

Abstract
Purpose
The aim of this study was to evaluate the ability of Porphyromonas gingivalis (P. gingivalis) to induce oxidation of high-density lipoprotein (HDL) and to determine whether the oxidized HDL induced by P. gingivalis exhibited altered antiatherogenic function or became proatherogenic.
Methods
P. gingivalis and THP-1 monocytes were cultured, and the extent of HDL oxidation induced by P. gingivalis was evaluated by a thiobarbituric acid-reactive substances (TBARS) assay. To evaluate the altered antiatherogenic and proatherogenic properties of P. gingivalis-treated HDL, lipid oxidation was quantified by the TBARS assay, and tumor necrosis factor alpha (TNF-α) levels and the gelatinolytic activity of matrix metalloproteinase (MMP)-9 were also measured. After incubating macrophages with HDL and P. gingivalis, Oil Red O staining was performed to examine foam cells.
Results
P. gingivalis induced HDL oxidation. The HDL treated by P. gingivalis did not reduce lipid oxidation and may have enhanced the formation of MMP-9 and TNF-α. P. gingivalis-treated macrophages exhibited more lipid aggregates than untreated macrophages.
Conclusions
P. gingivalis induced HDL oxidation, impairing the atheroprotective function of HDL and making it proatherogenic by eliciting a proinflammatory response through its interaction with monocytes/macrophages.
Keywords: Atherosclerosis, Cardiovascular diseases, Cholesterol, Periodontitis, Porphyromonas gingivalis
Graphical Abstract
INTRODUCTION
Chronic infectious diseases are known to increase the risk of atherosclerosis and cardiovascular disease [1]. Some reports have described a strong association between atherosclerosis and various infectious pathogens [2,3]. Therefore, infection-induced proatherogenic changes in the lipoprotein profile may be a mechanism underlying the increased risk of atherosclerosis in patients with chronic infections [4]. Several pathogens that cause chronic infections, such as Chlamydia pneumonia, Helicobacter pylori, and periodontal pathogens, may induce alterations in lipoprotein metabolism [5,6]. Periodontitis, a consequence of persistent bacterial infection and chronic inflammation, has been suggested to be a predictor of coronary heart disease [7]. Aggregatibacter actinomycetemcomitans and Porphyromonas gingivalis, the major periodontal pathogens, were found to accelerate lipid peroxidation and the progression of atherosclerosis in apolipoprotein E-deficient, spontaneously hyperlipidemic mice [2].
Elevated plasma levels of low-density lipoprotein (LDL) cholesterol are a key risk factor for atherosclerosis, a major cardiovascular disease. Oxidation of LDL is a risk factor for atherogenesis [8]. In contrast, high-density lipoprotein (HDL) cholesterol levels are inversely correlated with the risk of coronary artery disease. HDL prevents atherosclerosis by reversing the stimulatory effect of oxidized LDL on monocyte infiltration. Thus, oxidized LDL and HDL are antagonists in the development of cardiovascular disease [9]. HDL protects against atherosclerosis by removing excess cholesterol from macrophages through pathways involving reverse cholesterol transport. HDL also inhibits lipid oxidation, restores endothelial function, and promotes anti-inflammatory and antiapoptotic mechanisms. Such properties may contribute considerably to the capacity of HDL to inhibit atherosclerosis [10].
Infection and inflammation alter lipoprotein distribution and the composition of lipoprotein subclasses, causing dramatic changes in both HDL and LDL composition [11]. It has been suggested that systemic and vascular inflammation induces the conversion of HDL to a dysfunctional form that has an impaired antiatherogenic effect [10]. Structural changes in HDL altering its functionality, including oxidation, result in an increased risk of cardiovascular disease. Oxidized HDL not only loses important protective functions, but also acquires severe proinflammatory and proatherosclerotic properties [12].
The objectives of this study were to evaluate the ability of P. gingivalis, a major periodontal pathogen, to induce oxidation of HDL, and to evaluate whether P. gingivalis-oxidized HDL had impaired antiatherogenic function or became proatherogenic.
MATERIALS AND METHODS
Bacteria culture and HDL preparation
P. gingivalis (ATCC 33277, American Type Culture Collection, Manassas, VA, USA) was cultured anaerobically in tryptic soy broth (Difco Laboratories, Detroit, MI, USA) supplemented with hemin and menadione. HDL was purchased from Biovision (Biovision Inc., Milpitas, CA, USA) and was used at a concentration of 100 μg/mL.
THP-1 cell lines and culture conditions
Human monocytic THP-1 (TIB-202) cells were grown in RPMI 1640 medium (2 mM L-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 10 mM 4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid, and 1.0 mM sodium pyruvate) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin at 37°C in a humidified atmosphere containing 5% CO2. The THP-1 cells were infected with live P. gingivalis at a multiplicity of infection of 1:100 for 24 hours at 37°C in 5% CO2. HDL was incubated with THP-1 cells and P. gingivalis for 24 hours. HDL was oxidized with 0.5 μM CuSO4, a strong oxidizing agent, as a positive control. Untreated HDL, in conditions with no bacteria or monocytes, was used as a negative control. Each experiment was conducted in RPMI 1640 medium without human or fetal calf serum because those substances contain a high concentration of antioxidants that can hamper lipid oxidation assays.
Thiobarbituric acid-reactive substances (TBARS) assay
In order to evaluate the oxidation of HDL or LDL, a TBARS assay (R&D Systems, Minneapolis, MN, USA) was used. Oxidizing agents can alter lipid structure, resulting in the formation of malondialdehyde (MDA), which can be measured using the TBARS assay, in which aldehydes formed during lipoprotein oxidation react with thiobarbituric acid (TBA) to produce measurable fluorimetric MDA-TBA adducts. The assay used in this experiment was capable of detecting small quantities of TBARS. Monocytes were plated in 96-well microtiter plates at a density of 5×104 cells/well. Each well contained standard or sample (150 μL each) and 75 μL of TBA according to the manufacturer's instructions. The sample measured was the supernatant only. Baseline optical densities were measured at 532 nm using a microplate reader (Molecular Devices, Sunnyvale, CA, USA). The plates were incubated for 2 to 3 hours between 45°C and 50°C, and the final optical densities were measured. The baseline optical density of each well was subtracted from the final reading to compensate for the contribution of standards or sample to the final absorption. The concentrations of lipoprotein oxidation products were calculated by comparing them to a standard curve. In order to measure the antioxidant potential of P. gingivalis-treated HDL, LDL (100 μg/mL) was treated with CuSO4 (0.5 μM) for 24 hours to induce LDL oxidation, and then the samples were washed with phosphate-buffered saline (PBS). The cells were then seeded at 5×104 cells/well in a 96-well plate. The oxidized LDL and cells were treated with HDL and P. gingivalis.
Assay of tumor necrosis factor alpha (TNF-α)
The concentrations of TNF-α in cell culture supernatants were determined using enzyme-linked immunosorbent assay (ELISA) kits (eBioscience, San Diego, CA, USA) according to the manufacturer's protocol. Optical density was measured at 450 nm using an ELISA microplate reader (Molecular Devices).
Activity of matrix metalloproteinases (MMPs, gelatinases)
The activity of gelatinases secreted by the cells into the culture medium was determined by gelatin zymography. Gelatin (1 mg/mL) was polymerized with 7.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Cell culture supernatants normalized by protein concentration were electrophoresed at 120 V for 1.5 hours. The gels were treated with 2.5% Triton-X 100 for 30 minutes and subsequently incubated with substrate buffer (50 mM Tris-HCl, 5 mM CaCl2, 0.02% NaN3, pH 7.5) at 37°C for 48 hours. The gels were stained with Coomassie blue R-250 and imaged using an image scanner (Amersham Biosciences, Hong Kong, China). Gelatinolytic activity was quantified using a specialized program (Quantity One program, BioRad Laboratories India Pvt. Ltd, Haryana, India). The image was inverted for efficient quantification.
Oil Red O staining
Oil Red O staining was used to examine oxidized HDL-laden macrophagic cells. THP-1 cells treated with 100 ng/mL phorbol-12-myristate-13-acetate were cultured for 7 days to induce differentiation into macrophages. Macrophages were incubated with HDL and P. gingivalis for 24 hours, washed twice with PBS, and fixed with 4% paraformaldehyde. Intracellular lipid droplets were stained with Oil Red O solution (Sigma-Aldrich, St. Louis, MO, USA) for 1 hour and washed twice with distilled water for examination using light microscopy (×400 magnification).
Statistical analysis
Each experiment was repeated independently at least 3 times. Statistical analyses were carried out using SPSS Statistics for Windows, version 21.0 (IBM Corp., Armonk, NY, USA). For intergroup comparisons, statistical significance was determined by 1-way analysis of variance followed by the Tukey post hoc test. P values <0.05 were considered to indicate statistical significance.
RESULTS
Monocyte-mediated oxidation of HDL incubated with P. gingivalis
The intensity of HDL oxidation was quantified by measuring the concentration of TBARS. When THP-1 cells were incubated with HDL (100 μg/mL) for 24 hours, P. gingivalis-induced HDL oxidation was observed, in contrast to the HDL-only sample without P. gingivalis (Figure 1). The difference was statistically significant (P<0.05).
Figure 1.

Quantification of Porphyromonas gingivalis-induced HDL oxidation using a TBARS assay.
HDL: high-density lipoprotein, TBARS: thiobarbituric acid-reactive substances, Control: sample without HDL and Porphyromonas gingivalis, P.g.: Porphyromonas gingivalis.
a)Statistically significant (P<0.01).
Antioxidant ability of HDL and LDL incubated with P. gingivalis
In order to measure the antioxidant potential of P. gingivalis-treated HDL, lipid oxidation was measured by the TBARS assay (Figure 2). There was a significant reduction in TBARS when oxidized LDL was incubated with HDL; this result was independent of whether P. gingivalis was present (P<0.01). However, when P. gingivalis was present, in both the samples with oxidized LDL only and the samples with oxidized LDL and HDL, TBARS were present at a greater concentration than when P. gingivalis was not present (P<0.05). In the samples that did not contain HDL, there was an increase in TBARS when LDL was incubated with CuSO4 in the presence of P. gingivalis compared to samples without P. gingivalis (P<0.05).
Figure 2.

TBARS analysis of LDL incubated with CuSO4 and HDL in the presence and absence of Porphyromonas gingivalis.
LDL: low-density lipoprotein, HDL: high-density lipoprotein, Control: sample without LDL, HDL, and Porphyromonas gingivalis, P.g.: Porphyromonas gingivalis, TBARS: thiobarbituric acid-reactive substances.
a)Statistically significant (P<0.05); b)Statistically significant (P<0.01); c)Statistically significant (P<0.001).
Cytotoxic effects of HDL incubated with P. gingivalis
Under oxidative stress or proinflammatory stimuli, monocytes become activated and produce MMPs and proinflammatory cytokines such as TNF-α. Monocytes incubated with P. gingivalis in the presence of HDL produced significantly higher levels of TNF-α than monocytes treated with CuSO4. TNF-α levels were also higher in monocytes treated with HDL and P. gingivalis than in those treated with P. gingivalis alone (Figure 3A). The gelatinolytic activity of MMP-9 was higher in monocytes treated with HDL and P. gingivalis than in those treated with P. gingivalis alone (Figure 3B).
Figure 3.

Proinflammatory activity by Porphyromonas gingivalis-induced oxidized HDL. (A) Monocytes incubated with Porphyromonas gingivalis in the presence of HDL produced significantly higher levels of TNF-α than monocytes treated with CuSO4 or with HDL alone. (B) Monocytes incubated with Porphyromonas gingivalis and HDL showed higher MMP-9 activity than cells incubated with HDL alone.
HDL: high-density lipoprotein, TNF-α: tumor necrosis factor alpha, MMP: matrix metalloproteinase, P.g.: Porphyromonas gingivalis, Control: sample without Porphyromonas gingivalis.
a) Statistically significant (P<0.05); b)Statistically significant (P<0.001).
Microscopic analysis of foam cell formation by HDL incubated with P. gingivalis
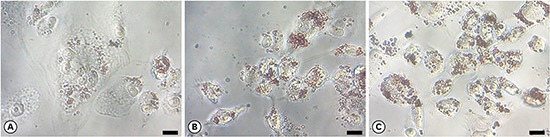
Using light microscopy (×400 magnification), lipid-laden cells exhibited more extensive Oil Red O staining than control cells (Figure 4). These data indicate a high accumulation of oxidized HDL. P. gingivalis-treated macrophages exhibited higher levels of lipid aggregates than macrophages not treated with P. gingivalis. Lipids aggregated to a greater extent in P. gingivalis-treated macrophages than in untreated macrophages, suggesting that macrophages treated with P. gingivalis and HDL engulfed a greater amount of oxidized HDL.
Figure 4.

Macrophage foam cell formation assessed by Oil Red O staining. The uptake of oxidized HDL by macrophages treated with (A) HDL only, (B) HDL oxidized with Porphyromonas gingivalis, and (C) HDL oxidized with CuSO4 was determined by light microscopy (scale bar=200 μm).
HDL: high-density lipoprotein.
DISCUSSION
Compelling evidence suggests that the circulating HDL levels are inversely related to the incidence of cardiovascular disease. HDL is believed to exert a protective function against the development of cardiovascular disease due to its role in mediating reverse cholesterol transport and preventing the oxidative modification of LDL, thus inhibiting cytotoxicity and the inflammatory responses triggered by cytokines, oxidized LDL, or oxidants [13]. HDL is susceptible to damaging structural modifications, including oxidation. Evidence has suggested that oxidized HDL exhibits a proatherogenic phenotype because oxidation markedly impairs the antiatherogenic function of HDL [14]. Thus, targeting defective HDL, rather than total circulating HDL, may be more effective for preventing and inhibiting atherosclerosis. Recent studies have also suggested that higher HDL levels may not always be cardioprotective because of the potential of HDL to become dysfunctional [15]. In order to investigate the mechanism linking the progression of atherosclerosis with periodontal bacteria, we investigated the ability of P. gingivalis to induce oxidation of HDL and whether the oxidized HDL induced by P. gingivalis showed altered antiatherogenic function or became proatherogenic.
Periodontal infection itself does not cause atherosclerosis, but rather accelerates it by inducing systemic inflammation and disrupting lipid metabolism. Chronic oral infection with P. gingivalis accelerates atheroma formation by shifting the lipid profile, with elevated levels of LDL and total cholesterol and reduced levels of HDL [16]. In atherosclerosis, macrophages stimulated by P. gingivalis drive the aggregation of fatty deposits in the vasculature and become foam cells, accelerating atheroma formation, in the presence of oxidized LDL [17]. In our previous study, we postulated that peptide 19 from P. gingivalis heat shock protein 60 has a distinct ability to induce native LDL oxidation as a plausible mechanism by which this peptide may drive epitope spreading to the neoantigen (i.e., oxidized LDL) in the pathogenesis of atherosclerosis [18]. In this study, we have found that P. gingivalis might also induce HDL oxidation. Our results suggest that P. gingivalis stimulates qualitative changes in the lipid profile by inducing oxidation of LDL and HDL.
High lipid oxidation was observed in both P. gingivalis-treated HDL and oxidized LDL compared to the corresponding samples without P. gingivalis. Our data suggest that P. gingivalis impairs antioxidant function. It is also important to confirm that when HDL is oxidized, it not only has a reduced antioxidant ability, but also exerts proatherogenic properties. Atherosclerosis can be further exacerbated by the progressive incorporation of cytotoxic byproducts of lipid and phospholipid oxidation into a changing monocyte population at the cellular level of the arterial endothelium [19]. Oxidized HDL elicits a proinflammatory response by stimulating the release of TNF-α and gelatinases/MMP-9 from monocytes [20]. TNF-α is emerging as a key cytokine that exerts adverse effects during atherosclerosis. It can augment the local inflammatory response by altering lipid homeostasis and increasing the uptake of cholesterol ester [21]. MMPs are capable of extensively degrading all components of the extracellular matrix. Several lines of evidence support the potential role of MMPs in human atherosclerosis and plaque disruption [22,23]. Our data support the proposal that P. gingivalis-treated HDL induces the release of TNF-alpha and MMP-9 in monocytes, thereby promoting proatherogenic effects. However, although the P. gingivalis-treated HDL sample showed higher TNF-α levels, we do not know whether this was caused by other products of P. gingivalis that were affected by P. gingivalis-induced oxidized HDL or directly by the P. gingivalis-induced oxidized HDL.
Oxidized LDL and HDL are endocytosed by macrophages to generate cholesterol-loaded foam cells. The formation of lipid-laden foam cells is an important pathophysiological process in atherosclerosis [24]. In this study, P. gingivalis-treated macrophages exhibited more lipid aggregates than macrophages not treated with P. gingivalis according to Oil Red O staining. The oxidized HDL fraction interacts with foam cells to enhance the delivery of free cholesterol to the cell surface. Thus, oxidized HDL increases the excess of arterial wall cholesterol [14]. In addition, oxidized HDL has been found in the intima of atheromatous plaques [25]. Oxidative modifications of HDL not only attenuate its beneficial properties, but also cause it to exert a direct proatherogenic effect.
In conclusion, this study suggests that P. gingivalis induces HDL oxidation, impairing the atheroprotective function of HDL and stimulating proatherogenic effects, via proinflammatory responses through its interaction with monocytes/macrophages.
Footnotes
Funding: This study was supported by the Dental Research Institute (PNUDH DRI-2016-04 awarded to Ji-Young Joo), Pusan National University Dental Hospital.
Author Contributions: Conceptualization: Ji-Young Joo; Data curation: Hyung-Joon Kim, Eun-Young Kwon; Formal analysis: Hyun-Joo Kim, Gil-Sun Cha; Funding acquisition: Ji-Young Joo; Investigation: Hyun-Joo Kim, Gil-Sun Cha, Ji-Young Joo; Methodology: Ju-Youn Lee, Jeomil Choi, Ji-Young Joo; Project administration: Ji-Young Joo; Writing - original draft: Hyun-Joo Kim, Gil-Sun Cha, Ji-Young Joo; Writing - review & editing: Hyung-Joon Kim, Eun-Young Kwon, Ju-Youn Lee, Jeomil Choi, Ji-Young Joo.
Conflict of Interest: No potential conflict of interest relevant to this article was reported.
References
- 1.Genco R, Offenbacher S, Beck J. Periodontal disease and cardiovascular disease: epidemiology and possible mechanisms. J Am Dent Assoc. 2002;133:14S–22S. doi: 10.14219/jada.archive.2002.0375. [DOI] [PubMed] [Google Scholar]
- 2.Koizumi Y, Kurita-Ochiai T, Oguchi S, Yamamoto M. Nasal immunization with Porphyromonas gingivalis outer membrane protein decreases P. gingivalis-induced atherosclerosis and inflammation in spontaneously hyperlipidemic mice. Infect Immun. 2008;76:2958–2965. doi: 10.1128/IAI.01572-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muhlestein JB, Anderson JL, Hammond EH, Zhao L, Trehan S, Schwobe EP, et al. Infection with Chlamydia pneumoniae accelerates the development of atherosclerosis and treatment with azithromycin prevents it in a rabbit model. Circulation. 1998;97:633–636. doi: 10.1161/01.cir.97.7.633. [DOI] [PubMed] [Google Scholar]
- 4.Lösche W, Karapetow F, Pohl A, Pohl C, Kocher T. Plasma lipid and blood glucose levels in patients with destructive periodontal disease. J Clin Periodontol. 2000;27:537–541. doi: 10.1034/j.1600-051x.2000.027008537.x. [DOI] [PubMed] [Google Scholar]
- 5.Laurila A, Bloigu A, Näyhä S, Hassi J, Leinonen M, Saikku P. Chronic Chlamydia pneumoniae infection is associated with a serum lipid profile known to be a risk factor for atherosclerosis. Arterioscler Thromb Vasc Biol. 1997;17:2910–2913. doi: 10.1161/01.atv.17.11.2910. [DOI] [PubMed] [Google Scholar]
- 6.Laurila A, Bloigu A, Näyhä S, Hassi J, Leinonen M, Saikku P. Association of Helicobacter pylori infection with elevated serum lipids. Atherosclerosis. 1999;142:207–210. doi: 10.1016/s0021-9150(98)00194-4. [DOI] [PubMed] [Google Scholar]
- 7.Pussinen PJ, Jauhiainen M, Vilkuna-Rautiainen T, Sundvall J, Vesanen M, Mattila K, et al. Periodontitis decreases the antiatherogenic potency of high density lipoprotein. J Lipid Res. 2004;45:139–147. doi: 10.1194/jlr.M300250-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Steinberg D. Low density lipoprotein oxidation and its pathobiological significance. J Biol Chem. 1997;272:20963–20966. doi: 10.1074/jbc.272.34.20963. [DOI] [PubMed] [Google Scholar]
- 9.Mertens A, Holvoet P. Oxidized LDL and HDL: antagonists in atherothrombosis. FASEB J. 2001;15:2073–2084. doi: 10.1096/fj.01-0273rev. [DOI] [PubMed] [Google Scholar]
- 10.Rosenson RS, Brewer HB, Jr, Ansell BJ, Barter P, Chapman MJ, Heinecke JW, et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat Rev Cardiol. 2016;13:48–60. doi: 10.1038/nrcardio.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khovidhunkit W, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C. Cholesterol efflux by acute-phase high density lipoprotein: role of lecithin:cholesterol acyltransferase. J Lipid Res. 2001;42:967–975. [PubMed] [Google Scholar]
- 12.Kameda T, Ohkawa R, Yano K, Usami Y, Miyazaki A, Matsuda K, et al. Effects of myeloperoxidase-induced oxidation on antiatherogenic functions of high-density lipoprotein. J Lipids. 2015;2015:592594. doi: 10.1155/2015/592594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Florentin M, Liberopoulos EN, Wierzbicki AS, Mikhailidis DP. Multiple actions of high-density lipoprotein. Curr Opin Cardiol. 2008;23:370–378. doi: 10.1097/HCO.0b013e3283043806. [DOI] [PubMed] [Google Scholar]
- 14.Francis GA. High density lipoprotein oxidation: in vitro susceptibility and potential in vivo consequences. Biochim Biophys Acta. 2000;1483:217–235. doi: 10.1016/s1388-1981(99)00181-x. [DOI] [PubMed] [Google Scholar]
- 15.Barter PJ, Baker PW, Rye KA. Effect of high-density lipoproteins on the expression of adhesion molecules in endothelial cells. Curr Opin Lipidol. 2002;13:285–288. doi: 10.1097/00041433-200206000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Maekawa T, Takahashi N, Tabeta K, Aoki Y, Miyashita H, Miyauchi S, et al. Chronic oral infection with Porphyromonas gingivalis accelerates atheroma formation by shifting the lipid profile. PLoS One. 2011;6:e20240. doi: 10.1371/journal.pone.0020240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miyakawa H, Honma K, Qi M, Kuramitsu HK. Interaction of Porphyromonas gingivalis with low-density lipoproteins: implications for a role for periodontitis in atherosclerosis. J Periodontal Res. 2004;39:1–9. doi: 10.1111/j.1600-0765.2004.00697.x. [DOI] [PubMed] [Google Scholar]
- 18.Joo JY, Cha GS, Chung J, Lee JY, Kim SJ, Choi J. Peptide 19 of Porphyromonas gingivalis heat shock protein is a potent inducer of low-density lipoprotein oxidation. J Periodontol. 2017;88:e58–e64. doi: 10.1902/jop.2016.160402. [DOI] [PubMed] [Google Scholar]
- 19.Soumyarani VS, Jayakumari N. Oxidized HDL induces cytotoxic effects: implications for atherogenic mechanism. J Biochem Mol Toxicol. 2014;28:481–489. doi: 10.1002/jbt.21588. [DOI] [PubMed] [Google Scholar]
- 20.Soumyarani VS, Jayakumari N. Oxidatively modified high density lipoprotein promotes inflammatory response in human monocytes-macrophages by enhanced production of ROS, TNF-α, MMP-9, and MMP-2. Mol Cell Biochem. 2012;366:277–285. doi: 10.1007/s11010-012-1306-y. [DOI] [PubMed] [Google Scholar]
- 21.Persson J, Nilsson J, Lindholm MW. Interleukin-1beta and tumour necrosis factor-alpha impede neutral lipid turnover in macrophage-derived foam cells. BMC Immunol. 2008;9:70. doi: 10.1186/1471-2172-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones CB, Sane DC, Herrington DM. Matrix metalloproteinases: a review of their structure and role in acute coronary syndrome. Cardiovasc Res. 2003;59:812–823. doi: 10.1016/s0008-6363(03)00516-9. [DOI] [PubMed] [Google Scholar]
- 23.Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116:59–69. doi: 10.1172/JCI25074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ren J, Jin W, Chen H. oxHDL decreases the expression of CD36 on human macrophages through PPARgamma and p38 MAP kinase dependent mechanisms. Mol Cell Biochem. 2010;342:171–181. doi: 10.1007/s11010-010-0481-y. [DOI] [PubMed] [Google Scholar]
- 25.Nakajima T, Origuchi N, Matsunaga T, Kawai S, Hokari S, Nakamura H, et al. Localization of oxidized HDL in atheromatous plaques and oxidized HDL binding sites on human aortic endothelial cells. Ann Clin Biochem. 2000;37:179–186. doi: 10.1258/0004563001899186. [DOI] [PubMed] [Google Scholar]