

Abstract
BACKGROUND
PTEN is well known to function as a tumor suppressor that antagonizes oncogenic signaling and maintains genomic stability. The PTEN gene is frequently deleted or mutated in human cancers and the wide cancer spectrum associated with PTEN deficiency has been recapitulated in a variety of mouse models of Pten deletion or mutation. Pten mutations are highly penetrant in causing various types of spontaneous tumors that often exhibit resistance to anticancer therapies including immunotherapy. Recent studies demonstrate that PTEN also regulates immune functionality.
OBJECTIVE
To understand the multifaceted functions of PTEN as both a tumor suppressor and an immune regulator.
METHODS
This review will summarize the emerging knowledge of PTEN function in cancer immunoediting. In addition, the mechanisms underlying functional integration of various PTEN pathways in regulating cancer evolution and tumor immunity will be highlighted.
RESULTS
Recent preclinical and clinical studies revealed the essential role of PTEN in maintaining immune homeostasis, which significantly expands the repertoire of PTEN functions. Mechanistically, aberrant PTEN signaling alters the interplay between the immune system and tumors, leading to immunosuppression and tumor escape.
CONCLUSION
Rational design of personalized anti-cancer treatment requires mechanistic understanding of diverse PTEN signaling pathways in modulation of the crosstalk between tumor and immune cells.
Keywords: PTEN, phosphoinositide 3-kinase, regulatory T cells, genome, epigenome, metabolism
Introduction
Cancer evolution is a joint consequence of genetic alterations, epigenetic aberration, and metabolic deregulation, which indicates disruption of multiple cellular autonomous machineries. In addition to tumor-intrinsic mechanisms, host-dependent immune surveillance shapes the entire process of tumor development, progression, and response to therapy. While the immune system can elicit protective anti-tumor responses, tumors often foster a tolerant microenvironment and induce immunosuppressive signals to diminish this activity. Understanding the basic mechanisms that control cancer immunoediting and how alteration of such regulatory mechanisms leads to cancer immune evasion is fundamentally important for designing new strategies for treating cancer.
The PTEN gene was discovered in 1997 by mapping of homologous deletion on chromosome 10q23-24 in multiple types of human cancer (Li et al., 1997; Steck et al., 1997). At that time, sequence motif analysis predicted the protein product of this potential tumor suppressor gene to be a protein phosphatase. A year later, PTEN was characterized as a lipid phosphatase that dephosphorylates phosphatidylinositol 3,4,5-trisphosphate (PIP3) by removing the 3-phosphate of the inositol ring (Maehama and Dixon, 1998). This lipid phosphatase function of PTEN antagonizes the catalytic activity of phosphoinositide 3-kinase (PI3K). Given the critical role of PI3K in regulating diverse cell behaviors and properties, PTEN’s antagonism of the PI3K/AKT signaling pathway contributes substantially to its multifaceted functions in cancer development, metabolism and immunity.
PTEN is a powerful tumor suppressor. Its potency in preventing tumorigenesis has been demonstrated by numerous Pten knockout and knockin mouse tumor models that faithfully phenocopied the wide spectrum of human cancers associated with PTEN mutations. Moreover, mechanistic studies revealed that PTEN regulates fundamental cellular processes to oppose oncogenesis. For example, PTEN controls energy metabolism (Garcia-Cao et al., 2012), cell motility (Tamura et al., 1998), genomic stability (Shen et al., 2007), and epigenome architecture (Chen et al., 2014). Although many cellular functions of PTEN rely on its canonical activity in suppressing PI3K signaling, growing evidence suggests that PTEN may protect the genome and prevent oncogenic transformation through PI3K-independent pathways.
In addition to controlling basic cellular functions, PTEN is also required for maintaining immune homeostasis. Following an earlier observation that germline deletion of Pten manifests autoimmune disorders (Di Cristofano et al., 1999), Mak and colleagues demonstrated impairment of central and peripheral immune tolerance using a T cell-specific Pten knockout mouse model (Suzuki et al., 2001). In the past few years have witnessed a dramatic explosion in the amount of evidence showing the important and diverse roles of PTEN in immune regulation. Here we summarize recent studies and current understanding of PTEN signaling in mediating mutual regulation between the immune system and evolving cancer and the implications for cancer treatment.
PTEN in immune homeostasis
The immune system continuously scans its surrounding microenvironment for pathogen associated molecular patterns, signaling molecules and antigens. Environmental stimuli provide a context for ongoing processes involving immune cell recalibration, regulation and activation, and these processes dynamically evolve with the changing milieu. Molecular pathways downstream from the signals converge at focal points, which ultimately tip the balance between a pro-inflammatory response and immune suppression. The PTEN molecule is an important focal point for integrating signals that promote immune tolerance.
PTEN signaling influences a broad array of immune cells of both the innate and adaptive compartments (Table 1). Macrophages that lack PTEN expression are prone to activation. Mice with Pten deletion in the myeloid lineage (PTENMyKO) demonstrated increased pathogenesis in a bleomycin-induced model of pulmonary fibrosis with a mixed M1/M2 population secreting pro-inflammatory and pro-fibrotic cytokines (Kral et al., 2016). Neutrophils with selective deletion of PTEN exhibited enhanced migration into an inflamed peritoneal cavity and increased engulfment of bacteria (Subramanian et al., 2007; Li et al., 2009). B cells with conditional Pten deletion also displayed a hyperactive phenotype with hyperproliferation and a lower threshold for activation through the B cell receptor. A systemic lupus erythematosus (SLE) mouse model linked pathogenic B cell activity to downregulation of Pten expression and increased Akt phosphorylation (Anzelon et al., 2003). PTEN also fundamentally modulates T cell development and function by promoting regulatory signals. In general, T cells have a plastic phenotype that facilitates remarkable flexibility in adapting their immune activity to the context of environmental signals. By modifying the activation status of multiple types of immune cells and modulating T cell phenotypes, PTEN exerts considerable influence over the direction of immune responses. The following section will focus on the role of PTEN in regulating T cell function and controlling the identity and functionality of regulatory T cells (Tregs).
Table 1.
Immune-specific Pten deletion and corresponding phenotypes
| Immune cells (for Pten deletion) | Phenotypes of Pten deficiency | References |
|---|---|---|
| T cells | Defective thymic negative selection; Impaired peripheral tolerance; Spontaneous CD4+ T cell activation; Autoantibody production; CD4+ T cell lymphoma | Suzuki et al., 2001 |
| Peripheral T cells | Increased proliferation and IL-2 production with TCR/CD28 stimulation; Augmented cytolytic activity | Locke et al., 2013* |
| CD4+ T cells | Impaired thymic selection; Increased proliferation; Activation of naïve cells w/o costimulaton; Autoimmunity; Lymphoma | Soone et al., 2012 |
| Regulatory T cells (Treg cells) | Autoimmune-lymphoproliferative disease; Excessive T helper type 1 (Th 1) responses; B cell activation; Excessive TFH cell and germinal center responses; Spontaneous inflammatory disease |
Huynh et al., 2015 Shrestha et al., 2015 |
| B cells | Hyperproliferation, resistance to apoptosis, increased migration, SLE | Anzelon et al., 2003 |
| Dendritic cells (DCs) | Expansion of CD8+ and CD103+ DCs | Jiao et al., 2014** |
| Neutrophils | Delayed apoptosis, augmented transendothelial migration, increased bacteria killing | Subramanian et al., 2007; Li et al., 2009 |
| Macrophages | Decreased TNF-α secretion, increased IL-10 production, increased phagocytosis | Kral et al., 2016 |
Additional refernces:
Locke et al. J Immunol, 2013 Aug 15; 191(4): 1677-85;
Jiao et al. J Immunol. 2014 Apr 1; 192(7): 3374-82.
Ample evidence has revealed a deterministic role of PTEN in maintaining immune homeostasis, especially in the function of CD4+ T cells. Studies modeling PTEN deficiency in various mouse models have elucidated sequelae spanning from thymic development to peripheral immune regulation. The phenotypic results of interrupting PTEN signaling in immunity reveal its comprehensive impact on central and peripheral tolerance. A murine model of global T cell Pten depletion resulted in impairment at several levels of immune homeostasis. Ptenflox/−;Cre-Lck mice demonstrated early onset lymphadenoapthy, splenomegaly and enlarged thymi (Suzuki et al., 2001). Negative thymic selection was defective with impaired deletion of transgenic T cells expressing the HY T cell receptor (TCR) in male mice. Splenic T cells from Ptenflox/− mice showed amplified cytokine production ex vivo in response to stimulation with anti-CD3 and anti-CD28 antibodies. These mice uniformly developed lymphoma by 5 months of age. Importantly, the activated T cells demonstrated markedly elevated levels of phospho-Akt relative to wild type T cells when activated with anti-CD3/CD28, implicating this downstream signaling pathway in the phenotypic dysregulation (Suzuki et al., 2001). Deletion of Pten in mature CD4+ T cells has milder consequences. Mice with a Ptenflox;Ox40Cre background did not develop spontaneous autoimmunity or lymphoma. However, CD4+ T helper cells responded to antigen stimulation with increased proliferation, higher amplitude of cytokine release and greater efficacy in augmenting CD8+ T cell responses (Soond et al., 2012). Taken together, these mouse models demonstrated that deficiency of Pten leads to a state of over-activation in affected T cells.
T cell activation directly signals via the downstream PTEN-PI3K axis. An αβ T cell receptor (TCR) initially recognizes and binds its cognate MHC:peptide antigen complex. Engagement of the TCR is at the cell surface activates Ras which leads to the activation of PI3K. This induces recruitment of AKT and PDK1 to the cell membrane where AKT is phosphorylated and mediates downstream transcriptional signaling (Chen et al., 2001). T cell activation requires two signals, one mediated by TCR engagement with an MHC/antigen complex, and the other by interaction of the co-receptor CD28 with co-stimulatory molecules B7-1 (CD80) and B7-2 (CD86). The CD28 constimulatory signal promotes AKT activation by PI3K and is essential for expression of IL-2 and its receptor as well as T cell activation and proliferation (Kane et al., 2001). Notably, CD4+ T cells with Pten deletion were able to achieve activation in the absence of CD28 costimulation. They also showed robust proliferation and IL-2 production in response to TCR activation in comparison with wild type T cells (Buckler et al., 2006). Usually, naïve T cells that receive TCR stimulation without costimulation undergo a deactivation transition to a state of anergy. However, T cells lacking Pten failed to be anergized in response to superantigen stimulation (Buckler et al., 2006). PTEN modulates T cell activation by founctioning as the central countermeasure to PI3K-mediated lipid phosphorylation.
PTEN supports Treg cell function
CD4+ T regulatory cells (Tregs), which express the canonical FOXP3 transcription factor, are an essential component of the adaptive immune system. Naturally occurring Tregs (nTregs) differentiate from common lymphoid progenitor cells during thymic development and emerge in the periphery as CD4+ T cells with unique αβ T cell receptors (Hsieh et al., 2012). Separately, an inducible population of naïve CD4+ T cells can also acquire expression of FOXP3 in the periphery (iTregs) (Josefowicz et al., 2012). Tregs express a characteristic program of molecules that inhibit effector immune responses. They release suppressive cytokines such as TGF-β and IL-10, act as a sink for the growth factor IL-2, and mediate contact inhibition of effector T (Teff) cells. Naïve CD4+ T cells in the periphery can reciprocally differentiate to either Th17 cells or Tregs depending on the environmental signals associated with activation, particularly the cytokine milieu. Several signaling pathways in Treg cells feed into the PTEN-PI3K axis, in order to render the downstream substrate, AKT, in a deactivated state. This includes maintaining dephosphorylation at Ser473 and Thr308 (Chen et al., 2001). Ex vivo analysis of human CD4+CD25+ and CD4+FOXP3+ T cells demonstrated an impaired ability to phosphorylate Ser473 in response to TCR stimulation. After inducing expression of a constitutively active form of AKT in Tregs, they acquired the ability to secrete pro-inflammatory cytokines, including IFN-γ and TNF-α (Crellin et al., 2007). Forced activation of AKT disrupts the regulatory program and lineage commitment of Tregs. Therefore, maintenance of deactivated AKT is an integral part of Tregs retaining a suppressive phenotype.
Tregs respond to inflammatory stimuli by upregulating PTEN, which promotes their regulatory function (Francisco et al., 2009). Immune checkpoint molecules contribute to this process when engaged by their ligands. Programmed death 1 (PD-1) is induced on activated T cells, including Tregs, through TCR engagement or stimulation with common γ-chain cytokines (Terawaki et al., 2011). PD-1 ligands (PD-L1 and PD-L2) are upregulated on antigen presenting cells (APCs), stromal cells and endothelial tissue in response to multiple inflammatory stimuli including common γ-chain cytokines, interferons, TNF-α, IL-10, IL-4 and GM-CSF (Eppihimer et al., 2002; Loke and Allison, 2003). Engagement of PD-1 decreases the expression and inhibits the function of Casein kinase 2 (CK2), a serine/threonine protein kinase that acts as a regulator of PTEN (Patsoukis et al., 2013). CK2 normally phosphorylates three amino acids in the C terminus of PTEN, which maintains its stability and inhibits its phosphatase activity (Torres and Pulido, 2001; Vazquez et al., 2000). Therefore, decreased levels of CK2 lead to increased enzymatic activity and turnover of PTEN. A second cell surface checkpoint molecule, Neuropilin-1 (NRP-1), is expressed at high levels on nTregs (Yadav et al., 2012). The ligand for NRP-1 is semaphorin-A, which is expressed on Teff cells and dendritic cells (DCs). Binding of semaphorin-A to NRP-1 promotes localization of PTEN at the immunologic synapse which helps limit downstream phosphorylation AKT (Fig. 1). NRP-1 signaling induces transcriptional alterations that favor Treg survival and stability (Delgoffe et al., 2013).
Figure 1.
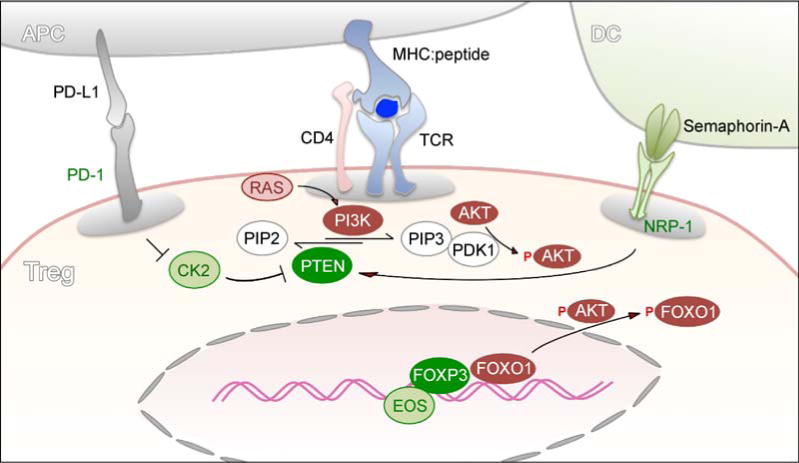
PTEN signaling in Treg cells. The canonical PTEN-PI3K-AKT signaling pathway plays a central role in maintaining the identity of Treg cells and their lineage stability. PTEN antagonizes PI3K-mediated generation of the second messenger, PIP3, and subsequent AKT phosphorylation. Upon TCR engagement, activated AKT phosphorylates FOXO proteins (such as FOXO1) and prevents them from entering the nucleus. Nuclear exclusion of FOXO proteins abrogates their transcriptional activity as a partner for FOXP3, leading to Treg instability. Checkpoint molecules that mediate the PD-1-CK2-PTEN pathway and the NRP1-PTEN pathway help maintain the level of PTEN in Tregs. The PD-1 suppressive signal reduces the expression of CK2, which prevents CK2-mediated PTEN degradation. NRP1 is highly expressed on natural Treg cells. Upon interaction of NRP1 with its ligand, semaphorin-A, PTEN is recruited to the immunological synapse to antagonize PI3K and promote Treg stability.
Development and maintenance of Treg cells requires expression of a core program of transcription factors, some of which are linked to the PTEN-PI3K axis. Changes in the levels or nuclear localization of these molecules can destabilize Tregs and alter their function. FOXO1 and FOXO3A are members of the forkhead box O family of transcription factors. In quiescent T cells, they are localized to the nucleus, but following TCR stimulation, they are exported to the cytosol (Riou et al., 2007; Ouyang et al., 2012). This transition is attributable to PI3K-mediated activation of AKT, which promotes phosphorylation and nuclear exclusion of FOXO proteins (Biggs et al., 1999; Brunet et al., 1999). Importantly, Tregs demonstrate attenuated nuclear clearance of FOXO1, even in the presence of low-level TCR stimulation. Continuous FOXO1 signaling is required to prevent Tregs from adopting an immunogenic phenotype and secreting IFN-γ (Ouyang et al., 2012). EOS, a member of the Ikaros family of molecules, is also a critical transcription factor for Treg maintenance. EOS acts as a corepressor with FOXP3 to suppress expression of an array of immune related genes, including IL-2 (Pan et al., 2009). A subpopulation of Tregs with the surface profile CD38+CD69+CD103− has demonstrated flexible expression of EOS with the ability to downregulate the gene when exposed to the pro-inflammatory cytokine IL-6. Plasticity of EOS-labile Tregs was shown to be important for licensing DCs to prime naïve CD8+ and CD4+ T cells (Sharma et al., 2013). While a direct mechanistic connection between PTEN and EOS has not been established, a correlation between Pten expression and Eos levels was shown in an in vivo tumor model with Pten-deficient Tregs. Importantly, Pten deletion resulted in downregulation of Eos in a large percentage of intratumoral Tregs with differentiation toward a pro-inflammatory phenotype (Sharma et al., 2015). As shown by the phenotypic drift of Tregs in murine knockout models, Pten plays a key role in preserving their functional integrity (Fig. 1).
Multiple signaling cascades and transcription factors that promote Treg cell function appear to rely on intact PTEN signaling. Turka and colleagues evaluated this phenomenon in a Pten-ΔTreg mouse model with deletion of the Pten gene in Foxp3 expressing cells. Pten-deficient Treg cells showed increased phosphorylation of Akt, both at baseline and with CD3/CD28 costimulation. Eighty percent of mice developed an autoimmune-lymphoproliferative disorder by 28 weeks of age. Tregs in the mutant mice proliferated at a higher rate than wild type Tregs, and they were defective in suppressing Th17 mediated autoimmunity in the setting of experimental autoimmune encephalomyelitis (EAE). These Tregs also displayed features suggestive of pathogenic conversion, including secretion of IL-17 and IFN-γ (Huynh et al., 2015). Other studies have illustrated the inherent plasticity of Treg cells. For example, a subset characterized by CD25loFOXP3+ has an inherent capacity to undergo pathogenic conversion into Th17 cells in the setting of autoimmune arthritis (Komatsu et al., 2014). Furthermore, loss of Pten expression impairs the ability of Treg cells to suppress Th1 and T follicular helper (Tfh) responses. Tfh cells are concentrated in lymphoid germinal centers and assist with B cell generation of plasma cells and memory B cells. Ptenfl/flFoxp3-Cre mice had increased germinal center formation and serum levels of IgG (Shrestha et al., 2015). At the molecular and phenotypic level, PTEN expression is linked with the phenotypic stability of Treg cells.
The relationship between T cell metabolism and phenotype is becoming more apparent, and emerging models demonstrate a stratified pattern of metabolic activity among Treg versus Teff cells. CD4+ Teff cells express high levels of the glucose transporter protein, GLUT1, and demonstrate correspondingly high levels of glycolytic activity. Tregs, however, are low expressors of GLUT1, and they preferentially utilize lipid oxidation. Transgenic mice that overexpressed GLUT1 were found to have increased levels of Teff cells, whereas stimulation of AMP-activated protein kinase (AMPK) increased Treg generation (Michalek et al., 2011). T cell metabolism is dynamic, and shifting utilization of various substrates is essential for T cell plasticity. When naïve CD4+ T cells are stimulated under Th17 polarizing conditions, HIF1α expression increases to promote glycolytic metabolism. A deficiency in HIF1α hinders Th17 differentiation (Shi et al., 2011). Conversely, PD-1 signaling leading to upregulation of PTEN and downstream inhibition of AKT was shown to promote expression of carnitine palmitoyltransferase 1A, a rate-limiting enzyme in fatty acid oxidation. This pathway effectively maintains resting levels of oxidative metabolism, which is characteristic of Treg and memory T cells (Patsoukis et al., 2015). The metabolic profile of individual immune cells has a deterministic impact on their phenotype (Newton et al., 2016). PTEN’s central role in promoting lipid oxidative metabolism is another fundamental way it sustains the Treg cell phenotype and maintains immune homeostasis.
PTEN as a tumor suppressor
PTEN is among the most frequently deleted or mutated genes in human cancer. The development of a wide spectrum of cancers has been recapitulated in mice by deleting Pten or introducing Pten mutations derived from cancer patients. These findings establish PTEN as a powerful tumor suppressor. The fundamental homeostatic role of PTEN has been demonstrated by prominent alterations of the metabolome, genome, and epigenome in its absence (Fig. 2).
Figure 2.
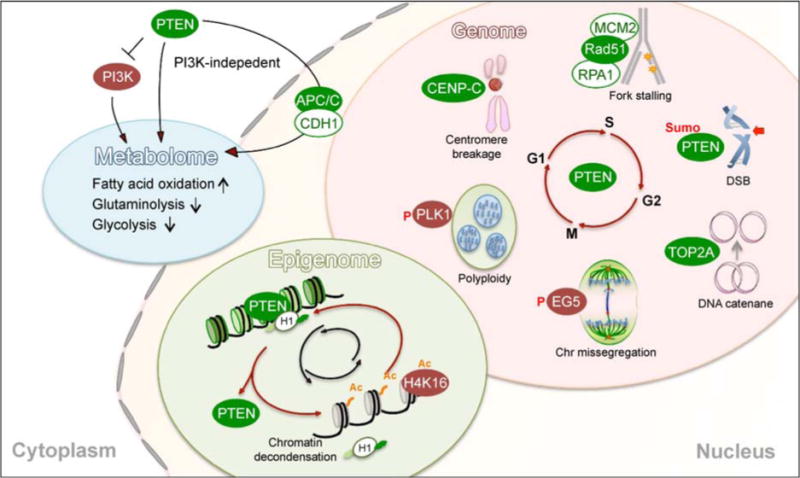
PTEN acts as a guardian of the genome, epigenome and metabolome to maintain cellular homeostasis. PTEN functions in both the cytoplasm and the nucleus to control multiple fundamental machineries for tumor suppression. PTEN regulates fatty acid oxidation, glycolysis and glutaminolysis in both PI3K–dependent and –independent manners. PTEN also functions in the nucleus to regulate multiple processes of genetic transmission during the cell cycle. When PTEN is mutated or inactivated, cells exhibit DNA replication and chromosome segregation defects, as well as structural aberrations such as centromere breakage, DSBs, chromosome entanglement, aneuploidy and polyploidy. Moreover, PTEN maintains proper chromatin organization through epigenetic regulation of histone modification.
PTEN has been shown to regulate fatty acid oxidation by inhibiting PI3K/AKT-mediated phosphorylation of FOXO1 and FOXO3a in brown adipose tissues, leading to increased energy expenditure (Ortega-Molina et al., 2012). Notably, this is the same PI3K/AKT/FOXO pathway previously described to maintain FOXP3 transcription activity and lineage stability in Treg cells. The distinct roles of the PTEN/PI3K/AKT/FOXO signaling axis in adipocytes and Tregs suggest that a conserved PTEN signaling pathway can be employed by different tissues to regulate diverse cellular functions and biological processes.
Multiple mechanisms are involved in PTEN regulation of cellular metabolism, some of which are independent of the PI3K pathway. Tumor cells exhibit an altered metabolism featuring increased glutaminolysis and a higher level of aerobic glycolysis. Pandolfi and colleagues demonstrated that PTEN reduces glutaminolysis and prevents the Warburg effect by utilizing both PI3K–dependent and –independent mechanisms. PTEN reduces glucose uptake by suppressing the PI3K/AKT pathway, whereas it enhances the stability of glutaminase by inhibiting proteasome-dependent protein degradation in a PI3K-independent manner (Garcia-Cao et al., 2012).
In addition to metabolic deregulation, genomic instability is another hallmark of cancer (Hanahan and Weinberg, 2011). Genomic stability relies on faithful transmission of DNA during each cell cycle. Recent studies demonstrated that PTEN controls multiple critical processes of genetic transmission, including DNA replication, DNA repair, and chromosome segregation. PTEN maintains the structural integrity of chromosomes, which is an integral part of its role as a guardian of the genome (Kritikou, 2007; Shen et al., 2007; Yin and Shen, 2008).
PTEN promotes accurate and efficient DNA replication and facilitates stalled fork restart in response to replication stress (Feng et al., 2015; He et al., 2015; Wang et al., 2015). When PTEN is absent, frequent interruption of DNA replication causes fork stalling and collapse. Moreover, lack of proper checkpoints results in cellular tolerance to stalled forks and premature cell cycle progression to mitosis with an unreplicated genome and damaged DNA (He et al., 2015). PTEN regulates DNA replication by recruiting important molecules such as replication protein A (RPA) and Rad51 to the replication fork. PTEN also restricts fork progression in response to replication stress by using a non-canonical protein phosphatase activity to dephosphorylate DNA helicase MCM2 (Feng et al., 2015). As all these molecular events occur in the nucleus, where dephosphorylation of PIP3 may not be the primary activity of PTEN, the regulation of DNA replication by nuclear PTEN is likely independent of the PI3K pathway.
Impaired DNA replication often results in accumulation of DNA catenanes, which must be resolved by DNA decatenation to license subsequent mitosis. PTEN has been shown to facilitate this process by maintaining the protein stability of topoisomerase IIα (TOP2A), a critical enzyme in DNA decatenation. In the absence of PTEN or TOP2A, cells exhibit a significantly higher tendency toward chromosomal entanglement (Kang et al., 2015). Cells lacking PTEN also harbor a higher basal level of DNA double strand breaks (DSBs), indicating defective DNA repair. Indeed, growing evidence has shown that PTEN is required for homology directed repair of DSBs (Shen et al., 2007; Mendes-Pereira et al., 2009; McEllin et al., 2010; Bassi et al., 2013). Interestingly, post-translational modification of PTEN by sumoylation assures its nuclear localization, which is essential for eliciting homologous recombination (HR)-dependent DNA repair (Bassi et al., 2013). Therefore, cells lacking PTEN demonstrate spontaneous accumulation of DNA damage and entanglement as a combined consequence of impaired DNA decatenation and repair (Fig. 2).
In addition to structural chromosome aberrations, aneuploidy and polyploidy frequently occur in cells lacking functional PTEN. A prior study showed that nuclear PTEN controls cellular senescence by interacting with an E3 ubiquitin ligase complex, APC-CDH1 (Song et al., 2011). In this report, loss of nuclear PTEN or the APC-CDH1 complex resulted in an enhanced retention of several mitotic kinases, suggesting a potential role of PTEN in regulating mitosis. The direct involvement of PTEN in mitotic control has been demonstrated recently. PTEN prevents chromosome missegregation and aneuploidy/polyploidy by regulating multiple critical mitotic proteins (He et al., 2016; Zhang et al., 2016; van Ree et al., 2016). Interestingly, PTEN utilizes distinct mechanisms in regulating EG5, a microtubule-based motor protein. PTEN maintains the optimal amount and activity of EG5 on the mitotic spindle by acting as a protein phosphatase (He et al., 2016), whereas PTEN can also recruit EG5 to centrosomes by using its PDZ binding domain (van Ree et al., 2016). These data suggest that PTEN may integrate multiple mechanisms to ensure proper behaviors of crucial mitotic proteins for faithful genetic transmission.
Both mitotic and non-mitotic processes of genetic transmission are executed in a chromatin environment that is undergoing dynamic condensation and decondensation. It is increasingly evident that PTEN controls global chromatin architecture. For example, the heterochromatin structure is markedly impaired following PTEN deletion (Chen et al., 2014; Gong et al., 2015). Mechanistic studies revealed that PTEN physically interacts with a major heterochromatin protein, HP1α, and the linker histone H1. Loss of PTEN results in release of these key components from chromatin, leading to impairment of normal chromatin compaction. The decondensed chromatin in PTEN-deficient cells is labeled with an epigenetic mark of histone H4 hyperacetylation at K16. Interestingly, chemical or genetic enforcement of H4K16 hyperacetylation interrupts the interaction between PTEN and histone H1, which provides a feed forward loop for progressive chromatin decondensation in response to PTEN dysfunction (Chen et al., 2014). The epigenetic deregulation of global chromatin architecture may impact all DNA-templated processes and contribute to the aberrant genetic transmission as described above in PTEN-deficient cells (Fig. 2).
In the absence of PTEN, metabolic, genetic and epigenetic aberrations creat intrinsic stress that impacts the cell’s fate and phenotype and alters its their response to therapeutic interventions. Conditional deletion of Pten in the mouse prostate triggers development of prostate cancer, which is accompanied by a cellular senescence phenotype (Chen et al., 2005). Consistently, loss of PTEN in glioma cells confers cellular senescence in response to ionizing radiation whereas PTEN-proficient cells undergo apoptosis under the same treatment (Lee et al., 2011). Together, these results suggest that PTEN plays a central role in switching cell fate in response to endogenous or exogenous genotoxic stress. Although senescence serves as a barrier to oppose tumor development and progression (Nardella et al., 2011), a recent study revealed that Pten null senescent tumor cells can promote growth of adjacent non-senescent tumor cells and result in chemoresistance (Toso et al., 2014). These effects are mediated by a mechanism associated with the senescence-associated secretory phenotype (SASP). Specifically, Pten null senescent tumors secrete multiple cytokines that signal immune inhibition. They also recruit immunosuppressive CD11b+ Gr-1+ myeloid cells to suppress CD8+ T cell proliferation, and thereby prevent adaptive tumor immunity. Further mechanistic study uncovered that these effects are largely mediated by activation of the Jak2/Stat3 pathway. Pharmacological inhibition of the Jak2/Stat3 pathway elicits a strong antitumor immune response and redirects Pten null senescent cells to apoptosis in response to chemotherapy (Toso et al., 2014). These observations demonstrate the pivotal role of PTEN signaling in mediating the functional interplay between tumor cells and the anti-tumor immune response.
Role of PTEN in anti-tumor immunity and immunoediting
As previously discussed, PTEN is ubiquitously expressed across tissue types, and its control over cell signaling has broad implications for cancer and immunity. PTEN is frequently mutated in sporadic cancers as well as hereditary tumor predisposition syndromes such as Cowden syndrome (CS), a typical PTEN hamartoma tumor syndrome (PHTS). Over 80% of patients with CS carry germline PTEN mutations, representing a unique clinical setting for the co-evolution of both tumors and the immune system deficient in PTEN. Germline deletion of Pten or knockin of Cowden-derived Pten mutations in mice closely models the tumor susceptibility seen in patients, corroborating the importance of PTEN in tumor suppression (Di Cristofano et al., 1998; Podsypanina et al., 1999; Stambolic et al., 2000; Sun et al., 2014; Papa et al., 2014). Interestingly, heterozygous germline Pten deletion not only results in a robust cancer phenotype but also disrupts immune regulation and predisposes to autoimmunity (Di Cristofano et al., 1999). A recent investigation of the immunological phenotype in PHTS patients provides a direct clinical correlate to these findings. Seventy nine patients with germline heterozygous mutations of PTEN were evaluated, and 43% of them were found to manifest some form of autoimmunity, lymphoid hyperplasia or both (Chen et al., 2016). Collectively, these findings implicate a dual role of PTEN in both tumor suppression and immune tolerance, and they underscore the relevance of exploring PTEN function in immune regulation.
The ability of PTEN to suppress hyperplasia and oncogenesis has been largely attributed to its cancer cell intrinsic effects. However, PTEN is also an essential modulator of tumor-extrinsic mechanisms. For example, PTEN plays a dual role in controlling cell motility and reshaping the tumor microenvironment. PTEN inhibits cell migration and focal adhesion by inactivating focal adhesion kinase (FAK) (Tamura et al., 1998). Hyperactivation of FAK can confer a fibrous tumor environment and has been recently demonstrated to contribute to the poor response of pancreatic cancer to immunotherapy. Inhibition of FAK diminishes fibrosis and renders previously unresponsive pancreatic ductal adenocarcinoma susceptible to T cell immunotherapy and PD-1 blockade (Jiang et al., 2016). There is also direct evidence that PTEN plays essential roles in the tumor stroma to modulate tumor progression and immune responses. Pten inactivation in stromal fibroblasts of mouse mammary glands promotes breast cancer development and progression by altering the transcriptome related to extra cellular matrix (ECM) and inducing an oncogenic secretome through a Pten-Ets2 pathway (Trimboli et al., 2009; Bronisz et al., 2012). Interestingly, loss of Pten in the tumor stroma not only increases ECM deposition, but also induces infiltration of F4/80-positive macrophages that is known to mediate peripheral tolerance. More significantly, simultaneous knockout of Ets2 in PtenloxP/loxP stromal cells diminishes F4/80-positive macrophages, and decreases tumor growth and progression (Trimboli et al., 2009). These studies suggest that PTEN dysfunction in both the tumor stroma or the tumor itself may reprogram the tumor microenvironment as well as the immune response (Fig. 3).
Figure 3.
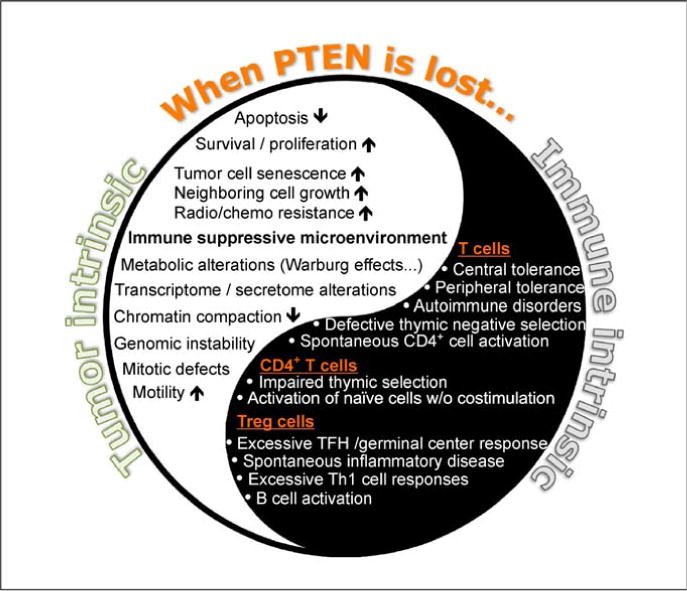
The Yin and Yang of PTEN-mediated cancer immunoediting: implication for targeted anti-tumor therapy. Alterations of PTEN and its signaling significantly affect immune regulation and the process of malignant transformation. PTEN is frequently mutated or inactivated during cancer evolution. Loss of PTEN leads to activation of multiple tumor-intrinsic oncogenic signals that can serve as therapeutic targets for anti-cancer treatment. On the other hand, PTEN inactivation disrupts immune tolerance and can enhance anti-tumor immunity. A thorough understanding of both the Yin and Yang of PTEN function at the immunological synapse is required to achieve the dual goal of promoting PTEN activity in tumors and suppressing PTEN signaling in regulatory immune cells.
There is a complex interplay between tumor progression and immunosurveillance, which is delineated by the cancer immunoediting model into a three-stage process: elimination, equilibrium, and escape (Dunn et al., 2002; Schreiber et al., 2011; Teng et al., 2015). The critical role of the immune system as an extrinsic tumor suppressor mechanism has been well recognized (Vesely et al., 2011), and numerous studies in patients have demonstrated an improved clinical outcome with the presence of tumor infiltrating lymphocytes (Galon et al., 2013). Nevertheless, cancer can utilize several immunosuppressive mechanisms to escape immune surveillance. Remarkable clinical success has been achieved for multiple cancers by inhibiting suppressive immune checkpoint receptors, such as CTLA-4 and PD-1, to activate anti-tumor T cells (Sharma and Allison, 2015). An improved understanding of tumor immunoediting is required to foster future development of treatments that target relevant immunosuppressive pathways.
In this context, the role of PTEN in guiding Treg cell function and modulating effector T cell activation deserves more attention. Munn and colleagues investigated a preclinical model of Pten inhibition. C57BL/6 mice with Treg-specific Pten deletion demonstrated reduced tumor growth when challenged with B16 melanoma. In wild type mice, utilization of a small molecule inhibitor of PTEN together with a single dose of cytoxan showed synergistic therapeutic efficacy of tumor control with associated activation of intratumoral dendritic cells and CD8+ T cells (Sharma et al., 2015). It is reasonable to deduce that pharmacologic inhibition of PTEN function removes a key regulatory signal that leads to destabilization of Tregs and potent anti-tumor immunity. These results suggest a potential translational approach for targeting PTEN to elicit intratumoral T cell responses.
PTEN’s role in regulating both oncogenesis and immunity places it at the interface between the tumor and immune system with different regulatory processes in the balance. Enhancement of immune cell function may come at the expense of downregulating a tumor suppressor and potentially enabling tumor progression. For example, an analysis of outcomes in melanoma patients with a BRAF mutation found that loss of PTEN expression correlated with poorer overall survival and decreased time to onset of melanoma brain metastases (Bucheit et al., 2014). Tumor modifications in the PTEN/PI3K/AKT axis cause significant biologic changes that have been shown to impact sensitivity to conventional treatments. HER-2 positive breast cancers with PTEN mutations or activation of PI3K/AKT were found to be resistant to treatment with the anti-HER2 antibody, trastuzumab (Dave et al., 2011). In esophageal cancer patients treated with chemoradiotherapy, increased genetic variations in AKT were associated with an increased risk of recurrence (Hildebrandt et al., 2009).
Loss of PTEN in tumor cells also impacts their interaction with the immune system. PTEN null prostate epithelium was found to secrete increased levels of CSF1 and IL-1β, which induced expansion of myeloid-derived suppressor cells and promoted tumor progression. Hwu and colleagues investigated the impact of PI3K/PTEN targeting on the tumor-immune interface. They observed that loss of PTEN expression in melanoma reduced CD8+ T cell mediated cytolysis of tumors in vitro. Tumors with reduced PTEN expression were also resistant to lymphocyte infiltration in vivo. Two key mechanisms connected to these findings were upregulation of VEGF expression and downregulation of autophagy by the tumor cells (Peng et al., 2016). In the setting of immunotherapy, it is important to recognize potential imbalances (both favorable and unfavorable) that result from inhibiting PTEN (Fig. 3). Downregulation of PTEN in T cells is favorable for generating anti-tumor immunity, but it also can induce tumor-intrinsic mechanisms that recruit myeloid-derived suppressor cells (MDSCs) and attenuate lymphocyte infiltration and T cell cytotoxic activity. Clinical studies have helped illustrate this concept. In patients with glioblastoma multiforme, expression of PD-L1 was increased in tumor samples with genetic deletions or mutations of PTEN. Selected tumor samples from these patients were also resistant to T cell killing when tested in vitro, whereas they demonstrated greater sensitivity to T cell killing following transfection with wild-type PTEN (Parsa et al., 2007). Results from cohorts of melanoma patients also showed trends suggestive of an immunosuppressive phenotype in tumors lacking PTEN. Patients with < 10% of tumor cells positive for PTEN by IHC showed significantly less volume response to anti-PD-1 therapy compared to patients with PTEN positive tumors (Parsa et al., 2007). Among a separate cohort of patients undergoing expansion of tumor infiltrating lymphocytes (TIL) for adoptive cell therapy, PTEN-negative tumors yielded a significantly lower rate of successful TIL growth compared to PTEN-positive tumors (Peng et al., 2016). Thus, both preclinical models and patient data indicate that loss or mutations of PTEN are associated with downregulation of anti-tumor immunity.
Ultimately, further investigation is needed to probe the translational utility of pharmacologically targeting PTEN for tumor treatment. One of the central unanswered questions is whether therapeutic gains from PTEN inhibition would, on balance, outweigh any collateral effects that are favorable to the tumor. Hwu et al. approached this problem by utilizing selective inhibition of PI3Kβ, an isoform that is not essential in the TCR activation pathway. With use of a small molecule inhibitor, they demonstrated decreased activation of AKT in tumor cells, increased T cell killing, and improved tumor treatment efficacy when combined with a PD-1 inhibitor. Beyond modulating PTEN directly, further investigation is also needed to elucidate downstream substrates as potential therapeutic targets. As previously described, there are multiple molecules in the PTEN pathway that promote cellular activation, metabolism and transcriptional regulation (Fig. 3). For example, diminished expression of FOXP3, EOS or FOXO proteins in Tregs can significantly abrogate their suppressive phenotype and even reprogram them with pro-inflammatory behavior. Further exploration of the activity of PTEN in T cells and tumor cells may reveal more specific targets for cancer immunotherapy.
Concluding remarks
Nearly 20 years of PTEN research has led to many new perspectives on this powerful tumor suppressor. Recognition of the multifaceted immune regulatory functions of PTEN and its role at the immunologic synapse represents a significant expansion from its originally elucidated role as a tumor suppressor. The majority of immune-related functions of PTEN have been attributed to its canonical activity that opposes PI3K signaling. Nevertheless, PTEN mediates a wide variety of biological activities beyond dephosphorylating PIP3. As a tumor suppressor, PTEN targets diverse molecules and pathways in both the cytoplasm and the nucleus to regulate the cancer-intrinsic metabolome, genome and epigenome. Some molecular mechanisms identified in cancer cells, may serve as common pathways in distinct cell systems and can be adopted by immune cells to regulate immune-related functions. Similarly, newly discovered PTEN immune functions may help clarify our understanding of previously uninterpretable tumor phenotypes.
Given the central position of PTEN in the setting of a tumor-immune interaction, further characterization of its signaling in immunoediting is expected to yield novel approaches to enhance tumor immunity. While the immune compartment has been extensively studied in tissue-specific Pten-deficient mice, few reports have investigated the interaction between the spontaneously developed tumors and infiltrating immune cells in Pten-deficient cancer models. Such studies would potentially inform a better understanding of the significance of PTEN deficiency in both tumor and immune cells. Collaboration among cancer biologists, immunologists, and oncologists will help uncover the genetic, epigenetic and metabolic mechanisms underlying the tumor-immune interaction and lead to new therapeutic strategies targeting the molecular and immune signature of each cancer patient.
Acknowledgments
Work in the authors’ laboratory is supported by NIH grant R01GM100478 and the Irma T. Hirschl/Monique Weill-Caulier Trust.
Footnotes
Compliance with ethics guidelines
The authors declare that they have no conflict of interest. This manuscript is a review article and does not involve a research protocol requiring approval by the relevant institutional review board or ethics committee.
References
- Anzelon AN, Wu H, Rickert RC. Pten inactivation alters peripheral B lymphocyte fate and reconstitutes CD19 function. Nat Immunol. 2003;4(3):287–294. doi: 10.1038/ni892. [DOI] [PubMed] [Google Scholar]
- Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B, Stambolic V. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science. 2013;341(6144):395–399. doi: 10.1126/science.1236188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci USA. 1999;96(13):7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronisz A, Godlewski J, Wallace JA, Merchant AS, Nowicki MO, Mathsyaraja H, Srinivasan R, Trimboli AJ, Martin CK, Li F, Yu L, Fernandez SA, Pécot T, Rosol TJ, Cory S, Hallett M, Park M, Piper MG, Marsh CB, Yee LD, Jimenez RE, Nuovo G, Lawler SE, Chiocca EA, Leone G, Ostrowski MC. Reprogramming of the tumour microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol. 2012;14(2):159–167. doi: 10.1038/ncb2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96(6):857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Bucheit AD, Chen G, Siroy A, Tetzlaff M, Broaddus R, Milton D, Fox P, Bassett R, Hwu P, Gershenwald JE, Lazar AJ, Davies MA. Complete loss of PTEN protein expression correlates with shorter time to brain metastasis and survival in stage IIIB/C melanoma patients with BRAFV600 mutations. Clin Cancer Res. 2014;20(21):5527–5536. doi: 10.1158/1078-0432.CCR-14-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler JL, Walsh PT, Porrett PM, Choi Y, Turka LA. Cutting edge: T cell requirement for CD28 costimulation is due to negative regulation of TCR signals by PTEN. J Immunol. 2006;177(7):4262–4266. doi: 10.4049/jimmunol.177.7.4262. [DOI] [PubMed] [Google Scholar]
- Chen HH, Handel N, Ngeow J, Muller J, Huhn M, Yang HT, Heindl M, Berbers RM, Hegazy AN, Kionke J, Travis S, Merkenschlager A, Kiess W, Wittekind C, Walker L, Ehl S, Yehia L, Sack U, Blaser R, Rensing-Ehl A, Reifenberger J, Keith J. Immune dysregulation in patients with PTEN hamartoma tumor syndrome: Analysis of FOXP3 regulatory T cells. J Allergy Clin Immunol. 2016;139(2):607–620. doi: 10.1016/j.jaci.2016.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Kim O, Yang J, Sato K, Eisenmann KM, McCarthy J, Chen H, Qiu Y. Regulation of Akt/PKB activation by tyrosine phosphorylation. J Biol Chem. 2001;276(34):31858–31862. doi: 10.1074/jbc.C100271200. [DOI] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Paolo Pandolfi P. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZH, Zhu M, Yang J, Liang H, He J, He S, Wang P, Kang X, McNutt MA, Yin Y, Shen WH. PTEN interacts with histone H1 and controls chromatin condensation. Cell Reports. 2014;8(6):2003–2014. doi: 10.1016/j.celrep.2014.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crellin NK, Garcia RV, Levings MK. Altered activation of AKT is required for the suppressive function of human CD4+CD25+ T regulatory cells. Blood. 2007;109(5):2014–2022. doi: 10.1182/blood-2006-07-035279. [DOI] [PubMed] [Google Scholar]
- Dave B, Migliaccio I, Gutierrez MC, Wu MF, Chamness GC, Wong H, Narasanna A, Chakrabarty A, Hilsenbeck SG, Huang J, Rimawi M, Schiff R, Arteaga C, Osborne CK, Chang JC. Loss of phosphatase and tensin homolog or phosphoinositol-3 kinase activation and response to trastuzumab or lapatinib in human epidermal growth factor receptor 2-overexpressing locally advanced breast cancers. J Clin Oncol. 2011;29(2):166–173. doi: 10.1200/JCO.2009.27.7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe GM, Woo SR, Turnis ME, Gravano DM, Guy C, Overacre AE, Bettini ML, Vogel P, Finkelstein D, Bonnevier J, Workman CJ, Vignali DAA. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature. 2013;501(7466):252–256. doi: 10.1038/nature12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP. Impaired Fas response and autoimmunity in Pten+/– mice. Science. 1999;285(5436):2122–2125. doi: 10.1126/science.285.5436.2122. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19(4):348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- Eppihimer MJ, Gunn J, Freeman GJ, Greenfield EA, Chernova T, Erickson J, Leonard JP. Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation. 2002;9(2):133–145. doi: 10.1038/sj/mn/7800123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Liang J, Li J, Li Y, Liang H, Zhao X, McNutt MA, Yin Y. PTEN Controls the DNA Replication Process through MCM2 in Response to Replicative Stress. Cell Reports. 2015;13(7):1295–1303. doi: 10.1016/j.celrep.2015.10.016. [DOI] [PubMed] [Google Scholar]
- Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206(13):3015–3029. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39(1):11–26. doi: 10.1016/j.immuni.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VC, Anastasiou D, Ito K, Sasaki AT, Rameh L, Carracedo A, Vander Heiden MG, Cantley LC, Pinton P, Haigis MC, Pandolfi PP. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell. 2012;149(1):49–62. doi: 10.1016/j.cell.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L, Govan JM, Evans EB, Dai H, Wang E, Lee SW, Lin HK, Lazar AJ, Mills GB, Lin SY. Nuclear PTEN tumor-suppressor functions through maintaining heterochromatin structure. Cell Cycle. 2015;14(14):2323–2332. doi: 10.1080/15384101.2015.1044174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- He J, Kang X, Yin Y, Chao KS, Shen WH. PTEN regulates DNA replication progression and stalled fork recovery. Nat Commun. 2015;6:7620. doi: 10.1038/ncomms8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Zhang Z, Ouyang M, Yang F, Hao H, Lamb KL, Yang J, Yin Y, Shen WH. PTEN regulates EG5 to control spindle architecture and chromosome congression during mitosis. Nat Commun. 2016;7:12355. doi: 10.1038/ncomms12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt MA, Yang H, Hung MC, Izzo JG, Huang M, Lin J, Ajani JA, Wu X. Genetic variations in the PI3K/PTEN/AKT/mTOR pathway are associated with clinical outcomes in esophageal cancer patients treated with chemoradiotherapy. J Clin Oncol. 2009;27(6):857–871. doi: 10.1200/JCO.2008.17.6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CS, Lee HM, Lio CW. Selection of regulatory T cells in the thymus. Nat Rev Immunol. 2012;12(3):157–167. doi: 10.1038/nri3155. [DOI] [PubMed] [Google Scholar]
- Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, Townamchai N, Gerriets VA, Rathmell JC, Sharpe AH, Bluestone JA, Turka LA. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol. 2015;16(2):188–196. doi: 10.1038/ni.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, Pachter JA, Wang-Gillam A, DeNardo DG. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22(8):851–860. doi: 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LP, Andres PG, Howland KC, Abbas AK, Weiss A. Akt provides the CD28 costimulatory signal for up-regulation of IL-2 and IFN-gamma but not TH2 cytokines. Nat Immunol. 2001;2(1):37–44. doi: 10.1038/83144. [DOI] [PubMed] [Google Scholar]
- Kang X, Song C, Du X, Zhang C, Liu Y, Liang L, He J, Lamb K, Shen WH, Yin Y. PTEN stabilizes TOP2A and regulates the DNA decatenation. Sci Rep. 2015;5:17873. doi: 10.1038/srep17873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, Tanaka S, Bluestone JA, Takayanagi H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. 2014;20(1):62–68. doi: 10.1038/nm.3432. [DOI] [PubMed] [Google Scholar]
- Kral JB, Kuttke M, Schrottmaier WC, Birnecker B, Warszawska J, Wernig C, Paar H, Salzmann M, Sahin E, Brunner JS, Österreicher C, Knapp S, Assinger A, Schabbauer G. Sustained PI3K Activation exacerbates BLM-induced Lung Fibrosis via activation of pro-inflammatory and pro-fibrotic pathways. Sci Rep. 2016;6:23034. doi: 10.1038/srep23034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kritikou E. PTEN- a new guardian of the genome. Nat Rev Mol Cell Biol. 2007;8(3):179. [Google Scholar]
- Lee JJ, Kim BC, Park MJ, Lee YS, Kim YN, Lee BL, Lee JS. PTEN status switches cell fate between premature senescence and apoptosis in glioma exposed to ionizing radiation. Cell Death Differ. 2011;18(4):666–677. doi: 10.1038/cdd.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275(5308):1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Li Y, Jia Y, Pichavant M, Loison F, Sarraj B, Kasorn A, You J, Robson BE, Umetsu DT, Mizgerd JP, Ye K, Luo HR. Targeted deletion of tumor suppressor PTEN augments neutrophil function and enhances host defense in neutropenia-associated pneumonia. Blood. 2009;113(20):4930–4941. doi: 10.1182/blood-2008-06-161414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci USA. 2003;100(9):5336–5341. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273(22):13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- McEllin B, Camacho CV, Mukherjee B, Hahm B, Tomimatsu N, Bachoo RM, Burma S. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. 2010;70(13):5457–5464. doi: 10.1158/0008-5472.CAN-09-4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, Waldman T, Lord CJ, Ashworth A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1(6–7):315–322. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186(6):3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer. 2011;11(7):503–511. doi: 10.1038/nrc3057. [DOI] [PubMed] [Google Scholar]
- Newton R, Priyadharshini B, Turka LA. Immunometabolism of regulatory T cells. Nat Immunol. 2016;17(6):618–625. doi: 10.1038/ni.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Molina A, Efeyan A, Lopez-Guadamillas E, Munoz-Martin M, Gomez-Lopez G, Canamero M, Mulero F, Pastor J, Martinez S, Romanos E, Mar Gonzalez-Barroso M, Rial E, Valverde AM, Bischoff JR, Serrano M. Pten positively regulates brown adipose function, energy expenditure, and longevity. Cell Metab. 2012;15(3):382–394. doi: 10.1016/j.cmet.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, Peng M, Chan P, Ma Q, Mo Y, Meijer D, Zhao K, Rudensky AY, Atwal G, Zhang MQ, Li MO. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature. 2012;491(7425):554–559. doi: 10.1038/nature11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan F, Yu H, Dang EV, Barbi J, Pan X, Grosso JF, Jinasena D, Sharma SM, McCadden EM, Getnet D, Drake CG, Liu JO, Ostrowski MC, Pardoll DM. Eos mediates Foxp3-dependent gene silencing in CD4 + regulatory T cells. Science. 2009;325(5944):1142–1146. doi: 10.1126/science.1176077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa A, Wan L, Bonora M, Salmena L, Song MS, Hobbs RM, Lunardi A, Webster K, Ng C, Newton RH, Knoblauch N, Guarnerio J, Ito K, Turka LA, Beck AH, Pinton P, Bronson RT, Wei W, Pandolfi PP. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell. 2014;157(3):595–610. doi: 10.1016/j.cell.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, Mischel PS, Stokoe D, Pieper RO. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, Li L, Boussiotis VA. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692. doi: 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol. 2013;33(16):3091–3098. doi: 10.1128/MCB.00319-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, Williams LJ, Deng W, Chen G, Mbofung R, Lazar AJ, Torres-Cabala CA, Cooper ZA, Chen PL, Tieu TN, Spranger S, Yu X, Bernatchez C, Forget MA, Haymaker C, Amaria R, McQuade JL, Glitza IC, Cascone T, Li HS, Kwong LN, Heffernan TP, Hu J, Bassett RL, Bosenberg MW, Woodman SE, Overwijk WW, Lizee G, Roszik J, Gajewski TF, Wargo JA, Gershenwald JE, Radvanyi L, Davies MA, Hwu P. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016;6(2):202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci USA. 1999;96(4):1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riou C, Yassine-Diab B, Van grevenynghe J, Somogyi R, Greller LD, Gagnon D, Gimmig S, Wilkinson P, Shi Y, Cameron MJ, Campos-Gonzalez R, Balderas RS, Kelvin D, Sekaly RP, Haddad EK. Convergence of TCR and cytokine signaling leads to FOXO3a phosphorylation and drives the survival of CD4+ central memory T cells. J Exp Med. 2007;204(1):79–91. doi: 10.1084/jem.20061681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- Sharma MD, Huang L, Choi JH, Lee EJ, Wilson JM, Lemos H, Pan F, Blazar BR, Pardoll DM, Mellor AL, Shi H, Munn DH. An inherently bifunctional subset of Foxp3+ T helper cells is controlled by the transcription factor eos. Immunity. 2013;38(5):998–1012. doi: 10.1016/j.immuni.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma MD, Shinde R, McGaha TL, Huang L, Holmgaard RB, Wolchok JD, Mautino MR, Celis E, Sharpe AH, Francisco LM, Powell JD, Yagita H, Mellor AL, Blazar BR, Munn DH. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci Adv. 2015;1(10):e1500845. doi: 10.1126/sciadv.1500845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128(1):157–170. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208(7):1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol. 2015;16(2):178–187. doi: 10.1038/ni.3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MS, Carracedo A, Salmena L, Song SJ, Egia A, Malumbres M, Pandolfi PP. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell. 2011;144(2):187–199. doi: 10.1016/j.cell.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soond DR, Garcon F, Patton DT, Rolf J, Turner M, Scudamore C, Garden OA, Okkenhaug K. Pten loss in CD4 T cells enhances their helper function but does not lead to autoimmunity or lymphoma. J Immunol. 2012;188(12):5935–5943. doi: 10.4049/jimmunol.1102116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V, Tsao MS, Macpherson D, Suzuki A, Chapman WB, Mak TW. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/− mice. Cancer Res. 2000;60(13):3605–3611. [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DHR, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15(4):356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Subramanian KK, Jia Y, Zhu D, Simms BT, Jo H, Hattori H, You J, Mizgerd JP, Luo HR. Tumor suppressor PTEN is a physiologic suppressor of chemoattractant-mediated neutrophil functions. Blood. 2007;109(9):4028–4037. doi: 10.1182/blood-2006-10-055319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Huang C, He J, Lamb KL, Kang X, Gu T, Shen WH, Yin Y. PTEN C-terminal deletion causes genomic instability and tumor development. Cell Reports. 2014;6(5):844–854. doi: 10.1016/j.celrep.2014.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Yamaguchi MT, Ohteki T, Sasaki T, Kaisho T, Kimura Y, Yoshida R, Wakeham A, Higuchi T, Fukumoto M, Tsubata T, Ohashi PS, Koyasu S, Penninger JM, Nakano T, Mak TW. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity. 2001;14(5):523–534. doi: 10.1016/s1074-7613(01)00134-0. [DOI] [PubMed] [Google Scholar]
- Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280(5369):1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- Teng MW, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest. 2015;125(9):3338–3346. doi: 10.1172/JCI80004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, Honjo T. IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. 2011;186(5):2772–2779. doi: 10.4049/jimmunol.1003208. [DOI] [PubMed] [Google Scholar]
- Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;276(2):993–998. doi: 10.1074/jbc.M009134200. [DOI] [PubMed] [Google Scholar]
- Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, Pinton S, Zhang J, Kalathur M, Civenni G, Jarrossay D, Montani E, Marini C, Garcia-Escudero R, Scanziani E, Grassi F, Pandolfi PP, Catapano CV, Alimonti A. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Reports. 2014;9(1):75–89. doi: 10.1016/j.celrep.2014.08.044. [DOI] [PubMed] [Google Scholar]
- Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, Thompson JC, Caserta E, Wang H, Chong JL, Naidu S, Wei G, Sharma SM, Stephens JA, Fernandez SA, Gurcan MN, Weinstein MB, Barsky SH, Yee L, Rosol TJ, Stromberg PC, Robinson ML, Pepin F, Hallett M, Park M, Ostrowski MC, Leone G. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature. 2009;461(7267):1084–1091. doi: 10.1038/nature08486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ree JH, Nam HJ, Jeganathan KB, Kanakkanthara A, van Deursen JM. Pten regulates spindle pole movement through Dlg1-mediated recruitment of Eg5 to centrosomes. Nat Cell Biol. 2016;18(7):814–821. doi: 10.1038/ncb3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20(14):5010–5018. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- Wang G, Li Y, Wang P, Liang H, Cui M, Zhu M, Guo L, Su Q, Sun Y, McNutt MA, Yin Y. PTEN regulates RPA1 and protects DNA replication forks. Cell Res. 2015;25(11):1189–1204. doi: 10.1038/cr.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, Anthony BA, Sverdrup FM, Head R, Kuster DJ, Ruminski P, Weiss D, V Schack D, Bluestone J Aon. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J Exp Med. 2012;209(10):1713–1722. S1711–1719. doi: 10.1084/jem.20120822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27(41):5443–5453. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Hou SQ, He J, Gu T, Yin Y, Shen WH. PTEN regulates PLK1 and controls chromosomal stability during cell division. Cell Cycle. 2016;15(18):2476–2485. doi: 10.1080/15384101.2016.1203493. [DOI] [PMC free article] [PubMed] [Google Scholar]