am Chem Soc. 2006;128:13046–13047. doi: 10.1021/ja065324n. [DOI] [PubMed] [Google Scholar]
- 23.Laschewsky A, Rekaï ED, Wischerhoff E. Tailoring of stimuli-responsive water soluble acrylamide and methacrylamide polymers. Macromol Chem Phys. 2001;202:276–286. doi: 10.1002/1521-3935(20010101)202:2<276::AID-MACP276>3.0.CO;2-1. [DOI] [Google Scholar]
- 24.Zhang S. Fabrication of novel biomaterials through molecular self-assembly. Nat Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 25.Rich A, Crick FH. The structure of collagen. Nature. 1955;176:915–916. doi: 10.1038/176915a0. [DOI] [PubMed] [Google Scholar]
- 26.O’Leary LER, Fallas JA, Bakota EL, Kang MK, Hartgerink JD. Multi-hierarchical self-assembly of a collagen mimetic peptide from triple helix to nanofibre and hydrogel. Nat Chem. 2011;3:821–828. doi: 10.1038/nchem.1123. [DOI] [PubMed] [Google Scholar]
- 27.Hartgerink JD, Beniash E, Stupp SI. Self-assembly and mineralization of peptide-amphiphile nanofibers. Science. 2001;294:1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 28.Yang Z, Xu B. Supramolecular hydrogels based on biofunctional nanofibers of self-assembled small molecules. J Mater Chem. 2007;17:2385–2393. doi: 10.1039/b702493b. [DOI] [Google Scholar]
- 29.Rowley JA, Madlambayan G, Mooney DJ. Alginate hydrogels as synthetic extracellular matrix materials. Biomaterials. 1999;20:45–53. doi: 10.1016/S0142-9612(98)00107-0. [DOI] [PubMed] [Google Scholar]
- 30.Braccini I, Pérez S. Molecular basis of C2+-induced gelation in alginates and pectins: The egg-box model revisited. Biomacromolecules. 2001;2:1089–1096. doi: 10.1021/bm010008g. [DOI] [PubMed] [Google Scholar]
- 31.Morton SW, et al. Scalable manufacture of built-to-order nanomedicine: Spray-assisted layer-by-layer functionalization of PRINT nanoparticles. Adv Mater. 2013;25:4707–4713. doi: 10.1002/adma.201302025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leijten J, et al. Advancing Tissue Engineering: A Tale of Nano-, Micro-, and Macroscale Integration. Small. 2016;12:2130–2145. doi: 10.1002/smll.201501798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bekturov EA, Bimendina LA. Speciality Polymers. Springer; 1981. pp. 99–147. [Google Scholar]
- 34.Hennink WE, van Nostrum CF. Novel crosslinking methods to design hydrogels. Adv Drug Deliv Rev. 2002;54:13–36. doi: 10.1016/S0169-409X(01)00240-X. [DOI] [PubMed] [Google Scholar]
- 35.DeForest CA, Polizzotti BD, Anseth KS. Sequential click reactions for synthesizing and patterning three-dimensional cell microenvironments. Nat Mater. 2009;8:659–664. doi: 10.1038/nmat2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeForest CA, Anseth KS. Cytocompatible click-based hydrogels with dynamically tunable properties through orthogonal photoconjugation and photocleavage reactions. Nat Chem. 2011;3:925–931. doi: 10.1038/nchem.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Azagarsamy MA, Anseth KS. Bioorthogonal click chemistry: An indispensable tool to create multifaceted cell culture scaffolds. ACS Macro Lett. 2013;2:5–9. doi: 10.1021/mz300585q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Illeperuma WRK, Suo Z, Vlassak JJ. Hybrid hydrogels with extremely high stiffness and toughness. ACS Macro Lett. 2014;3:520–523. doi: 10.1021/mz5002355. [DOI] [PubMed] [Google Scholar]
- 39.Annabi N, et al. Highly elastic and conductive human-based protein hybrid hydrogels. Adv Mater. 2016;28:40–49. doi: 10.1002/adma.201503255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuk H, Zhang T, Parada GA, Liu X, Zhao X. Skin-inspired hydrogel-elastomer hybrids with robust interfaces and functional microstructures. Nat Commun. 2016;7:12028. doi: 10.1038/ncomms12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun J-Y, et al. Highly stretchable and tough hydrogels. Nature. 2012;489:133–136. doi: 10.1038/nature11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaharwar AK, Peppas NA, Khademhosseini A. Nanocomposite hydrogels for biomedical applications. Biotechnol Bioeng. 2014;111:441–453. doi: 10.1002/bit.25160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bodugoz-Senturk H, Macias CE, Kung JH, Muratoglu OK. Poly(vinyl alcohol)-acrylamide hydrogels as load-bearing cartilage substitute. Biomaterials. 2009;30:589–596. doi: 10.1016/j.biomaterials.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 44.Feinberg AW. Biological soft robotics. Annu Rev Biomed Eng. 2015;17:243–265. doi: 10.1146/annurev-bioeng-071114-040632. [DOI] [PubMed] [Google Scholar]
- 45.Annabi N, et al. Elastomeric recombinant protein-based biomaterials. Biochem Eng J. 2013;77:110–118. doi: 10.1016/j.bej.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y-N, et al. A highly elastic and rapidly crosslinkable elastin-like polypeptide-based hydrogel for biomedical applications. Adv Funct Mater. 2015;25:4814–4826. doi: 10.1002/adfm.201501489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Annabi N, et al. Engineered cell-laden human protein-based elastomer. Biomaterials. 2013;34:5496–5505. doi: 10.1016/j.biomaterials.2013.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haraguchi K, Takehisa T. Nanocomposite hydrogels: A unique organic–inorganic network structure with extraordinary mechanical, optical, and swelling/de-swelling properties. Adv Mater. 2002;14:1120–1124. doi: 10.1002/1521-4095(20020816)14:16<1120::AID-ADMA1120>3.0.CO;2-9. [DOI] [Google Scholar]
- 49.Li J, Suo Z, Vlassak JJ. Stiff, strong, and tough hydrogels with good chemical stability. J Mater Chem B Mater Biol Med. 2014;2:6708–6713. doi: 10.1039/C4TB01194E. [DOI] [PubMed] [Google Scholar]
- 50.Bin Imran A, et al. Extremely stretchable thermosensitive hydrogels by introducing slide-ring polyrotaxane cross-linkers and ionic groups into the polymer network. Nat Commun. 2014;5:5124. doi: 10.1038/ncomms6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao X. Multi-scale multi-mechanism design of tough hydrogels: Building dissipation into stretchy networks. Soft Matter. 2014;10:672–687. doi: 10.1039/C3SM52272E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaharwar AK, et al. Bioactive silicate nanoplatelets for osteogenic differentiation of human mesenchymal stem cells. Adv Mater. 2013;25:3329–3336. doi: 10.1002/adma.201300584. [DOI] [PubMed] [Google Scholar]
- 53.Gong JP, Katsuyama Y, Kurokawa T, Osada Y. Double-network hydrogels with extremely high mechanical strength. Adv Mater. 2003;15:1155–1158. doi: 10.1002/adma.200304907. [DOI] [Google Scholar]
- 54.Henderson KJ, Zhou TC, Otim KJ, Shull KR. Ionically cross-linked triblock copolymer hydrogels with high strength. Macromolecules. 2010;43:6193–6201. doi: 10.1021/ma100963m. [DOI] [Google Scholar]
- 55.Sun TL, et al. Physical hydrogels composed of polyampholytes demonstrate high toughness and viscoelasticity. Nat Mater. 2013;12:932–937. doi: 10.1038/nmat3713. [DOI] [PubMed] [Google Scholar]
- 56.Gonzalez MA, et al. Strong, tough, stretchable, and self-adhesive hydrogels from intrinsically unstructured proteins. Adv Mater. 2017;29:1604743. doi: 10.1002/adma.201604743. [DOI] [PubMed] [Google Scholar]
- 57.Oh JY, et al. Intrinsically stretchable and healable semiconducting polymer for organic transistors. Nature. 2016;539:411–415. doi: 10.1038/nature20102. [DOI] [PubMed] [Google Scholar]
- 58.Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv Mater. 2006;18:1345–1360. doi: 10.1002/adma.200501612. [DOI] [Google Scholar]
- 59.Yuk H, Zhang T, Lin S, Parada GA, Zhao X. Tough bonding of hydrogels to diverse non-porous surfaces. Nat Mater. 2016;15:190–196. doi: 10.1038/nmat4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gaharwar AK, et al. Shear-thinning nanocomposite hydrogels for the treatment of hemorrhage. ACS Nano. 2014;8:9833–9842. doi: 10.1021/nn503719n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paul A, et al. Injectable graphene oxide/hydrogel-based angiogenic gene delivery system for vasculogenesis and cardiac repair. ACS Nano. 2014;8:8050–8062. doi: 10.1021/nn5020787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Phadke A, et al. Rapid self-healing hydrogels. Proc Natl Acad Sci USA. 2012;109:4383–4388. doi: 10.1073/pnas.1201122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cui J, del Campo A. Multivalent H-bonds for self-healing hydrogels. Chem Commun (Camb) 2012;48:9302–9304. doi: 10.1039/c2cc34701f. [DOI] [PubMed] [Google Scholar]
- 64.Geisler IM, Schneider JP. Evolution-based design of an injectable hydrogel. Adv Funct Mater. 2012;22:529–537. doi: 10.1002/adfm.201102330. [DOI] [Google Scholar]
- 65.Appel EA, et al. Self-assembled hydrogels utilizing polymer-nanoparticle interactions. Nat Commun. 2015;6:6295. doi: 10.1038/ncomms7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakahata M, Takashima Y, Yamaguchi H, Harada A. Redox-responsive self-healing materials formed from host-guest polymers. Nat Commun. 2011;2:511. doi: 10.1038/ncomms1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang M, et al. Self-healing supramolecular gels formed by crown ether based host-guest interactions. Angew Chem Int Ed. 2012;124:7117–7121. doi: 10.1002/ange.201203063. [DOI] [PubMed] [Google Scholar]
- 68.Avery RK, et al. An injectable shear-thinning biomaterial for endovascular embolization. Sci Transl Med. 2016;8:365ra156. doi: 10.1126/scitranslmed.aah5533. [DOI] [PubMed] [Google Scholar]
- 69.Xavier JR, et al. Bioactive nanoengineered hydrogels for bone tissue engineering: A growth-factor-free approach. ACS Nano. 2015;9:3109–3118. doi: 10.1021/nn507488s. [DOI] [PubMed] [Google Scholar]
- 70.Tibbitt MW, Anseth KS. Dynamic microenvironments: The fourth dimension. Sci Transl Med. 2012;4:160ps24. doi: 10.1126/scitranslmed.3004804. [DOI] [PubMed] [Google Scholar]
- 71.Loessner D, et al. Functionalization, preparation and use of cell-laden gelatin methacryloyl-based hydrogels as modular tissue culture platforms. Nat Protoc. 2016;11:727–746. doi: 10.1038/nprot.2016.037. [DOI] [PubMed] [Google Scholar]
- 72.Schuurman W, et al. Gelatin-methacrylamide hydrogels as potential biomaterials for fabrication of tissue-engineered cartilage constructs. Macromol Biosci. 2013;13:551–561. doi: 10.1002/mabi.201200471. [DOI] [PubMed] [Google Scholar]
- 73.Wylie RG, et al. Spatially controlled simultaneous patterning of multiple growth factors in three-dimensional hydrogels. Nat Mater. 2011;10:799–806. doi: 10.1038/nmat3101. [DOI] [PubMed] [Google Scholar]
- 74.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Photodegradable hydrogels for dynamic tuning of physical and chemical properties. Science. 2009;324:59–63. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.DeForest CA, Tirrell DA. A photoreversible protein-patterning approach for guiding stem cell fate in three-dimensional gels. Nat Mater. 2015;14:523–531. doi: 10.1038/nmat4219. [DOI] [PubMed] [Google Scholar]
- 76.Khetan S, et al. Degradation-mediated cellular traction directs stem cell fate in covalently crosslinked three-dimensional hydrogels. Nat Mater. 2013;12:458–465. doi: 10.1038/nmat3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang C, Tibbitt MW, Basta L, Anseth KS. Mechanical memory and dosing influence stem cell fate. Nat Mater. 2014;13:645–652. doi: 10.1038/nmat3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sakiyama-Elbert SE, Panitch A, Hubbell JA. Development of growth factor fusion proteins for cell-triggered drug delivery. FASEB J. 2001;15:1300–1302. doi: 10.1096/fj.00-0564fje. [DOI] [PubMed] [Google Scholar]
- 79.Lutolf MP, et al. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. Proc Natl Acad Sci USA. 2003;100:5413–5418. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Purcell BP, et al. Injectable and bioresponsive hydrogels for on-demand matrix metalloproteinase inhibition. Nat Mater. 2014;13:653–661. doi: 10.1038/nmat3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Watarai A, et al. TGFβ functionalized starPEG-heparin hydrogels modulate human dermal fibroblast growth and differentiation. Acta Biomater. 2015;25:65–75. doi: 10.1016/j.actbio.2015.07.036. [DOI] [PubMed] [Google Scholar]
- 82.Taubenberger AV, et al. 3D extracellular matrix interactions modulate tumour cell growth, invasion and angiogenesis in engineered tumour microenvironments. Acta Biomater. 2016;36:73–85. doi: 10.1016/j.actbio.2016.03.017. [DOI] [PubMed] [Google Scholar]
- 83.Zieris A, et al. Biohybrid networks of selectively desulfated glycosaminoglycans for tunable growth factor delivery. Biomacromolecules. 2014;15:4439–4446. doi: 10.1021/bm5012294. [DOI] [PubMed] [Google Scholar]
- 84.Zhang L, Liang H, Jacob J, Naumov P. Photogated humidity-driven motility. Nat Commun. 2015;6:7429. doi: 10.1038/ncomms8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Breger JC, et al. Self-folding thermo-magnetically responsive soft microgrippers. ACS Appl Mater Interfaces. 2015;7:3398–3405. doi: 10.1021/am508621s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu Q, Bauer JM, Moore JS, Beebe DJ. Responsive biomimetic hydrogel valve for microfluidics. Appl Phys Lett. 2001;78:2589–2591. doi: 10.1063/1.1367010. [DOI] [Google Scholar]
- 87.Ma C, et al. Supramolecular Lego assembly towards three-dimensional multi-responsive hydrogels. Adv Mater. 2014;26:5665–5669. doi: 10.1002/adma.201402026. [DOI] [PubMed] [Google Scholar]
- 88.Wu ZL, et al. Three-dimensional shape transformations of hydrogel sheets induced by small-scale modulation of internal stresses. Nat Commun. 2013;4:1586. doi: 10.1038/ncomms2549. [DOI] [PubMed] [Google Scholar]
- 89.Kokkinis D, Schaffner M, Studart AR. Multimaterial magnetically assisted 3D printing of composite materials. Nat Commun. 2015;6:8643. doi: 10.1038/ncomms9643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao X, et al. Active scaffolds for on-demand drug and cell delivery. Proc Natl Acad Sci USA. 2011;108:6–72. 7. doi: 10.1073/pnas.1007862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim S, Laschi C, Trimmer B. Soft robotics: A bioinspired evolution in robotics. Trends Biotechnol. 2013;31:287–294. doi: 10.1016/j.tibtech.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 92.Kuribayashi-Shigetomi K, Onoe H, Takeuchi S. Cell origami: Self-folding of three-dimensional cell-laden microstructures driven by cell traction force. PLOS ONE. 2012;7:e51085. doi: 10.1371/journal.pone.0051085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nawroth JC, et al. A tissue-engineered jellyfish with biomimetic propulsion. Nat Biotechnol. 2012;30:792–797. doi: 10.1038/nbt.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Park S-J, et al. Phototactic guidance of a tissue-engineered soft-robotic ray. Science. 2016;353:158–162. doi: 10.1126/science.aaf4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shin SR, et al. Carbon-nanotube-embedded hydrogel sheets for engineering cardiac constructs and bioactuators. ACS Nano. 2013;7:2369–2380. doi: 10.1021/nn305559j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shin SR, et al. Aligned carbon nanotube-based flexible gel substrates for engineering bio-hybrid tissue actuators. Adv Funct Mater. 2015;25:4486–4495. doi: 10.1002/adfm.201501379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shin SR, et al. Reduced graphene oxide-gelMA hybrid hydrogels as scaffolds for cardiac tissue engineering. Small. 2016;12:3677–3689. doi: 10.1002/smll.201600178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang YS, et al. From cardiac tissue engineering to heart-on-a-chip: Beating challenges. Biomed Mater. 2015;10:034006. doi: 10.1088/1748-6041/10/3/034006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Saladin KS, Miller L. Anatomy and Physiology. McGraw-Hill; 1998. [Google Scholar]
- 100.Ma X, et al. Deterministically patterned biomimetic human iPSC-derived hepatic model via rapid 3D bioprinting. Proc Natl Acad Sci USA. 2016;113:2206–2211. doi: 10.1073/pnas.1524510113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Du Y, Lo E, Ali S, Khademhosseini A. Directed assembly of cell-laden microgels for fabrication of 3D tissue constructs. Proc Natl Acad Sci USA. 2008;105:9522–9527. doi: 10.1073/pnas.0801866105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Du Y, et al. Sequential assembly of cell-laden hydrogel constructs to engineer vascular-like microchannels. Biotechnol Bioeng. 2011;108:1693–1703. doi: 10.1002/bit.23102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Du Y, et al. Surface-directed assembly of cell-laden microgels. Biotechnol Bioeng. 2010;105:655–662. doi: 10.1002/bit.22552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Oliveira NM, et al. Hydrophobic hydrogels: Towards construction of floating (bio)microdevices. Chem Mater. 2016;28:3641–3648. doi: 10.1021/acs.chemmater.5b04445. [DOI] [Google Scholar]
- 105.Seeman NC. Nucleic acid junctions and lattices. J Theor Biol. 1982;99:237–247. doi: 10.1016/0022-5193(82)90002-9. [DOI] [PubMed] [Google Scholar]
- 106.Wei B, Dai M, Yin P. Complex shapes self-assembled from single-stranded DNA tiles. Nature. 2012;485:623–626. doi: 10.1038/nature11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Delebecque CJ, Lindner AB, Silver PA, Aldaye FA. Organization of intracellular reactions with rationally designed RNA assemblies. Science. 2011;333:470–474. doi: 10.1126/science.1206938. [DOI] [PubMed] [Google Scholar]
- 108.Yin P, et al. Programming DNA tube circumferences. Science. 2008;321:824–826. doi: 10.1126/science.1157312. [DOI] [PubMed] [Google Scholar]
- 109.Chworos A, et al. Building programmable jigsaw puzzles with RNA. Science. 2004;306:2068–2072. doi: 10.1126/science.1104686. [DOI] [PubMed] [Google Scholar]
- 110.Ke Y, Ong LL, Shih WM, Yin P. Three-dimensional structures self-assembled from DNA bricks. Science. 2012;338:1177–1183. doi: 10.1126/science.1227268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Qi H, et al. DNA-directed self-assembly of shape-controlled hydrogels. Nat Commun. 2013;4:2275. doi: 10.1038/ncomms3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Todhunter ME, et al. Programmed synthesis of three-dimensional tissues. Nat Methods. 2015;12:975–981. doi: 10.1038/nmeth.3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Murphy SV, Atala A. 3D bioprinting of tissues and organs. Nat Biotechnol. 2014;32:773–785. doi: 10.1038/nbt.2958. [DOI] [PubMed] [Google Scholar]
- 114.Malda J, et al. 25th anniversary article: Engineering hydrogels for biofabrication. Adv Mater. 2013;25:5011–5028. doi: 10.1002/adma.201302042. [DOI] [PubMed] [Google Scholar]
- 115.Hull CW. Google Patents. 1986. [Google Scholar]
- 116.Tumbleston JR, et al. Additive manufacturing. Continuous liquid interface production of 3D objects. Science. 2015;347:1349–1352. doi: 10.1126/science.aaa2397. [DOI] [PubMed] [Google Scholar]
- 117.Liu J, Hwang HH, Wang P, Whang G, Chen S. Direct 3D-printing of cell-laden constructs in microfluidic architectures. Lab Chip. 2016;16:1430–1438. doi: 10.1039/C6LC00144K. [DOI] [PubMed] [Google Scholar]
- 118.Colosi C, et al. Microfluidic bioprinting of heterogeneous 3D tissue constructs using low-viscosity bioink. Adv Mater. 2016;28:677–684. doi: 10.1002/adma.201503310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang YS, et al. Bioprinting 3D microfibrous scaffolds for engineering endothelialized myocardium and heart-on-a-chip. Biomaterials. 2016;110:45–59. doi: 10.1016/j.biomaterials.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Billiet T, Gevaert E, De Schryver T, Cornelissen M, Dubruel P. The 3D printing of gelatin methacrylamide cell-laden tissue-engineered constructs with high cell viability. Biomaterials. 2014;35:49–62. doi: 10.1016/j.biomaterials.2013.09.078. [DOI] [PubMed] [Google Scholar]
- 121.Bertassoni LE, et al. Hydrogel bioprinted microchannel networks for vascularization of tissue engineering constructs. Lab Chip. 2014;14:2202–2211. doi: 10.1039/C4LC00030G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kolesky DB, et al. 3D bioprinting of vascularized, heterogeneous cell-laden tissue constructs. Adv Mater. 2014;26:3124–3130. doi: 10.1002/adma.201305506. [DOI] [PubMed] [Google Scholar]
- 123.Kolesky DB, Homan KA, Skylar-Scott MA, Lewis JA. Three-dimensional bioprinting of thick vascularized tissues. Proc Natl Acad Sci USA. 2016;113:3179–3184. doi: 10.1073/pnas.1521342113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hong S, et al. 3D printing of highly stretchable and tough hydrogels into complex, cellularized structures. Adv Mater. 2015;27:4035–4040. doi: 10.1002/adma.201501099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bhattacharjee T, et al. Writing in the granular gel medium. Sci Adv. 2015;1:e1500655. doi: 10.1126/sciadv.1500655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Highley CB, Rodell CB, Burdick JA. Direct 3D printing of shear-thinning hydrogels into self-healing hydrogels. Adv Mater. 2015;27:5075–5079. doi: 10.1002/adma.201501234. [DOI] [PubMed] [Google Scholar]
- 127.Hinton TJ, et al. Three-dimensional printing of complex biological structures by freeform reversible embedding of suspended hydrogels. Sci Adv. 2015;1:e1500758. doi: 10.1126/sciadv.1500758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kang H-W, et al. A 3D bioprinting system to produce human-scale tissue constructs with structural integrity. Nat Biotechnol. 2016;34:312–319. doi: 10.1038/nbt.3413. [DOI] [PubMed] [Google Scholar]
- 129.Khalil S, Nam J, Sun W. Multi-nozzle deposition for construction of 3D biopolymer tissue scaffoldsnull. Rapid Prototyping J. 2005;11:9–17. doi: 10.1108/13552540510573347. [DOI] [Google Scholar]
- 130.Hardin JO, Ober TJ, Valentine AD, Lewis JA. Microfluidic printheads for multimaterial 3D printing of viscoelastic Inks. Adv Mater. 2015;27:3279–3284. doi: 10.1002/adma.201500222. [DOI] [PubMed] [Google Scholar]
- 131.Liu W, et al. Rapid continuous multimaterial extrusion bioprinting. Adv Mater. 2017;29:1604630. doi: 10.1002/adma.201604630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ober TJ, Foresti D, Lewis JA. Active mixing of complex fluids at the microscale. Proc Natl Acad Sci USA. 2015;112:12293–12298. doi: 10.1073/pnas.1509224112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Peppas NA, Khademhosseini A. Make better, safer biomaterials. Nature. 2016;540:335–337. doi: 10.1038/540335a. [DOI] [PubMed] [Google Scholar]
- 134.Caló E, Khutoryanskiy VV. Biomedical applications of hydrogels: A review of patents and commercial products. Eur Polym J. 2015;65:252–267. doi: 10.1016/j.eurpolymj.2014.11.024. [DOI] [Google Scholar]
- 135.Kamata H, Akagi Y, Kayasuga-Kariya Y, Chung UI, Sakai T. “Nonswellable” hydrogel without mechanical hysteresis. Science. 2014;343:873–875. doi: 10.1126/science.1247811. [DOI] [PubMed] [Google Scholar]
- 136.Visser J, et al. Reinforcement of hydrogels using three-dimensionally printed microfibres. Nat Commun. 2015;6:6933. doi: 10.1038/ncomms7933. [DOI] [PubMed] [Google Scholar]
- 137.Blaeser A, et al. Controlling shear stress in 3D bioprinting is a key factor to balance printing resolution and stem cell integrity. Adv Healthc Mater. 2016;5:326–333. doi: 10.1002/adhm.201500677. [DOI] [PubMed] [Google Scholar]
- 138.Raviv D, et al. Active printed materials for complex self-evolving deformations. Sci Rep. 2014;4:7422. doi: 10.1038/srep07422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tibbits S. 4D printing: Multi-material shape change. Architect Des. 2014;84:116–121. [Google Scholar]
- 140.Gladman AS, Matsumoto EA, Nuzzo RG, Mahadevan L, Lewis JA. Biomimetic 4D printing. Nat Mater. 2016;15:413–418. doi: 10.1038/nmat4544. [DOI] [PubMed] [Google Scholar]
- 141.Ge Q, et al. Multimaterial 4D printing with tailorable shape memory polymers. Sci Rep. 2016;6:31110. doi: 10.1038/srep31110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Miao S, et al. 4D printing smart biomedical scaffolds with novel soybean oil epoxidized acrylate. Sci Rep. 2016;6:27226. doi: 10.1038/srep27226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li YC, Zhang YS, Akpek A, Shin SR, Khademhosseini A. 4D bioprinting: The next-generation technology for biofabrication enabled by stimuli-responsive materials. Biofabrication. 2016;9:012001. doi: 10.1088/1758-5090/9/1/012001. [DOI] [PubMed] [Google Scholar]