

Abstract
Arrestins specifically bind active phosphorylated G protein-coupled receptors (GPCRs). Receptor binding induces the release of the arrestin C-tail, which in non-visual arrestins contains high-affinity binding sites for clathrin and its adaptor AP2. Thus, serving as a physical link between the receptor and key components of the internalization machinery of the coated pit is the best-characterized function of non-visual arrestins in GPCR trafficking. However, arrestins also regulate GPCR trafficking less directly by orchestrating their ubiquitination and deubiquitination. Several reports suggest that arrestins play additional roles in receptor trafficking. Non-visual arrestins appear to be required for the recycling of internalized GPCRs, and the mechanisms of their function in this case remain to be elucidated. Moreover, visual and non-visual arrestins were shown to directly bind N-ethylmaleimide-sensitive factor, an important ATPase involved in vesicle trafficking, but neither molecular details nor the biological role of these interactions is clear. Considering how many different proteins arrestins appear to bind, we can confidently expect the elucidation of additional trafficking-related functions of these versatile signaling adaptors.
1. ARRESTINS AND GPCR TRAFFICKING
Preferential binding of arrestins to active phosphorylated receptors was discovered about 30 years ago.1 The finding that arrestin binding suppresses receptor coupling to cognate G proteins was made soon after in the visual system.2 The mechanism turned out to be remarkably simple: direct competition between arrestin and G protein for overlapping sites.3,4 For some time, it appeared that the only function arrestins have is to bind active phosphorylated G protein-coupled receptors (GPCRs), precluding receptor interactions with G proteins by direct competition.3,4 The first described non-GPCR binding partners of arrestins were trafficking proteins: clathrin in 19965 and clathrin adaptor AP2 a few years later.6 These data demonstrated that arrestins play an essential role not only in GPCR desensitization7 but also in receptor endocytosis,8 via trafficking signals added by receptor-bound arrestins. The discovery that arrestins are ubiquitinated upon receptor binding and regulate ubiquitination of GPCRs9 revealed yet another mechanism, whereby arrestins regulate receptor trafficking indirectly. Here, we discuss several known mechanisms of arrestin effects on GPCR trafficking and highlight observations that suggest that there are many other mechanisms that still remain to be elucidated.
2. NON-VISUAL ARRESTINS MEDIATE GPCR INTERNALIZATION VIA COATED PITS
Arrestins promote GPCR internalization by virtue of recruitment of clathrin and AP2 via fairly well-mapped binding sites in the C-tail of non-visual arrestins5,6,10,11 (Fig. 1). Interestingly, the C-tail in the basal conformation of all arrestins is anchored to the N-domain,12–16 whereas receptor binding triggers its release.17–19 The expression of separated arrestin C-tail carrying these sites inhibits GPCR internalization, apparently by winning the competition with the arrestin–receptor complexes for clathrin and AP2.20 This finding provided the first clear evidence of functional significance of shielding of the arrestin C-tail in the basal conformation and its release upon receptor binding. In free arrestins, the C-tail is anchored to the body of the molecule, which makes it inaccessible, preventing its competition with the receptor-bound arrestins for the components of internalization machinery (reviewed in Ref. 21).
Figure 1.
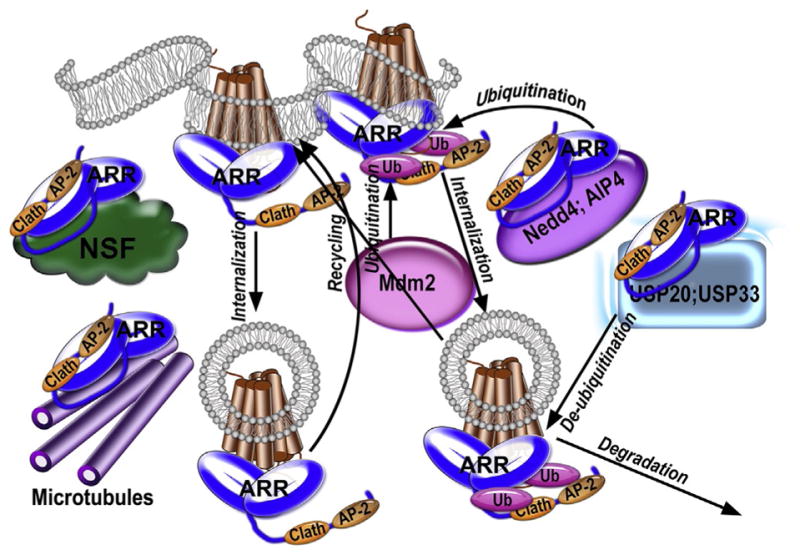
Arrestins play many roles in GPCR trafficking. Arrestins (ARR) bind active phos-phorylated GPCRs (shown as a seven-helix bundle). Receptor binding induces the release of the arrestin C-tail, which carries binding sites for clathrin (Clath) and adaptor protein-2 (AP2). The interactions of these sites with clathrin and AP2 promote receptor internalization via coated pits. Arrestins also recruit ubiquitin ligases Mdfm2, Nedd4, and AIP4 to the complex, which favors ubiquitination of both non-visual arrestins and at least some GPCRs. Arrestins also recruit certain deubiquitination enzymes (USP20 and USP33 are shown), facilitating receptor deubiquitination. The role of arrestin interactions with microtubules, centrosome, and N-ethylmaleimide-sensitive factor (NSF) in trafficking of GPCRs and/or other proteins remains to be elucidated.
Another known mechanism of arrestin recruitment to the coated pit is its direct binding to phosphoinositides, which was reported to be necessary for GPCR internalization.22 Since resident coated pit protein AP2 is also recruited to this part of the membrane via phosphoinositide binding,23 one might think that as soon as the arrestin–receptor complex is formed, it has no choice but to move to the coated pit. However, this does not appear to be the case. In muscarinic M2 receptor, which was among the first shown to bind arrestins,24 two Ser/Thr clusters in the third cytoplasmic loop were identified as critical for arrestin binding and receptor desensitization.25 Yet the elimination of these clusters, and even dominant-negative dynamin K44A mutant that blocks the internalization of β2AR in the same cells, did not prevent M2 endocytosis, suggesting that M2 receptor does not use coated pits and internalizes in an arrestin-independent manner.25 Interestingly, overexpression of non-visual arrestins can redirect some M2 to coated pits,25 suggesting that this receptor can use more than one route. Many other GPCRs were shown to have that choice. For example, chemo-kine receptor CCR5 uses both phosphorylation- and arrestin-dependent and -independent pathways.26 Cysteinyl leukotriene type 1 receptor internalizes normally in mouse embryonic fibroblasts lacking both non-visual arrestins, yet arrestin expression facilitates its internalization,27 apparently directing it to the arrestin-dependent pathway, which is usually not preferred, similar to M2 receptor.25 Metabotropic glutamate receptor mGluR1a constitutively internalizes via arrestin-independent mechanism, whereas its agonist-dependent internalization appears to be mediated by arrestin-2.28 Endogenous and overexpressed serotonin 5HT4 receptor internalizes via arrestin-dependent pathway, but the deletion of Ser/Thr cluster targeted by G protein-coupled receptor kinases (GRKs) redirects it to an alternative pathway and even facilitates its internalization.29
Thus, it appears that the ability of GPCRs to use more than one internalization pathway is a general rule, rather than an exception, likely representing one of the many backup mechanisms cells usually have. Many receptors have recognizable internalization motifs in their sequence, so arrestin binding simply adds new ones. The relative strength of these motifs, as well as the arrestin expression levels, likely determines the pathway(s) each receptor chooses in a particular cell. The dominant internalization pathway of a particular receptor is not necessarily the same in different cell types, or even at different functional states of the same cell (reviewed in Ref. 8). Variety, rather than uniformity, characterizes the world of GPCR signaling and trafficking.30
3. VISUAL ARRESTINS AND TRAFFICKING PROTEINS
In vertebrate rod photoreceptors, rhodopsin is localized on the discs, which are detached from the plasma membrane31 and therefore are topologically equivalent to vesicles with internalized non-visual GPCRs. Thus, vertebrate rhodopsin is not supposed to be internalized. Indeed, arrestin-1, which is the prevalent arrestin isoform in both rods and cones,32 does not have conventional clathrin- or AP2-binding elements in its C-tail.33 However, sequence comparison of arrestin-1 and non-visual subtypes shows that in the region homologous to AP2-binding motif in arrestin-2 and -3, only one positive charge is missing.34 Therefore, it is hardly surprising that arrestin-1 also binds AP2, albeit with ~30 times lower affinity.34 Constitutively active rhodopsin–K296E is a naturally occurring mutant that causes autosomal dominant retinitis pigmentosa in humans, apparently due to constitutive phosphorylation and formation of a stable complex with arrestin-1.35 The concentration of rhodopsin in the outer segment of rods reaches ~3 mM.31 Rods also express roughly 8 arrestin molecules per 10 rhodopsins,36–38 so the concentrations of both proteins and their complex formed in bright light are very high. It turns out that at these concentrations even low affinity matters: the presence of WT arrestin-1 facilitates rod death in animals expressing rhodopsin–K296E, with visible accumulation of AP2 in the outer segment, where it is not observed in normal mice.34 In contrast, truncated arrestin-1 lacking the C-tail containing the low-affinity AP2-binding site protects photoreceptors in these animals and preserves their function.34 Thus, in rod and cone photoreceptors, both of which express very high levels of arrestin-1,32 even relatively low-affinity interactions, which would not matter in other cells, with submicromolar concentrations of both non-visual arrestins,39,40 can become biologically relevant.
Interestingly, the localization of rhodopsin on invaginations of the plasma membrane in flies, in contrast to detached discs in vertebrate rods, is one of the many differences between vertebrate and invertebrate photo-receptors. Another difference directly follows from this localization: Drosophila rhodopsin is internalized, like “normal” vertebrate GPCRs, via clathrin- and AP2-mediated mechanism.41 In fly photoreceptors, arrestin is evenly distributed, whereas in dark-adapted vertebrate rods, it is concentrated in the inner segment, with fairly small fraction in the outer segment, where rhodopsin resides.36–38 However, in both types of photoreceptors upon illumination, arrestin translocates to rhodopsin-containing membranes.36–38,42–45 Like non-visual arrestins, and in contrast to vertebrate visual arrestin,22 visual arrestin in Drosophila has high-affinity phosphoinositide-binding site.43 It was proposed that due to phosphoinositide binding, Drosophila arrestin translocates to rhodopsin on phosphoinositide-rich vesicles moved with the help of Drosophila myosin III (NINAC).42 The participation of NINAC in metarhodopsin inactivation in Drosophila was independently confirmed,46 but arrestin translocation was found to be largely driven by its binding to rhodopsin in flies,44 just like in mice.45 Thus, the internalization of invertebrate rhodopsin apparently follows the same rules as many non-visual GPCRs: active receptor recruits arrestin via direct binding,47 which then links it to the key components of the coated pit.5,6,41
4. UBIQUITINATION AND DEUBIQUITINATION IN GPCR CYCLING AND SIGNALING
Monoubiquitination of many proteins regulates their trafficking and signaling, rather than proteasomal degradation.48 Two GPCRs, β2AR9 and chemokine receptor CXCR4,49 were shown to be ubiquitinated in response to agonist activation. Arrestin ubiquitination upon receptor binding, as well as the role of arrestin in GPCR ubiquitination, was discovered a few years later than the interactions of non-visual arrestins with clathrin and AP2.9 It appears that arrestin ubiquitination by Mdm2 prolongs the life of the arrestin–receptor complex.50 As only receptor-bound arrestins facilitate ERK1/2 activation,51,52 it is natural that arrestin ubiquitination increases ERK1/2 activation induced by GPCR stimulation.53 Slow deubiquitination of the receptor-bound arrestin prolongs the dwell time of the complex inside the cell and slows down receptor recycling.50 However, receptor or arrestin ubiquitination per se does not appear to be necessary for arrestin-dependent internalization: virtually complete suppression of agonist-induced ubiquitination of arrestin-2 does not appreciably affect endocytosis of β2AR.54 Arrestin-2 recruits ubiquitin ligase AIP4 to ubiq-uitinate CXCR4, which affects endosomal sorting of this receptor.55 Receptor-bound arrestin-3 recruits yet another ubiquitin ligase, Nedd4, which ubiquitinates β2AR, and this receptor modification is required for lysosomal targeting of internalized β2AR,56 although arrestin domain-containing protein 3 was also suggested as the mediator of the interaction of Nedd4 with β2AR.57,58 Finally, both non-visual arrestins bind a fourth ubiquitin ligase, parkin.54 Interestingly, parkin binding enhances arrestin interactions with Mdm2, but paradoxically strongly reduces arrestin ubiquitination in response to receptor activation.54 The possible role of parkin in receptor modification remains to be elucidated. To further complicate matters, arrestins were found to recruit deubiquitinating enzymes USP20 and USP33 to β2AR, which facilitate receptor recycling and resensitization.59,60
To summarize, it is clear that arrestins bind several ubiquitin ligases and recruit them at least to some GPCRs. Both arrestins and GPCRs are ubiquitinated upon receptor stimulation. Receptor ubiquitination appears to play a role in sorting and lysosomal targeting, whereas the ubiquitination of arrestins likely affects their affinity for receptors. However, arrestin-mediated recruitment of some deubiquitinating enzymes suggests that their role in GPCR trafficking is more complex and includes postendocytotic steps. Interestingly, the role of arrestins in recruiting deubiquitinases was shown on β2AR,59,60 which appears to contradict the idea that arrestins bound to this particular receptor dissociate from it very quickly.61 Thus, the biological functions of arrestin-assisted ubiquitination and deubiquitination of GPCRs and similar modifications of non-visual arrestins need to be further clarified. One should also keep in mind that the role of the same processes in trafficking of different GPCRs is not necessarily the same: the very fact that animals have so many members of this superfamily suggests that variety, rather than uniformity, is the key.30
5. FASTER CYCLING PREVENTS RECEPTOR DOWNREGULATION
With very few exceptions, the fate of internalized receptors is not predetermined: they can be recycled back to the plasma membrane and reused, or sent to lysosomes and destroyed.7 The latter process leads to the reduction of overall receptor number, usually termed downregulation. We do not know how the choice between recycling and elimination is made, but it appears that the intensity and/or duration of signaling can tip the scales one way or another. In the process of internalization and recycling, most receptors transition through several functional states. First, in case of GPCRs that internalize via arrestin-dependent pathway, after phosphorylation by GRKs and arrestin binding receptors, move into coated vesicles and then to endosomes. The internal pH in endosomes is much lower than on the extracellular side of the membrane.62 It is likely (but remains unproven) that acidification facilitates the dissociation of the ligand. The loss of the bound agonist and consequent transition into inactive state is the only conceivable mechanism of subsequent release of bound arrestins: both non-visual subtypes demonstrate lower binding to inactive phosphoreceptors,63–65 even though the difference is not as dramatic as in the case of visual arrestin-1.66,67 Arrestin dissociation is necessary to make receptor-attached phosphates accessible to phosphatases,68 so it must precede receptor dephosphorylation. Since both non-visual arrestins require at least two phosphates for high-affinity binding,63 dephosphorylation has to be a multistep process. It must be completed, as it appears that only fully dephosphorylated receptors are recycling competent.69,70 One conceivable model is that only certain functional states of the receptor can be diverted to lysosomes and destroyed; and the other is that every state can be transported to lysosomes, so that the longer the time that a GPCR spends in the endosomal compartment, the higher the probability that it will be transported to lysosomes and destroyed.
Similar to visual arrestin-1, both non-visual arrestins can be made to bind active unphosphorylated GPCRs by mutations destabilizing the main phosphate sensor, the polar core, by mutations detaching the C-tail from the body of the molecule, or by C-tail deletions.64,65,71 The effect of two different arrestin-2 mutants, one activated by polar core mutation and the other by the C-tail detachment, on cycling of β2AR was tested in cells.72 Since these forms of arrestin-2 bind the same active receptor as GRKs, they actually compete with GRKs and suppress receptor phosphorylation both in vitro, in the system reconstituted from purified proteins, and in cells.72 It turned out that in cells, these preactivated arrestin-2 mutants bind unphosphorylated β2AR and induce its internalization. Interestingly, unphosphorylated β2AR internalized in complex with these mutants recycles very rapidly, much faster than in the presence of WT arrestin-2 that only binds phosphorylated receptor.72 Importantly, the expression of phosphorylation-independent arrestin-2 mutants protected the receptor from downregulation, so that, in sharp contrast to cells expressing WT arrestin-2, even after 24 h of agonist exposure virtually no β2AR was lost.72 This was the first study of the effect of the nature of the arrestin–receptor complex on the fate of internalized receptor. It did not answer all questions. The results can be interpreted in the context of both models: (1) as an indication that rapid cycling reduces the chances of the receptor to be diverted to lysosomes, or (2) as a suggestion that only phosphorylated forms of the receptor are diverted to that compartment and destroyed. The use of nonphosphorylatable β2AR mutants in similar experiments is necessary to resolve this issue.
6. ARRESTINS IN RECEPTOR RECYCLING AND VESICLE TRAFFICKING: QUESTIONS WITHOUT ANSWERS
The mechanism whereby arrestin-2 and -3 participate in GPCR internalization is fairly well established: the C-tail of both non-visual arrestins is released upon receptor binding,19 which increases the accessibility of clathrin and AP2-binding sites in this element.10,73,74 In addition, arrestins appear to recruit ubiquitin ligases to GPCRs, and receptor ubiquitination plays a role in receptor sorting.9,55,56 Yet it is still unclear how arrestins participate in other steps of GPCR trafficking. N-Formyl-peptide receptor binds arrestin-2 and -3 in an activation- and phosphorylation-dependent manner,75,76 yet it was reported to internalize in the absence of both non-visual arrestins.77 However, in mouse embryonic fibroblasts lacking both non-visual arrestins, internalized N-formyl-peptide receptor does not recycle.77 The receptor travels to the perinuclear recycling compartment and gets stuck there, but its recycling can be rescued by the expression of either arrestin-2 or -3.77 These data suggest that, as far as N-formyl-peptide receptor recycling is concerned, the two non-visual arrestins are functionally redundant. Yet we do not have many clues how exactly are arrestins involved in GPCR recycling. One conceivable scenario is that arrestins bind to this receptor after internalization and recruit deubiquitinating enzymes necessary for recycling, as was shown in the case of β2AR,59,60 but this leaves open the question why arrestins do not bind it before endocytosis, similar to β2AR.5,6,9 Existing evidence does not suggest any good answers to this question.
Another issue that needs experimental clarification is arrestin binding to the N-ethylmaleimide-sensitive factor (NSF), an ATPase involved in vesicle trafficking. Arrestin-2 binding to NSF was discovered 15 years ago,78 but its functional significance in case of non-visual arrestins remains unclear. Interestingly, a few years ago, visual arrestin-1 was shown to interact with NSF in photoreceptors.79 It appears that in rods, arrestin-1 is necessary to maintain proper NSF function and normal level of neurotransmitter release.79 However, the molecular mechanism of this arrestin-1 effect remains to be elucidated.
7. CONCLUSIONS AND FUTURE DIRECTIONS
The role of non-visual arrestins in recruiting GPCRs to coated pits and facilitation of receptor internalization via this pathway is fairly well established. The case of ubiquitin modification of receptors and arrestins is less straightforward: arrestins seem to recruit enzymes responsible for ubiquitination and deubiquitination of GPCRs. These modifications play distinct roles in receptor trafficking, but the exact role of non-visual arrestins, which are also ubiquitinated in response to receptor stimulation, remains to be elucidated. The functions of non-visual arrestins in complex trafficking itineraries of individual GPCR subtypes might be different. How arrestins affect the recycling of internalized GPCRs, and how exactly arrestin binding regulates NSF function and vesicle trafficking, remains even less clear (Fig. 1). Cytoskeleton is intimately involved in trafficking of many proteins. Arrestins were shown to bind microtubules80–82 and a very specialized structure containing polymerized tubulin, the centro-some.83 However, the role of these interactions in the transport of receptors and/or other molecules within the cell still needs to be defined. Most likely, recent finding that non-visual arrestins recruit clathrin to microtubules targeting focal adhesions, thereby facilitating integrin internalization and focal adhesion disassembly,84 is only the tip of the iceberg.
ABBREVIATIONS
- AIP4
atrophin-1-interacting protein 4
- AP2
adaptor protein 2
- β2AR
β2-adrenergic receptor
- GPCR
G protein-coupled receptor
- GRK
G protein-coupled receptor kinase
- Nedd4
neural precursor cell expressed developmentally down-regulated protein 4
Footnotes
We use systematic names of arrestin proteins: arrestin-1 (historic names S-antigen, 48 kDa protein, visual or rod arrestin), arrestin-2 (β-arrestin or β-arrestin1), arrestin-3 (β-arrestin2 or hTHY-ARRX), and arrestin-4 (cone or X-arrestin; for unclear reasons, its gene is called “arrestin 3” in the HUGO database).
References
- 1.Kuhn H, Hall SW, Wilden U. Light-induced binding of 48-kDa protein to photorecep-tor membranes is highly enhanced by phosphorylation of rhodopsin. FEBS Lett. 1984;176:473–478. doi: 10.1016/0014-5793(84)81221-1. [DOI] [PubMed] [Google Scholar]
- 2.Wilden U, Hall SW, Kühn H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc Natl Acad Sci USA. 1986;83:1174–1178. doi: 10.1073/pnas.83.5.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilden U. Duration and amplitude of the light-induced cGMP hydrolysis in vertebrate photoreceptors are regulated by multiple phosphorylation of rhodopsin and by arrestin binding. Biochemistry. 1995;34:1446–1454. doi: 10.1021/bi00004a040. [DOI] [PubMed] [Google Scholar]
- 4.Krupnick JG, Gurevich VV, Benovic JL. Mechanism of quenching of photo-transduction. Binding competition between arrestin and transducin for pho-sphorhodopsin. J Biol Chem. 1997;272:18125–18131. doi: 10.1074/jbc.272.29.18125. [DOI] [PubMed] [Google Scholar]
- 5.Goodman OB, Jr, Krupnick JG, Santini F, et al. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383(6599):447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 6.Laporte SA, Oakley RH, Zhang J, et al. The 2-adrenergic receptor/arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci USA. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carman CV, Benovic JL. G-protein-coupled receptors: turn-ons and turn-offs. Curr Opin Neurobiol. 1998;8:335–344. doi: 10.1016/s0959-4388(98)80058-5. [DOI] [PubMed] [Google Scholar]
- 8.Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharmacol Ther. 2006;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science. 2001;294:1307–1313. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]
- 10.Kim YM, Benovic JL. Differential roles of arrestin-2 interaction with clathrin and adaptor protein 2 in G protein-coupled receptor trafficking. J Biol Chem. 2002;277:30760–30768. doi: 10.1074/jbc.M204528200. [DOI] [PubMed] [Google Scholar]
- 11.Kang DS, Kern RC, Puthenveedu MA, von Zastrow M, Williams JC, Benovic JL. Structure of an arrestin2-clathrin complex reveals a novel clathrin binding domain that modulates receptor trafficking. J Biol Chem. 2009;284:29860–29872. doi: 10.1074/jbc.M109.023366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirsch JA, Schubert C, Gurevich VV, Sigler PB. The 2.8 Å crystal structure of visual arrestin: a model for arrestin’s regulation. Cell. 1999;97(2):257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- 13.Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Crystal structure of beta-arrestin at 1.9 Å: possible mechanism of receptor binding and membrane translocation. Structure. 2001;9(9):869–880. doi: 10.1016/s0969-2126(01)00644-x. [DOI] [PubMed] [Google Scholar]
- 14.Sutton RB, Vishnivetskiy SA, Robert J, et al. Crystal structure of cone arrestin at 2.3 Å: evolution of receptor specificity. J Mol Biol. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 15.Zhan X, Gimenez LE, Gurevich VV, Spiller BW. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual arrestins. J Mol Biol. 2011;406:467–478. doi: 10.1016/j.jmb.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milano SK, Pace HC, Kim YM, Brenner C, Benovic JL. Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry. 2002;41(10):3321–3328. doi: 10.1021/bi015905j. [DOI] [PubMed] [Google Scholar]
- 17.Hanson SM, Francis DJ, Vishnivetskiy SA, et al. Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc Natl Acad Sci USA. 2006;103:4900–4905. doi: 10.1073/pnas.0600733103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vishnivetskiy SA, Francis DJ, Van Eps N, et al. The role of arrestin alpha-helix I in receptor binding. J Mol Biol. 2010;395:42–54. doi: 10.1016/j.jmb.2009.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhuo Y, Vishnivetskiy SA, Zhan X, Gurevich VV, Klug CS. Identification of receptor binding-induced conformational changes in non-visual arrestins. J Biol Chem. 2014;289(30):20991–21002. doi: 10.1074/jbc.M114.560680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orsini MJ, Benovic JL. Characterization of dominant negative arrestins that inhibit beta2-adrenergic receptor internalization by distinct mechanisms. J Biol Chem. 1998;273(51):34616–34622. doi: 10.1074/jbc.273.51.34616. [DOI] [PubMed] [Google Scholar]
- 21.Gurevich VV, Gurevich EV. The new face of active receptor bound arrestin attracts new partners. Structure. 2003;11:1037–1042. doi: 10.1016/s0969-2126(03)00184-9. [DOI] [PubMed] [Google Scholar]
- 22.Gaidarov I, Krupnick JG, Falck JR, Benovic JL, Keen JH. Arrestin function in G protein-coupled receptor endocytosis requires phosphoinositide binding. EMBO J. 1999;18:871–881. doi: 10.1093/emboj/18.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaidarov I, Keen JH. Phosphoinositide-AP-2 interactions required for targeting to plasma membrane clathrin-coated pits. J Cell Biol. 1999;146:755–764. doi: 10.1083/jcb.146.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gurevich VV, Richardson RM, Kim CM, Hosey MM, Benovic JL. Binding of wild type and chimeric arrestins to the m2 muscarinic cholinergic receptor. J Biol Chem. 1993;268(23):16879–16882. [PubMed] [Google Scholar]
- 25.Pals-Rylaarsdam R, Gurevich VV, Lee KB, Ptasienski J, Benovic JL, Hosey MM. Internalization of the m2 muscarinic acetylcholine receptor: arrestin-independent and -dependent pathways. J Biol Chem. 1997;272:23682–23689. doi: 10.1074/jbc.272.38.23682. [DOI] [PubMed] [Google Scholar]
- 26.Kraft K, Olbrich H, Majoul I, Mack M, Proudfoot A, Oppermann M. Characterization of sequence determinants within the carboxyl-terminal domain of chemokine receptor CCR5 that regulate signaling and receptor internalization. J Biol Chem. 2001;276:34408–34418. doi: 10.1074/jbc.M102782200. [DOI] [PubMed] [Google Scholar]
- 27.Naik S, Billington CK, Pascual RM, et al. Regulation of cysteinyl leukotriene type 1 receptor internalization and signaling. J Biol Chem. 2005;280:8722–8732. doi: 10.1074/jbc.M413014200. [DOI] [PubMed] [Google Scholar]
- 28.Dale LB, Bhattacharya M, Seachrist JL, Anborgh PH, Ferguson SS. Agonist-stimulated and tonic internalization of metabotropic glutamate receptor 1a in human embryonic kidney 293 cells: agonist-stimulated endocytosis is beta-arrestin1 isoform-specific. Mol Pharmacol. 2001;60:1243–1253. doi: 10.1124/mol.60.6.1243. [DOI] [PubMed] [Google Scholar]
- 29.Barthet G, Gaven F, Framery B, et al. Uncoupling and endocytosis of 5-HT4 receptors: distinct molecular events with different GRK2 requirements. J Biol Chem. 2005;280:27924–27934. doi: 10.1074/jbc.M502272200. [DOI] [PubMed] [Google Scholar]
- 30.Gurevich VV, Gurevich EV. Rich tapestry of G protein-coupled receptor signaling and regulatory mechanisms. Mol Pharmacol. 2008;74(2):312–316. doi: 10.1124/mol.108.049015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pugh EN, Jr, Lamb TD. Phototransduction in vertebrate rods and cones: molecular mechanisms of amplification, recovery and light adaptation. In: Stavenga DG, DeGrip WJ, Pugh EN Jr, editors. Handbook of Biological Physics. Molecular Mechanisms in Visual Transduction. Amsterdam: Elsevier; 2000. pp. 183–255. [Google Scholar]
- 32.Nikonov SS, Brown BM, Davis JA, et al. Mouse cones require an arrestin for normal inactivation of phototransduction. Neuron. 2008;59:462–474. doi: 10.1016/j.neuron.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gurevich EV, Gurevich VV. Arrestins are ubiquitous regulators of cellular signaling pathways. Genome Biol. 2006;7:236. doi: 10.1186/gb-2006-7-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moaven H, Koike Y, Jao CC, Gurevich VV, Langen R, Chen J. Visual arrestin interaction with clathrin adaptor AP-2 regulates photoreceptor survival in the vertebrate retina. Proc Natl Acad Sci USA. 2013;110(23):9463–9468. doi: 10.1073/pnas.1301126110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li T, Franson WK, Gordon JW, Berson EL, Dryja TP. Constitutive activation of phototransduction by K296E opsin is not a cause of photoreceptor degeneration. Proc Natl Acad Sci USA. 1995;92(8):3551–3555. doi: 10.1073/pnas.92.8.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanson SM, Gurevich EV, Vishnivetskiy SA, Ahmed MR, Song X, Gurevich VV. Each rhodopsin molecule binds its own arrestin. Proc Natl Acad Sci USA. 2007;104:3125–3128. doi: 10.1073/pnas.0610886104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song X, Vishnivetskiy SA, Seo J, Chen J, Gurevich EV, Gurevich VV. Arrestin-1 expression in rods: balancing functional performance and photoreceptor health. Neuroscience. 2011;174:37–49. doi: 10.1016/j.neuroscience.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strissel KJ, Sokolov M, Trieu LH, Arshavsky VY. Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J Neurosci. 2006;26:1146–1153. doi: 10.1523/JNEUROSCI.4289-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gurevich EV, Benovic JL, Gurevich VV. Arrestin2 and arrestin3 are differentially expressed in the rat brain during postnatal development. Neuroscience. 2002;109:421–436. doi: 10.1016/s0306-4522(01)00511-5. [DOI] [PubMed] [Google Scholar]
- 40.Gurevich EV, Benovic JL, Gurevich VV. Arrestin2 expression selectively increases during neural differentiation. J Neurochem. 2004;91:1404–1416. doi: 10.1111/j.1471-4159.2004.02830.x. [DOI] [PubMed] [Google Scholar]
- 41.Orem NR, Xia L, Dolph PJ. An essential role for endocytosis of rhodopsin through interaction of visual arrestin with the AP-2 adaptor. J Cell Sci. 2006;119(Pt 15):3141–3148. doi: 10.1242/jcs.03052. [DOI] [PubMed] [Google Scholar]
- 42.Lee SJ, Montell C. Light-dependent translocation of visual arrestin regulated by the NINAC myosin III. Neuron. 2004;43:95–103. doi: 10.1016/j.neuron.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 43.Lee SJ, Xu H, Kang LW, Amzel LM, Montell C. Light adaptation through phosphoinositide-regulated translocation of Drosophila visual arrestin. Neuron. 2003;39:121–132. doi: 10.1016/s0896-6273(03)00390-8. [DOI] [PubMed] [Google Scholar]
- 44.Satoh AK, Xia H, Yan L, Liu CH, Hardie RC, Ready DF. Arrestin translocation is stoichiometric to rhodopsin isomerization and accelerated by phototransduction in Drosophila photoreceptors. Neuron. 2010;67(6):997–1008. doi: 10.1016/j.neuron.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nair KS, Hanson SM, Mendez A, et al. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46:555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu CH, Satoh AK, Postma M, Huang J, Ready DF, Hardie RC. Ca2+-dependent metarhodopsin inactivation mediated by calmodulin and NINAC myosin III. Neuron. 2008;59(5):778–789. doi: 10.1016/j.neuron.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barak LS, Ferguson SS, Zhang J, Caron MG. A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J Biol Chem. 1997;272:27497–27500. doi: 10.1074/jbc.272.44.27497. [DOI] [PubMed] [Google Scholar]
- 48.Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315:201–205. doi: 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- 49.Marchese A, Benovic JL. Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J Biol Chem. 2001;276:45509–45512. doi: 10.1074/jbc.C100527200. [DOI] [PubMed] [Google Scholar]
- 50.Shenoy SK, Lefkowitz RJ. Trafficking patterns of beta-arrestin and G protein-coupled receptors determined by the kinetics of beta-arrestin deubiquitination. J Biol Chem. 2003;278:14498–14506. doi: 10.1074/jbc.M209626200. [DOI] [PubMed] [Google Scholar]
- 51.Coffa S, Breitman M, Hanson SM, et al. The effect of arrestin conformation on the recruitment of c-Raf1, MEK1, and ERK1/2 activation. PLoS One. 2011;6:e28723. doi: 10.1371/journal.pone.0028723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luttrell LM, Roudabush FL, Choy EW, et al. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci USA. 2001;98(5):2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shenoy SK, Barak LS, Xiao K, et al. Ubiquitination of beta-arrestin links seven-transmembrane receptor endocytosis and ERK activation. J Biol Chem. 2007;282:29549–29562. doi: 10.1074/jbc.M700852200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahmed MR, Zhan X, Song X, Kook S, Gurevich VV, Gurevich EV. Ubiquitin ligase parkin promotes Mdm2-arrestin interaction but inhibits arrestin ubiquitination. Biochemistry. 2011;50:3749–3763. doi: 10.1021/bi200175q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bhandari D, Trejo J, Benovic JL, Marchese A. Arrestin-2 interacts with the ubiquitin-protein isopeptide ligase atrophin-interacting protein 4 and mediates endosomal sorting of the chemokine receptor CXCR4. J Biol Chem. 2007;282:36971–36979. doi: 10.1074/jbc.M705085200. [DOI] [PubMed] [Google Scholar]
- 56.Shenoy SK, Xiao K, Venkataramanan V, Snyder PM, Freedman NJ, Weissman AM. NEDD4 mediates agonist-dependent ubiquitination, lysosomal targeting and degradation of the beta 2 adrenergic receptor. J Biol Chem. 2008;283:22166–22176. doi: 10.1074/jbc.M709668200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nabhan JF, Pan H, Lu Q. Arrestin domain-containing protein 3 recruits the NEDD4 E3 ligase to mediate ubiquitination of the beta2-adrenergic receptor. EMBO Rep. 2010;11:605–611. doi: 10.1038/embor.2010.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han SO, Kommaddi RP, Shenoy SK. Distinct roles for β-arrestin2 and arrestin-domain-containing proteins in β2 adrenergic receptor trafficking. EMBO Rep. 2013;14(2):164–171. doi: 10.1038/embor.2012.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berthouze M, Venkataramanan V, Li Y, Shenoy SK. The deubiquitinases USP33 and USP20 coordinate beta2 adrenergic receptor recycling and resensitization. EMBO J. 2009;28(12):1684–1696. doi: 10.1038/emboj.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shenoy SK, Modi AS, Shukla AK, et al. Beta-arrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase Mdm2. Proc Natl Acad Sci USA. 2009;106:6650–6655. doi: 10.1073/pnas.0901083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, barrestin1, and barrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- 62.Van Dyke RW. Acidification of lysosomes and endosomes. Subcell Biochem. 1996;27:331–360. doi: 10.1007/978-1-4615-5833-0_10. [DOI] [PubMed] [Google Scholar]
- 63.Gurevich VV, Dion SB, Onorato JJ, et al. Arrestin interaction with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, β2-adrenergic, and m2 muscarinic cholinergic receptors. J Biol Chem. 1995;270:720–731. doi: 10.1074/jbc.270.2.720. [DOI] [PubMed] [Google Scholar]
- 64.Celver J, Vishnivetskiy SA, Chavkin C, Gurevich VV. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J Biol Chem. 2002;277(11):9043–9048. doi: 10.1074/jbc.M107400200. [DOI] [PubMed] [Google Scholar]
- 65.Kovoor A, Celver J, Abdryashitov RI, Chavkin C, Gurevich VV. Targeted construction of phosphorylation-independent β-arrestin mutants with constitutive activity in cells. J Biol Chem. 1999;274:6831–6834. doi: 10.1074/jbc.274.11.6831. [DOI] [PubMed] [Google Scholar]
- 66.Gurevich VV, Benovic JL. Cell-free expression of visual arrestin. Truncation mutagen-esis identifies multiple domains involved in rhodopsin interaction. J Biol Chem. 1992;267:21919–21923. [PubMed] [Google Scholar]
- 67.Gurevich VV, Benovic JL. Visual arrestin interaction with rhodopsin: sequential multi-site binding ensures strict selectivity towards light-activated phosphorylated rhodopsin. J Biol Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- 68.Palczewski K, McDowell H, Jakes S, Ingebritsen TS, Hargrave PA. Regulation of rhodopsin dephosphorylation by arrestin. J Biol Chem. 1989;264:15770–15773. [PubMed] [Google Scholar]
- 69.Hsieh C, Brown S, Derleth C, Mackie K. Internalization and recycling of the CB1 can-nabinoid receptor. J Neurochem. 1999;73:493–501. doi: 10.1046/j.1471-4159.1999.0730493.x. [DOI] [PubMed] [Google Scholar]
- 70.Morrison KJ, Moore RH, Carsrud ND, et al. Repetitive endocytosis and recycling of the beta 2-adrenergic receptor during agonist-induced steady state redistribution. Mol Pharmacol. 1996;50:692–699. [PubMed] [Google Scholar]
- 71.Gurevich VV, Pals-Rylaarsdam R, Benovic JL, Hosey MM, Onorato JJ. Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J Biol Chem. 1997;272:28849–28852. doi: 10.1074/jbc.272.46.28849. [DOI] [PubMed] [Google Scholar]
- 72.Pan L, Gurevich EV, Gurevich VV. The nature of the arrestin × receptor complex determines the ultimate fate of the internalized receptor. J Biol Chem. 2003;278:11623–11632. doi: 10.1074/jbc.M209532200. [DOI] [PubMed] [Google Scholar]
- 73.Xiao K, Shenoy SK, Nobles K, Lefkowitz RJ. Activation-dependent conformational changes in {beta}-arrestin 2. J Biol Chem. 2004;279(53):55744–55753. doi: 10.1074/jbc.M409785200. [DOI] [PubMed] [Google Scholar]
- 74.Nobles KN, Guan Z, Xiao K, Oas TG, Lefkowitz RJ. The active conformation of beta-arrestin1: direct evidence for the phosphate sensor in the N-domain and conformational differences in the active states of beta-arrestins1 and -2. J Biol Chem. 2007;282(29):21370–21381. doi: 10.1074/jbc.M611483200. [DOI] [PubMed] [Google Scholar]
- 75.Key TA, Foutz TD, Gurevich VV, Sklar LA, Prossnitz ER. N-Formyl peptide receptor phosphorylation domains differentially regulate arrestin and agonist affinity. J Biol Chem. 2003;278:4041–4047. doi: 10.1074/jbc.M204687200. [DOI] [PubMed] [Google Scholar]
- 76.Bennett TA, Maestas DC, Prossnitz ER. Arrestin binding to the G protein-coupled N-formyl peptide receptor is regulated by the conserved “DRY” sequence. J Biol Chem. 2000;275:24590–24594. doi: 10.1074/jbc.C000314200. [DOI] [PubMed] [Google Scholar]
- 77.Vines CM, Revankar CM, Maestas DC, et al. N-Formyl peptide receptors internalize but do not recycle in the absence of arrestins. J Biol Chem. 2003;278(43):41581–41584. doi: 10.1074/jbc.C300291200. [DOI] [PubMed] [Google Scholar]
- 78.McDonald PH, Cote NL, Lin FT, Premont RT, Pitcher JA, Lefkowitz RJ. Identification of NSF as a beta-arrestin1-binding protein. Implications for beta2-adrenergic receptor regulation. J Biol Chem. 1999;274:10677–10680. doi: 10.1074/jbc.274.16.10677. [DOI] [PubMed] [Google Scholar]
- 79.Huang SP, Brown BM, Craft CM. Visual arrestin 1 acts as a modulator for N-ethylmaleimide-sensitive factor in the photoreceptor synapse. J Neurosci. 2010;30:9381–9391. doi: 10.1523/JNEUROSCI.1207-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hanson SM, Cleghorn WM, Francis DJ, et al. Arrestin mobilizes signaling proteins to the cytoskeleton and redirects their activity. J Mol Biol. 2007;368(2):375–387. doi: 10.1016/j.jmb.2007.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hanson SM, Francis DJ, Vishnivetskiy SA, Klug CS, Gurevich VV. Visual arrestin binding to microtubules involves a distinct conformational change. J Biol Chem. 2006;281:9765–9772. doi: 10.1074/jbc.M510738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nair KS, Hanson SM, Kennedy MJ, Hurley JB, Gurevich VV, Slepak VZ. Direct binding of visual arrestin to microtubules determines the differential subcellular localization of its splice variants in rod photoreceptors. J Biol Chem. 2004;279:41240–41248. doi: 10.1074/jbc.M406768200. [DOI] [PubMed] [Google Scholar]
- 83.Shankar H, Michal A, Kern RC, Kang DS, Gurevich VV, Benovic JL. Non-visual arrestins are constitutively associated with the centrosome and regulate centrosome function. J Biol Chem. 2010;285(11):8316–8329. doi: 10.1074/jbc.M109.062521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cleghorn WM, Branch KM, Kook S, et al. Arrestins regulate cell spreading and motility via focal adhesion dynamics. Mol Biol Cell. 2015;26(4):622–635. doi: 10.1091/mbc.E14-02-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]