

How individual Aβ species accumulate in Alzheimer’s disease has not been fully characterized. Shinohara et al. report that N-terminally truncated Aβ42 species accumulate differently from full-length Aβ42 across brain areas as well as disease stages, suggesting distinct underlying mechanisms and roles in the disease.
Keywords: Alzheimer’s disease, amyloid-β, neuropathology, apoE, tau
Abstract
Accumulation of amyloid-β peptides is a dominant feature in the pathogenesis of Alzheimer’s disease; however, it is not clear how individual amyloid-β species accumulate and affect other neuropathological and clinical features in the disease. Thus, we compared the accumulation of N-terminally truncated amyloid-β and full-length amyloid-β, depending on disease stage as well as brain area, and determined how these amyloid-β species respectively correlate with clinicopathological features of Alzheimer’s disease. To this end, the amounts of amyloid-β species and other proteins related to amyloid-β metabolism or Alzheimer’s disease were quantified by enzyme-linked immunosorbent assays (ELISA) or theoretically calculated in 12 brain regions, including neocortical, limbic and subcortical areas from Alzheimer’s disease cases (n = 19), neurologically normal elderly without amyloid-β accumulation (normal ageing, n = 13), and neurologically normal elderly with cortical amyloid-β accumulation (pathological ageing, n = 15). We observed that N-terminally truncated amyloid-β42 and full-length amyloid-β42 accumulations distributed differently across disease stages and brain areas, while N-terminally truncated amyloid-β40 and full-length amyloid-β40 accumulation showed an almost identical distribution pattern. Cortical N-terminally truncated amyloid-β42 accumulation was increased in Alzheimer’s disease compared to pathological ageing, whereas cortical full-length amyloid-β42 accumulation was comparable between Alzheimer’s disease and pathological ageing. Moreover, N-terminally truncated amyloid-β42 were more likely to accumulate more in specific brain areas, especially some limbic areas, while full-length amyloid-β42 tended to accumulate more in several neocortical areas, including frontal cortices. Immunoprecipitation followed by mass spectrometry analysis showed that several N-terminally truncated amyloid-β42 species, represented by pyroglutamylated amyloid-β11-42, were enriched in these areas, consistent with ELISA results. N-terminally truncated amyloid-β42 accumulation showed significant regional association with BACE1 and neprilysin, but not PSD95 that regionally associated with full-length amyloid-β42 accumulation. Interestingly, accumulations of tau and to a greater extent apolipoprotein E (apoE, encoded by APOE) were more strongly correlated with N-terminally truncated amyloid-β42 accumulation than those of other amyloid-β species across brain areas and disease stages. Consistently, immunohistochemical staining and in vitro binding assays showed that apoE co-localized and bound more strongly with pyroglutamylated amyloid-β11-x fibrils than full-length amyloid-β fibrils. Retrospective review of clinical records showed that accumulation of N-terminally truncated amyloid-β42 in cortical areas was associated with disease onset, duration and cognitive scores. Collectively, N-terminally truncated amyloid-β42 species have spatiotemporal accumulation patterns distinct from full-length amyloid-β42, likely due to different mechanisms governing their accumulations in the brain. These truncated amyloid-β species could play critical roles in the disease by linking other clinicopathological features of Alzheimer’s disease.
Introduction
Alzheimer’s disease is neuropathologically characterized by the accumulation of amyloid-β peptides mostly ending at residues 40/42 and tau proteins, which respectively appear as amyloid plaques and neurofibrillary tangles. Although both neuropathological features contribute to the disease, the widely accepted amyloid cascade hypothesis indicates that it is the accumulation of amyloid-β, in particular amyloid-β42, that initiates Alzheimer’s disease pathogenesis (Karran et al., 2011). Thus, it is critical to investigate how individual amyloid-β species accumulate in the brain in order to understand the pathogenic mechanisms involved in Alzheimer’s disease.
Previous studies investigating the regional distribution of amyloid-β accumulation in different stages of the disease have provided important clues as to the mechanism involved in amyloid-β accumulation or its associated features in human brains (Klunk et al., 2004; Buckner et al., 2005; Jack et al., 2008; Vlassenko et al., 2010). Most of these spatiotemporal studies were based on amyloid PET imaging. However, by uniquely addressing the regional distribution of amyloid-β accumulation and molecules related to amyloid-β metabolism through quantitative biochemical assays, we recently observed significant regional associations among amyloid-β40/42 accumulation, synaptic markers, apolipoprotein E (apoE, encoded by APOE) and amyloid precursor protein (APP) in the normal ageing, pathological ageing (presumable early stage of disease) and Alzheimer’s disease. Our findings not only complement previous studies, but also provide further pathogenic insight into amyloid-β40/42 accumulation in the brain during the development of Alzheimer’s disease (Shinohara and Bu, 2013; Shinohara et al., 2013, 2014).
Although our previous studies focused on the accumulation of full-length amyloid-β40/42, it is also known that N-terminally truncated (N-truncated) amyloid-β40/42 accumulates in Alzheimer’s disease brains (Masters et al., 1985; Mori et al., 1992; Miller et al., 1993; Naslund et al., 1994; Saido et al., 1996; Miravalle et al., 2005; Portelius et al., 2010; Moore et al., 2012). In addition to the well-studied pyroglutamylated amyloid-βpE3-40/42 and amyloid-βpE11-40/42, many different species of N-truncated amyloid-β40/42 that begin at residues 2 to 11 are found in Alzheimer’s disease brains (Naslund et al., 1994; Miravalle et al., 2005; Portelius et al., 2010; Moore et al., 2012). Although these N-truncated amyloid-β species could be generated during maturation processes of amyloid plaques, consisted of primarily full-length amyloid-β, through enzymatic or non-enzymatic pathways (Guntert et al., 2006; Portelius et al., 2015; Lyons et al., 2016), such concepts have not been validated in vivo partly because N-truncated amyloid-β species are rare in animal models (Kawarabayashi et al., 2001; Kalback et al., 2002; Schieb et al., 2011). N-truncated amyloid-β species are more likely to aggregate and be more neurotoxic than full-length amyloid-β and thus might be good targets for therapy (Pike et al., 1995; Demattos et al., 2012; Bayer and Wirths, 2014; Cynis et al., 2016). Several proteases were found to be involved in the generation of each N-terminal truncation of amyloid-β (Portelius et al., 2008; Bayer and Wirths, 2014; Schonherr et al., 2016). However, despite these findings, N-truncated amyloid-β accumulation in brains has not yet been fully characterized for its relationships to spatiotemporal aspects of the disease as well as clinical outcome.
In this study, by examining 12 regions of post-mortem brains from the normal ageing, pathological ageing and Alzheimer’s disease cases, we compared N-truncated amyloid-β40/42 species to full-length amyloid-β40/42 in their accumulation and association with other Alzheimer’s disease-related molecules, and assessed their relationship with other neuropathological changes and clinical features. Our findings provide critical insights into the origins and roles of N-truncated amyloid-β accumulation in the pathogenesis of Alzheimer’s disease.
Materials and methods
Human brain tissue
Post-mortem brain tissues were obtained through the Mayo Clinic Brain Bank, whose operating procedures were approved by the Mayo Institutional Review Board. In this study, we biochemically analysed the same cohorts of normal ageing (n = 13, average age = 82.9 ± 10.8 years), pathological ageing (n = 15, average age = 92.7 ± 5.9 years) and patients with sporadic Alzheimer’s disease (n = 19, average age = 84.7 ± 7.8 years) that we previously reported (Shinohara et al., 2014). These subjects are also described in more detail in Supplementary Table 1. For immunohistochemical analysis, we examined six additional Alzheimer’s disease cases with similar clinicopathological features from the Mayo Clinic Brain Bank (Supplementary Table 1).
Sample preparation
Grey matter of five neocortical areas [dorsolateral prefrontal (BA9), orbitofrontal (BA12), inferior temporal (BA20), inferior parietal (BA39/40), and primary visual (BA17)], three limbic areas [posterior cingulate (BA31), entorhinal (BA28), and amygdala], and four subcortical areas [striatum (caudate), thalamus, hypothalamus, and cerebellum] were dissected and kept frozen until protein extraction was performed. These 12 brain areas are also described in Supplementary Table 2. Brain lysates were prepared by a three-step extraction method based on differential solubility in detergents (Triton™ X-100) and chaotropic agents (guanidine hydrochloride) as previously described (Shinohara et al., 2013). In brief, after removal of meninges and blood vessels, 100–200 mg of frozen brain tissue was homogenized with a Polytron® homogenizer (KINEMATICA) in an ice-cold Tris-buffered saline (TBS) containing protease inhibitor cocktail (PIC; Roche Diagnostics). After centrifugation at 100 000g for 60 min at 4°C, the supernatant was aliquoted and stored at −80°C (referred to as TBS fraction or TBS). The residual pellet was rehomogenized in TBS plus 1% Triton™ X-100 with PIC, incubated with mild agitation for 1 h at 4°C and centrifuged as above. The resultant supernatant was aliquoted and stored at −80°C (referred to as TX fraction or TX). The residual pellet was rehomogenized in TBS plus 5 M guanidine hydrochloride, pH 7.6, and incubated with mild agitation for 12–16 h at room temperature. After centrifugation as above, the resultant supernatant (referred to as GuHCl fraction or GuHCl) was diluted with nine volumes of TBS, aliquoted and stored at −80°C.
Quantification of proteins
Levels of total protein, full-length amyloid-β40 (amyloid-β1-40) and amyloid-β42 (amyloid-β1-42), APP, APP-CTFβ (C99), β-site cleaving enzyme 1 (BACE1), presenilin-1 (PS1), apoE, insulin degrading enzyme (IDE), neprilysin, low-density lipoprotein receptor (LDLR), LDLR-related protein 1 (LRP1), glial fibrillary acidic protein (GFAP), synaptophysin, postsynaptic density 95 (PSD95), and tau were determined by ELISA or enzymatic assays as previously described (Shinohara et al., 2013, 2014). Levels of total amyloid-β40 (amyloid-βx-40) and amyloid-β42 (amyloid-βx-42) were determined by ELISA using an end-specific monoclonal antibody for capture (13.1.1 for amyloid-βx-40 and 2.1.3 for amyloid-βx-42) and a biotin-conjugated 266 antibody for detection (epitope: 16–23 amino acids of amyloid-β, kindly provided by Dr Ronald B. DeMattos, Lilly Research Laboratories, Indianapolis, IN). Standard amyloid-β1-40 or amyloid-β1-42 peptides (AnaSpec) were used with amyloid-β1-40 and amyloid-βx-40 ELISAs and with amyloid-β1-42 and amyloid-βx-42 ELISAs, respectively. Levels of N-truncated amyloid-β40 (amyloid-βt-40) and amyloid-β42 (amyloid-βt-42) were calculated by subtracting amyloid-β1-40 and amyloid-β1-42 levels from total amyloid-βx-40 and amyloid-βx-42 levels, respectively. Levels of pyroglutamylated amyloid-βpE3-42 were determined by ELISA using 2.1.3 capture antibody and a biotin-conjugated anti-amyloid-βpE3-x detection antibody (Synaptic Systems). Amyloid-βpE3-42 peptides (AnaSpec) were used as standards. Levels of pyroglutamylated amyloid-βpE11-42 were determined by ELISA using 2.1.3 capture antibody and a biotin-conjugated anti- amyloid-βpE11-x detection antibody (Synaptic Systems). Amyloid-βpE11-42 peptides (AnaSpec) were used as standards. Colorimetric quantification was performed on a Synergy HT plate reader (BioTek) using horseradish peroxidase (HRP)-linked streptavidin (Vector) and 3,3′,5,5′-tetramethylbenzidine substrate (Sigma). Levels of specific proteins were determined on one of the three fractions, based on their abundance among these fractions (Shinohara et al., 2014). Specifically, the TBS fraction was used to measure levels of cytosolic/secreted proteins and molecules (i.e. IDE, GFAP, and PSD95). The TX fraction was used to measure levels of membrane proteins (i.e. APP, APP-CTFβ, BACE1, PS1, NEP, LDLR, LRP1, and synaptophysin). GuHCl fraction was used to measure levels of pathological aggregated tau proteins. amyloid-β and apoE levels were determined in all fractions.
Immunoprecipitation followed by mass spectrometry
Immunoprecipitation followed by mass spectrometry (IP/MS) was performed according to the method of Moore et al. (2012) with some modifications. In brief, magnetic protein A/G beads (Thermo Fisher Scientific) were initially blocked by 4% Block ACE solution (Bio-Rad) and then incubated with 2.1.3 antibody (for amyloid-βx-42) for 1 h at room temperature with constant shaking. The beads were then washed with TBS and incubated with appropriately diluted brain GuHCl fraction for 12–16 h at 4°C with constant shaking. Bound beads were washed sequentially with 0.1% and 0.05% Triton™ X-100 in 20 mM KCl solution followed by water. Samples were eluted with a mixture of 75% acetonitrile, 24.9% water and 0.1% formic acid, mixed in equal volume with saturated sinapinic acid in 50% acetonitrile, 49.5% water and 0.5% trifluoroacetic acid, and spotted (2 µl) onto a ProteinChip® Gold Array (Bio-Rad) for analysis with a Bio-Rad Protein Chip System Series 4000 mass spectrometer (Enterprise Edition).
Immunohistochemistry and image analysis
Double immunofluorescence staining with rabbit polyclonal anti-amyloid-β1-x antibody (IBL-America) or anti-amyloid-βpE11-x antibody (Synaptic Systems) and mouse monoclonal anti-apoE antibody (clone D6E10; Abcam) was performed on amygdala sections from six Alzheimer’s disease cases (Supplementary Table 1). The deparaffinized and rehydrated sections were pretreated with 95% formic acid for 30 min and then steamed in distilled water for 30 min. Next, sections were blocked with Protein Block plus Serum Free (Dako) for 1 h and incubated with either rabbit polyclonal anti-amyloid-β1-x or anti-amyloid-βpE11-x antibody, and mouse monoclonal anti-apoE antibody (20 µg/ml) diluted by Antibody Diluent containing Background-Reducing Components (Dako) overnight at 4°C. Sections were washed three times with PBS at room temperature and then incubated with Alexa Fluor® 568-conjugated goat anti-rabbit IgG and Alexa Fluor® 488-conjugated goat anti-mouse IgG (1:500, Thermo Fisher Scientific) diluted by Antibody Diluent with Background Reducing Component (Dako) for 1.5 h at room temperature in a light protected chamber. Sections were again washed three times with PBS at room temperature, incubated with 1% Sudan Black for 2 min, washed with distilled water, and mounted with Vectashield mounting media with DAPI (Vector Laboratories). Images were taken with a Zeiss Axio Imager Z1 fluorescent microscope (Carl Zeiss MicroImaging) under ×20 field. More than five areas in the amygdala section of each case were randomly chosen based on apoE signal, and co-localization between apoE and amyloid-β signals was analysed via Person’s correlation coefficient with Costes’s automatic threshold by using the ImageJ Plug-in (Bolte and Cordelieres, 2006) and averaged over all areas. The co-localizations between apoE and amyloid-β1-x signals and between apoE and amyloid-βpE11-x signals across all cases were compared by paired t-test.
In vitro amyloid-β fibrillization and apoE binding assays
In vitro amyloid-β fibril formation assays were performed according to the method of Hori et al. (2007) with some modifications. In brief, synthetic amyloid-β peptides (AnaSpec) were fist solubilized in 1,1,1,3,3,3-hexafluoro-2-propanol (Sigma) and lyophilized (Freeze Dry System, Labconco). Amyloid-β peptides were then resolubilized in PBS containing 2% dimethyl sulfoxide at 25 µM, filtered through a 0.22 -µm pore filter (Millipore), and incubated at 37°C for the indicated times. After incubation, some aliquots were put immediately on ice to prevent further fibril formation, followed by addition of thioflavin T (Sigma) at 20 µM. Fluorescence (λex = 450 nm and λem = 482 nm) was then measured with a Synergy H1 plate reader (BioTek). For apoE binding assay, a 50-µl aliquot of amyloid-β fibrils incubated for 72 h was transferred to Nunc MaxiSorp® 96 well plates (Thermo Fisher Scientific) and incubated overnight at 4°C. Plates were then blocked by Block Ace (Bio-Rad) for 2 h, washed with PBS, and incubated with the indicated concentration of apoE3 recombinant proteins (Fitzgerald) overnight at 4°C. After washing with PBS, plates were incubated with biotin-conjugated goat anti-apoE antibody (Meridian Life Science) for 2 h. Colorimetric quantification was performed on a Synergy HT plate reader using HRP-linked streptavidin and 3,3’,5,5’-tetramethylbenzidine substrate.
Antemortem clinical symptoms
We reviewed autopsy case records from the brain bank database at Mayo Clinic, Jacksonville. The data were obtained retrospectively by medical record review. Demographic data including age at onset, disease duration, Dementia Rating Scale (DRS) and Kokmen Short Test of Mental Status were retrieved from available medical records. Twelve of 19 patients had DRS records at multiple time points. Annual changes of DRS were calculated by averaging DRS changes between two adjacent time points. Twelve of 19 patients were also assessed by Kokmen Test within 6 years before death. The last score of the Kokmen Test was used for analysis in this study.
Data analysis of neuroanatomical distribution of amyloid-β accumulation
All values measured by ELISAs were first normalized by total protein levels in the sample. Comparisons of cortical amyloid-β levels (averaged value across seven cortical areas) among multiple cohorts were performed by Kruskal-Wallis one-way ANOVA on ranks, with all pairwise comparisons using Dunn’s test. To assess region-specific difference, protein levels were normalized by the average value within an individual to adjust for the influence of difference between individuals. Comparisons of such normalized values between different amyloid-β species in each brain area were performed by Wilcoxon signed-rank test. The non-parametric Spearman rank correlation coefficient was used to summarize the degree of correlation between median levels of each protein (normalized value within individual) across 12 brain regions (Shinohara et al., 2013). All statistical analyses were performed by JMP Pro (version 10.0.0; SAS, Cary, NC) or Prism (version 6.05; GraphPad Software, Inc., La Jolla, CA). P-values < 0.05 were considered significant.
Results
Distinct accumulation of N-truncated and full-length amyloid-β42 across disease stages and brain areas
First, we compared levels of accumulated (i.e. GuHCl fraction) full-length amyloid-β40/42, total amyloid-β40/42 and N-truncated amyloid-β40/42 in the brain cortical areas among normal ageing, pathological ageing and Alzheimer’s disease cases. Compared to pathological ageing, Alzheimer’s disease had more accumulated amyloid-β40 irrespective of full-length, total or N-truncated forms, as shown by large differences in the accumulation of these amyloid-β40 between pathological ageing and Alzheimer’s disease (Fig. 1A). This observation is consistent with a previous study (Wang et al., 1999). Interestingly, amyloid-β42 showed a different pattern of accumulation that depended on the class of amyloid-β42 forms. Although full-length amyloid-β42 levels were comparable between pathological ageing and Alzheimer’s disease, levels of N-truncated amyloid-β42 were significantly increased in Alzheimer’s disease compared to pathological ageing (Fig. 1B). These trends persisted even after adjustment for age (Supplementary Fig. 1). We also confirmed increased levels of N-truncated amyloid-β42 in individual brain areas of Alzheimer’s disease relative to pathological ageing, except for some subcortical areas, where levels of N-truncated amyloid-β42 were very low compared to those in cortical areas. In contrast, full-length amyloid-β42 levels in Alzheimer’s disease were mostly comparable to those in pathological ageing in all areas (Supplementary Fig. 2). These results suggest that N-truncated amyloid-β42 accumulation increases with advanced disease (Alzheimer’s disease), while full-length amyloid-β42 accumulation almost reaches a plateau in the earlier stage of the disease (pathological ageing).
Figure 1.

Comparison of accumulated amyloid-β averaged across seven cortical areas in normal ageing, pathological ageing and Alzheimer’s disease cases. (A) Levels of full-length amyloid-β40 (Aβ1-40), total amyloid-β40 (Aβx-40), and N-truncated amyloid-β40 (Aβt-40) in GuHCl fraction are averaged across seven cortical areas within each individual and compared between normal ageing, pathological ageing and Alzheimer’s disease. (B) Levels of full-length amyloid-β42 (Aβ1-42), total amyloid-β42 (Aβx-42), and N-truncated amyloid-β42 (Aβt-42) in GuHCl fraction are averaged across seven cortical areas within each individual and compared between normal ageing, pathological ageing and Alzheimer’s disease. Data represent mean ± standard error (SE) *P < 0.05, **P < 0.01, ***P < 0.001; Dunn’s test. AD = Alzheimer’s disease; NA = normal ageing; PA = pathological ageing.
Next, we compared the regional distribution of full-length amyloid-β and N-truncated amyloid-β accumulation (i.e. GuHCl fraction) in Alzheimer’s disease cases by analysing normalized values within individuals, which enabled us to address neuroanatomical aspects in Alzheimer’s disease (Shinohara et al., 2013, 2014). Although some areas, especially subcortical areas, showed an imbalance between full-length amyloid-β40 and N-truncated amyloid-β40 accumulation (Fig. 2A), they generally showed a similar regional distribution as shown by the strong correlation between their regional distributions (Fig. 2C). On the other hand, full-length amyloid-β42 and N-truncated amyloid-β42 accumulation showed a significantly different regional distribution. While several cortical areas (orbitofrontal cortex, dorsolateral prefrontal cortex, inferior parietal cortex and posterior cingulate cortex) and subcortical areas (striatum, hypothalamus, and thalamus) were more liable to accumulate full-length amyloid-β42 than N-truncated amyloid-β42, other cortical areas (inferior temporal cortex and primary visual cortex) and to a greater extent some limbic areas (entorhinal cortex and amygdala) were more liable to accumulate N-truncated amyloid-β42 than full-length amyloid-β42 (Fig. 2B). This significantly different regional distribution was confirmed by the weak correlation between their regional distributions (Fig. 2D), and the analysis comparing absolute value of these amyloid-β species in each area (Supplementary Fig. 3). Though this study did not focus on amyloid-β in soluble fractions (TBS or TX), whose levels were very low and thus sometimes difficult to measure (∼0.1% of amyloid-β levels in the GuHCl fraction) (Shinohara et al., 2013, 2014), similar trends were observed between full-length and N-truncated amyloid-β42 distributions in such fractions (data not shown). In the pathological ageing, it was not clear whether there was a difference in the regional distribution of full-length amyloid-β42 accumulation and N-truncated amyloid-β42 accumulation by analysing each area, probably due to large variations between cases (Supplementary Fig. 4A and B). However, while there was a significant regional association between full-length and N-truncated amyloid-β40 accumulation, those of full-length and N-truncated amyloid-β42 accumulation was very weak (Supplementary Fig. 4C and D), as seen in Alzheimer’s disease cases. Taken together, these results demonstrated that N-truncated amyloid-β42 shows a distinct regional pattern of accumulation that differs from full-length amyloid-β42. This regional difference does not apply to amyloid-β40 accumulation, and it could occur in the earlier disease stage (pathological ageing), but is more prominent in the symptomatic stage (Alzheimer’s disease).
Figure 2.

Regional distribution of full-length and N-truncated amyloid-β40/42 in the neocortical, limbic and subcortical areas of Alzheimer’s disease cases. After normalization within each individual of the Alzheimer’s disease group, full-length amyloid-β40 (1-40) levels and N-truncated amyloid-β40 (t-40) levels (A) or full-length amyloid-β42 (1-42) levels and N-truncated amyloid-β42 (t-42) levels (B) in GuHCl fraction are plotted by 12 brain areas with a box-and-whisker diagram. *P < 0.05, **P < 0.01, ***P < 0.001; Wilcoxon signed-rank test. (C) Amyloid-βt-40 levels are plotted against amyloid-β1-40 levels in GuHCl fraction in each brain area of Alzheimer’s disease cases. (D) Amyloid-βt-42 levels are plotted against amyloid-β1-42 levels in GuHCl fraction in each brain area of Alzheimer’s disease cases. Values are medians with 25 and 75 percentiles. Correlation coefficient (r) and P-value were acquired by Spearman rank correlation test. Blue text = neocortical areas; purple text = limbic areas; and orange text = subcortical areas. Aβ = amyloid-β; AM = amygdala; CB = cerebellum; DF = dorsolateral prefrontal cortex; EC = entorhinal cortex; HT = hypothalamus; IP = inferior parietal cortex; IT = inferior temporal cortex; OF = orbitofrontal cortex; PC = posterior cingulate cortex; ST = striatum; TL = thalamus; VC = primary visual cortex.
We then further characterized the specific species of N-truncated amyloid-β42 in the GuHCl fraction by using IP/MS. Three regions, representing varying abundance of N-truncated amyloid-β42 relative to full-length amyloid-β42 (amygdala: higher amounts of N-truncated amyloid-β42, but lesser amounts of full-length amyloid-β42; dorsolateral prefrontal cortex: moderate amounts of N-truncated amyloid-β42 despite higher amounts of full-length amyloid-β42; and striatum: very low amounts of N-truncated amyloid-β42 despite high amounts of full-length amyloid-β42) were analysed from five Alzheimer’s disease cases whose amyloid-β levels were close to the median of the entire cohort. We detected several species of N-truncated amyloid-β42, including amyloid-βpE3-42, amyloid-β4-42, amyloid-βpE11-42 with this technique, particularly in the amygdala (Fig. 3A). When the relative abundance of N-truncated amyloid-β42 to full-length amyloid-β42 in each area was analysed, there were many species of N-truncated amyloid-β42 that accumulated more in the amygdala than in dorsolateral prefrontal cortex and striatum, consistent with ELISA results. Of note, amyloid-βpE11-42, which might be the most abundant amyloid-β42 isoform in the amygdala according to the intensity of the mass spectrometry peak, showed more accumulation in dorsolateral prefrontal cortex than in striatum. In addition, some of the other species (amyloid-β9-42 and amyloid-β11-42) showed similar differences (Fig. 3B). Consistently, a pilot study using a few cases of pathological ageing showed a similar trend in relative abundance of N-truncated amyloid-β42 species across these areas (data not shown). These results supported our initial findings regarding the relative abundance of N-truncated amyloid-β42 in these areas.
Figure 3.

Accumulation of individual N-truncated amyloid-β42 species. (A) Representative IP-mass spectra of full-length and N-truncated amyloid-β42 in the dorsolateral prefrontal cortex (DF), amygdala (AM), and striatum (ST) of an Alzheimer’s disease case. Note some undefined peaks in addition to those predicted for amyloid-β42 species. (B) Ratios of each N-truncated amyloid-β42 species to full-length amyloid-β42 calculated from the peak intensity of IP-MS in the dorsolateral prefrontal cortex, amygdala, and striatum regions from five Alzheimer’s disease cases. Data represent mean ± SE *P < 0.05, **P < 0.01; paired t-tests with Holm-Sidak’s correction for multiple comparisons. (C) Levels of amyloid-βpE3-42 and amyloid-βpE11-42 in GuHCl fraction are averaged across seven cortical areas within each individual and compared between normal ageing (NA), pathological ageing (PA) and Alzheimer’s disease (AD). *P < 0.05, **P < 0.01, ***P < 0.001; Dunn’s test. (D) Amyloid-βpE3-42 levels are plotted against amyloid-βt-42 levels in GuHCl fraction in each brain area of Alzheimer’s disease cases. (E) Amyloid-βpE11-42 levels are plotted against amyloid-βt-42 levels in GuHCl fraction in each brain area of Alzheimer’s disease cases. Values are medians with 25 and 75 percentiles. Correlation coefficient (r) and P-value were acquired by Spearman rank correlation test. Blue text = neocortical areas; purple text = limbic areas; and orange text = subcortical areas. Aβ = amyloid-β; AM = amygdala; CB = cerebellum; DF = dorsolateral prefrontal cortex; EC = entorhinal cortex; HT = hypothalamus; IP = inferior parietal cortex; IT = inferior temporal cortex; OF = orbitofrontal cortex; PC = posterior cingulate cortex; ST = striatum; TL = thalamus; VC = primary visual cortex.
Based on these results, we next determined the levels of representative N-truncated amyloid-β42 species by using ELISAs specific for amyloid-βpE3-42 and amyloid-βpE11-42 (Supplementary Fig. 5). Accumulated (i.e. GuHCl fraction) amyloid-βpE3-42 and amyloid-βpE11-42 levels in the cortical areas tended to be both increased in pathological ageing, compared to normal ageing, and significantly increased in Alzheimer’s disease compared to both normal ageing and pathological ageing (Fig. 3C). These trends persisted after adjustment for age (Supplementary Fig. 6), and were similar to that observed for total N-truncated amyloid-β42 in Fig. 1B. We also confirmed such trends in individual brain areas, except for some subcortical areas where levels of these truncated amyloid-β42 species were lower compared to cortical areas (Supplementary Fig. 7). Of note, while amyloid-βpE3-42 accumulation showed moderate regional association with total N-truncated amyloid-β42 accumulation in Alzheimer’s disease cases (Fig. 3D), amyloid-βpE11-42 accumulation showed very strong regional association with total N-truncated amyloid-β42 accumulation (Fig. 3E). The regional correlations between these pyroglutamylated amyloid-β42 accumulation and full-length or total N-truncated amyloid-β42 accumulation are summarized in Supplementary Table 3. The relatively similar distributions of amyloid-βpE11-42 and total N-truncated amyloid-β42 accumulation were also confirmed by comparisons in each area (Supplementary Fig. 8). Of note, after subtracting these pyroglutamylated amyloid-β42 from total N-truncated amyloid-β42 levels, the remaining N-truncated amyloid-β42 accumulations showed distribution similar to those of total N-truncated amyloid-β42 (Supplementary Table 3). In pathological ageing, the regional distributions of total N-truncated amyloid-β42 accumulation were also more similar to those of amyloid-βpE11-42 rather than those of amyloid-βpE3-42 (Supplementary Table 4). These results showed that amyloid-βpE11-42, one of the most abundant N-truncated amyloid-β42 species, as well as other non-pyroglutamylated amyloid-β42 species reflect the region-specific accumulations of total N-truncated amyloid-β42, while amyloid-βpE3-42 showed less regional correlation with total N-truncated amyloid-β42.
Regional associations between N-truncated amyloid-β42 accumulation and molecules related to amyloid-β metabolism
By analysing the relationships between the regional distribution of full-length amyloid-β accumulation in Alzheimer’s disease or the normal controls and those of molecules related to amyloid-β metabolism in the normal controls, our previous study addressed the pathomechanism of amyloid-β accumulation during the development of Alzheimer’s disease (Shinohara et al., 2013, 2014). However, our current findings that (i) the levels of N-truncated amyloid-β42 accumulation were increased in Alzheimer’s disease; and (ii) the distinct regional accumulation of N-truncated amyloid-β42 relative to full-length amyloid-β42 was more apparent in Alzheimer’s disease, compared to the pathological ageing cases, suggest that the mechanism contributing to the formation of such region-specific distribution of N-truncated amyloid-β42 might be more apparent in the later stages of disease. Thus, we analysed regional associations between N-truncated amyloid-β42 accumulation and molecules related to amyloid-β metabolism within Alzheimer’s disease cases. The results are summarized in Table 1. While the regional distribution of full-length amyloid-β42 accumulation showed strong positive and negative associations with those of PSD95 and apoE in the soluble TX fraction, respectively, total N-truncated amyloid-β42 accumulation did not show significant regional associations with PSD95 and apoE. However, total N-truncated amyloid-β42 accumulation showed significant positive and inverse associations with BACE1 and neprilysin, respectively, as shown in Table 1 or Fig. 4A. The regional distribution of amyloid-βpE11-42 also showed similar significant associations with BACE1 and neprilysin, as shown in Table 1 or Fig. 4B. When compared to the regional distribution in normal ageing, total N-truncated amyloid-β42 or amyloid-βpE11-42 accumulation in Alzheimer’s disease also did not associate with PSD95 or apoE, but negatively associated with neprilysin (Supplementary Table 5). These results at least suggest that the mechanisms involving N-truncated amyloid-β42 accumulation are different from those of full-length amyloid-β42 accumulation.
Table 1.
Regional associations between full-length or N-truncated amyloid-β42 accumulation and proteins related to amyloid-β metabolism, or accumulation of tau and apoE in Alzheimer’s disease
| Amyloid-β1-42 | Amyloid-βt-42 | Amyloid-βpE3-42 | Amyloid-βpE11-42 | |||||
|---|---|---|---|---|---|---|---|---|
| r | P-value | r | P-value | r | P-value | r | P-value | |
| Soluble (TBS or TX) fractions | ||||||||
| ApoE TBS | −0.49 | 0.106 | −0.52 | 0.082 | −0.71 | 0.010 | −0.55 | 0.065 |
| ApoE TX | −0.81 | 0.001 | −0.33 | 0.302 | −0.67 | 0.017 | −0.37 | 0.236 |
| APP | 0.55 | 0.067 | −0.51 | 0.092 | −0.04 | 0.914 | −0.36 | 0.251 |
| APP-CTFβ | 0.00 | 1.000 | −0.20 | 0.541 | −0.08 | 0.795 | −0.19 | 0.565 |
| BACE1 | −0.01 | 0.966 | 0.77 | 0.004 | 0.52 | 0.080 | 0.75 | 0.005 |
| PS1 | 0.24 | 0.443 | −0.23 | 0.463 | −0.04 | 0.897 | −0.28 | 0.370 |
| Neprilysin | 0.00 | 1.000 | −0.62 | 0.030 | −0.27 | 0.391 | −0.61 | 0.036 |
| IDE | 0.52 | 0.080 | −0.37 | 0.240 | 0.02 | 0.948 | −0.33 | 0.293 |
| LRP1 | 0.30 | 0.342 | −0.18 | 0.571 | −0.13 | 0.697 | −0.10 | 0.758 |
| LDLR | −0.38 | 0.217 | −0.46 | 0.134 | −0.54 | 0.071 | −0.53 | 0.076 |
| GFAP | −0.62 | 0.031 | 0.12 | 0.712 | −0.36 | 0.255 | 0.08 | 0.800 |
| Synaptophysin | 0.57 | 0.051 | 0.40 | 0.194 | 0.74 | 0.006 | 0.39 | 0.208 |
| PSD95 | 0.86 | <0.001 | −0.20 | 0.541 | 0.36 | 0.245 | −0.09 | 0.792 |
| Insoluble (GuHCl) fraction | ||||||||
| Tau GuHCl | 0.39 | 0.208 | 0.69 | 0.012 | 0.57 | 0.051 | 0.79 | 0.002 |
| ApoE GuHCl | 0.11 | 0.729 | 0.97 | <0.001 | 0.69 | 0.014 | 0.97 | <0.001 |
Correlation coefficient (r) and P-values were acquired by the non-parametric Spearman rank test comparing the median value of normalized levels of the indicated amyloid-β species in the GuHCl fraction with the median value of normalized levels of proteins related to amyloid-β metabolism in soluble fractions (TBS or TX), or tau and apoE in insoluble (GuHCl) fraction in 19 Alzheimer’s disease cases across 12 brain regions. Significant correlations are highlighted in bold.
Figure 4.

Associations between N-truncated amyloid-β42 accumulation and proteins related to amyloid-β metabolism, or accumulation of tau and apoE across brain areas or disease stages. Amyloid-βt-42 levels or amyloid-βpE11-42 levels in GuHCl fraction are plotted against neprilysin (NEP) levels (A) and BACE1 levels (B) in each brain area of Alzheimer’s disease cases. Amyloid-βt-42 levels or amyloid-βpE11-42 levels in GuHCl fraction are plotted against tau levels (C) or apoE levels (D) in GuHCl fraction in each brain area of Alzheimer’s disease cases. Values are median with 25 and 75 percentiles. Amyloid-βt-42 levels or amyloid-βpE11-42 levels in GuHCl fraction are plotted against tau levels (E) or apoE levels (F) in GuHCl fraction averaged across seven cortical areas of individual pathological ageing or Alzheimer’s disease cases. Correlation coefficient (r) and P-value were acquired by Spearman rank correlation test. PA = pathological ageing. Blue text = neocortical areas; purple text = limbic areas; and orange text = subcortical areas. Aβ = amyloid-β; AM = amygdala; CB = cerebellum; DF = dorsolateral prefrontal cortex; EC = entorhinal cortex; HT = hypothalamus; IP = inferior parietal cortex; IT = inferior temporal cortex; OF = orbitofrontal cortex; PC = posterior cingulate cortex; ST = striatum; TL = thalamus; VC = primary visual cortex.
Associations between N-truncated amyloid-β42 accumulation and tau or apoE accumulation across brain areas and disease stages
In addition to tau accumulation in the form of neurofibrillary tangles, Alzheimer’s disease-related neuropathology includes apoE accumulation, which co-deposits with amyloid-β plaques (Namba et al., 1991; Nishiyama et al., 1997), probably reflecting its pathogenic roles as the strongest risk gene for Alzheimer’s disease (Kanekiyo et al., 2014). Interestingly, previous neuropathological studies and our recent biochemical study showed that there are regional differences between amyloid-β deposits and tau accumulation or apoE accumulation (Braak and Braak, 1991; Yamaguchi et al., 1994; Thal et al., 2002; Shinohara et al., 2014). However, as such previous studies neither fully distinguished each amyloid-β species nor considered atypical anatomical accumulation of N-truncated amyloid-β42, we analysed the regional associations between N-truncated amyloid-β42 accumulation and tau or apoE accumulation (GuHCl fraction) within Alzheimer’s disease cases, as summarized in Table 1 and Supplementary Table 6. Of note, tau accumulation showed better regional associations with total N-truncated amyloid-β42 and amyloid-βpE11-42 accumulation among several amyloid-β species (also shown in Fig. 4C). More interestingly, the regional distribution of apoE accumulation strongly associated with the distribution of total N-truncated amyloid-β42 and amyloid-βpE11-42 accumulation (also shown in Fig. 4D). Stronger association between tau or apoE accumulation and N-truncated amyloid-β42 accumulation among several amyloid-β species were also observed across different disease stages with or without including normal ageing or pathological ageing in the brain cortical areas (Supplementary Table 7, also shown in Fig. 4E and F) as well as the amygdala (Supplementary Table 8). These results suggest the existence of a specific mechanism linking between N-truncated amyloid-β42 accumulation and these Alzheimer’s disease-related neuropathologies.
Strong co-localization and binding between N-truncated amyloid-β42 and apoE
As the association between N-truncated amyloid-β42 and apoE accumulation across brain areas and disease stages was unexpectedly very strong, we next characterized the morphological relationship between apoE and amyloid-β deposits detected by anti-amyloid-β1-x and amyloid-βpE11-x antibodies. The amygdala area of Alzheimer’s disease cases was analysed. We first confirmed that most parenchymal amyloid-like apoE deposits were positive for amyloid-β42 (data not shown). While some portions of amyloid-β1-x signals overlapped with accumulated apoE signals, other portions of amyloid-β1-x signals existed in areas surrounding apoE signals or without apoE signals (Fig. 5A). On the other hand, major portions of amyloid-βpE11-x signals overlapped with apoE signals (Fig. 5B). By comparing their co-localization across six different cases, we confirmed that apoE signals overlapped more strongly with amyloid-βpE11-x signals than with amyloid-β1-x signals (Fig. 5C), indicating that their strong co-deposition underlies the strong spatiotemporal associations between apoE and amyloid-βpE11-42, and probably total N-truncated amyloid-β42.
Figure 5.

Co-localization and in vitro binding assay of apoE with N-truncated amyloid-β. Amygdala sections of an Alzheimer’s disease case were double-stained using anti-apoE antibody (green) and anti-amyloid-β1-x antibody (red, A) or anti amyloid-βpE11-x antibody (red, B). (C) Co-localization of apoE signal with each amyloid-β signal were compared across different Alzheimer’s disease cases (n = 6). ***P < 0.001; paired t-test. Scale bar = 50 µm. (D and E) In vitro binding assay of apoE with fibrillar amyloid-β1-42 and amyloid-βpE11-42. Bovine serum albumin (BSA) was used as the negative control. (D) One representative experiment with three technical repeats. (E) Combined data from four independent experiments with 6 µg/ml apoE. Data represent mean ± SD. ***P < 0.001; Tukey-Krammer test. Aβ = amyloid-β.
To test the hypothesis that apoE has higher binding affinity to N-truncated amyloid-β42 rather than full-length amyloid-β42, we performed in vitro binding assays. Synthetic amyloid-β1-42 peptides and amyloid-βpE11-42 peptides were first incubated to form stable fibrils. Fibril formation measured by the thioflavin-T fluorescence assay revealed two features: (i) the times to reach 50% of the maximum fluorescence intensity of amyloid-β1-42 peptides and amyloid-βpE11-42 peptides were 6.9 ± 1.4 and 14.0 ± 1.8 h, respectively; and (ii) the fluorescence intensity of stable amyloid-β1-42 fibrils was higher than that of stable amyloid-βpE11-42 fibrils (Supplementary Fig. 9). These results suggest that amyloid-βpE11-42 aggregates less readily than amyloid-β1-42 peptides. When both fibril incubations had reached plateau values at 72 h, we assessed their binding to apoE. Interestingly, despite less apparent aggregation, amyloid-βpE11-42 fibrils showed higher binding to apoE in an apoE concentration-dependent manner, compared to amyloid-β1-42 fibrils (Fig. 5D and E). These results indicate that amyloid-βpE11-42 aggregates are more likely than amyloid-β1-42 aggregates to recruit apoE into their deposits.
Associations with antemortem clinical features
As our results indicate that N-truncated amyloid-β42 accumulation could be closely related to several features of Alzheimer’s disease neuropathology, we then analysed the association of various amyloid-β species with antemortem disease features as summarized in Table 2. Despite the limitation of retrospective analysis, tau accumulation in cortical areas significantly associated with rapid disease progression, measured by annual DRS change, while apoE accumulation significantly associated with a worse score in the Kokmen Short Test of Mental Status, indicating the potential relevance of these accumulations in Alzheimer’s disease and also the utility of our retrospective analysis. Interestingly, compared to the accumulation of full-length amyloid-β42, or full-length or total N-truncated amyloid-β40, amyloid-βpE11-42 or total N-truncated amyloid-β42 accumulation in cortical areas tended to show more significant associations with earlier age of onset or shorter disease duration, respectively. Of note, rapid disease progression showed a trend (but not significant) toward association with total N-truncated amyloid-β42 accumulation in cortical areas, while its associations with full-length amyloid-β40/42 were minimal. Amyloid-βpE11-42 accumulation also associated with a worse score in the Kokmen Short Test of Mental Status. These significant associations are visualized in Supplementary Fig. 10. Subcortical, including amygdala, accumulation of N-truncated amyloid-β42 did not significantly associate with these clinical features (Supplementary Table 9). Of note, after subtracting levels of pyroglutamylated amyloid-β42 from total N-truncated amyloid-β42, the remaining N-truncated amyloid-β42 accumulation still tended to show significant associations with some of these clinical features (Table 2). This observation suggests important roles of cortical accumulation of non-pyroglutamylated species of N-truncated amyloid-β42, in addition to pyroglutamylated amyloid-β42, in the clinical manifestation of Alzheimer’s disease.
Table 2.
Retrospective analysis for associations between full-length or N-truncated amyloid-β accumulation in cortical areas and antemortem clinical features
| Age at onset | Disease duration | DRS change | Kokmen Test | |||||
|---|---|---|---|---|---|---|---|---|
| r | P-value | r | P-value | r | P-value | r | P-value | |
| Amyloid-β1-40 | −0.20 | 0.424 | 0.46 | 0.057 | 0.39 | 0.204 | 0.05 | 0.878 |
| Amyloid-βx-40 | −0.21 | 0.404 | 0.48 | 0.043 | 0.36 | 0.255 | −0.07 | 0.819 |
| Amyloid-βt-40 | −0.22 | 0.380 | 0.52 | 0.028 | 0.31 | 0.329 | −0.23 | 0.479 |
| Amyloid-β1-42 | 0.15 | 0.562 | 0.29 | 0.235 | 0.07 | 0.822 | 0.72 | 0.009 |
| Amyloid-βx-42 | −0.01 | 0.963 | −0.26 | 0.288 | −0.41 | 0.188 | 0.10 | 0.755 |
| Amyloid-βt-42 | −0.12 | 0.627 | −0.51 | 0.030 | −0.49 | 0.103 | −0.31 | 0.332 |
| Amyloid-βpE3-42 | −0.44 | 0.065 | 0.25 | 0.319 | −0.37 | 0.235 | −0.29 | 0.360 |
| Amyloid-βpE11-42 | −0.57 | 0.013 | 0.29 | 0.247 | 0.08 | 0.816 | −0.59 | 0.046 |
| Amyloid-βt-42 − pE3-42 − pE11-42 | 0.05 | 0.835 | −0.55 | 0.018 | −0.51 | 0.094 | −0.14 | 0.657 |
| Tau GuHCl | −0.24 | 0.332 | −0.02 | 0.932 | −0.72 | 0.008 | 0.11 | 0.730 |
| ApoE GuHCl | −0.32 | 0.201 | 0.40 | 0.102 | 0.19 | 0.552 | −0.64 | 0.025 |
Correlation coefficient (r) and P-value were acquired by the Pearson test comparing levels of indicated amyloid-β species, tau or apoE in the GuHCl fraction averaged across seven cortical areas with age at onset (n = 18), disease duration (n = 18), annual change of DRS (n = 12) and the last score of the Kokmen Short Test of Mental Status (n = 12) in Alzheimer’s disease cases. Significant correlations are highlighted in bold.
Discussion
Although accumulating evidence indicates that N-truncated amyloid-β could play important roles in Alzheimer’s disease (Bayer and Wirths, 2014), information about how N-truncated amyloid-β accumulates in the brain and associates with other clinicopathological features of Alzheimer’s disease is limited. While biochemically analysing 12 different brain regions as previously reported (Shinohara et al., 2013, 2014), we unexpectedly noticed that the regional distribution of N-terminally truncated amyloid-β42 and full-length amyloid-β42 was markedly different; N-terminally truncated amyloid-β42 species were likely to accumulate more in some brain areas, especially some limbic areas (amygdala and entorhinal cortex), while full-length amyloid-β42 tended to accumulate more in neocortical areas, especially frontal cortices (Fig. 2). These results were confirmed by IP/MS analysis and a species-specific ELISA for pyroglutamylated amyloid-β11-42 (Fig. 3). Our findings are significant given that regional distribution of amyloid-β provides an important clue to understanding the pathomechanism of amyloid-β accumulation in the brain (Table 1) (Buckner et al., 2005; Vlassenko et al., 2010; Shinohara et al., 2013, 2014). Moreover, associations with several clinicopathological features supports critical roles of N-terminally truncated amyloid-β in the disease (Figs 4, 5 and Table 2). A model summarizes associations of accumulating full-length and N-truncated amyloid-β42, depending on brain area and disease stage is shown in Fig. 6.
Figure 6.
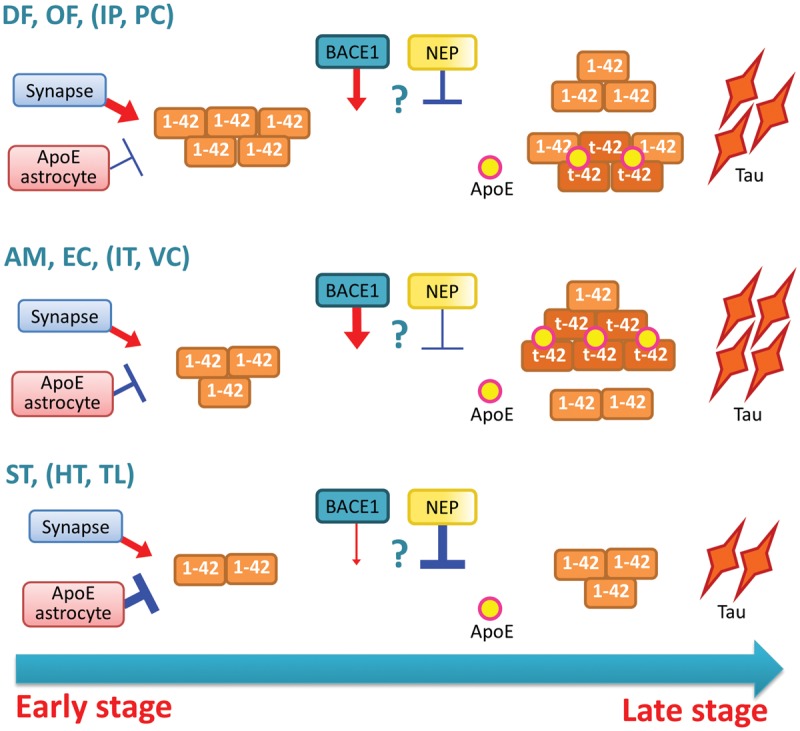
Model depicting the changes of full-length and N-truncated amyloid-β42 accumulation depending on disease stage and brain areas. In the early stage of disease, full-length amyloid-β42 (1-42) accumulates in a region-specific manner, where frontal cortices [dorsolateral prefrontal (DF) and orbitofrontal (OF)], and some other cortical areas, including inferior parietal cortex (IP) and posterior cingulate cortex (PC), tend to show more accumulation. The development of this region specificity may involve two factors: an enhancement by synaptic processes and a suppression by astrocytes or their secreted apoE (Shinohara et al., 2013). During disease progression, N-truncated amyloid-β42 (t-42) accumulates in a distinct region-specific manner compared to full-length amyloid-β42 accumulation, where some limbic areas [amygdala (AM) and entorhinal cortex (EC)] and other cortical areas, including inferior temporal cortex (IT) and primary visual cortex (VC), tend to show more accumulation. Of note, in subcortical areas, such as striatum (ST), hypothalamus (HT) and thalamus (TL), N-truncated amyloid-β42 accumulation is very low compared to the considerable amounts of full-length amyloid-β42. Such spatiotemporal specificity might be formed by processes distinct those involved in full-length amyloid-β42 accumulation. BACE1 and neprilysin might contribute to spatiotemporal accumulation of N-truncated amyloid-β42, although additional or alternative factors are likely to exist. Tau and apoE accumulations associate with N-truncated amyloid-β42 accumulation, and their cortical accumulation could reflect more advanced stage of disease, as manifested in several clinical outcomes.
Previously reported studies analysed individual amyloid-β species in different brain areas and led to some inconsistencies among these studies, such as the relative abundance of individual amyloid-β species or their changes across disease stages. Portelius et al. (2010) characterized the accumulation of individual amyloid-β species in three areas (cortex, hippocampus, and cerebellum) by IP/MS, but did not observe significant differences in the regional pattern of each amyloid-β species across these three areas. However, by analysing 12 brain areas, the current study clearly demonstrates the distinct regional pattern between N-truncated amyloid-β42 and full-length amyloid-β42 accumulations. Of note, this trend appears not to be simply attributed to the brain anatomy, as one limbic area (posterior cingulate cortex) and two neocortical areas (primary visual cortex and inferior temporal cortex), respectively showed an opposite trend with other limbic or neocortical areas (Fig. 2B). Also, accumulations of amyloid-βpE11-42 and to a lesser extent amyloid-βpE3-42 show regional distributions distinct from that of full-length amyloid-β42, though our results suggest that other amyloid-β species truncated at residues 4 to 11 might also follow such distinct pattern of N-truncated amyloid-β42. Thus, additional characterization of individual amyloid-β species by specific ELISAs in more brain areas will be useful to further define their regional accumulation.
It is noteworthy that while full-length amyloid-β42 levels were comparable between Alzheimer’s disease and the pathological ageing, levels of N-truncated amyloid-β42, including amyloid-βpE3-42 and amyloid-βpE11-42, were increased in Alzheimer’s disease compared to the pathological ageing (Figs 1 and 3). Such differences were observed across neocortical and limbic areas (Supplementary Fig. 2). By analysing the temporal cortex, Portelius et al. (2015) also recently observed that levels of N-truncated amyloid-β were increased in Alzheimer’s disease, while levels of full-length amyloid-β were comparable to those in pathological ageing. Though it is still debated whether pathological ageing is an early phase of Alzheimer’s disease or a status resisting amyloid-β toxicity (Murray and Dickson, 2014), these results would highlight the role of N-truncated amyloid-β in the development of Alzheimer’s disease. Moreover, given that pathological ageing is an early phase of Alzheimer’s disease, these results suggest that full-length amyloid-β42 accumulation occurs in the earlier phase, while N-truncated amyloid-β42 continues to accumulate until the later phase of the disease. Our observation might be consistent with the notion that diffuse plaques, an early form of amyloid-β accumulation consisting mostly of full-length amyloid-β42, constitute a major part of amyloid plaques in the pathological ageing, while N-truncated amyloid-β species exist in more mature plaques that are more abundant in Alzheimer’s disease (Iwatsubo et al., 1996; Dickson, 1997; Guntert et al., 2006).
Although further clarification is needed of the biological and pathological events that contribute to the spatiotemporal accumulation of N-truncated amyloid-β42 in the brain, its significant regional association with BACE1 and inverse association with neprilysin, but no significant association with PSD95, provide some clues that specific mechanisms distinct from those involved in the accumulation of the full-length amyloid-β exist in the brain. Our results are consistent with the possibilities that (i) increased BACE1 could lead to de novo production of amyloid-β11-40/42, and also enhance truncation of amyloid-β through its degradation (Liu et al., 2002; Saido and Leissring, 2012); and (ii) lower neprilysin reduces degradation of amyloid-β prior to or potentially after its aggregation, resulting in accumulation of several N-truncated amyloid-β species (Wang et al., 2006). Although it is still under debate regarding the levels and activities of these enzymes (Miners et al., 2010), increased BACE1 levels/activities and decreased neprilysin levels/activities were previously observed in brains of patients with Alzheimer’s disease (Wang et al., 2006; Vassar et al., 2009). These pathogenic events could together promote the accumulation of N-truncated amyloid-β species during disease progression. However, further evidence suggests that additional or alternative processes are also likely to be involved in spatiotemporal N-truncated amyloid-β42 accumulation: (i) regional associations between N-truncated amyloid-β42 accumulation and BACE1 or neprilysin are relatively weak compared to those observed between the accumulations of N-truncated amyloid-β and apoE or between full-length amyloid-β accumulation and PSD95 or soluble apoE (Table 1) (Shinohara et al., 2013, 2014); (ii) several N-truncated amyloid-β species accumulate that are not direct products of BACE1-cleaved APP or neprilysin-degraded amyloid-β; (iii) atypical regional distribution is specific to amyloid-β42 but not amyloid-β40; and (iv) N-terminal truncation of amyloid-β is likely to occur during maturation of initial amyloid deposits, consisting of full-length amyloid-β, probably excluding the possibility that de novo produced N-truncated amyloid-β accumulate during disease progression. Glutaminyl cyclase catalyses pyroglutamate modification and may contribute to pyroglutamylated amyloid-β accumulation in the brain (Hartlage-Rubsamen et al., 2011; Becker et al., 2013). However, glutaminyl cyclase might not be the sole enzyme responsible for the spatiotemporal accumulation of N-truncated amyloid-β42 in the brain based on the following observations: (i) non-pyroglutamylated amyloid-β42 can also contribute to the N-truncated amyloid-β that show a regional distribution distinct from that of full-length amyloid-β42; and (ii) the regional distribution of amyloid-βpE3-42 is not entirely similar to that of amyloid-βpE11-42.
We observed strong spatiotemporal associations of tau and to a greater extent apoE with amyloid-βpE11-42, compared to full-length amyloid-β42. Although some previous studies have observed that N-truncated (pyroglutamylated) amyloid-β were associated with tau, emphasizing their role in the disease (Nussbaum et al., 2012; Mandler et al., 2014), few studies addressed their relationship with apoE. Previously, Thal et al. (2005) observed weaker overlaps between apoE and full-length amyloid-β42 accumulation, but they concluded that apoE masks the N-terminal epitope of amyloid-β. Although our study does not directly rule out such a possibility, our in vitro assay indicates that apoE likely binds to the accumulated N-truncated amyloid-β42 more strongly than to the accumulated full-length amyloid-β42. Interestingly, the stronger binding was observed despite lower aggregation propensity of amyloid-βpE11-42, suggesting that some mechanism other than amyloid aggregation determines the binding affinity between amyloidogenic proteins and apoE. As the apoE-binding site may exist in the residue 12–28 region of amyloid-β (Strittmatter et al., 1993), removal of the N-terminal segment might facilitate binding with apoE. Alternatively, pyroglutamylated amyloid-β is more hydrophobic and hence more lipophilic, and thus more likely to recruit apoE into its deposits (Schlenzig et al., 2009). Such atypical properties of N-terminal truncated amyloid-β could result in greater neurotoxicity, relative to full-length amyloid-β (Bayer and Wirths, 2014). As apoE accumulation alone associated with a worse score in the Kokmen Short Test of Mental Status (Table 2), additional investigation is needed into how the interaction between amyloid-β and apoE accumulation affects the neurotoxicity of N-terminal truncated amyloid-β and whether apoE accumulation itself could be utilized as a clinical biomarker for disease severity.
Limited information is available regarding the effects N-truncated amyloid-β42 accumulation on clinical outcomes. In particular, to our knowledge, the effects of N-truncated amyloid-β42 other than amyloid-βpE3-40/42 have not previously been reported. Despite the limitations of retrospective analysis, we observed that total N-truncated amyloid-β42 or amyloid-βpE11-42 accumulation in cortical areas is associated with four important clinical features; earlier disease onset, shorter disease duration until death, rapid disease progression, and a worse cognitive score. Importantly, neither full-length amyloid-β40/42 nor N-truncated amyloid-β40 is inversely associated with these clinical features, though some positive associations exist (Table 2), possibly highlighting the critical role of N-truncated amyloid-β42 accumulation in the disease. We also observed that earlier disease onset is associated with a worse cognitive score (Supplementary Fig. 11). These results are consistent with a notion that earlier disease onset, severe cognitive impairment and shorter disease duration are associated with each other (Larson et al., 2004; Todd et al., 2013). Thus, although additional studies are necessary to demonstrate the causal link, our results suggest that Alzheimer’s disease-related clinical outcomes are impacted to a greater extent by N-truncated amyloid-β42 accumulation than other amyloid-β accumulations. Of note, we observed (i) some differences between total N-truncated amyloid-β42 and amyloid-βpE11-42; and (ii) still significant associations after subtracting pyroglutamylated amyloid-β42 levels from total N-truncated amyloid-β42 levels, indicating the need to further define the specific species of N-truncated amyloid-β42 most strongly associated with each clinical feature in the future study.
Several important implications for future studies can be drawn from the current findings. First, neuropathological amyloid-β assessment does not necessarily differentiate antibodies against the N-terminal epitope of amyloid-β (Hyman et al., 2012). Such ambiguity should be carefully considered, as it might produce variable results regarding spatiotemporal amyloid-β accumulation as well as pathological roles of amyloid-β accumulation in the disease. Second, it would be useful to determine to what extent amyloid-specific ligands used for clinical imaging can distinguish full-length amyloid-β and N-terminal truncated amyloid-β accumulation in order to better interpret the results from current amyloid imaging studies. Third, few studies to date assess how individual N-truncated amyloid-β40/42 levels change in CSF/plasma in different disease stages and how they are associated with the levels of other biomarkers and clinical outcomes, relative to full length amyloid-β40/42 levels. Finally, APP transgenic mouse models accumulate much less N-truncated amyloid-β, especially amyloid-β that is truncated after residue 3 (Kawarabayashi et al., 2001; Kalback et al., 2002; Schieb et al., 2011). New animal models accumulating N-truncated amyloid-β species similar to those in human brains may better recapitulate neuropathological disease features, including apoE and tau accumulation. Such models will also be valuable for developing and testing therapies.
Supplementary Material
Acknowledgements
We thank Ms. Mary D. Davis and Ms. Lindsey M. Felton for the careful reading of this manuscript, Drs Jungsu Kim and Jaekwang Kim for sharing equipment and helpful discussion, and Dr Takahisa Kanekiyo and other Bu laboratory members for helpful discussion.
Funding
This research was supported by grants from the National Institutes of Health (NIH) (R37AG027924, RF1 AG051504, and P01 NS074969-Project 3 to G.B.); Cure Alzheimer’s Fund (to G.B.); Mayo Clinic Alzheimer’s Disease Research Center (Alzheimer’s diseaseRC) (PI: R.C.P., P50 AG016574) (to D.W.D, G.B. and M.S.); BrightFocus Foundation (to M.S.). The authors also acknowledge the many individuals who contribute to the Mayo Clinic Alzheimer Disease Research Center and Mayo Clinic Study of Aging (PI: R.C.P., U01 AG006786), and support from the GHR Foundation and Mayo Foundation for Education and Research without whose contributions this study would not have been possible.
Conflict of interest
R.C.P. has been serving as a consultant for Roche, Inc., Merck, Inc., Genentech, Inc., Biogen, Inc. and Eli Lilly and company. All other authors declare that they have no conflicts of interest.
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- DRS
Dementia Rating Scale
- GuHCl
guanidine hydrochloride
References
- Bayer TA, Wirths O. Focusing the amyloid cascade hypothesis on N-truncated Abeta peptides as drug targets against Alzheimer's disease. Acta Neuropathol 2014; 127: 787–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A, Kohlmann S, Alexandru A, Jagla W, Canneva F, Bauscher C, et al. Glutaminyl cyclase-mediated toxicity of pyroglutamate-beta amyloid induces striatal neurodegeneration. BMC Neurosci 2013; 14: 1471–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 2006; 224(Pt 3): 213–32. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82: 239–59. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, et al. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci 2005; 25: 7709–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cynis H, Frost JL, Crehan H, Lemere CA. Immunotherapy targeting pyroglutamate-3 Abeta: prospects and challenges. Mol Neurodegener 2016; 11: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demattos RB, Lu J, Tang Y, Racke MM, Delong CA, Tzaferis JA, et al. A plaque-specific antibody clears existing beta-amyloid plaques in Alzheimer's disease mice. Neuron 2012; 76: 908–20. [DOI] [PubMed] [Google Scholar]
- Dickson DW. The pathogenesis of senile plaques. J Neuropathol Exp Neurol 1997; 56: 321–39. [DOI] [PubMed] [Google Scholar]
- Guntert A, Dobeli H, Bohrmann B. High sensitivity analysis of amyloid-beta peptide composition in amyloid deposits from human and PS2APP mouse brain. Neuroscience 2006; 143: 461–75. [DOI] [PubMed] [Google Scholar]
- Hartlage-Rubsamen M, Morawski M, Waniek A, Jager C, Zeitschel U, Koch B, et al. Glutaminyl cyclase contributes to the formation of focal and diffuse pyroglutamate (pGlu)-Abeta deposits in hippocampus via distinct cellular mechanisms. Acta Neuropathol 2011; 121: 705–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori Y, Hashimoto T, Wakutani Y, Urakami K, Nakashima K, Condron MM, et al. The Tottori (D7N) and English (H6R) familial Alzheimer disease mutations accelerate Abeta fibril formation without increasing protofibril formation. J Biol Chem 2007; 282: 4916–23. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 2012; 8: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwatsubo T, Saido TC, Mann DM, Lee VM, Trojanowski JQ. Full-length amyloid-beta (1-42) and amino-terminally modified and truncated amyloid-beta 42 deposit in diffuse plaques. Am J Pathol 1996; 149: 1823–30. [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain 2008; 131: 665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalback W, Watson MD, Kokjohn TA, Kuo YM, Weiss N, Luehrs DC, et al. APP transgenic mice Tg2576 accumulate Abeta peptides that are distinct from the chemically modified and insoluble peptides deposited in Alzheimer's disease senile plaques. Biochemistry 2002; 41: 922–8. [DOI] [PubMed] [Google Scholar]
- Kanekiyo T, Xu H, Bu G. ApoE and Abeta in Alzheimer's disease: accidental encounters or partners? Neuron 2014; 81: 740–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran E, Mercken M, Strooper BD. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 2011; 10: 698–712. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci 2001; 21: 372–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol 2004; 55: 306–19. [DOI] [PubMed] [Google Scholar]
- Larson EB, Shadlen MF, Wang L, McCormick WC, Bowen JD, Teri L, et al. Survival after initial diagnosis of Alzheimer disease. Ann Intern Med 2004; 140: 501–9. [DOI] [PubMed] [Google Scholar]
- Liu K, Doms RW, Lee VM. Glu11 site cleavage and N-terminally truncated A beta production upon BACE overexpression. Biochemistry 2002; 41: 3128–36. [DOI] [PubMed] [Google Scholar]
- Lyons B, Friedrich M, Raftery M, Truscott R. Amyloid plaque in the human brain can decompose from abeta(1-40/1-42) by spontaneous nonenzymatic processes. Anal Chem 2016; 88: 2675–84. [DOI] [PubMed] [Google Scholar]
- Mandler M, Walker L, Santic R, Hanson P, Upadhaya AR, Colloby SJ, et al. Pyroglutamylated amyloid-beta is associated with hyperphosphorylated tau and severity of Alzheimer's disease. Acta Neuropathol 2014; 128: 67–79. [DOI] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 1985; 82: 4245–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DL, Papayannopoulos IA, Styles J, Bobin SA, Lin YY, Biemann K, et al. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer′s disease. Arch Biochem Biophys 1993; 301: 41–52. [DOI] [PubMed] [Google Scholar]
- Miners JS, van Helmond Z, Kehoe PG, Love S. Changes with age in the activities of beta-secretase and the Abeta-degrading enzymes neprilysin, insulin-degrading enzyme and angiotensin-converting enzyme. Brain Pathol 2010; 20: 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miravalle L, Calero M, Takao M, Roher AE, Ghetti B, Vidal R. Amino-terminally truncated Abeta peptide species are the main component of cotton wool plaques. Biochemistry 2005; 44: 10810–21. [DOI] [PubMed] [Google Scholar]
- Moore BD, Chakrabarty P, Levites Y, Kukar TL, Baine AM, Moroni T, et al. Overlapping profiles of Abeta peptides in the Alzheimer's disease and pathological aging brains. Alzheimers Res Ther 2012; 4: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H, Takio K, Ogawara M, Selkoe DJ. Mass spectrometry of purified amyloid beta protein in Alzheimer's disease. J Biol Chem 1992; 267: 17082–6. [PubMed] [Google Scholar]
- Murray ME, Dickson DW. Is pathological aging a successful resistance against amyloid-beta or preclinical Alzheimer’s disease? Alzheimers Res Ther 2014; 6: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res 1991; 541: 163–6. [DOI] [PubMed] [Google Scholar]
- Naslund J, Schierhorn A, Hellman U, Lannfelt L, Roses AD, Tjernberg LO, et al. Relative abundance of Alzheimer A beta amyloid peptide variants in Alzheimer disease and normal aging. Proc Natl Acad Sci USA 1994; 91: 8378–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama E, Iwamoto N, Ohwada J, Arai H. Distribution of apolipoprotein E in senile plaques in brains with Alzheimer's disease: investigation with the confocal laser scan microscope. Brain Res 1997; 750: 20–4. [DOI] [PubMed] [Google Scholar]
- Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-beta. Nature 2012; 485: 651–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike CJ, Overman MJ, Cotman CW. Amino-terminal deletions enhance aggregation of beta-amyloid peptides in vitro. J Biol Chem 1995; 270: 23895–8. [DOI] [PubMed] [Google Scholar]
- Portelius E, Bogdanovic N, Gustavsson MK, Volkmann I, Brinkmalm G, Zetterberg H, et al. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer's disease. Acta Neuropathol 2010; 120: 185–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portelius E, Lashley T, Westerlund A, Persson R, Fox NC, Blennow K, et al. Brain amyloid-beta fragment signatures in pathological ageing and Alzheimer's disease by hybrid immunoprecipitation mass spectrometry. Neurodegener Dis 2015; 15: 50–7. [DOI] [PubMed] [Google Scholar]
- Portelius E, Zetterberg H, Gobom J, Andreasson U, Blennow K. Targeted proteomics in Alzheimer's disease: focus on amyloid-beta. Expert Rev Proteomics 2008; 5: 225–37. [DOI] [PubMed] [Google Scholar]
- Saido T, Leissring MA. Proteolytic degradation of amyloid beta-protein. Cold Spring Harb Perspect Med 2012; 2: a006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saido TC, Yamao-Harigaya W, Iwatsubo T, Kawashima S. Amino- and carboxyl-terminal heterogeneity of beta-amyloid peptides deposited in human brain. Neurosci Lett 1996; 215: 173–6. [DOI] [PubMed] [Google Scholar]
- Schieb H, Kratzin H, Jahn O, Möbius W, Rabe S, Staufenbiel M, et al. β-Amyloid peptide variants in brains and cerebrospinal fluid from amyloid precursor protein (APP) transgenic mice: comparison with human Alzheimer amyloid. J Biol Chem 2011; 286: 33747–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenzig D, Manhart S, Cinar Y, Kleinschmidt M, Hause G, Willbold D, et al. Pyroglutamate formation influences solubility and amyloidogenicity of amyloid peptides. Biochemistry 2009; 48: 7072–8. [DOI] [PubMed] [Google Scholar]
- Schonherr C, Bien J, Isbert S, Wichert R, Prox J, Altmeppen H, et al. Generation of aggregation prone N-terminally truncated amyloid beta peptides by meprin beta depends on the sequence specificity at the cleavage site. Mol Neurodegener 2016; 11: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, Bu G. What can we learn from regional vulnerability to amyloid-β accumulation in nondemented individuals? Neurodegener Dis Manag 2013; 3: 187–9. [Google Scholar]
- Shinohara M, Fujioka S, Murray ME, Wojtas A, Baker M, Rovelet-Lecrux A, et al. Regional distribution of synaptic markers and APP correlate with distinct clinicopathological features in sporadic and familial Alzheimer's disease. Brain 2014; 137(Pt 5): 1533–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, Petersen RC, Dickson DW, Bu G. Brain regional correlation of amyloid-beta with synapses and apolipoprotein E in non-demented individuals: potential mechanisms underlying regional vulnerability to amyloid-beta accumulation. Acta Neuropathol 2013; 125: 535–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA 1993; 90: 8098–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Capetillo-Zarate E, Schultz C, Rub U, Saido TC, Yamaguchi H, et al. Apolipoprotein E co-localizes with newly formed amyloid beta-protein (Abeta) deposits lacking immunoreactivity against N-terminal epitopes of Abeta in a genotype-dependent manner. Acta Neuropathol 2005; 110: 459–71. [DOI] [PubMed] [Google Scholar]
- Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002; 58: 1791–800. [DOI] [PubMed] [Google Scholar]
- Todd S, Barr S, Roberts M, Passmore AP. Survival in dementia and predictors of mortality: a review. Int J Geriatr Psychiatry 2013; 28: 1109–24. [DOI] [PubMed] [Google Scholar]
- Vassar R, Kovacs DM, Yan R, Wong PC. The β-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J Neurosci 2009; 29: 12787–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlassenko AG, Vaishnavi SN, Couture L, Sacco D, Shannon BJ, Mach RH, et al. Spatial correlation between brain aerobic glycolysis and amyloid-beta (Abeta) deposition. Proc Natl Acad Sci USA 2010; 107: 17763–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D-S, Dickson DW, Malter JS. β-amyloid degradation and Alzheimer's disease. J Biomed Biotechnol 2006; 2006: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Dickson DW, Trojanowski JQ, Lee VMY. The levels of soluble versus insoluble brain amyloid-β distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol 1999; 158: 328–37. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Ishiguro K, Sugihara S, Nakazato Y, Kawarabayashi T, Sun X, et al. Presence of apolipoprotein E on extracellular neurofibrillary tangles and on meningeal blood vessels precedes the Alzheimer beta-amyloid deposition. Acta Neuropathol 1994; 88: 413–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.