

Abstract
Mycobacterium fortuitum is a natural fish pathogen. It induces apoptosis in headkidney macrophages (HKM) of catfish, Clarias sp though the mechanism remains largely unknown. We observed M. fortuitum triggers calcium (Ca2+) insult in the sub-cellular compartments which elicits pro-apototic ER-stress factor CHOP. Alleviating ER-stress inhibited CHOP and attenuated HKM apoptosis implicating ER-stress in the pathogenesis of M. fortuitum. ER-stress promoted calpain activation and silencing the protease inhibited caspase-12 activation. The study documents the primal role of calpain/caspase-12 axis on caspase-9 activation in M. fortuitum-pathogenesis. Mobilization of Ca2+ from ER to mitochondria led to increased mitochondrial Ca2+ (Ca2+)m load,, mitochondrial permeability transition (MPT) pore opening, altered mitochondrial membrane potential (ΔΨm) and cytochrome c release eventually activating the caspase-9/-3 cascade. Ultra-structural studies revealed close apposition of ER and mitochondria and pre-treatment with (Ca2+)m-uniporter (MUP) blocker ruthenium red, reduced Ca2+ overload suggesting (Ca2+)m fluxes are MUP-driven and the ER-mitochondria tethering orchestrates the process. This is the first report implicating role of sub-cellular Ca2+ in the pathogenesis of M. fortuitum. We summarize, the dynamics of Ca2+ in sub-cellular compartments incites ER-stress and mitochondrial dysfunction, leading to activation of pro-apoptotic calpain/caspase-12/caspase-9 axis in M. fortuitum-infected HKM.
Introduction
M. fortuitum is a rapidly growing, atypical, non-tubercular mycobacteria affecting wide range of animals including humans1–3. In fish, it is one of the etiologic agents causing piscine-tuberculosis or mycobacteriosis, a fatal disease characterized by the development of gray-white nodular structures and presence of single or multiple granulomatous lesions on several parenchymal organs 1. Despite its diverse host trophism and zoonotic importance, our knowledge on pathogenic mechanisms and virulence factors expressed by M. fortuitum is incomplete.
Alterations in cytosolic calcium (Ca2+)c levels play crucial role in microbial pathogenesis and disease outcome with reports suggesting pro-and anti-apoptotic roles of Ca2+ on mycobacteria-infected macrophages4, 5. Once Ca2+ is mobilized, it either interacts with various Ca2+-binding proteins or gets sequestered into the ER6. Calcium influx or depletion from the ER induces ER-stress6, 7. The ability to mount ER-stress response is critical for cell survival, but chronic or unresolved ER stress can lead to expression of pro-apoptotic C/EBP homologous protein (CHOP)8. Though prolonged ER-stress has been linked to mycobacterial pathogenesis9–14, it has not been reported in M. fortuitum.
To mitigate stress, the ER releases Ca2+ ((Ca2+)ER) through ER-membrane resident inositol-1,4,5-trisphosphate receptors (IP3R) and ryanodine receptors (RYRs)15. The (Ca2+)ER is either pumped out of the cell through specific channels or taken up by mitochondria through specific uniport transporter like M1CU1 and VDAc, the latter being facilitated by the known proximity between the two organelles16, 17.
Calcium overload to mitochondria leads to mitochondrial structure-function alterations eventually releasing the pro-apoptotic cytochrome c to the cytosol17. Activation of caspases, a family of cysteine-dependent aspartate-directed proteases, is central to apoptosis and caspase-12 appears to be the prime caspase involved in ER-stress induced apoptosis18. Calpains are Ca2+-activated non-lysosomal cysteine proteases which exist in two isoforms, calpain-1 and calpain-219. Each calpain consists of an 80 kDa catalytic subunit and a common 28 kDa subunit19. The role for calpain in promoting mycobacteria-induced apoptosis is still under investigation10, 11, 20. Several reports suggested the role of calpains in the activation of caspase-1221, 22 implicating the plurality of Ca2+ involvement in apoptosis.
The fish immune system is well-developed and comprised of both innate and adaptive immunity. However, unlike other vertebrates, the head kidney (HK) represents the main immunocompetent organ and HKM are important constituents of fish innate immunity23. We recently demonstrated the role of caspase-8 in M. fortuitum infection induced HKM apoptosis24. However, the interaction of caspase-12 and caspase-9 is not reported in M. fortuitum pathogenesis. In the present study we investigated the the role of caspase-12 and caspase-9 in M. fortuitum pathogenesis. Our results for the first time implicate Ca2+ dynamics between ER and mitochondria important for M. fortuitum induced apoptosis. We suggest that ER-stress espouses apoptosis of M. fortuitum-infected HKM and activation of calpain/caspase-12/caspase-9 axis crucial for initiating the apoptotic cascade.
Results
M. fortuitum-induced intracellular Ca2+ imbalance lead to CHOP- mediated HKM apoptosis
Previously, we reported that the imbalance in (Ca2+)c triggers apoptosis in M. fortuitum-infected fish macrophages24. Here, we studied the dynamics of (Ca2+)c in the two sub-cellular compartments, ER and mitochondria.
ER is the main storehouse of intracellular Ca2+ and under stressed condition (Ca2+)ER is released through IP3R and RYRs located on the ER-membrane. CHOP is a marker for ER-stress6, 7 and our preliminary results suggested significant CHOP mRNA expression at 2 h (Fig. 1b) and protein at 24 h (data not shown) in M. fortuitum-infected HKM. The HKM were pre-treated with 2-APB and Dant, specific inhibitors for IP3R and RYR respectively25, infected with M. fortuitum and the changes in CHOP expression and apoptosis studied at 24 h p.i. We observed decreased expression of CHOP (Fig. 1a) and HKM apoptosis (Figure S1) which suggested positive co-relation between (Ca2+)ER depletion and CHOP expression in M. fortuitum infected HKM. In the same line, we observed declined expression of CHOP in presence of intracellular Ca2+ chealator BAPTA/AM (Fig. 1a).
Fig. 1. M. fortuitum induces CHOP- mediated HKM apoptosis.

a HKM pre-treated or transfected with indicated inhibitors or siRNAs respectively prior to the infection with M. fortuitum and the CHOP protein expression was studied by confocal microscope using FITC-conjugated secondary antibody. The images are representative of three independent experiments and observed under confocal microscope ( × 40). b HKM were infected with M. fortuitum and CHOP mRNA expression was quantified by qPCR at indicated time p.i. c HKM were transfected with CHOP-siRNA or scrambled siRNA prior to infection with M. fortuitum and CHOP mRNA expression was quantified. Vertical bars represent mean ± SE (n = 3).*P < 0.05, compared to HKM; γP < 0.05, compared to HKM + Sc; •P < 0.05, compared to HKM + MF + Sc. HKM, control headkidney macrophage; HKM + MF, HKM infected with M. fortuitum; HKM + Sc + MF, HKM transfected with scrambled siRNA followed by M. fortuitum infection; HKM + CHOP-siRNA + MF, HKM transfected with CHOP-siRNA infected with M. fortuitum; HKM + 4-PBA + MF, HKM + 2-APB + MF, HKM + Dant + MF, HKM + BAPTA/AM + MF, HKM pre-treated with 4-PBA, 2-APB, Dant, BAPTA/AM respectively followed by M. fortuitum infection
Pre-treatment of HKM with general ER-stress inhibitor 4-PBA down-regulated CHOP expression (Fig. 1a), attenuated caspase-3 activity and HKM apoptosis (Figure S1). These findings were confirmed using CHOP-siRNA. Transfection with CHOP-siRNA down-regulated CHOP expression at mRNA (Fig. 1c) and protein level (Fig. 1a) besides attenuating M. fortuitum-induced HKM apoptosis (Figure S1). Our results for the first time implicated ER-stress induced CHOP in M. fortuitum-induced apoptosis and corroborate with earlier studies suggesting the pro-apoptotic role of CHOP in mycobacterial pathogenesis.
Mobilization of (Ca2+)ER into mitochondria led to mitochondrial dysfunction
Mitochondrial dysfunction due to Ca2+ overload is keystone in determining the fate of mycobacteria-infected macrophages26, 27. HKM were stained with Rhod-2/AM and Mito-Tracker Green FM and observed under the confocal microscope. The increase in Rhod-2 AM fluroscence clearly indicates increased (Ca2+)m uptake following 1 h of adding the bacteria with peak fluroscence recorded at 6 h p.i (Figure S2). Calcium influx to the mitochondria occurs through MUP16 and to explore this HKM were pre-treated with MUP blocker ruthenium red (RR) and (Ca2+)m dynamics monitored at 6 h p.i. We observed Rhod-2/AM fluroscence intensity was reversed in presence of RR (Fig. 2a) suggesting (Ca2+)m influx in M. fortuitum-infected HKM is uniporter driven.
Fig. 2. Close apposition of ER and mitochondria leads to mitochondrial-Ca2+ overload in M. fortuitum infected HKM.

a HKM pre-treated with or without indicated inhibitors were infected with M. fortuitum and mitochondrial-Ca2+ uptake studied 6 h p.i. by Rhod-2/AM and Mitotracker green marker. The images are representative of three independent experiments and observed under confocal microscope ( × 40). b Transmission electron microscopy of uninfected HKM (B1), M. fortuitum infected HKM at 6 h p.i. (B2) and 24 h p.i. (B3, B4). The images are representative of three independent experiments. HKM, control headkidney macrophage; HKM + MF, HKM infected with M. fortuitum; HKM + RR + MF, HKM + BAPTA/AM + MF, HKM + Dant + MF, HKM + 2-APB + MF, HKM pre-treated with RR, BAPTA/AM, Dant and 2-APB respectively and infected with M. fortuitum. Yellow arrow,mitochondrion; Red arrow, ER
To investigate the mobilization of (Ca2+)ER to mitochondria HKM were pre-treated separately with Dant, 2-APB and (Ca2+)m uptake monitored. The decrease in Rhod-2/AM fluorescence in presence of Dant and 2-APB (Fig. 2a), clearly proved the mobility of Ca2+ from ER to mitochondria. We reasoned, for the uptake of (Ca2+)ER through MUP, the two organelles ought to come in close proximity thereby facilitating the process. M. fortuitum-infected HKM were examined by TEM at 6 h and 24 h p.i. (Fig. 2b) and we observed spatial change in the sub-cellular organization with mitochondria in close apposition with ER, which appeared more evident in the HKM collected at 24 h p.i. (Fig. 2B3,B4).
The elevation in (Ca2+)m reduces mitochondrial membrane potential (ΔΨm)28. We monitored the changes in ΔΨm in M. fortuitum-infected HKM at different time points using the JC-1 dye. The increase in green fluorescence29 indicated time-dependent reduction in ΔΨm in infected HKM (Figure S3). Pre-treatment of HKM with specific inhibitors RR, Dant and 2-APB restored ΔΨm (Fig. 3a), which confirmed the uptake of (Ca2+)ER on mitochondrial dysfunction. The loss in ΔΨm leads to the formation of MPT28. The cell-permeant, green-fluorescent, lipophilic dye DiOC6 accumulates in mitochondria and its release is a reliable indicator for ΔΨm loss and MPT pore opening30. When we compared DiOC6 fluoroscence levels in uninfected and M. fortuitum-infected HKM, significant loss in fluoroscence levels was noted in the infected HKM (Fig. 3b) which suggested M. fortuitum infection leads to loss in ΔΨm and MPT formation. Among several molecules released via MPT, pro-apoptotic cytochrome c is important. Hence, the next step was studying cytochrome c release in M. fortuitum-infected HKM. Our confocal microscopy images suggests the transloction of cytochrome c to the cytosol of M. fortuitum-infected HKM (Fig. 3c). Pre-treatment with MPT inhibitor CsA restored ΔΨm (Fig. 3a), retained DiOC6 (data not shown) and inhibited cytochrome c release (Fig. 3c) in infected HKM. To this we concluded that acquisition of (Ca2+)ER impairs mitochondrial functioning triggering the apoptosis of M. fortuitum-infected HKM.
Fig. 3. Alteration in cytosolic Ca2+ homeostasis induces mitochondrial dysfunction triggering M. fortuitum-induced HKM apoptosis.

a The alteration in ΔΨm was studied using JC-1 dye. HKM pre-treated with or without indicated inhibitors were infected with M. fortuitum and at 12 h p.i. ΔΨm studied by confocal microscopy ( × 40). b MPT formation was studied using DiOC6 dye. HKM were infected with M. fortuitum for indicated time period and the relative fluorescence intensity of DiOC6 plotted. c HKM pre-treated with or without indicated inhibitors were infected with M. fortuitum and the cytosolic translocation of cytochrome c studied by immunoflurescence using TRITC- tagged secondary antibody at 24 h p.i. The nuclei were stained with DAPI. The images are representative of three independent experiments and observed under confocal microscope ( × 40). Vertical bars represent mean ± SE (n = 3).*P < 0.05, compared to HKM. HKM, control head kidney macrophage; HKM + MF, HKM infected with M. fortuitum; HKM + RR + MF, HKM + CsA + MF, HKM + BAPTA/AM + MF, HKM + Dant + MF, HKM + 2-APB + MF, HKM pre-treated with RR, CsA, BAPTA/AM, Dant, 2-APB respectively and infected with M. fortuitum
Cytosolic Ca2+ imbalance activates the calpain/caspase-12 axis
Calpains are implicated in apoptosis induced by several mycobacteria11, 20. In absence of earlier reports, we studied the role of calpain in M. fortuitum-induced HKM apoptosis. Realtime primers for the common 28-kDa regulatory subunit gene (CAPNS1) were designed and qPCR results demonstrated maximum CAPNS1-mRNA expression at 1 h p.i. (Fig. 4a). We followed this by measuring calpain activity using specific kit and noted maximum calpain activity at 2 h p.i. (Fig. 4c) and selected these two time points for subsequent studies.
Fig. 4. Activation of calpain/caspase-12 axis is consequent to ER-stress in M. fortuitum infected HKM.

a HKM were infected with M. fortuitum and at indicated time p.i. CAPNS1-mRNA expression was quantified by qPCR. b HKM were transfected separately with CAPNS1-siRNA or scrambled siRNA and CAPNS1-mRNA expression was quantified by qPCR. c HKM were infected with M. fortuitum and at indicated time p.i. calpain activity was measured using assay kits. d HKM transfected separately with CAPNS1-siRNA, scrambled siRNA or pre-treated with indicated inhibitors were infected with M. fortuitum and calapin activity measured at 2 h p.i. e HKM pre-treated with indicated inhibitors or transfected with CAPNS1-siRNA were infected with M. fortuitum and caspase-12 activity was studied at 24 h p.i. using specific kit and observed under confocal microscope ( × 40). The images are representative of three independent experiments. Vertical bars represent mean ± SE (n = 3).*P < 0.05, compared to HKM; γP < 0.05, compared to HKM + Sc; #P < 0.05, compared to HKM + MF; •P < 0.05, compared to HKM + Sc + MF. HKM, control head kidney macrophage; HKM + Sc, HKM transfected with scrambled siRNA; HKM + MF, HKM infected with M. fortuitum; HKM + Sc + MF, HKM transfected with scrambled siRNA followed by M. fortuitum infection; HKM + CAPNS1-siRNA + MF, HKM transfected with CAPNS1-siRNA then infected with M. fortuitum; HKM + PD150606 + MF, HKM + Calpain1i + MF, HKM + Calpain2i + MF, HKM + 4-PBA + MF, HKM + BAPTA/AM + MF, HKM + Z-ATAD-FMK + MF, HKM pre-treated with PD150606, Calpain1i, Calpain2i, 4-PBA, BAPTA/AM, Z-ATAD-FMK respectively and infected with M. fortuitum
Pre-treatment with BAPTA/AM attenuated calpain activity (Fig. 4d) indicating calpain activation to be Ca2+-dependent in M. fortuitum-infected HKM. Transfection studies were carried out with CAPNS1-siRNA and the results from RNAi studies demonstrated significant reduction in M. fortuitum induced calpain-mRNA expression (Fig. 4b), -protein activity (Fig. 4d) and HKM apoptosis (Figure S1). HKM were pre-treated with pan-calpain inhibitor PD150606 and the changes in calpain and caspase-3 activity and HKM apoptosis studied. We observed significant attenuation in calpain activity (Fig. 4d), caspase-3 activity and HKM apoptosis (Figure S1) in presence of PD150606. The inactive analog PD145305 had no effect on calpain and caspase-3 activation as well as HKM apoptosis (data not shown). Calpains exists in two isoforms and our interest was to identify their relative involvements in M. fortuitum-induced HKM apoptosis. In this direction, HKM pre-treated separately with calpain 1i and calpain 2i were infected with M. fortuitum and caspase-3 activation and apoptosis monitored. We observed calpain 1i and calpain 2i were equally effective in inhibiting calpain activation (Fig. 4d), caspase-3 activation and HKM apoptosis (Figure S1) suggesting both calpain isoforms contribute equivalently to M. fortuitum-pathogenesis.
ER-stress can induce the activation of calpains31. To correlate ER-stress with calpain activity the HKM were pre-treated with 4-PBA and calpain activity studied in the infected cells. We observed that 4-PBA pre-treatment led to significant reduction in calpain activiation (Fig. 4d) in M. fortuitum-infected HKM. These findings suggested ER-stress contribute towards pro-apoptotic calpain activation in M. fortuitum infected HKM.
We followed this by studying ER-stress induced caspase-12 activation. HKM were infected with M. fortuitum and caspase-12 expression monitored under the confocal microscope at 24 h p.i., the end point of the study. We observed significant caspase-12 activity in infected HKM. Pre-treatment with caspase-12 inhibitor Z-ATAD-FMK and 4-PBA inhibited caspase-12 activity (Fig. 4e) and attenuated M. fortuitum-induced HKM apoptosis (Figure S1). Thus, we concluded that ER-stress leads to pro-apoptotic caspase-12 activation in M. fortuitum-infected HKM.
Earlier studies suggested the involvement of calpains in caspase-12 activation21. To investigate this, the HKM were treated with PD150606, Calpain 1i, Calpain 2i or transfected with CAPNS1-siRNA prior to M. fortuitum infection and caspase-12 activity studied. We observed the down-regulation in caspase-12 activity in presence of PD150606, Calpain 1i, Calpain 2i and CAPNS1-siRNA respectively (Fig. 4e). PD145305 failed to inhibit caspase-12 activation (data not shown), suggesting the role of calpain on caspase-12 activation in M. fortuitum-infected HKM. Based on these findings we concluded that M. fortuitum-induced ER-stress lead to the activation of pro-apoptotic calpain/caspase-12 axis in HKM.
Caspase-12 and cytochrome c instigate caspase-9 activation to expedite M. fortuitum induced-HKM apoptosis
The release of cytochrome c leads to activation of caspase-9 and cellular apoptosis17. We set out to determine the role of cytochrome c-caspase-9 axis in M. fortuitum-induced HKM apoptosis. Enhanced caspase-9 activity was noted in M. fortuitum infected HKM (Fig. 5) and pre-treatment with caspase-9 inhibitor Z-LEHD-FMK significantly attenuated caspase-3 activity and HKM apoptosis (Figure S1) implicating the involvement of caspase-9 in M. fortuitum pathogenesis. We extended our study and noted that caspase-9 activity was attenuated in the presence of MPT inhibitor CsA (Fig. 5) which ensured cytochrome c released due to MPT is critical for caspase-9 activation in M. fortuitum-infected HKM.
Fig. 5. Cytochrome c and caspase-12 independently induce caspase-9 activation in M. fortuitum infected HKM.
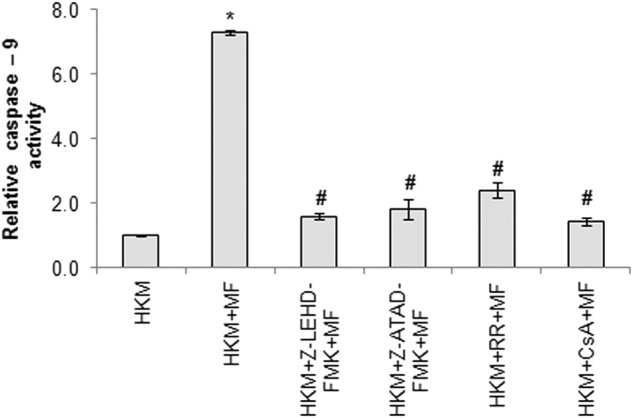
HKM pre-treated with or without indicated inhibitors were infected with M. fortuitum and caspase-9 activity measured 24 h p.i. Vertical bars represent mean ± SE (n = 3).*P < 0.05, compared to HKM; #P < 0.05, compared to HKM + MF. HKM, control headkidney macrophage; HKM + MF, HKM infected with M. fortuitum; HKM + Z-LEHD-FMK + MF, HKM + Z-ATAD-FMK + MF, HKM + RR + MF, HKM + CsA + MF, HKM pre-treated with Z-LEHD-FMK, ZATAD-FMK, RR, CsA respectively and infected with M. fortuitum
In an earlier study it was noted that caspase-12 influences caspase-9 further intensifying the apoptotic cascade32. To probe this, we pre-treated the HKM with the caspase-12 inhibitor Z-ATAD-FMK and assayed caspase-9 activity. It is evident from Fig. 5 that caspase-9 activity was attenuated in Z-ATAD-FMK pre-treated HKM. Based on these observations we propose that both cytochrome c and caspase-12 mediated pathways intersect at caspase-9 to expedite HKM death induced by M. fortuitum.
Discussion
In the present study, we report that the cross-talk between ER and mitochondria aggravates down-stream apoptotic signaling in M. fortuitum infected HKM wherein, Ca2+ dynamics in the sub-cellular compartments plays a crucial role on expediting the death program.
The pro-apoptotic transcription factor CHOP is marker for ER-stress7. The expression of CHOP was significantly reduced in presence of BAPTA/AM, an intracellular Ca2+ chelator implicating alteration in intracellular Ca2+ homeostasis is closely related to ER-stress generation in M. fortuitum infected macrophages. We hypothesize, elevated cytoslic Ca2+ induces protein misfolding affecting protein loading in ER and BAPTA/AM might reduce misfolded proteins thus attenuating ER-stress induced by M. fortuitum. 4-PBA is low molecular weight chemical chaperone which has several biological effects of which inhibiting ER-stress is important33. It helps in stabilizing protein conformation thereby improving the folding capacity of ER and represses UPR33, 34. We hypothesized 4-PBA would prevent HKM apoptosis through inhibition of ER-stress induced CHOP expression. We observed that alleviating ER-stress with 4-PBA down-regulated CHOP expression coupled with decline in caspase-3 activity and HKM apoptosis. Besides, CHOP-siRNA suppressed caspae-3 activity and HKM apoptosis. CHOP has ‘versatile role’ in ER-stress mediated apoptosis. It down-regulates BCL-2 anti-apoptotic proteins at transcriptional level triggering the mitochondrial apoptotic cascade35. It has been also reported CHOP induces the transcription of ER oxidoreductin 1α (ERO1α) leading to hypoxic milieu in ER, that enhances downstream death signaling35, 36. In this context, it would be interesting to study the versatility of CHOP in M. fortuitum induced apoptosis.
Based on these observations, we propose that the ER-overload and unresolved ER-stress induced by M. fortuitum is a crucial trigger to induce HKM apoptosis. The role of cytosolic Ca2+ on ER-stress generation and macrophage apoptosis has been reported in several mycobacteria10, 11 and our results extends this to M. fortuitum, suggesting it to be common virulence trait for different mycobacterial species. This is the first report suggesting that M. fortuitum can induce ER-stress with pathological implications in host. Keeping the diverse host trophism of M. fortuitum in view it would be interesting to see whether same pathogenic mechanisms are employed by the bacteria to induce pathogenesis across species barrier.
Our results with 2-APB and Dant showed significant suppression in CHOP expression implicating the definite role of (Ca2+)ER depletion in in M. fortuitum pathogenesis. The next step was to look for the likely down-stream targets induced by Ca2+ and calpain appeared attractive. Calpains are non-lysosomal cysteine proteases consisting of 80-kDa catalytic subunit and a common 28-kDa regulatory subunit, calpain small-1 (CAPNS1), encoded by CAPNS1 gene required for functioning37, 38. We observed over-expression of CAPNS1 mRNA and higher calpain activity and inhibiting the protease activity resulted in down regulation of caspase-3 and M. fortuitum induced HKM apoptosis. The presence of the two different tissue isoforms, calpain-1 and -2 is well documented in fish39, 40. Our results demonstrated that both isoforms are important for inducing M. fortuitum induced HKM apoptosis. Alleviating ER-stress with 4-PBA significantly reduced calpain activity suggesting calpain activation consequent to ER-stress generation in M. fortuitum infected HKM. Although the involvement of ER-calpain axis has been reported in the pathogensis of both atypical11 and typical mycobacteria20 our results constitute the first report in M. fortuitum suggesting calpain activation as an evolutionary conserved virulence attribute for mycobacteria.
We posited that the efflux of ER-Ca2+ into the cytoplasm through IP3 and RYR activates calpain to initiate downstream effects. Hence, calpain activity was studied in presence of 2-APB and Dant. Importantly, 2-APB and Dant, failed to completely abrogate calpain activity emphasizing Ca2+ efflux in M. fortuitum infected HKM involves multitude of pathways.
The activation of caspase-12 as a marker for ER-stress has recently been demonstrated in fish41. It has been observed that calpain cleaves the ER-resident pro-caspase-12 to active caspase-12 further intensifying the apoptotic cascade. Our results for the first time showed calpain-induced caspase-12 activation to be an important step in the pathogenesis of M. fortuitum. It was suggested that calpain-2 activation leads to cleavage of caspase-1221. We did not observe any difference in the ability of either calpain isoforms on caspase-12 activation suggesting caspase-12 to be substrate for both calpain-1 and -2 in M. fortuitum pathogenesis. We are currently studying the mechanisms of calpain dependency on caspase-12 activation in M. fortuitum pathogenesis.
An obligatory step in the ER-stress pathway is mitochondrial dysfunctioning with Ca2+ playing an active role on initiating the process16, 17. The participation of Ca2+ in M. fortuitum induced HKM apoptosis prompted us to explore ER-mitochondrial cross-talk in the pathogenesis induced by the bacterium. We observed overflow of (Ca2+)ER to mitochondria of M. fortuitum infected HKM. Mitochondrial calcium overload alters mitochondrial membrane permeability and leads to opening of MPT. Our results showed that pre-treatment with MPT inhibitor CsA inhibited ΔΨm dissipation, caspase-3 activation and HKM apoptosis suggesting MPT formation to be associated with M. fortuitum pathogenesis. It has been reported that a close physical contact is pre-requisite for the mobility of Ca2+ from ER to mitochondria and MUP are the likely “hotspots” through which Ca2+ enters the mitochondria6. Ultra-structural studies depicted close apposition between the two organelles with mitochondria docked onto the ER. We used the specific MUP inhibitor RR and observed diminished mitochondrial Ca2+ uptake with concomitant decline in caspase-3 activity and HKM apoptosis. Similar reduction in mitochondrial Ca2+ load and MPT formation were also noted when the HKM were pre-treated with 2-APB and Dant. We propose that the interim association between the two organelles facilitates the efficient transfer of (Ca2+)ER via MUPs leading to mitochondrial dysfunctioning and apoptosis of HKM.
MPT formation leads to overproduction of superoxide anions and release of pro-apoptotic cytochrome c into the cytosol. Increased amount of superoxide has been frequented with apoptosis induced by mycobacterial pathogens10, 11. Our preliminary results suggested the role of superoxide anions on M. fortuitum induced HKM apoptosis24. We hypothesize that MPT formation contributes to the overall process of apoptosis through the release of superoxide anions in the infected HKM. Blocking MPT formation by CsA significantly down-regulated cytochrome c release in cytosol suggesting close association between MPT formation and cytochrome c release in M. fortuitum infected HKM. We detected significant caspase-9 activity in M. fortuitum infected HKM which could be inhibited in presence of CsA suggesting MPT formation and cytochrome c release having a significant role on caspase-9 activation in M. fortuitum infected HKM. Earlier studies suggested a role of caspase-12 on activating the caspase-9/-3 axis31, 42. We observed, inhibiting caspase-12 significantly down-regulated the activation of caspase-9/3 axis, suggesting that caspase-9 can be activated by multiple pathways in M. fortuitum infected HKM. The involvement of caspase-9 is well documented in pathogenesis induced by several mycobacteria12, 43–45 our study extends this to M. fortuitum.
Mycobacteria-induced cell death depends on several factors including nature of bacterial strains, MOI, host cell types and durations of infection46–48. There are also reports suggesting mycobacteria causes caspase-dependent apoptotic and caspase-independent necrotic death depending on varied conditions of infection48. In this study, we observed caspase mediated apoptosis of catfish macrophages at 24 h p.i. and caspase-12/caspase-9 axis playing crucial role in triggering the process. The role of caspase-12 in ER-stress induced apoptosis is contentious with studies suggesting caspase-12 not to be part of UPR-induced apoptosis49. There are also reports that ER-stress induces caspase-independent necrosis48. In these studies, chemical stressers (tunicamycin, thapsigargin etc) have been used to induce and study the consequences of ER-stress, which may not be akin to pathogen induced stress. Nonetheless, presence of late apoptotic (AV+PI+) and necrotic (AV−PI+) subsets in M. fortuitum infected HKM along with inability of caspase inhibitors to completely abrogate cell death suggests a subset of HKM might be undergoing caspase-independent death. In this context, it would be interesting to study CHOP mediated necroptosis or necrosis in the immunopathogenesis of M. fortuitum at later time points or with higher MOI.
Cytochrome c also binds to IP3 receptors on the ER facilitating (Ca2+)ER release50. We believe that besides its direct involvement in caspase-9 mediated apoptosis cytochrome c also contributes towards mitochondrial Ca2+ influx necessary for induction of HKM apoptosis. MPT formation leads to overproduction of superoxide anions and release of pro-apoptotic cytochrome c into the cytososl and increased amount of superoxide anions has been implicated in apoptosis induced by mycobacterial pathogens10, 11. We hypothesize that MPT formation also contributes to the overall process of apoptosis through the release of superoxide anion in the infected HKM.
The role of mitochondria in piscine mycobacteriosis is not clear. Recently it has been reported in M. marinum-zebrafish model that by modulating mitochondrial permeability transition pore formation mycobacteria induced programmed necrosis (necroptosis)51. We believe that these contradictions likely underline the complex and dynamic nature of the mycobacterial pathogenesis.
To conclude, our results established that Ca2+ dynamics of in sub-cellular compartments lead to ER-stress generation and mitochondrial dysfunctioning in M. fortuitum-infected HKM. We propose that altered cytosloic-Ca2+ triggers ER-stress accompanied with (Ca2+)ER release. ER-Ca2+, besides activating the calapin-caspase-12 axis also induces mitochondrial dysfunctioning; the two pathways converge at caspase-9 initiating caspase-3 mediated HKM apoptosis (Fig. 6). These findings would be useful for understanding the pathogenesis of M. fortuitum as well as controlling mycobacteriosis.
Fig. 6. Overview of the work.

M. fortuitum-induced alteration in Ca2+ homeostasis aggravates ER-stress. This leads to (i) activation of the calpain/ caspase-12 axis and (ii) mitochondrial dysfunction resulting in the cytosloic translocation of cytochrome c. The two pathways converge at caspase-9 leading to apoptosis of piscine macrophages
Materials and methods
Bacterial strains and growth conditions
Mycobacterium fortuitum (Strain MTCC 993) purchased from Microbial Type Culture Collection and Gene Bank (MTCC), Chandigarh, India were grown at 30 °C in standard Middlebrook 7H9 broth (HiMedia). The identity of the isolates was confirmed by AFB staining and 16 S rDNA sequencing. As the bacteria are sensitive to amikacin 50 µg/mL of the same was added to eradicate the extracellular bacteria24.
Isolation of HKM and infection with M. fortuitum
All animal experiments were approved by the Animal Ethics Committee, University of Delhi (DU/ZOOL/IAEC-R/2013/34) and carried out in accordance with the protocols approved by The Prevention of Cruelty to Animals Act, Govt. of India. The methods for catfish (Clarias sp) maintenance and the protocols for obtaining HKM and infecting them with M. fortuitum (multiplicity of infection (MOI 10)) has been described earlier24.
Inhibitors used
Intrcellular Ca2+ chelator (1, 2-Bis (2-aminophenoxy) ethane-N,N,N′,N′-tetraacetic acid tetrakis (acetoxymethyl ester), BAPTA/AM, 5 μM)), ER-stress alleviator (4-Phenyl butyric acid, 4-PBA, 10 µM), IP3 receptor antagonist (2-Aminoethyl diphenylborinate, 2-APB, 100 µM), calpain 1 inhibitor (N-acetyl-leucyl-leucyl-norleucinal, Calpain 1i, 50 µM), calpain 2 inhibitor (N-acetyl-leucyl-leucyl-methioninal, Calpain 2i, 50 µM), mitochondrial uniporter inhibitor (Ruthenium Red, RR, 20 µM), were purchased from Sigma. Pan-calpain inhibitor ([3-(4-iodophenyl)-2-mercapto-(Z)-2-propenoic acid], PD150606, 50 μΜ), negative control for calpain inhibitor (2-mercapto-3-phenypropionic acid, PD145305, 50 μM), rynodine receptor blocker (Dantrolene, Dant, 20 µM) were purchased from Calbiochem. MPTP blocker (Cyclosporin A, CsA, 5 µM) was from US Biological. Caspase-12 inhibitor (Z-ATAD-FMK, 10 μΜ) and caspase-9 inhibitor (Z-LEHD-FMK, 10 μΜ) were purchased from Biovision. Cytotoxicity test was done to determine the concentration of inhibitors used (data not shown). The inhibitors were added to the cell culture 1 h prior to the M. fortuitum infection and maintained throughout the experiment. The viability of HKM treated with the indicated concentrations of the inhibitors remained maintained at all-time points as checked by the trypan blue (0.4%) dye exclusion method. The concentrations of different inhibitors used for the study also had no effect on bacterial growth per se when added to Middlebrook 7H9 or complete-RPMI.
siRNA Transfection
The siRNA transfection was carried out using HiPerFect Transfection Reagent (Qiagen), as described earlier24, 52. Transfection efficiency was confirmed by Real-Time PCR, protein and apoptosis assays. Five nano mole each of targeted CHOP [SENSE AUGAAGACUUGCAAGAUAUdTdT & ANTISENSE AUAUCUUGCAAGUCUUCAUdTdT], CAPNS1 [SENSE CAUGGACUUCGACAACUACdTdT & ANTISENSE GUAGUUGUCGAAGUCCAUGdTdT], and siRNA Universal negative CONTROL (Sigma) were used for this study.
RNA isolation, cDNA synthesis, cloning, amplification, sequencing and quantative real-time PCR
HKM (2 × 107) transfected separately with or without targeted or scrambled siRNA were infected with M. fortuitum and at indicated time p.i. the total RNA was isolated using TRIZOL (Sigma). cDNA was prepared from 1 μg of DNase treated (RNase-free) RNA using first strand cDNA synthesis kit as per manufacturer’s instructions (MBI Fermentas). Degenerate primers were designed using the homologous stretch across fish for CHOP and all vertebrates for the common calpain small sub-unit (CAPNS1) as the template (Table S1). The cDNA was amplified; the amplicons extracted using HiPura gel extraction kit (HiMedia), cloned into pGEM-T EASY vector (Promega) and sequenced (Macrogen). The sequences obtained (Table S2) were aligned to nBLAST and submitted to EMBL or NCBI database. The sequence for CHOP (accession number EMBL-LK054407) showed 80 % identity with CHOP-mRNA sequence of zebrafish (Danio rerio) and the sequence for CAPNS1 (accession number NCBI-KM242108) showed 80 % sequence identity with CAPNS1 sub-unit of Atlantic salmon (Salmo salar).
The quantification of CHOP and CAPNS1 mRNA were performed using SYBR green PCR Master Mix (Applied Biosystems) by Real-Time PCR (ABI ViiA, Applied Biosystems) as described earlier. The gene specific real-time primers for CHOP (FP:5′- GTTGGAGGCGTGGTATGAAG-3′; RP:5′-GAAACTCCGGCTCTTTCTCG-3′) and CAPNS1 (FP:5′-ACGGGAAAACTGGGGTTCG-3′; RP:5′-TGCTTATAGACAGCCTGCCAC-3′) have been used. Expression levels of target genes were analyzed by comparative ΔΔCT method using β-actin as the internal control (endogenous control) and uninfected HKM (0 h) was used as the calibrator24.
Apoptosis study
HKM (1 × 106) transfected or pre-treated with or without indicated concentrations of targeted or scrambled siRNAs or specific inhibitors were infected with or without M. fortuitum (MOI 10) and apoptosis studied at 24 h p.i. by Hoechst 33342 (Sigma) and annexinV-FITC & propidium iodide (AV-PI, BD-Pharmingen) staining in fluroscence microscope ( × 40, Nikon Eclipse 400) as described earlier24.
Immunofluorescence studies
HKM (5 × 106) transfected or pre-treated with or without targeted or scrambled siRNAs and specific inhibitors were infected with or without M. fortuitum. At the indicated time p.i. the HKM were washed and fixed in methanol and were incubated in blocking and permeabilizing solution (PBS, 2 mg/mL BSA, 0.2 mg/mL saponin) for 1 h at room temperature. The cells were washed and incubated with primary antibodies; CHOP (mouse, 1:100, Cell Signalling Technology) and cytochrome c (mouse, 1:100, Biovision) separately overnight at 4 °C. The HKM were washed in PBST (PBS containing 0.1 % Tween-20) and stained with FITC or TRITC conjugated secondary antibodies (1: 250) for 3 h at 30 °C and visualized under confocal microscope ( × 40 oil immersion, 1.30 NA, Nikon Eclipse A1Rsi-TiE-300)41. Nuclei were stained with DAPI (1 μg/mL) before mounting on microslide.
Imaging analysis of (Ca2+)m uptake
The HKM (2 × 106) pre-treated with or without specific inhibitors were infected with or without M. fortuitum. At indicated time p.i. the cells were washed, loaded simultaneously with Rhod-2/AM and mitotracker green (50 nM, Molecular Probes), incubated at 30 °C for 30 min then washed, mounted on microslide with cover slips using fluoroshield and visualized under confocal microscope ( × 40 oil immersion, 1.30 NA, Nikon Eclipse A1Rsi-TiE-300).
Measurement of ΔΨm
The changes in ΔΨm and the induction of the mitochondrial permeability transition (MPT) were studied using JC1 (Cayman) and DiOC6 (Sigma) dyes respectively. In case of JC-1, cells with a high ΔΨm were those forming J-aggregates and in case of DiOC6, high ΔΨm was attributed to cells with a high fluorescence signal.
HKM (2 × 106) pre-treated with or without indicated concentrations of different inhibitors were infected with M. fortuitum for the indicated time p.i. and were loaded with DiOC6 (100 nM) during the last 30 min of infection then lysed in deionized water, and the reduction in the accumulation of DiOC6 was read in a fluorimeter (HT synergy) at excitation and emission wavelengths of 488 and 500 nm respectively. The relative change in fluorescence was plotted.
In parallel study, HKM (2 × 106) pre-treated with or without indicated concentrations of different inhibitors were infected with or without M. fortuitum for indicated time periods. The cell pellet was harvested, washed with phosphate buffered saline and loaded with JC-1 (20 µM, Cayman) for 20 min. The cells were washed, mounted in microslide with cover slips using fluoroshield and the red/green fluorescence was digitized at indicated time p.i. using confocal microscope ( × 40 oil immersions, 1.30 NA, Nikon Eclipse A1Rsi-TiE-300).
Transmission electron microscopy
HKM (2 × 107) uninfected or infected with M. fortuitum for the indicated time period were washed and fixed with 2.5 % glutaraldehyde (Polaron, Biorad) in 0.1 M phosphate buffer (pH 7.4). The fixed HKM were processed as reported earlier24 and examined under Tecnai 12 Bio-twin transmission electron microscope (FEI, 80 kV).
Calpain assay
Calpain activity was studied using a fluorogenic activity assay kit (Calbiochem). Briefly, HKM (1 × 105) pre-treated with or without targeted or scrambled siRNAs and specific inhibitors were infected with or without M. fortuitum. The HKM were washed at indicated time p.i., lysed, calpain activity measured at 360 nm excitation and 460 nm emission respectively and the relative calpain activity plotted.
Caspase assay (caspase-12, caspase-9 and caspase-3)
Caspase-12, caspase-9 and Caspase-3 were studied using specific assay kits according to the instructions of the manufacturer (Biovision) and using the reagents supplied with the kit. Briefly, HKM (1 × 106) pre-treated with or without indicated concentrations of different inhibitors or targeted or scrambled siRNAs were infected with or without M. fortuitum for 24 h. The cells were washed and FITC-tagged caspase-12 inhibitor Z-ATAD-FMK was added into each culture and left for 30 mins at 30 °C followed by washing in fluorescein wash buffer. The HKM were mounted on microslides with cover slips using fluoroshield and analyzed under confocal microscope ( × 40 oil immersion)41.
For caspase-9 and caspase-3 assays the HKM were washed, re-suspended in 50 µl of lysis buffer and incubated on ice for 10 min. The cell lysate was collected by centrifugation at 10,000 × g for 5 min at 4 °C. To 50 μl of cell lysate, 50 μl of 2 × reaction buffer containing DTT (10 mM), PMSF (5 mM) and specific substrates were added, the mixture was incubated at 37 °C for 5 h and the absorbance read at 405 nm in a micro-plate reader (HT synergy)40.
Statistical analysis
Mean ± SE were calculated for each parameter considered in the present study in the different groups of fish. Pair wise comparison was done by employing t-test: two samples using unequal variance to determine the statistical significance between the groups. The value of p < 0.05 was considered statistically significant.
Electronic supplementary material
Acknowledgements
This work was supported by NFBSFARA/ICAR Project Grant (RNAi-2014), University of Delhi Doctoral Research Programme (Dean (R)/R&D/2012/917) and UGC (MRP-MAJOR-ZOOL-2013-36692). DD, PK, AS, and GV were supported by ICAR Fellowship (Govt of India), CSIR Fellowship (Govt of India), UGC Fellowship (Govt of India), CSIR Fellowship (Govt of India) respectively. We thank A.K. Pal for maintenance of fish.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by A. Rufini
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41420-018-0034-9.
References
- 1.Talaat AM, Trucksis M, Kane AS, Reimschuessel R. Pathogenicity of Mycobacterium fortuitum and Mycobacterium smegmatis to goldfish, Carassius auratus. Vet. Microbiol. 1999;66:151–164. doi: 10.1016/S0378-1135(99)00002-4. [DOI] [PubMed] [Google Scholar]
- 2.Bercovier H, Vincent V. Mycobacterial infections in domestic and wild animals due to Mycobacterium marinum, M. fortuitum, M. chelonae, M. porcinum, M. farcinogenes, M. smegmatis, M. scrofulaceum, M. xenopi, M. kansasii, M. simiae and M. genavense. Rev. Sci. Tech. Int. Epiz. 2001;20:265–290. doi: 10.20506/rst.20.1.1269. [DOI] [PubMed] [Google Scholar]
- 3.Smith MB, Schnadig VJ, Boyars MC, Woods GL. Clinical and pathologic features of Mycobacterium fortuitum infections an emerging pathogen in patients with AIDS. Am. J. Clin. Pathol. 2001;116:225–232. doi: 10.1309/HF2V-E8WV-PX4Q-CHQH. [DOI] [PubMed] [Google Scholar]
- 4.Rojas M, García LF, Nigou J, Puzo G, Olivier M. Mannosylated lipoarabinomannan antagonizes Mycobacterium tuberculosis-induced macrophage apoptosis by altering Ca+ 2-dependent cell signaling. J. Infect. Dis. 2000;182:240–251. doi: 10.1086/315676. [DOI] [PubMed] [Google Scholar]
- 5.Mehto S, et al. Mycobacterium tuberculosis and human immunodeficiency virus type 1 cooperatively modulate macrophage apoptosis via toll like Receptor 2 and calcium homeostasis. PLoS ONE. 2015;10:e0131767. doi: 10.1371/journal.pone.0131767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 7.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Marciniak SJ, et al. CHOPinduces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seimon TA, et al. Induction of ER stress in macrophages of tuberculosis granulomas. PLoS ONE. 2010;5:e12772. doi: 10.1371/journal.pone.0012772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi JA, et al. Mycobacterial HBHA induces endoplasmic reticulum stress-mediated apoptosis through the generation of reactive oxygen species and cytosolic Ca2+in murine macrophage RAW 264.7 cells. Cell Death Dis. 2013;4:e957. doi: 10.1038/cddis.2013.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim YJ, et al. Mycobacterium kansasii-induced death of murine macrophages involves endoplasmic reticulum stress responses mediated by reactive oxygen species generation or calpain activation. Apoptosis. 2013;18:150–159. doi: 10.1007/s10495-012-0792-4. [DOI] [PubMed] [Google Scholar]
- 12.Cui Y, et al. Mycobacterium bovis induces endoplasmic reticulum stress mediated-apoptosis by activating IRF3 in a murine macrophage cell line. Front. Cell Infect. Microbiol. 2016;6:182. doi: 10.3389/fcimb.2016.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim YJ, et al. Roles of endoplasmic reticulum stress-mediated apoptosis in M1-polarized macrophages during mycobacterial infections. Sci. Rep. 2016;6:37211. doi: 10.1038/srep37211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jo SH, et al. Calreticulin modulates the intracellular survival of mycobacteria by regulating ER-stress-mediated apoptosis. Oncotarget. 2017;8:58686–58698. doi: 10.18632/oncotarget.17419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 16.Hajnóczky G, et al. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Cal. 2006;40:553–560. doi: 10.1016/j.ceca.2006.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27:6407–6418. doi: 10.1038/onc.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress‐induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suzuki K, Hata S, Kawabata Y, Sorimachi H. Structure, activation, and biology of calpain. Diabetes. 2004;53:S12–S18. doi: 10.2337/diabetes.53.2007.S12. [DOI] [PubMed] [Google Scholar]
- 20.Francis RJ, Butler RE, Stewart GR. Mycobacterium tuberculosis ESAT-6 is a leukocidin causing Ca2+influx, necrosis and neutrophil extracellular trap formation. Cell Death Dis. 2014;5:e1474. doi: 10.1038/cddis.2014.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakagawa T, Yuan J. Cross-talk between two cysteine protease families Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez JA, et al. Calpain and caspase processing of caspase-12 contribute to the ER stress-induced cell death pathway in differentiated PC12 cells. Apoptosis. 2010;15:1480–1493. doi: 10.1007/s10495-010-0526-4. [DOI] [PubMed] [Google Scholar]
- 23.Secombes CJ, Fletcher TC. The role of phagocytes in the protective mechanisms of fish. Annu. Rev. Fish. Dis. 1992;2:53–71. doi: 10.1016/0959-8030(92)90056-4. [DOI] [Google Scholar]
- 24.Datta D, et al. Calcium and superoxide-mediated pathways converge to induce nitric oxide-dependent apoptosis in Mycobacterium fortuitum-infected fish macrophages. PLoS ONE. 2016;111:e0146554. doi: 10.1371/journal.pone.0146554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Landowski TH, Megli CJ, Nullmeyer KD, Lynch RM, Dorr RT. Mitochondrial-mediated disregulation of Ca2+ is a critical determinant of Velcade (PS-341/bortezomib) cytotoxicity in myeloma cell lines. Can. Res. 2005;65:3828–3836. doi: 10.1158/0008-5472.CAN-04-3684. [DOI] [PubMed] [Google Scholar]
- 26.Duan L, Gan H, Golan DE, Remold HG. Critical role of mitochondrial damage in determining outcome of macrophage infection with Mycobacterium tuberculosis. J. Immunol. 2002;169:5181–5187. doi: 10.4049/jimmunol.169.9.5181. [DOI] [PubMed] [Google Scholar]
- 27.Chen M, Gan H, Remold HG. A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J. Immunol. 2006;176:3707–3716. doi: 10.4049/jimmunol.176.6.3707. [DOI] [PubMed] [Google Scholar]
- 28.Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (ΔΨm) in apoptosis; an update. Apoptosis. 2003;8:115–128. doi: 10.1023/A:1022945107762. [DOI] [PubMed] [Google Scholar]
- 29.Sakamuru S, et al. Application of a homogenous membrane potential assay to assess mitochondrial function. Physiol. Genom. 2012;44:495–503. doi: 10.1152/physiolgenomics.00161.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodrigues CM, et al. Ursodeoxycholic acid prevents cytochrome c release in apoptosis by inhibiting mitochondrial membrane depolarization and channel formation. Cell Death Differ. 1999;6:842–854. doi: 10.1038/sj.cdd.4400560. [DOI] [PubMed] [Google Scholar]
- 31.Wu J, Kaufman RJ. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 2006;13:374–384. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- 32.Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–380. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- 33.Carlisle RE, et al. 4 Phenylbutyrate inhibits tunicamycin-induced acute kidney injury via CHOP/GADD153 repression. PLoS ONE. 2014;9:e84663. doi: 10.1371/journal.pone.0084663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kubota K, et al. Suppressive effects of 4‐phenylbutyrate on the aggregation of Pael receptors and endoplasmic reticulum stress. J. Neurochem. 2006;97:1259–1268. doi: 10.1111/j.1471-4159.2006.03782.x. [DOI] [PubMed] [Google Scholar]
- 35.Iurlaro R, Munoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016;283:2640–2652. doi: 10.1111/febs.13598. [DOI] [PubMed] [Google Scholar]
- 36.Marciniak SJ, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol. Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 38.Arthur JSC, Elce JS, Hegadorn C, Williams K, Greer PA. Disruption of the murine calpain small subunit gene, Capn4: calpain is essential for embryonic development but not for cell growth and division. Mol. Cell Biol. 2000;20:4474–4481. doi: 10.1128/MCB.20.12.4474-4481.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lepage SE, Bruce AE. Characterization and comparative expression of zebrafish calpain system genes during early development. Dev. Dyn. 2008;237:819–829. doi: 10.1002/dvdy.21459. [DOI] [PubMed] [Google Scholar]
- 40.Banerjee C, Goswami R, Verma G, Datta M, Mazumder S. Aeromonas hydrophila induced headkidney macrophage apoptosis in Clarias batrachus involves the activation of calpain and is caspase-3 mediated. Dev. Comp. Immunol. 2012;37:323–333. doi: 10.1016/j.dci.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 41.Banerjee C, et al. Ameliorating ER-stress attenuates Aeromonas hydrophila-induced mitochondrial dysfunctioning and caspase mediated HKM apoptosis in Clarias batrachus. Sci. Rep. 2014;4:5820. doi: 10.1038/srep05820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. C Hem. 2002;277:34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- 43.Sánchez, A., Espinosa, P., García, T. & Mancilla, R. The 19 kDa Mycobacterium tuberculosis lipoprotein (LpqH) induces macrophage apoptosis through extrinsic and intrinsic pathways: a role for the mitochondrial apoptosis-inducing factor. Clin Dev Immunol. https://doi.org/10.1155/2012/950503 (2012). [DOI] [PMC free article] [PubMed]
- 44.Cui Y, et al. Mycobacterium bovis induces endoplasmic reticulum stress mediated-apoptosis by activating IRF3 in a murine acrophage cell line. Front Cell Infect. Microbiol. 2016;6:182. doi: 10.3389/fcimb.2016.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee KI, et al. Mycobacterium avium MAV2054 protein induces macrophage apoptosis by targeting mitochondria and reduces intracellular bacterial growth. Sci. Rep. 2016;6:37804. doi: 10.1038/srep37804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kasahara K, et al. Expression of chemokines and induction of rapid cell death in human blood neutrophils by Mycobacterium tuberculosis. J. Infect. Dis. 1998;178:127–137. doi: 10.1086/515585. [DOI] [PubMed] [Google Scholar]
- 47.Kelly DM, ten Bokum AMC, O’Leary SM, O’Sullivan MP, Keane J. Bystander macrophage apoptosis after Mycobacterium tuberculosisH37Ra infection. Infect. Immun. 2008;76:351–360. doi: 10.1128/IAI.00614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rachel E, et al. The balance of apoptotic and necrotic cell death in Mycobacterium tuberculosis infected macrophages is not dependent on bacterial virulence. PLoS ONE. 2012;7:e47573. doi: 10.1371/journal.pone.0042446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Obeng EA, Boise LH. Caspase-12 and caspase-4 are not required for caspase-dependent endoplasmic reticulum stress-induced apoptosis. J. Biol. Chem. 2005;280:29578–29587. doi: 10.1074/jbc.M502685200. [DOI] [PubMed] [Google Scholar]
- 50.Boehning D, et al. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 2003;5:1051–1061. doi: 10.1038/ncb1063. [DOI] [PubMed] [Google Scholar]
- 51.Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. 2013;153:521–534. doi: 10.1016/j.cell.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Banerjee C, et al. Role of Calmodulin-Calmodulin Kinase II, cAMP/Protein Kinase A and ERK 1/2 on Aeromonas hydrophila-induced apoptosis of headkidney macrophages. PLoS Pathog. 2014;10:e1004018. doi: 10.1371/journal.ppat.1004018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.