

Mutations in autophagy genes cause childhood-onset multisystem diseases with predominant nervous system involvement and provide a ‘genetic window’ into autophagy's role in neurobiology and disease. In this review, Ebrahimi-Fakhari et al. examine the clinical and molecular spectrum of ‘congenital disorders of autophagy’, a novel class of inborn errors of metabolism.
Keywords: autophagy, inborn errors of metabolism, mammalian target of rapamycin (mTOR), neurodevelopment, neurodegeneration

Mutations in autophagy genes cause childhood-onset multisystem diseases with predominant nervous system involvement and provide a ‘genetic window’ into autophagy's role in neurobiology and disease. In this review, Ebrahimi-Fakhari et al. examine the clinical and molecular spectrum of ‘congenital disorders of autophagy’, a novel class of inborn errors of metabolism.
Abstract
Single gene disorders of the autophagy pathway are an emerging, novel and diverse group of multisystem diseases in children. Clinically, these disorders prominently affect the central nervous system at various stages of development, leading to brain malformations, developmental delay, intellectual disability, epilepsy, movement disorders, and neurodegeneration, among others. Frequent early and severe involvement of the central nervous system puts the paediatric neurologist, neurogeneticist, and neurometabolic specialist at the forefront of recognizing and treating these rare conditions. On a molecular level, mutations in key autophagy genes map to different stages of this highly conserved pathway and thus lead to impairment in isolation membrane (or phagophore) and autophagosome formation, maturation, or autophagosome-lysosome fusion. Here we discuss ‘congenital disorders of autophagy’ as an emerging subclass of inborn errors of metabolism by using the examples of six recently identified monogenic diseases: EPG5-related Vici syndrome, beta-propeller protein-associated neurodegeneration due to mutations in WDR45, SNX14-associated autosomal-recessive cerebellar ataxia and intellectual disability syndrome, and three forms of hereditary spastic paraplegia, SPG11, SPG15 and SPG49 caused by SPG11, ZFYVE26 and TECPR2 mutations, respectively. We also highlight associations between defective autophagy and other inborn errors of metabolism such as lysosomal storage diseases and neurodevelopmental diseases associated with the mTOR pathway, which may be included in the wider spectrum of autophagy-related diseases from a pathobiological point of view. By exploring these emerging themes in disease pathogenesis and underlying pathophysiological mechanisms, we discuss how congenital disorders of autophagy inform our understanding of the importance of this fascinating cellular pathway for central nervous system biology and disease. Finally, we review the concept of modulating autophagy as a therapeutic target and argue that congenital disorders of autophagy provide a unique genetic perspective on the possibilities and challenges of pathway-specific drug development.
Introduction
Inborn errors of metabolism comprise a large group of single gene disorders that affect various aspects of cellular metabolism. Based on the specific pathway involved, such conditions can be categorized as disorders of carbohydrate metabolism, amino or organic acid metabolism, fatty acid oxidation and mitochondrial metabolism, lysosomal diseases, peroxisomal disorders, and others. While these conditions are traditionally considered ‘enzymopathies’ due to the interruption of specific pathways with resulting accumulation of toxic or interfering intermediate products and/or reduced ability to synthesize an essential metabolite, the recent introduction of next-generation sequencing into diagnostics has resulted in the identification and characterization of novel inborn errors of metabolism.
One such cellular pathway for inborn errors of metabolism that has emerged in recent years is the autophagy pathway, with a number of genetic defects leading to inherited multisystem diseases with prominent nervous system involvement. The prominent involvement of the CNS, peripheral nerves and skeletal muscles, often in the same patient, puts neurologists and neurogeneticists at the forefront for diagnosing and treating ‘congenital disorders of autophagy’. Putative single gene disorders of the autophagy pathway provide a ‘genetic window’ into the role of autophagy in humans and will inform our understanding of this fascinating pathway and its implications for neurobiology and disease. The latter is particularly important, given that dysregulated autophagy has been implicated in many other inherited and sporadic diseases, including neurodegenerative and neurodevelopmental diseases (Pan et al., 2008a; Ebrahimi-Fakhari et al., 2012, 2014; Nixon, 2013; Ebrahimi-Fakhari and Sahin, 2015), myopathies (Ravenscroft et al., 2015), cardiovascular diseases (Lavandero et al., 2013), cancer (Galluzzi et al., 2015), infectious diseases (Huang and Brumell, 2014), and metabolic diseases (Christian et al., 2013), all of which also affect the CNS.
We herein discuss ‘congenital disorders of autophagy’ as an emerging subclass of inborn errors of metabolism by using the examples of six monogenic diseases of autophagy that affect the brain (Table 1). We also highlight associations between defective autophagy and other inborn errors of metabolism such as lysosomal storage diseases and neurodevelopmental diseases, such as those associated with the mTOR pathway.
Table 1.
Congenital disorders of autophagy
| Disease (# OMIM) | Gene | Affected stage of the autophagy pathway | Neurological manifestations |
|---|---|---|---|
|
EPG5 |
|
|
|
WDR45 |
|
|
|
SNX14 |
|
|
|
SPG11 and ZFYVE26 |
|
|
|
TECPR2 |
|
|
The autophagy pathway
Autophagy (Greek for ‘self-eating’) is the umbrella term for essential intracellular pathways that deliver cytosolic cargo to lysosomes for degradation (reviewed in Ravikumar et al., 2010; Klionsky et al., 2011; Kaushik and Cuervo, 2012; Feng et al., 2014) (see Supplementary Table 1 for a glossary of autophagy-related molecules and processes). At least three subtypes of autophagy have been characterized according to the mechanism of cargo delivery: microautophagy, chaperone-mediated autophagy, and macroautophagy (Fig. 1). Microautophagy uses inward invagination of the lysosomal membrane to deliver cytoplasmic cargo directly to the lysosome for degradation. Chaperone-mediated autophagy selectively recognizes targeting motifs (KFERQ-like motifs) on cytosolic proteins and translocates targeted proteins across the lysosomal membrane with the help of chaperone-proteins and the lysosomal adapter protein LAMP-2A. Macroautophagy uses the de novo formation of double-membrane vesicles to sequester parts of the cytosol containing proteins, lipids, and organelles. The basic principles underlying autophagy are highly conserved from yeast to mammals, highlighting the fundamental importance of this pathway for cellular homeostasis and survival. Beyond the turnover of macromolecules and organelles, recent studies have implicated a role for autophagy in related cell functions such as membrane trafficking, regulation of energy metabolism (Efeyan et al., 2015), adaptive immunity (Gomes and Dikic, 2014), and cell death (Green and Levine, 2014).
Figure 1.

The three main subtypes of autophagy and their implications for multiple tissues and organ systems. Autophagy mainly comprises of three subtypes: microautophagy, chaperone-mediated autophagy and macroautophagy. These are distinguished based on the route and mechanism of cargo delivery to lysosomes, the final degrading organelles. Research over the last decades has elucidated a number of cell and tissue-specific functions that depend on or are critically influenced by autophagy. These functions are present in many organ systems, including the nervous system. The variety of functions of autophagy in different organ systems under physiological conditions is emphasized by the broad clinical manifestations in congenital disorders of autophagy, which manifest as multisystem diseases with prominent CNS pathology. CMA = chaperone-mediated autophagy.
In this review, we focus on macroautophagy (hereafter referred to as autophagy), the ‘bulk’ degradation process that is characterized by the formation of double-membrane bound autophagic vesicles, which undergo a dynamic stepwise maturation process: initiation, nucleation of an isolation membrane, elongation of evolving vesicles, closure, maturation, and finally fusion with late endosomes or lysosomes (Figs 1 and 2). The unique double-membrane autophagic vesicle is named the autophagosome, while the fusion product of autophagosomes and lysosomes is referred to as the autolysosome. Although autophagy has been generally thought of as a non-specific degradation pathway, several specific forms of autophagy have been identified in recent years. Examples of specific forms of autophagy include the selective removal of damaged mitochondria (or ‘mitophagy’; Ashrafi and Schwarz, 2013), peroxisomes (or ‘pexophagy’; Oku and Sakai, 2010), ribosomes (or ‘ribophagy’; Suzuki, 2013), aberrant protein aggregates (or ‘aggrephagy’; Yamamoto and Simonsen, 2011), and lipid droplets (or ‘lipophagy’; Liu and Czaja, 2013). Post-translational modifications such as ubiquitination or phosphorylation are critical for recruiting and tailoring the autophagy machinery to each specific cargo (Okamoto, 2014) and thus might serve as a target for developing therapeutic approaches. Ubiquitination seems to be particularly important to label cargo for autophagy, and a number of autophagy receptors with an ubiquitin-binding domain (such as p62/SQSTM1, NBR1, NDP52 or optineurin) have been identified, adding a level of plasticity and precision to the autophagy machinery (Stolz et al., 2014). p62/SQSTM1 is of particular interest because it is not only an important adapter protein for ubiquitinated cargo (Wurzer et al., 2015), but also a substrate for autophagic degradation (Bjorkoy et al., 2005; Komatsu et al., 2007a). Cells deficient in autophagy therefore often accumulate protein aggregates that contain p62, making this a widely used assay to detect autophagy deficits (Klionsky et al., 2012).
Figure 2.

Macroautophagy: an overview of the molecular pathway and mutations associated with congenital disorders of autophagy. Macroautophagy is a step-wise process resulting in the formation of double-membrane-bound autophagic vesicles that engulf their cargo before fusing with lysosomes. The principle stages of macroautophagy include: (I) initiation; (II) nucleation of an isolation membrane (also called phagophore); (III) elongation of evolving autophagic vesicles; (IV) engulfment of cargo and closure of the autophagosomal membrane; (V) autophagosome maturation; (VI) fusion with late endosomes or lysosomes; and finally (VII) degradation of cargo through lysosomal hydrolases. The last step yields basic metabolites that are then recycled. Mutations in congenital disorders of autophagy and single gene disorders associated with deficits in the regulation of autophagy impair different stages of the pathway. Through interfering with the beclin 1 complex, hereditary spastic paraplegia-associated recessive mutations in ZFYVE26 (SPG15) impair early stages such as the formation of the isolation membrane. Mutations in TECPR2 (SPG49) were recently shown to be critically involved in maintaining endoplasmic reticulum exit sites that may serve as scaffolds for the formation of early autophagosome intermediates. X-linked WDR45 mutations cause beta-propeller protein-associated neurodegeneration (BPAN) and have been found to potentially interfere with the elongation of nascent autophagic vesicles. Autosomal-recessive EPG5 mutations in Vici syndrome as well as SNX14 mutations in SNX14-associated autosomal-recessive cerebellar ataxia and intellectual disability syndrome impact the late stages of the autophagy pathway through impairing autophagosome-lysosome fusion. Mutations in SPG11 (SPG11) lead to a defect in autophagic lysosome reformation. Autophagy-associated diseases also affect different stages of autophagy regulation. An example for mTOR-associated neurodevelopmental diseases, loss-of-function mutations in TSC1 or TSC2 in tuberous sclerosis complex lead to constitutive activation of mTORC1 and thus block autophagic flux at multiple stages. Locating the defect to the late stages of the pathway, lysosomal storage diseases impact lysosomal metabolism and thus block upstream steps in the autophagy pathway. Impaired crosstalk between the lysosomal pathway and autophagosome biogenesis might also impact the coordinated regulation of both compartments. AR-CAID = autosomal-recessive cerebellar ataxia and intellectual disability syndrome; LSD = lysosomal storage disease; TSC = tuberous sclerosis complex.
Autophagy is constitutively active to some extent in all cells at any given time in order to maintain a homeostatic balance between catabolic and anabolic processes. To dynamically adjust autophagic activity to the intracellular metabolic state and the extracellular environment, numerous signalling pathways converge to regulate autophagy initiation (Fig. 2), many of these via mTORC1 (mammalian target of rapamycin complex 1; Lipton and Sahin, 2014) or AMPK (AMP-activated protein kinase; Alers et al., 2012). Maintenance of baseline autophagic flux is essential to neurons, skeletal and cardiac muscle. This probably explains the increased vulnerability of these tissues to deficits in autophagy and may account for the prominent neurological and neuromuscular manifestations in primary disorders of autophagy. Specific differences in autophagy regulation and baseline autophagic flux in highly differentiated, post-mitotic cells such as neurons remain to be explored in detail, and might help to explain the preferential degeneration of specific neuronal subtypes such as cerebellar Purkinje cells or long-projecting cortical neurons that form projecting or commissural connections. On a similar note, while the role of autophagy in neuronal physiology is increasingly appreciated, the contribution of defective glial autophagy to neurodegenerative phenotypes in mice (Hara et al., 2006; Komatsu et al., 2006, 2007b; Nishiyama et al., 2007) and humans (see below) with defective autophagy remains relatively unexplored.
Following pioneering work in yeast, many proteins and mechanisms in mammalian autophagy have been uncovered. Multiple autophagy-related proteins (Atg) coordinate the different steps of autophagosome formation and turnover (Fig. 2, see Supplementary Table 1 for a glossary of autophagy-related molecules and processes discussed here). The induction step in the autophagy cascade is critically determined by the phosphorylation status of the ULK1-ATG13-FIP200 complex, which drives the nucleation of the isolation membrane (Wong et al., 2013) (Fig. 2). Under normal nutrient conditions, active mTORC1 phosphorylates ULK1 (unc-51 like autophagy activating kinase 1) and ATG13 to repress ULK1 kinase activity, thereby inhibiting autophagy (Ganley et al., 2009; Hosokawa et al., 2009; Jung et al., 2009; Kim et al., 2011). On the other hand, cell stress induced by starvation, amino acid deprivation, or growth factor withdrawal inhibits mTORC1 activity, leading to autophagy induction (Wong et al., 2013).
AMPK, the second kinase at the heart of autophagy regulation, is a major positive regulator of autophagy under stress conditions. Under conditions of low intracellular energy (when the ration of AMP to ATP is high), activated AMPK induces autophagy both by phosphorylating ULK1 (at a different site than mTORC1) and by inhibiting mTORC1 (Egan et al., 2011; Kim et al., 2011; Di Nardo et al., 2014). Both AMPK and mTOR also control cell growth and metabolism, thereby coupling these processes to autophagy.
In the next step of the autophagic cascade, formation of an isolation membrane is initiated by a second protein complex, the beclin 1-Atg14L-VPS34 kinase complex, which enriches the initiation site with phosphatidylinositol-3-phosphate to recruit interacting regulatory proteins such as Atg9 (Funderburk et al., 2010) (Fig. 2). Atg9 serves as an important adapter molecule that recruits membranes and lipids to expand the isolation membrane (Mari et al., 2010; Orsi et al., 2012; Yamamoto et al., 2012). At a regulatory level, phosphorylation of beclin 1 by Akt inhibits autophagy (Wang et al., 2012b), while phosphorylation at a different residue by AMPK or ULK1 promotes its integration into the beclin 1-Atg14L-VPS34 kinase complex and initiates autophagy (Kim et al., 2013; Russell et al., 2013). Likewise, phosphorylation of Atg9 by ULK1 is required for the efficient recruitment of additional factors to the formation site and subsequent expansion of the isolation membrane (Papinski et al., 2014).
The precise subcellular location where the isolation membrane is formed remains controversial, with the endoplasmic reticulum, Golgi complex, plasma membrane, recycling endosomes, and mitochondrial membrane being putative candidates (Lamb et al., 2013). It seems likely that membranes are derived from multiple sources and perhaps in a cell type- or even location-specific manner, e.g. it is conceivable that the source of autophagic membranes might be different in distal neurites compared to the cell body. Elongation of evolving autophagosomes requires two ubiquitin-like conjugation systems: the ATG5-ATG12-ATG16L1 and the LC3 (microtubule-associated protein light chain 3 or Atg8; encoded by MAP1LC3A)–phosphatidylethanolamine conjugation system (Mizushima et al., 2011) (Fig. 2); the former functions as an E3 ligase that mediates the lipidation of LC3, whereas lipidated LC3 and its family members GATE16 (GABARAPL2) and GABARAP are coupled to the autophagosomal membrane where they support its elongation and closure. The lipidated form of LC3, LC3-II, is of particular interest as it localizes to the inner and outer autophagosomal membrane, making this protein a widely used marker to identify and quantify autophagosome formation and turnover (Klionsky et al., 2012).
Fusion with lysosomes, the final step in the life cycle of autophagosomes, often occurs in the perinuclear region, where the bulk of lysosomes usually reside. Autophagic vesicles, however, almost certainly form everywhere in the cell and thus have to be shipped towards the perinuclear region by dynein-dependent retrograde transportation on microtubules. This process is particularly important for neurons with their long processes (Maday et al., 2012; Cheng et al., 2015). Local autophagic degradation, for example of damaged mitochondria (Ashrafi et al., 2014), might be another important contributor to homeostasis in remote cellular regions such as distal axons, where most of the axonal autophagosomes form (Maday and Holzbaur, 2014). Lysosomal enzymes efficiently degrade the sequestered cargo, and the basic building blocks are recycled back to the cytosol for reuse. After cargo degradation, lysosomal components are also retrieved from autolysosomes to replenish the lysosomal pool (Yu et al., 2010).
Not surprisingly, lysosomal metabolism is intimately coupled to autophagy regulation. This is achieved on the transcriptional level through a recently described pathway that involves the master regulator of both lysosomal and autophagic vesicle biogenesis, the basic helix-loop-helix leucine-zipper transcription factor EB (TFEB). TFEB regulates the expression of the coordinated lysosomal expression and regulation (CLEAR) network of genes involved in lysosomal biogenesis and autophagy (Sardiello et al., 2009; Palmieri et al., 2011; Settembre et al., 2011). Interestingly, mTORC1 associates and phosphorylates TFEB at the lysosomal membrane under conditions of nutrient sufficiency and thus prevents its translocation to the nucleus. In response to cellular energy depletion, however, mTORC1-dependent phosphorylation of TFEB is impaired, triggering the transcription of genes that encode proteins required for autophagosome formation and autophagic flux (Martina et al., 2012; Roczniak-Ferguson et al., 2012). Given its central role in regulating the autophagy-lysosomal pathway, TFEB has gained significant attention as a potential therapeutic target for diseases where defective autophagy and/or lysosomal dysfunction have been implicated, such as hepatic α1-antitrypsin deficiency (Pastore et al., 2013), Pompe disease (Spampanato et al., 2013), Parkinson’s disease (Decressac et al., 2013) or Huntington’s disease (Tsunemi et al., 2012).
Congenital disorders of autophagy
The emergence of rapid and more widely available next-generation DNA sequencing technology has led to the identification of the genetic basis of many rare diseases, including inborn errors of metabolism. Single gene disorders affecting the autophagy pathway are increasingly identified, and genetic variants in autophagy genes are found to contribute to a number of major diseases. Here we discuss six ‘congenital disorders of autophagy’ (Table 1) that predominantly affect the brain and argue for a novel subclass of inborn errors of metabolism.
Vici syndrome: EPG5 mutations
First described by Dionisi-Vici et al. (1988), Vici syndrome (OMIM #242840) is a rare autosomal recessive multisystem disease with ∼50 published cases to date (Dionisi Vici et al., 1988; del Campo et al., 1999; Chiyonobu et al., 2002; Miyata et al., 2007; Al-Owain et al., 2010; McClelland et al., 2010; Rogers et al., 2011; Finocchi et al., 2012; Ozkale et al., 2012; Said et al., 2012; Cullup et al., 2013, 2014; Ehmke et al., 2014; Filloux et al., 2014; Tasdemir et al., 2015; Byrne et al., in press). Vici syndrome is classically characterized by a set of five cardinal features that include agenesis of the corpus callosum, bilateral cataracts, hypertrophic and/or dilated cardiomyopathy, combined immunodeficiency, and skin, hair and retinal hypopigmentation (Fig. 3). These five manifestations in addition to three features recently identified to occur in the majority of Vici patients, namely acquired microcephaly, failure to thrive and profound developmental delay, allow a clinical diagnosis that is confirmed by a positive genetic test with a specificity and sensitivity of >90% (Byrne et al., in press). Congenital midline defects such as a cleft lip or palate, thymic aplasia or congenital deficiency of the thymus, and hypospadia may also occur but are less frequent. Additional general findings include facial dysmorphism, dysphagia, recurrent pulmonary and mucocutaneous infections secondary to immunodeficiency, and neonatal-onset hypotonia secondary to myopathy. Chronic anaemia, renal tubular acidosis, liver dysfunction, and lung hypoplasia have also been described in a few cases (Fig. 3).
Figure 3.

Vici syndrome: a multisystem disease. Mutations in the autophagy gene EPG5 cause Vici syndrome, a paradigm for multisystem diseases associated with defective autophagy. The eight cardinal features of Vici syndrome consist of agenesis of the corpus callosum, acquired microcephaly, bilateral cataracts, hypertrophic or dilated cardiomyopathy, combined immunodeficiency, and skin, hair or retinal hypopigmentation, failure to thrive and profound developmental delay. CNS manifestations are manifold and include congenital brain malformations such as agenesis of the corpus callosum with colpocephaly, cerebellar hypoplasia, hypoplasia/atrophy of the brainstem, abnormalities of the septum pellucidum, opercular hypoplasia and probably age-dependent abnormal T2 signal in the thalami. Dysgenesis of the falx, non-lissencephalic cortical dysplasia, polymicrogyria of the cerebral hemispheres, or bilateral schizencephaly have also been reported in a few individuals. Delayed myelination and diffuse white matter atrophy are commonly reported. Pointing to the potential presence of a neurodegenerative phenotype in addition to prominent deficits in brain development, dilated ventricles, Purkinje cell loss, and diffuse cerebral atrophy have been described in a subset of patients.
The neurological phenotype is broad and, in addition to the prominent feature of agenesis of the corpus callosum (often with colpocephaly), which occurs in almost all patients, involves other brain malformations such as non-lissencephalic cortical or cerebellar vermis dysplasia, pontine hypoplasia, abnormalities of the septum pellucidum, abnormal T2 signal diffusely in the thalami, and myelination defects (Table 1 and Fig. 3). Of these secondary features, reduced white matter bulk and under-opercularization of the Sylvian fissures are found in the majority of patients. Post-mortem examination of a single case confirmed agenesis of the corpus callosum as well as prominent hypoplasia of the pons and cortico-spinal tracts. Neurological sequelae are manifold and include progressive postnatal microcephaly, profound developmental delay and intellectual disability, motor impairment, nystagmus, sensorineuronal deafness, and seizures (Table 1 and Fig. 3). In infancy, the latter frequently evolve into epileptic encephalopathy that is often refractory to treatment. Interestingly, the reported loss of previously acquired skills as well as the occurrence of progressive microcephaly in Vici patients who survive the neonatal period, suggest a neurodegenerative component in addition to the prominent neurodevelopmental defects. This notion is corroborated by a neurodegenerative phenotype in Epg5-deficient Drosophila melanogaster (CG14299; Byrne et al., in press) and transgenic mice (Zhao et al., 2013a) as discussed below. The disease is usually fatal in infancy or early childhood with recurrent, severe infections and/or progressive heart failure likely being the main cause of mortality.
Using whole-exome sequencing in affected individuals, Cullup et al. (2013) identified recessive mutations in the EPG5 gene on chromosome 18q12.3 as the genetic cause of Vici syndrome. EPG5 consists of 44 exons encoding a protein of 2579 amino acids at maximum. The mutations identified so far map to almost the entire gene with no clear mutational hotspot. Most mutations are predicted to lead to a truncated protein product, thus favouring a loss-of-function mechanism. EPG5 is the human homologue of the metazoan-specific autophagy gene Epg5 (ectopic P-granule autophagy protein 5), which encodes the key regulatory autophagy protein Epg5. Like many autophagy-related genes, Epg5 first emerged in a Caenorhabditis elegans-based genetic screening for modifiers of autophagic substrate degradation. Mutant epg-5 in C. elegans, as well as a gene knockdown in mammalian cells, was found to result in the accumulation of dysfunctional non-degenerative autolysosomes, arguing that EPG5 is critically involved in the late stages of the autophagy cascade such as autophagosome-lysosome fusion or proteolysis within autolysosomes (Tian et al., 2010; Zhao et al., 2013a).
Further delineating the role of EPG5, characterization of autophagy in skeletal muscle tissue and fibroblasts from Vici patients revealed an accumulation of LC3-positive autophagic vesicles, and of the autophagy linker-proteins NBR1 and p62. This suggests a block in autophagic flux, a finding further corroborated by the accumulation of K63-polyubiquitinated proteins, which are usually targeted for autophagic degradation. Evidence for a block at the late stages of autophagy, namely during autophagosome–lysosome fusion, was provided by showing a reduction in the co-localization of both organelles (Cullup et al., 2013). In retrospect, these findings are consistent with earlier studies in available muscle biopsies from Vici patients that found abundant vacuoles and dense bodies suggesting a lysosomal origin (Al-Owain et al., 2010; McClelland et al., 2010; Cullup et al., 2013).
In summary, EPG5 deficiency in Vici syndrome leads to a critical impairment of the late stages of the autophagy pathway (Table 1 and Fig. 2). Global reduction in autophagic flux leads to impaired development and function in a multitude of tissues and thus to a multisystem disease (Fig. 3). Recurrent infections are a significant source of morbidity and mortality in Vici syndrome. This might be secondary to the fact that defective autophagy impairs immunity and the clearance of intracellular pathogens in particular, as this has been recently found to critically depend on intact autophagy (Jo et al., 2013). Despite the convincing role for EPG5 mutations in human cases, Epg5 knockout mice do not fully recapitulate the phenotype found in Vici syndrome and interestingly resemble instead key neuropathological features of the neurodegenerative disease amyotrophic lateral sclerosis (Zhao et al., 2013a). Neurodegeneration mainly occurs in cortical pyramidal neurons and motor neurons of the spinal cord and is accompanied by accumulation of p62-positive and ubiquitinated protein aggregates. Not surprisingly, knockout mice display progressive motor deficits and die around 12 months of age. Muscle atrophy, fibrillations and sharp waves on electromyography indicate active denervation. Interestingly, in addition to the degenerative changes secondary to denervation, glycogen accumulation is found in skeletal muscle of Epg5 deficient mice arguing for a ‘storage disease phenotype’ similar to glycogen accumulation observed in muscle and other tissues from patients with Vici syndrome (Al-Owain et al., 2010; McClelland et al., 2010; Cullup et al., 2013). Although no overt developmental brain malformations were discovered, Epg5 deficient mice were consistently found to have a reduced thickness of the corpus callosum with a partial or complete agenesis in a few knockout animals (Zhao et al., 2013b). Thus, while Epg5 knockout mice show some phenotypic similarities to patients with Vici syndrome, including corpus callosum dysgenesis and myopathy, core clinical features, however, are not recapitulated. Nevertheless, Vici syndrome, a multisystem disease in humans, is an important example for the concept of ‘congenital disorders of autophagy’ as it highlights the consequences of dysfunctional autophagy for organ development and the complex role of autophagy in CNS development.
Beta-propeller protein-associated neurodegeneration: WDR45 mutations
Neurodegeneration with brain iron accumulation (NBIA) is a clinically and genetically heterogeneous group of single gene disorders characterized by dysregulation of iron metabolism leading to iron deposits in the basal ganglia, particularly in the globus pallidus and substantia nigra (Horvath, 2013; Meyer et al., 2015). A recently identified subtype within this group is beta-propeller protein-associated neurodegeneration (BPAN), a disease that had been previously summarized under the term ‘static encephalopathy of childhood with neurodegeneration in adulthood’ (SENDA) syndrome, based on its distinct pattern of clinical and MRI findings and its characteristic natural history (Fig. 4). BPAN accounts for ∼1–2 % of all NBIA cases and is the only form of the disease with an X-linked dominant inheritance pattern, although thus far only patients carrying de novo mutations have been reported (Haack et al., 2012; Saitsu et al., 2013; Nishioka et al., 2015).
Figure 4.
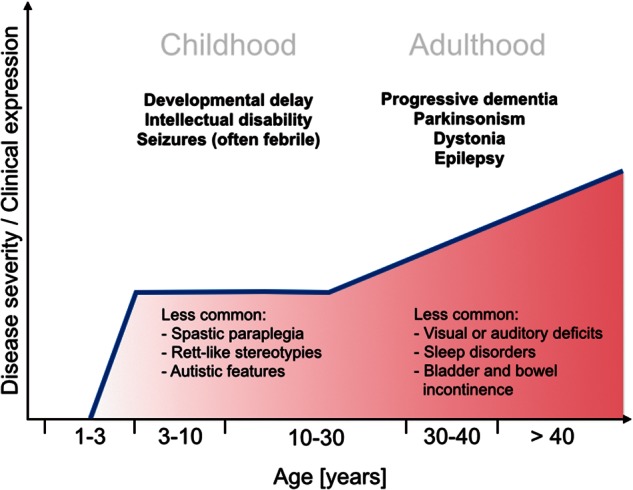
BPAN: natural history of a biphasic disease. Patients with WDR45-mutation associated BPAN commonly present with a distinct biphasic clinical course. Initial manifestations in infancy or early childhood consist of global developmental delay, intellectual disability, and seizures, which tend to progress in the later stages of the disease. Cerebellar ataxia, spasticity, Rett-like hand stereotypies, and autistic features have also been reported in a subset of patients. The disease advances in adolescence or early adulthood when dystonia, parkinsonism, and progressive cognitive decline become prominent. Other manifestations often include bladder and bowel incontinence, disordered sleep, and visual and auditory deficits.
Clinically, BPAN is a biphasic disease that begins with global developmental delay in infancy or early childhood (Table 1 and Fig. 4) (Hayflick et al., 2013; Nishioka et al., 2015). Seizures of various types including generalized tonic-clonic but also focal, absence, atonic or myoclonic seizures are frequent initial presentations, often in the context of a febrile illness. Spastic paraparesis is another commonly reported early finding. Symptoms remain relatively static until the second phase of the disease suddenly begins in adolescence or early adulthood with progressive cognitive decline, dementia, dystonia, and parkinsonism dominating the clinical picture. The latter mainly consists of bradykinesia, rigidity and postural instability while tremor is not commonly seen. In a number of cases, these symptoms have responded to levodopa (Hayflick et al., 2013). Seizures, too, usually progress in the adult phase of the disease. Sleep abnormalities, neuropsychiatric symptoms within the autistic and affective spectrum, eye movement abnormalities, optic nerve atrophy, sensorineural hearing loss and Rett-like hand stereotypies have also been reported in a subset of patients (Hayflick et al., 2013; Ohba et al., 2014; Rathore et al., 2014; Verhoeven et al., 2014). Distinctive MRI changes allow a diagnosis in many cases (Kruer et al., 2012): iron accumulation is detectable on regular T2 sequences in the substantia nigra even at an early disease stage, while detecting iron deposits in the globus pallidum requires other techniques such as gradient-echo or T2*-weighted sequences. Hyperintensity on T1-weighted axial images with a central band of hypointense signal has been reported as a pathognomonic feature of BPAN (Kruer et al., 2012; Ichinose et al., 2014; Ozawa et al., 2014). Cerebral atrophy, or less commonly cerebellar atrophy, and basal ganglia calcification (Van Goethem et al., 2014) can also be seen. Post-mortem examination of two adult cases of BPAN revealed extensive and widespread accumulation of hyperphosphorylated tau protein in the form of neurofibrillary tangles, arguing that BPAN shares neuropathological features with classic degenerative tauopathies such as Alzheimer’s disease (Hayflick et al., 2013; Paudel et al., 2015). BPAN is inevitably fatal with current therapeutic strategies being targeted at symptomatic relief of parkinsonian features, dystonia and seizures.
Using exome sequencing, two independent groups recently uncovered mutations in the WDR45 gene as the genetic cause of BPAN (Haack et al., 2012; Saitsu et al., 2013). WDR45, also known as WIPI4, is located on the X-chromosome and is one of the four mammalian homologues of the core autophagy gene epg-6 in C. elegans (Proikas-Cezanne et al., 2004; Lu et al., 2011). WDR45 encodes a WD repeat protein, a superfamily of proteins that is characterized by repeating units with a conserved core of ∼40 amino acids that terminate with tryptophan-aspartic acid (WD) residues. WD40 proteins have a highly symmetrical beta-propeller tertiary structure that enables them to regulate the assembly of multiprotein complexes by providing a stable anchoring platform for simultaneous and reversible protein–protein interactions (Stirnimann et al., 2010). Based on these properties, WD-repeat proteins are key components of many essential biological functions and pathways, including autophagy, signal transduction pathways, transcriptional regulation, cell cycle control, apoptosis, and vesicular trafficking (Stirnimann et al., 2010).
WDR45, the WD-repeat protein mutated in BPAN, interacts with autophagy proteins Atg2 and Atg9 to regulate key steps during autophagosome formation and elongation (Behrends et al., 2010; Lu et al., 2011). Hence, depletion of WDR45 in mammalian cells leads to the accumulation of autophagosomes and early autophagic vesicles (Behrends et al., 2010; Lu et al., 2011). Investigating the effect of WDR45 mutations in lymphoblast cell lines derived from BPAN patients, Saitsu et al. (2013) found that levels of WDR45 protein were strongly diminished, suggesting that the mutant protein is structurally unstable and undergoes rapid degradation (Saitsu et al., 2013). Subsequently, autophagic flux is impaired, leading to an accumulation of abnormal and immature autophagic vacuoles. The latter was confirmed by showing that accumulating abnormal autophagosome precursors stain for both Atg9A and LC3, indicating improper autophagosome formation, since, under normal conditions, Atg9A only transiently associates with the autophagosome formation site and is absent from mature LC3-positive autophagosomes (Orsi et al., 2012). In summary, findings in cells from BPAN patients indicate that autophagy is perturbed at an early stage (Table 1 and Fig. 2), suggesting that autophagy dysfunction might account for the neurological sequelae of the disease. Interestingly, although BPAN is clinically thought to primarily affect the brain, WDR45 is expressed in many human tissues with the highest expression found in skeletal muscle (Proikas-Cezanne et al., 2004). A muscle phenotype, however, remains to be formally investigated. The selective degeneration of neuronal populations may be explained by cell-type-specific differences in autophagy and tolerance to changes in this pathway. Neurons as non-dividing, highly differentiated cells heavily rely on intracellular degradation pathways to maintain homeostasis and normal structure and function. This is exemplified by brain-specific knockout mice of core autophagy genes showing a striking neurodegenerative phenotype similar to that found in ageing-related neurodegenerative diseases (Hara et al., 2006; Komatsu et al., 2006). Because mice globally deficient in autophagy die shortly after birth (Kuma et al., 2004; Komatsu et al., 2005), WDR45 mutations in affected human males might be inevitably lethal at an early stage. Recently reported conditional CNS-specific WDR45 knockout mice (Nes-WDR45fl/Y) show axonal pathology with swollen axons and accumulation of autophagy substrates p62 and ubiquitin. Although neither neurodegeneration nor iron deposition are prominent phenotypes, Nes-WDR45fl/Y mice, at the behavioural level, exhibit subtle motor coordination deficits and poor learning and memory, suggesting deficits in neuronal circuit formation or neurotransmission (Zhao et al., 2015). Another interesting lead into the pathogenesis of BPAN comes from the recent appreciation of the role of autophagy in iron metabolism. The bioavailability of intracellular iron is critically controlled through the delivery of ferritin to autophagosomes and lysosomes for degradation (ferritinophagy), allowing release of iron into the cytoplasm (Kidane et al., 2006; Asano et al., 2011; Mancias et al., 2014). Hence, deficits in ferritinophagy could disturb iron homeostasis, potentially contributing to the iron storage phenotype seen in NBIA that seems to confer selectivity for vulnerable brain regions.
In summary, BPAN, the second discussed congenital disorder of autophagy, is of particular importance as this disease, for the first time, confirms that genetic deficits in the autophagy pathway are indeed associated with early-onset neurodegeneration in humans (Saitsu et al., 2013). This further supports a role for autophagy in the pathogenesis of an expanding list of sporadic ageing-related neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and others (Ebrahimi-Fakhari et al., 2012; Nixon, 2013) and may provide novel insights into the role of autophagy in iron metabolism.
SNX14-associated autosomal-recessive cerebellar ataxia and intellectual disability syndrome
The childhood-onset autosomal-recessive cerebellar ataxias are a group of heterogeneous inherited diseases characterized by progressive cerebellar atrophy and prominent Purkinje cell degeneration. Next-generation sequencing has allowed a genetic and molecular resolution of a number of entities and holds great promise to facilitate an early diagnosis in many cases (Nemeth et al., 2013; Pyle et al., 2015). Recently, two independent groups identified truncating loss-of-function mutations in the sorting nexin gene, SNX14, as the cause of a clinically distinct autosomal-recessive cerebellar ataxia syndrome (Thomas et al., 2014; Akizu et al., 2015). Patients with bi-allelic SNX14 mutations present with globally delayed development, hypotonia, absent speech, progressive cerebellar atrophy and ataxia, seizures, and a storage disease phenotype consisting of coarse facial features, macroglossia, hypertrychosis, kyphoscoliosis, sensorineural hearing loss, and hepatosplenomegaly in a few cases (Table 1) (Sousa et al., 2014; Thomas et al., 2014; Akizu et al., 2015). On clinical grounds, these manifestations suggest overlapping disease mechanisms with lysosomal storage diseases with prominent involvement of the cerebellum such as Niemann-Pick disease type C (NP-C) or Tay-Sachs disease. Indeed, mutant SNX14 patient fibroblasts contain enlarged lysosomes (Thomas et al., 2014; Akizu et al., 2015), providing a rationale to further investigate the role of SNX14 in lysosomal turnover.
SNX14 encodes a protein of the sorting nexin family that contains two putative transmembrane domains (Mas et al., 2014). Through the RGS (regulator of G protein signalling) domain, SNX14 attenuates Gαs-coupled G protein-coupled receptor signalling. Through its PX (phox homology) domain SNX14 binds membrane phosphatidylinositol residues and is involved in intracellular trafficking. Phospholipids on membranes of different organelles provide a platform for recruiting factors that mediate membrane turnover and fusion. With regards to the autophagy pathway, this is important for key membrane fusion events such as the fusion of phosphatidylinositol 3-phosphate coated autophagosomes with phosphatidylinositol (3,5)-bisphosphate carrying lysosomes (Dall’Armi et al., 2013). The importance of phospholipids for autophagy is exemplified by recent studies that show key regulatory functions in the regulation of canonical (Burman and Ktistakis, 2010) and non-canonical autophagy (Vicinanza et al., 2015). In addition, cardiolipins, a subtype of phospholipids localized to the inner mitochondrial membrane, serve as a crucial signal for autophagic degradation when exposed on the surface of depolarized mitochondria (Chu et al., 2013). Binding to their respective phospholipid signature, SNX14 co-localizes with the lysosome and is enriched in autophagosome containing cell fractions (Akizu et al., 2015). In induced pluripotent stem cell (iPSC)-derived neuronal cells from patients with SNX14 mutations, lysosomes are increased in size and autophagosome clearance is impaired (Akizu et al., 2015). This pattern closely resembles findings in certain lysosomal storage diseases (Ebrahimi-Fakhari et al., 2014). Knockdown of snx14 in a zebrafish model confirmed these observations and linked them to progressive Purkinje cell loss, suggesting neuronal cell death secondary to impaired autophagy (Akizu et al., 2015). Purkinje cells are exquisitely sensitive to autophagy impairment and perturbations of lysosomal metabolism (Hara et al., 2006; Komatsu et al., 2006), which may be attributable to their size, high metabolic activity, and complex dendritic architecture. Not surprisingly, post-mortem neuropathological assessment showed a near complete absence of Purkinje cells in an affected individual with bi-allelic SNX14 mutations (Akizu et al., 2015).
SNX14-associated autosomal-recessive cerebellar ataxia is thus a further disease that links autophagy to neurodegeneration and highlights the importance of autophagy for Purkinje cell function and survival. Given the emerging links between cerebellar circuit dysfunction and syndromic forms of autism-spectrum disorder (Tsai et al., 2012), it is interesting to note that the majority of reported patients with SNX14 mutations show autistic-like behaviour (Akizu et al., 2015).
Hereditary spastic paraplegia: SPG11 (SPG11), SPG15 (ZFYVE26) and SPG49 (TECPR2)
Hereditary spastic paraplegia (HSP) is a clinically and genetically heterogeneous group of neurodegenerative diseases that occurs at a prevalence of about 3–10 cases per 100 000 individuals (Salinas et al., 2008; Blackstone, 2012). All forms of HSP share the unifying feature of distal axonal degeneration in the corticospinal tracts (Lo Giudice et al., 2014). In some cases, ascending spinal tracts such as the gracile fasciculus and the spinocerebellar tracts may also be affected. Clinically, degeneration of the corticospinal tracts leads to progressive weakness and spasticity of the lower limbs. Additional manifestations in the various forms of HSP described to date range from congenital brain abnormalities such as agenesis of the corpus callosum or cerebellar dysplasia, to signs of neuronal dysfunction and neurodegeneration such as cognitive impairment, ataxia, optic nerve atrophy, epilepsy, and peripheral neuropathy (Lo Giudice et al., 2014). Given the unifying feature of axonal degeneration of the long tracts, the HSPs serve as a genetically tractable model for identifying pathways that ensure axonal function and survival. Covering all modes of inheritance, more than 70 disease loci and 50 spastic paraplegia genes have been identified to date. These genes map to multiple cellular pathways and processes, including endoplasmic reticulum function, vesicle formation, membrane trafficking, mitochondrial function, lipid metabolism, myelination, axonal transport and autophagy (Blackstone, 2012; Lo Giudice et al., 2014; Noreau et al., 2014). Here, we discuss SPG11, SPG15 and SPG49, three autosomal recessive forms of HSP characterized by autophagy dysfunction.
SPG11 (OMIM #604360) and SPG15 (OMIM #270700) are the most prevalent forms of autosomal recessive HSP with thin corpus callosum (Goizet et al., 2009; Schule et al., 2009; Pensato et al., 2014). The clinical phenotype in SPG11 and SPG15 is often indistinguishable (Table 1) and an accurate diagnosis therefore requires genetic testing (Schule et al., 2009). Patients with SPG11 or SPG15 often present with Kjellin syndrome, in which early-onset spastic paraplegia is accompanied by intellectual disability, pigmentary retinopathy, cerebellar dysfunction, and distal amyotrophy in the first or second decade of life (Kjellin, 1959). Parkinsonism is also sometimes observed.
Following linkage to the SPG11 locus on chromosome 15q (Martinez Murillo et al., 1999; Shibasaki et al., 2000), the gene for SPG11, SPG11, and its protein product spatacsin were identified (Stevanin et al., 2007, 2008).
For SPG15, Hughes et al. (2001) were first to identify the causative gene locus on chromosome 14q from two Irish families with autosomal-recessive Kjellin syndrome before Hanein et al. (2008) pinpointed the causal gene to ZFYVE26, which encodes the zinc-finger protein spastizin. Over 100 mutations in SPG11 and >20 mutations in ZFYVE26 have been identified in patients, the majority of which are truncating, nonsense mutations (Goizet et al., 2009; Pensato et al., 2014).
Spastizin is a 285 kDa protein comprising of a zinc finger domain, a FYVE domain, and a leucine zipper domain (Hanein et al., 2008). FYVE-domain containing proteins bind phosphatidylinositol 3-phosphate, suggesting a likely function of spastizin in membrane trafficking (Kutateladze and Overduin, 2001). Spastizin is expressed most abundantly in motor cortex, hippocampus, cerebellum, pons and spinal cord, particularly during development (Hanein et al., 2008; Murmu et al., 2011; Khundadze et al., 2013). Intracellularly, spastizin has been observed to co-localize with a variety of structures and organelles, including endosomes and the endoplasmic reticulum and, to a likely lesser extent, microtubules, mitochondria, and the nucleus (Hanein et al., 2008; Murmu et al., 2011; Khundadze et al., 2013; Vantaggiato et al., 2013).
Zfyve26 knockout mice develop normally but by 12 months of age acquire a spastic and ataxic gait disorder accompanied by neuron loss in the motor cortex and the cerebellum, thus recapitulating the clinical phenotype of patients with HSP (Khundadze et al., 2013). Zebrafish models generated by morpholino-mediated knockdown of zfyve26 also show motor neuron growth impairment and morphological abnormalities (Martin et al., 2012). Interestingly, knockdown of SPG11 results in a strikingly similar phenotype (Martin et al., 2012), providing genetic evidence that spastizin and spatacsin might act as part of the same pathway as previously suggested by physical interaction of the two proteins (Slabicki et al., 2010) and overlapping clinical features in SPG15 and SPG11 patients (Boukhris et al., 2008, Schule et al., 2009).
At the cellular level, high-density LAMP1-positive membrane-bound vesicles and lipopigment accumulate in neurons of Zfyve26 knockout mice and precede neurodegenerative changes (Khundadze et al., 2013). Enlarged lysosomes are readily visible in fibroblasts derived from SPG15 patients (Renvoise et al., 2014), similarly suggesting deficits in the autophagy-lysosomal and/or endosomal-lysosomal pathway. Vantaggiato et al. (2013) recently provided an important lead by identifying an interaction between spastizin and the beclin 1-UVRAG-Rubicon complex of the autophagy pathway (Vantaggiato et al., 2013) (Fig. 2). As discussed above, beclin 1 has a multifaceted role in autophagy: in association with Atg14L and Ambra1, the beclin 1-VPS34-VPS15 class III phosphatidylinositol 3-kinase complex is involved in autophagosome formation by regulating phosphatidylinositol 3-phosphate synthesis; separately, when interacting with cofactors UVRAG and Bif-1, beclin 1-VPS34-VPS15 induces autophagosome maturation and endosome fusion. Finally, Rubicon, in complex with UVRAG and beclin 1-VPS34-VPS15, functions as a negative regulator of autophagy (Liang et al., 2006; Itakura et al., 2008; Matsunaga et al., 2009; Zhong et al., 2009; Kang et al., 2011). Spastizin interacts with the beclin 1-UVRAG-Rubicon multiprotein complex and is thus involved in autophagosome maturation (Vantaggiato et al., 2013, 2014). Mutations in spastizin disrupt its interaction with beclin 1 and inhibit autophagosome maturation, leading to an accumulation of immature autophagosomes in fibroblasts and lymphoblasts from SPG15 patients (Vantaggiato et al., 2013). RNA-interference-mediated knockdown of spastizin in cultured neurons was shown to yield similar results. In addition, reduced co-localization of LC3 and LAMP1 was noted in basal and autophagy-promoting conditions, indicating a block in autophagosome-lysosome fusion (Vantaggiato et al., 2013). Recently, pathogenic alterations of autophagy and lysosomal function in SPG15 have been further confirmed by showing that spastizin and the SPG11 protein spatacsin are instrumental for the reformation of autophagic lysosomes, a recycling pathway that generates new lysosomes (Chang et al., 2014; Varga et al., 2015). This mechanism of lysosome biogenesis maintains a pool of lysosomes that are competent to fuse with autophagosomes in order to create new autolysosomes (Yu et al., 2010). Spastizin forms a complex with spatacsin and is targeted to lysosomes through spastizin’s FYVE domain. Loss of either spastizin or spatacsin results in accumulation of enlarged autolysosomes, which is reversible by reintroducing wild-type spastizin. Similarly, accumulation of autophagosomes and depletion of free lysosomes is seen, suggesting a block at the autophagic lysosome reformation stage (Chang et al., 2014; Varga et al., 2015). Taken together, disruption of the critical steps of autophagosome maturation and lysosomal biogenesis contributes to the neurodegeneration seen in SPG15 and related forms of HSP such as SPG11.
Oz-Levi et al. (2012) identified a new form of apparently autosomal-recessive HSP, SPG49 (OMIM #615031), in three Jewish Bukharian families. Affected individuals present with distinct dysmorphic features, delayed development and muscular hypotonia at around 2 years of age before phenotypically progressing to intellectual disability, spastic paraplegia, rigidity, dysarthria and ataxia (Oz-Levi et al., 2012). Severe central apnoea and gastrooesophageal reflux disease are additional features of the disease. Exome sequencing revealed a single and likely causal variant (c.3416delT) in the tectonin β-propeller containing protein 2 (TECPR2) gene (also KIAA0329), resulting in a premature stop codon (Oz-Levi et al., 2012). The truncated protein resulting from the putative pathogenic c.3416delT mutation undergoes rapid degradation, indicating a loss-of-function mechanism. Through high-throughput proteomic analysis of the autophagy pathway, TECPR2 has been established to be a binding partner of the mammalian Atg8 protein family, including LC3, and a probable positive regulator of autophagosome formation (Behrends et al., 2010). Using fibroblasts of affected SPG49 patients and siRNA-mediated knockdown of TECPR2 in cultured cell lines, loss of TECPR2 was found to result in a decreased number of autophagosomes and reduced delivery of LC3 and p62 for lysosomal degradation. These results suggest that SPG49 pathology involves a significant, though not entirely complete, impairment of the autophagy pathway (Oz-Levi et al., 2012). Providing insights into the mechanism of defective autophagy in SPG49, a recent study showed that TECPR2 is involved in maintaining functional endoplasmic reticulum exit sites, which may serve as scaffolds for the formation of autophagosomes (Stadel et al., 2015).
Autophagy in mTOR-associated neurodevelopmental diseases
Pathways that monitor nutrient or amino acid supply (mTORC1 pathway) and cellular ATP-levels (AMPK pathway) critically control autophagy. A central regulator of cellular metabolism, the ubiquitously expressed serine/threonine kinase mTORC1 facilitates anabolic processes that supply the basic building blocks for cell growth, differentiation and proliferation. Not surprisingly, mTORC1 also blocks catabolic pathways such as autophagy via transcriptional and post-translational mechanisms. Conversely, conditions known to induce autophagy, such as for example starvation or growth factor deprivation, effectively reduce mTORC1 activity. Critical targets of mTORC1 that mediate its effect on autophagy are TFEB, regulating autophagy at the transcriptional level, and the ULK1/2 complex, AMBRA1 and the ATG14L-associated VPS34 complex, proteins that are involved in the autophagy initiation step as discussed above (Fig. 2). mTORC1 is important for disorders of autophagy for two reasons: firstly, genetic deficits of the mTOR pathway will almost certainly provoke changes in autophagy, and therefore ‘mTORpathies’ can be regarded as ‘congenital disorders of autophagy regulation,’ and secondly and perhaps most importantly, mTORC1 inhibitors, including drugs currently approved for a variety of conditions, are clinically available inducers of autophagy. An example for an mTOR-associated neurodevelopmental disease with defective autophagy is tuberous sclerosis complex (TSC). In this multisystem disease, loss-of-function mutations in TSC1 or TSC2 lead to a constitutive activation of the mTORC1 pathway (Lipton and Sahin, 2014; DiMario et al., 2015). Overactive mTORC1 is the key pathogenic molecular mechanism in TSC and provides the scientific rationale for the use of mTORC1 inhibitors to treat this disease (Julich and Sahin, 2014; Ebrahimi-Fakhari and Sahin, 2015). Autophagy deficits in TSC have been implicated as contributors to epileptogenesis (McMahon et al., 2012), brain malformations (Yasin et al., 2013), tumour formation (Liang et al., 2014), autism, and neurocognitive deficits (Tang et al., 2014) by impacting neuronal metabolism and synaptic signalling. Understanding autophagy dysfunction as a downstream event of TSC1 or TSC2 mutations is important and might lead to novel therapeutic targets. mTOR-associated diseases therefore highlight the close ties of the autophagy pathway to other fundamental signalling cascades and thus provide an opportunity to investigate the role of dysregulated autophagy in neurodevelopmental diseases.
Autophagy in lysosomal storage diseases
Progressive accumulation of undigested macromolecules in lysosomes is the hallmark feature of lysosomal storage diseases, a group of nearly 60 different diseases caused by mutations in genes encoding for lysosomal enzymes or membrane proteins (Boustany, 2013; Platt, 2014). Not surprisingly, impaired lysosomal metabolism has many effects on upstream pathways that mediate cargo delivery to the lysosomes. Recent studies have documented impaired autophagy in patient samples and disease models of many different lysosomal storage diseases (Lieberman et al., 2012; Ebrahimi-Fakhari et al., 2014). These deficits in autophagy are likely independent and significant contributors to neurodegeneration and other neuronal and non-neuronal disease manifestations, as convincingly illustrated in Niemann-Pick disease Type C (NP-C) models, where a block in autophagic flux and an accumulation of autophagosomes secondary to impaired maturation has been documented (Elrick et al., 2012; Maetzel et al., 2014; Sarkar et al., 2014). Interestingly, re-expression of NPC1, the protein mutated in NP-C, restored autophagy deficits in NPC1-deficient cells, and pharmacological stimulation of autophagy had a cell-protective effect. The latter concept was recently confirmed in iPSC-derived hepatic and neuronal cells from NP-C patients, where mTOR-independent autophagy inducers, identified through a large-scale screening approach for small molecules, promoted cell viability (Maetzel et al., 2014). Combining compounds that mobilize lysosomal cholesterol, such as 2-hydroxypropyl-β-cyclodextrin (Vite et al., 2015) with inducers of autophagy is thus a promising novel therapeutic approach. Glycogen storage disease type II or Pompe disease, although primarily a severe myopathy, is another important case for the potential therapeutic role of autophagy. Genetic approaches that employ TFEB to enhance autophagic flux have shown great promise in proof-of-principle in vitro and in vivo studies (Spampanato et al., 2013). Future investigations for small molecules or effective genetic strategies that would render TFEB a clinically approachable target will prove or disprove the clinical utility of these findings for lysosomal storage diseases.
Unifying features in congenital disorders of autophagy
Major differences in the clinical manifestations of different congenital disorders of autophagy are obvious. This clinical heterogeneity may result from the fact that genes and proteins mutated in individual diseases map to different stages of the pathway and may lead to different degrees of residual autophagic activity. Clinical manifestations are also likely influenced by tissue-specific expression of autophagy proteins during and after development, differences in susceptibility of tissues to deficits in autophagy, a different degree of autophagic flux and metabolic activity in different cell types, and compensatory mechanisms or the existence of genetic or environmental modifiers. Identification and molecular delineation of novel congenital deficits of autophagy in humans will provide an understanding of these factors and will thus provide unique insights into the role of autophagy in human development and disease. Despite a variable clinical expression, a few unifying features are appreciable in congenital disorders of autophagy (Box 1). This includes, for example, a clinical manifestation in multiple organ systems with predominant involvement of the nervous system, an onset in childhood or adolescence, a progressive disease course with neurodegenerative features, and a ‘storage phenotype’ (Box 1). An involvement of the long white matter tracts as seen, for example, with agenesis or progressive thinning of the corpus callosum is another intriguing common denominator. How disruption of neuronal autophagy affects higher brain functions remains an open question and has implications for complex disease manifestations such as intellectual disability or epilepsy.
Box 1.
Unifying clinical characteristics of congenital disorders of autophagy
• Disease onset in early childhood/adolescence
• Prominent nervous system involvement
• Multiple brain regions involved
• Long white-matter tracts (corpus callosum and cortico-spinal tract) and the cerebellum (Purkinje cells) are often affected
• Neurodegenerative phenotype with developmental regression and cognitive decline
• Storage disease phenotype
• Delayed development / intellectual disability, muscular hypotonia, seizures and movement disorders are common
• Often multisystem disease with additional non-neuronal manifestations (including myopathy and ophthalmic manifestations)
• Progressive disease course, often fatal
Restoring deficits in autophagy as a therapeutic approach
Pharmacological approaches that modulate autophagy have received increasing attention as a potential therapy for a broad range of autophagy-associated diseases (Table 2) (Ebrahimi-Fakhari et al., 2012; Rubinsztein et al., 2012; Nixon, 2013). Many small molecule inducers of autophagy act through the mTOR pathway (e.g. rapamycin and rapalogs), but mTOR-independent regulators also exist (e.g. carbamazepine or lithium, which signal through inositol 1,4,5-trisphosphate; Table 2). Target specificity is a tremendous challenge for drug development, and many available compounds have autophagy-independent off-target effects. Not surprisingly, a number of known autophagy modulators are currently in clinical trials or in clinical use for indications other than autophagy induction. It is imperative to learn how these compounds affect autophagy and whether existing drugs with an extensive clinical safety profile could be readily used to target autophagy. This concerns, for example, the use of the antiepileptic drug carbamazepine (Hidvegi et al., 2010; Maetzel et al., 2014) or lysosomotropic agents such as chloroquine (Amaravadi et al., 2011). Although the diseases discussed in this review are rare, the extent to which they might inform us about therapeutic targets in the autophagy pathway is significant. Based on current evidence, enhancing residual autophagy function or bypassing molecular deficits appear to be rational therapeutic approaches for congenital disorders of autophagy. The latter might be achieved by targeting the stage of the autophagy pathway that is specifically disrupted in each given disease. Understanding the biology of congenital disorders of autophagy will help to design these proposed stage-specific autophagy-targeted therapies.
Table 2.
Modulators of autophagy in neuronal disease models
| Compound | Proposed target and mechanism | Model system and selected references |
|---|---|---|
| Bafilomycin A1 |
|
|
| Carbamazepine |
|
|
| (Hydroxy-) Chloroquine |
|
|
| Clonidine |
|
Neuronal cell model: D. melanogaster; zebrafish Williams et al., 2008 |
| Latrepirdine |
|
Neuronal cell model, mouse models: Steele et al., 2013a, 2013b |
| Lithium |
|
|
| Nilotinib |
|
Mouse models: Hebron et al., 2013; Lonskaya et al., 2014 |
| Rapamycin and rapalogs |
|
|
| Resveratrol |
|
|
| Rilmenidine |
|
|
| SMER28 and other SMER |
|
Neuronal cell model: Tian et al., 2011; D. melanogaster: Sarkar et al., 2007b |
| Trehalose |
|
|
| Valproate |
|
Neuronal cell model, D. melanogaster: Williams et al., 2008 |
| Verapamil (and other Ca2+-channel blockers) |
|
Neuronal cell model, D. melanogaster; zebrafish: Williams et al., 2008 |
IPSCs have emerged as a valuable preclinical model to study rare diseases, given that reliable animal models for these conditions are often not readily available. In addition, several protocols exist that enable differentiation into relevant cell types such as neuronal cells. These will be instrumental for understanding disease phenotypes arising from different genetic mutations in individual patients and will provide a very useful platform for high throughput screening approaches that aim to identify molecules that restore autophagy function in a given disease. Recent studies have described the generation of human iPSC lines from patients with a range of inherited diseases (Park et al., 2008; Ebert et al., 2009; Lee et al., 2009; Marchetto et al., 2010) and proof-of-principle screening studies for modulators of autophagy using iPSC are emerging (Maetzel et al., 2014).
The transition from cellular and mouse models to humans will contest the potential for harnessing autophagy-based therapies. To translate potential targets into therapies, a challenge that will have to be overcome is the development of in vivo biomarkers to detect changes in autophagy in response to therapeutics or disease progression. This will be most effective when stage-specific methods or surrogate biomarkers become available to confirm target engagement, which will be essential to interpret outcomes in clinical trials. As with most congenital childhood-onset diseases with a clear neurodevelopmental phenotype, another concern is the optimal timing to start treatment. Along the same lines, it remains to be seen whether existing lesions, such as for example iron storage in the basal ganglia of BPAN patients, are reversible by correcting autophagy deficits.
Conclusions
Genetic disruption of the autophagy–lysosomal pathway, a fundamental metabolic pathway, results in a new and diverse group of multisystem diseases. We therefore argue that based on their shared genetic and molecular characteristics, these should be collectively termed ‘congenital disorders of autophagy’, a novel subclass within the field of inborn errors of metabolism. In the past 2 years alone, next-generation sequencing technologies have allowed for the identification of a number of pathogenic mutations in core autophagy genes and will likely continue to reveal novel single gene disorders of this crucial cellular pathway. Bridging the gap between the identification of causative genes to the delineation of molecular disease mechanisms is an upcoming challenge. Development of disease models, such as those generated through iPSC technology, will allow us to gain insights into potential therapeutic targets and will yield novel therapies. Insights into congenital diseases of autophagy may be important to a wide spectrum of more common sporadic diseases with defective autophagy.
Supplementary Material
Acknowledgements
The authors thank R. Friedman (Boston Children’s Hospital, Translational Neuroscience Center) for help with database queries.
Funding
D.E.-F. acknowledges support from the Graduate Academy of the University of Heidelberg, the Young Investigator Award Program at Ruprecht-Karls-University Heidelberg Faculty of Medicine, the Daimler and Benz Foundation (Daimler und Benz Stiftung, Ladenburg, Germany) and the Reinhard-Frank Foundation (Reinhard-Frank-Stiftung, Hamburg, Germany). A.S. is supported by a scholarship from the German National Academic Foundation (Studienstiftung des Deutschen Volkes e.V.). L.W. is supported by the Young Investigator Award Program at Ruprecht-Karls-University Heidelberg Faculty of Medicine. J.L. received support from the Medical Scientist Training Program from the National Institute of General Medical Sciences (T32GM007753). G.F.H. received generous support from the Dietmar Hopp Stiftung (St. Leon-Rot, Germany). H.J. acknowledges support from the Myotubular Trust, United Kingdom. M.S. is supported by NIH (U01 NS082320, P20 NS080199, U54NS092090, P30 HD018655), Nancy Lurie Marks Family Foundation, Boston Children’s Hospital Translational Research Program. The Developmental Synaptopathies Consortium (U54NS092090) is a part of the National Center for Advancing Translational Sciences (NCATS) Rare Diseases Clinical Research Network (RDCRN). RDCRN is an initiative of the Office of Rare Diseases Research (ORDR), NCATS, funded through collaboration between NCATS, National Institute of Mental Health, National Institute of Neurological Disorders and Stroke and National Institute of Child Health and Human Development.
The funding agencies had no role in the design, preparation or writing of this manuscript.
Conflict of interest
D.E-F. and L.W. report receiving travel grants from Actelion Pharmaceuticals for attending an international scientific meeting in 2014. M.S. receives research funding from Novartis, Shire and Roche. A.S., J.L., S.B., G.F.H. and H.J. report no conflict of interest.
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- AMPK
AMP-activated protein kinase
- Atg
autophagy-related gene/protein
- BPAN
beta-propeller protein-associated neurodegeneration
- HSP
hereditary spastic paraplegia
- iPSC
induced pluripotent stem cell
- mTOR
mammalian target of rapamycin
- mTORC1
mammalian target of rapamycin complex 1
References
- Akizu N, Cantagrel V, Zaki MS, Al-Gazali L, Wang X, Rosti RO, et al. Nat Genet 2015; 47: 528–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, Humaidan H, Al-Hindi H, Al-Homoud I, et al. Vici syndrome associated with unilateral lung hypoplasia and myopathy. Am J Med Genet A 2010; 152A: 1849–53. [DOI] [PubMed] [Google Scholar]
- Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32: 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17: 654–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano T, Komatsu M, Yamaguchi-Iwai Y, Ishikawa F, Mizushima N, Iwai K. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol Cell Biol 2011; 31: 2040–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol 2014; 206: 655–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 2013; 20: 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bains M, Florez-McClure ML, Heidenreich KA. Insulin-like growth factor-I prevents the accumulation of autophagic vesicles and cell death in Purkinje neurons by increasing the rate of autophagosome-to-lysosome fusion and degradation. J Biol Chem 2009; 284: 20398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466: 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171: 603–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstone C. Cellular pathways of hereditary spastic paraplegia. Annu Rev Neurosci 2012; 35: 25–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B, Smith DA, Mooney D, Jung SS, Walsh DM, Platt FM. Macroautophagy is not directly involved in the metabolism of amyloid precursor protein. J Biol Chem 2010; 285: 37415–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukhris A, Stevanin G, Feki I, Denis E, Elleuch N, Miladi MI, et al. Hereditary spastic paraplegia with mental impairment and thin corpus callosum in Tunisia: SPG11, SPG15, and further genetic heterogeneity. Arch Neurol 2008; 65: 393–402. [DOI] [PubMed] [Google Scholar]
- Boustany RM. Lysosomal storage diseases–the horizon expands. Nat Rev Neurol 2013; 9: 583–98. [DOI] [PubMed] [Google Scholar]
- Burman C, Ktistakis NT. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett 2010; 584: 1302–12. [DOI] [PubMed] [Google Scholar]
- Byrne S, Jansen L, U-King-Im JM, Siddiqui A, Lidov H, Bodi I, et al. EPG5-related Vici syndrome: a paradigm of neurodevelopmental disorders with defective autophagy. Brain, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo K, Nassif M, Valenzuela V, Rojas F, Matus S, Mercado G, et al. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 2013; 9: 1308–20. [DOI] [PubMed] [Google Scholar]
- Chang J, Lee S, Blackstone C. Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J Clin Invest 2014; 124: 5249–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JW, Choi H, Cotman SL, Jung YK. Lithium rescues the impaired autophagy process in CbCln3(Deltaex7/8/Deltaex7/8) cerebellar cells and reduces neuronal vulnerability to cell death via IMPase inhibition. J Neurochem 2011; 116: 659–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng XT, Zhou B, Lin MY, Cai Q, Sheng ZH. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J Cell Biol 2015; 209: 377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiyonobu T, Yoshihara T, Fukushima Y, Yamamoto Y, Tsunamoto K, Nishimura Y, et al. Sister and brother with Vici syndrome: agenesis of the corpus callosum, albinism, and recurrent infections. Am J Med Genet 2002; 109: 61–6. [DOI] [PubMed] [Google Scholar]
- Christian P, Sacco J, Adeli K. Autophagy: Emerging roles in lipid homeostasis and metabolic control. Biochim Biophys Acta 2013; 1831: 819–24. [DOI] [PubMed] [Google Scholar]
- Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 2013; 15: 1197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes CJ, Qin K, Cook J, Solanki A, Mastrianni JA. Rapamycin delays disease onset and prevents PrP plaque deposition in a mouse model of Gerstmann-Straussler-Scheinker disease. J Neurosci 2012; 32: 12396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullup T, Dionisi-Vici C, Kho AL, Yau S, Mohammed S, Gautel M, et al. Clinical utility gene card for: Vici Syndrome. Eur J Hum Genet 2014; 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullup T, Kho AL, Dionisi-Vici C, Brandmeier B, Smith F, Urry Z, et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet 2013; 45: 83–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall'Armi C, Devereaux KA, Di Paolo G. The role of lipids in the control of autophagy. Curr Biol 2013; 23: R33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci USA 2013; 110: E1817–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Campo M, Hall BD, Aeby A, Nassogne MC, Verloes A, Roche C, et al. Albinism and agenesis of the corpus callosum with profound developmental delay: Vici syndrome, evidence for autosomal recessive inheritance. Am J Med Genet 1999; 85: 479–85. [DOI] [PubMed] [Google Scholar]
- Di Nardo A, Wertz MH, Kwiatkowski E, Tsai PT, Leech JD, Greene-Colozzi E, et al. Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1. Hum Mol Genet 2014; 23: 3865–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMario FJ, Jr, Sahin M, Ebrahimi-Fakhari D. Tuberous sclerosis complex. Pediatr Clin North Am 2015; 62: 633–48. [DOI] [PubMed] [Google Scholar]
- Dionisi Vici C, Sabetta G, Gambarara M, Vigevano F, Bertini E, Boldrini R, et al. Agenesis of the corpus callosum, combined immunodeficiency, bilateral cataract, and hypopigmentation in two brothers. Am J Med Genet 1988; 29: 1–8. [DOI] [PubMed] [Google Scholar]
- Duarte-Silva S, Neves-Carvalho A, Soares-Cunha C, Teixeira-Castro A, Oliveira P, Silva-Fernandes A, et al. Lithium chloride therapy fails to improve motor function in a transgenic mouse model of Machado-Joseph disease. Cerebellum 2014; 13: 713–27. [DOI] [PubMed] [Google Scholar]
- Ebert AD, Yu J, Rose FF, Jr., Mattis VB, Lorson CL, Thomson JA, et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009; 457: 277–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, Rockenstein E, Masliah E, Hyman BT, et al. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J Neurosci 2011; 31: 14508–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, Sahin M. Autism and the synapse: emerging mechanisms and mechanism-based therapies. Curr Opin Neurol 2015; 28: 91–102. [DOI] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, Wahlster L, Hoffmann GF, Kolker S. Emerging role of autophagy in pediatric neurodegenerative and neurometabolic diseases. Pediatr Res 2014; 75: 217–26. [DOI] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, Wahlster L, McLean PJ. Protein degradation pathways in Parkinson's disease: curse or blessing. Acta Neuropathol 2012; 124: 153–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature 2015; 517: 302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331: 456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmke N, Parvaneh N, Krawitz P, Ashrafi MR, Karimi P, Mehdizadeh M, et al. First description of a patient with Vici syndrome due to a mutation affecting the penultimate exon of EPG5 and review of the literature. Am J Med Genet A 2014; 164A: 3170–5. [DOI] [PubMed] [Google Scholar]
- Elrick MJ, Yu T, Chung C, Lieberman AP. Impaired proteolysis underlies autophagic dysfunction in Niemann-Pick type C disease. Hum Mol Genet 2012; 21: 4876–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 2014; 24: 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filloux FM, Hoffman RO, Viskochil DH, Jungbluth H, Creel DJ. Ophthalmologic Features of Vici Syndrome. J Pediatr Ophthalmol Strabismus 2014; 51: 214–20. [DOI] [PubMed] [Google Scholar]
- Finocchi A, Angelino G, Cantarutti N, Corbari M, Bevivino E, Cascioli S, et al. Immunodeficiency in Vici syndrome: a heterogeneous phenotype. Am J Med Genet A 2012; 158A: 434–9. [DOI] [PubMed] [Google Scholar]
- Fornai F, Longone P, Cafaro L, Kastsiuchenka O, Ferrucci M, Manca ML, et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 2008; 105: 2052–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol 2010; 20: 355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J 2015; 34: 856–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284: 12297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goizet C, Boukhris A, Maltete D, Guyant-Marechal L, Truchetto J, Mundwiller E, et al. SPG15 is the second most common cause of hereditary spastic paraplegia with thin corpus callosum. Neurology 2009; 73: 1111–9. [DOI] [PubMed] [Google Scholar]
- Gomes LC, Dikic I. Autophagy in antimicrobial immunity. Mol Cell 2014; 54: 224–33. [DOI] [PubMed] [Google Scholar]
- Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014; 157: 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack TB, Hogarth P, Kruer MC, Gregory A, Wieland T, Schwarzmayr T, et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet 2012; 91: 1144–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanein S, Martin E, Boukhris A, Byrne P, Goizet C, Hamri A, et al. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am J Hum Genet 2008; 82: 992–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441: 885–9. [DOI] [PubMed] [Google Scholar]
- Hayflick SJ, Kruer MC, Gregory A, Haack TB, Kurian MA, Houlden HH, et al. beta-Propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain 2013; 136(Pt 6): 1708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Koprich JB, Wang Y, Yu WB, Xiao BG, Brotchie JM, et al. Treatment with trehalose prevents behavioral and neurochemical deficits produced in an AAV alpha-synuclein rat model of Parkinson's disease. Mol Neurobiol 2015. [DOI] [PubMed] [Google Scholar]
- Hebron ML, Lonskaya I, Moussa CE. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson's disease models. Hum Mol Genet 2013; 22: 3315–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 2010; 329: 229–32. [DOI] [PubMed] [Google Scholar]
- Horvath R. Brain iron takes off: a new propeller protein links neurodegeneration with autophagy. Brain 2013; 136(Pt 6): 1687–91. [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20: 1981–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol 2014; 12: 101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CA, Byrne PC, Webb S, McMonagle P, Patterson V, Hutchinson M, et al. SPG15, a new locus for autosomal recessive complicated HSP on chromosome 14q. Neurology 2001; 56: 1230–3. [DOI] [PubMed] [Google Scholar]
- Ichinose Y, Miwa M, Onohara A, Obi K, Shindo K, Saitsu H, et al. Characteristic MRI findings in beta-propeller protein-associated neurodegeneration (BPAN). Neurol Clin Pract 2014; 4: 175–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19: 5360–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Yu JT, Zhu XC, Zhang QQ, Cao L, Wang HF, et al. Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology 2014; 85: 121–30. [DOI] [PubMed] [Google Scholar]
- Jo EK, Yuk JM, Shin DM, Sasakawa C. Roles of autophagy in elimination of intracellular bacterial pathogens. Front Immunol 2013; 4: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julich K, Sahin M. Mechanism-based treatment in tuberous sclerosis complex. Pediatr Neurol 2014; 50: 290–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20: 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18: 571–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 2012; 22: 407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khundadze M, Kollmann K, Koch N, Biskup C, Nietzsche S, Zimmer G, et al. A hereditary spastic paraplegia mouse model supports a role of ZFYVE26/SPASTIZIN for the endolysosomal system. PLoS Genet 2013; 9: e1003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidane TZ, Sauble E, Linder MC. Release of iron from ferritin requires lysosomal activity. Am J Physiol Cell Physiol 2006; 291: C445–55. [DOI] [PubMed] [Google Scholar]
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013; 152: 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13: 132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjellin K. Familial spastic paraplegia with amyotrophy, oligophrenia, and central retinal degeneration. Arch Neurol 1959; 1: 133–40. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8: 445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Baehrecke EH, Brumell JH, Chu CT, Codogno P, Cuervo AM, et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 2011; 7: 1273–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klucken J, Poehler AM, Ebrahimi-Fakhari D, Schneider J, Nuber S, Rockenstein E, et al. Alpha-synuclein aggregation involves a bafilomycin A 1-sensitive autophagy pathway. Autophagy 2012; 8: 754–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441: 880–4. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007a; 131: 1149–63. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169: 425–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr, Iwata J, Kominami E, et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci USA 2007b; 104: 14489–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruer MC, Boddaert N, Schneider SA, Houlden H, Bhatia KP, Gregory A, et al. Neuroimaging features of neurodegeneration with brain iron accumulation. AJNR Am J Neuroradiol 2012; 33: 407–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432: 1032–6. [DOI] [PubMed] [Google Scholar]
- Kutateladze T, Overduin M. Structural mechanism of endosome docking by the FYVE domain. Science 2001; 291: 1793–6. [DOI] [PubMed] [Google Scholar]
- Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 2013; 14: 759–74. [DOI] [PubMed] [Google Scholar]
- Lavandero S, Troncoso R, Rothermel BA, Martinet W, Sadoshima J, Hill JA. Cardiovascular autophagy: Concepts, controversies, and perspectives. Autophagy 2013; 9: 1455–66. [DOI] [PubMed] [Google Scholar]
- Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 2009; 461: 402–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhang S, Zhang X, Li T, Tang Y, Liu H, et al. Autophagy enhancer carbamazepine alleviates memory deficits and cerebral amyloid-beta pathology in a mouse model of Alzheimer's disease. Curr Alzheimer Res 2013; 10: 433–41. [DOI] [PubMed] [Google Scholar]
- Li Y, Guo Y, Wang X, Yu X, Duan W, Hong K, et al. Trehalose decreases mutant SOD1 expression and alleviates motor deficiency in early but not end-stage amyotrophic lateral sclerosis in a SOD1-G93A mouse model. Neuroscience 2015; 298: 12–25. [DOI] [PubMed] [Google Scholar]
- Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol 2006; 8: 688–99. [DOI] [PubMed] [Google Scholar]
- Liang N, Zhang C, Dill P, Panasyuk G, Pion D, Koka V, et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J Exp Med 2014; 211: 2249–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy 2012; 8: 719–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton JO, Sahin M. The neurology of mTOR. Neuron 2014; 84: 275–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Czaja MJ. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ 2013; 20: 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Giudice T, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A. Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms. Exp Neurol 2014; 261: 518–39. [DOI] [PubMed] [Google Scholar]
- Lonskaya I, Hebron ML, Desforges NM, Schachter JB, Moussa CE. Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J Mol Med (Berl) 2014; 92: 373–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Yang P, Huang X, Hu W, Guo B, Wu F, et al. The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev Cell 2011; 21: 343–57. [DOI] [PubMed] [Google Scholar]
- Maday S, Holzbaur EL. Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell 2014; 30: 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Wallace KE, Holzbaur EL. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol 2012; 196: 407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maetzel D, Sarkar S, Wang H, Abi-Mosleh L, Xu P, Cheng AW, et al. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Reports 2014; 2: 866–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One 2011; 6: e25416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014; 509: 105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010; 143: 527–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, Reggiori F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol 2010; 190: 1005–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Yanicostas C, Rastetter A, Naini SM, Maouedj A, Kabashi E, et al. Spatacsin and spastizin act in the same pathway required for proper spinal motor neuron axon outgrowth in zebrafish. Neurobiol Dis 2012; 48: 299–308. [DOI] [PubMed] [Google Scholar]
- Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012; 8: 903–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Murillo F, Kobayashi H, Pegoraro E, Galluzzi G, Creel G, Mariani C, et al. Genetic localization of a new locus for recessive familial spastic paraparesis to 15q13-15. Neurology 1999; 53: 50–6. [DOI] [PubMed] [Google Scholar]
- Mas C, Norwood SJ, Bugarcic A, Kinna G, Leneva N, Kovtun O, et al. Structural basis for different phosphoinositide specificities of the PX domains of sorting nexins regulating G-protein signaling. J Biol Chem 2014; 289: 28554–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009; 11: 385–96. [DOI] [PubMed] [Google Scholar]
- McClelland V, Cullup T, Bodi I, Ruddy D, Buj-Bello A, Biancalana V, et al. Vici syndrome associated with sensorineural hearing loss and evidence of neuromuscular involvement on muscle biopsy. Am J Med Genet A 2010; 152A: 741–7. [DOI] [PubMed] [Google Scholar]
- McMahon J, Huang X, Yang J, Komatsu M, Yue Z, Qian J, et al. Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J Neurosci 2012; 32: 15704–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies FM, Huebener J, Renna M, Bonin M, Riess O, Rubinsztein DC. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 2010; 133(Pt 1): 93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer E, Kurian MA, Hayflick SJ. Neurodegeneration with brain iron accumulation: genetic diversity and pathophysiological mechanisms. Annu Rev Genomics Hum Genet 2015; 16:257–79. [DOI] [PubMed] [Google Scholar]
- Miyata R, Hayashi M, Sato H, Sugawara Y, Yui T, Araki S, et al. Sibling cases of Vici syndrome: sleep abnormalities and complications of renal tubular acidosis. Am J Med Genet A 2007; 143: 189–94. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27: 107–32. [DOI] [PubMed] [Google Scholar]
- Murmu RP, Martin E, Rastetter A, Esteves T, Muriel MP, El Hachimi KH, et al. Cellular distribution and subcellular localization of spatacsin and spastizin, two proteins involved in hereditary spastic paraplegia. Mol Cell Neurosci 2011; 47: 191–202. [DOI] [PubMed] [Google Scholar]
- Nemeth AH, Kwasniewska AC, Lise S, Parolin Schnekenberg R, Becker EB, Bera KD, et al. Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model. Brain 2013; 136(Pt 10): 3106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K, Oyama G, Yoshino H, Li Y, Matsushima T, Takeuchi C, et al. High frequency of beta-propeller protein-associated neurodegeneration (BPAN) among patients with intellectual disability and young-onset parkinsonism. Neurobiol Aging 2015; 36: 2004.e9–15. [DOI] [PubMed] [Google Scholar]
- Nishiyama J, Miura E, Mizushima N, Watanabe M, Yuzaki M. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy 2007; 3: 591–6. [DOI] [PubMed] [Google Scholar]
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med 2013; 19: 983–97. [DOI] [PubMed] [Google Scholar]
- Noreau A, Dion PA, Rouleau GA. Molecular aspects of hereditary spastic paraplegia. Exp Cell Res 2014; 325: 18–26. [DOI] [PubMed] [Google Scholar]
- Ohba C, Nabatame S, Iijima Y, Nishiyama K, Tsurusaki Y, Nakashima M, et al. De novo WDR45 mutation in a patient showing clinically Rett syndrome with childhood iron deposition in brain. J Hum Genet 2014; 59: 292–5. [DOI] [PubMed] [Google Scholar]
- Okamoto K. Organellophagy: Eliminating cellular building blocks via selective autophagy. J Cell Biol 2014; 205: 435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oku M, Sakai Y. Peroxisomes as dynamic organelles: autophagic degradation. FEBS J 2010; 277: 3289–94. [DOI] [PubMed] [Google Scholar]
- Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 2012; 23: 1860–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz-Levi D, Ben-Zeev B, Ruzzo EK, Hitomi Y, Gelman A, Pelak K, et al. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am J Hum Genet 2012; 91: 1065–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Koide R, Nakata Y, Saitsu H, Matsumoto N, Takahashi K, et al. A novel WDR45 mutation in a patient with static encephalopathy of childhood with neurodegeneration in adulthood (SENDA). Am J Med Genet A 2014; 164a: 2388–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkale M, Erol I, Gumus A, Ozkale Y, Alehan F. Vici syndrome associated with sensorineural hearing loss and laryngomalacia. Pediatr Neurol 2012; 47: 375–8. [DOI] [PubMed] [Google Scholar]
- Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 2011; 20: 3852–66. [DOI] [PubMed] [Google Scholar]
- Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain 2008a; 131(Pt 8): 1969–78. [DOI] [PubMed] [Google Scholar]
- Pan T, Kondo S, Zhu W, Xie W, Jankovic J, Le W. Neuroprotection of rapamycin in lactacystin-induced neurodegeneration via autophagy enhancement. Neurobiol Dis 2008b; 32: 16–25. [DOI] [PubMed] [Google Scholar]
- Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A, et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell 2014; 53: 471–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, et al. Disease-specific induced pluripotent stem cells. Cell 2008; 134: 877–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore N, Blomenkamp K, Annunziata F, Piccolo P, Mithbaokar P, Maria Sepe R, et al. Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Mol Med 2013; 5: 397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paudel R, Li A, Wiethoff S, Bandopadhyay R, Bhatia K, de Silva R, et al. Neuropathology of Beta-propeller protein associated neurodegeneration (BPAN): a new tauopathy. Acta Neuropathol Commun 2015; 3: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensato V, Castellotti B, Gellera C, Pareyson D, Ciano C, Nanetti L, et al. Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain 2014; 137(Pt 7): 1907–20. [DOI] [PubMed] [Google Scholar]
- Platt FM. Sphingolipid lysosomal storage disorders. Nature 2014; 510: 68–75. [DOI] [PubMed] [Google Scholar]
- Proikas-Cezanne T, Waddell S, Gaugel A, Frickey T, Lupas A, Nordheim A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 2004; 23: 9314–25. [DOI] [PubMed] [Google Scholar]
- Pyle A, Smertenko T, Bargiela D, Griffin H, Duff J, Appleton M, et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain 2015; 138(Pt 2): 276–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathore GS, Schaaf CP, Stocco AJ. Novel mutation of the WDR45 gene causing beta-propeller protein-associated neurodegeneration. Mov Disord 2014; 29: 574–5. [DOI] [PubMed] [Google Scholar]
- Ravenscroft G, Laing NG, Bonnemann CG. Pathophysiological concepts in the congenital myopathies: blurring the boundaries, sharpening the focus. Brain 2015; 138(Pt 2): 246–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90: 1383–435. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36: 585–95. [DOI] [PubMed] [Google Scholar]
- Renvoise B, Chang J, Singh R, Yonekawa S, FitzGibbon EJ, Mankodi A, et al. Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11. Ann Clin Transl Neurol 2014; 1: 379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 2012; 5: ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Navarro JA, Rodriguez L, Casarejos MJ, Solano RM, Gomez A, Perucho J, et al. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis 2010; 39: 423–38. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Aufmuth B, Monesson S. Vici syndrome: a rare autosomal recessive syndrome with brain anomalies, cardiomyopathy, and severe intellectual disability. Case Rep Genet 2011; 2011: 421582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose C, Menzies FM, Renna M, Acevedo-Arozena A, Corrochano S, Sadiq O, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington's disease. Hum Mol Genet 2010; 19: 2144–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11: 709–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013; 15: 741–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said E, Soler D, Sewry C. Vici syndrome–a rapidly progressive neurodegenerative disorder with hypopigmentation, immunodeficiency and myopathic changes on muscle biopsy. Am J Med Genet A 2012; 158A: 440–4. [DOI] [PubMed] [Google Scholar]
- Saitsu H, Nishimura T, Muramatsu K, Kodera H, Kumada S, Sugai K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet 2013; 45: 445–9, 449e1. [DOI] [PubMed] [Google Scholar]
- Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol 2008; 7: 1127–38. [DOI] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science 2009; 325: 473–7. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 2007a; 282: 5641–52. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005; 170: 1101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Maetzel D, Korolchuk VI, Jaenisch R. Restarting stalled autophagy a potential therapeutic approach for the lipid storage disorder, Niemann-Pick type C1 disease. Autophagy 2014; 10: 1137–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, et al. Small molecules enhance autophagy and reduce toxicity in Huntington's disease models. Nat Chem Biol 2007b; 3: 331–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer V, Lavenir I, Ozcelik S, Tolnay M, Winkler DT, Goedert M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain 2012; 135(Pt 7): 2169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schule R, Schlipf N, Synofzik M, Klebe S, Klimpe S, Hehr U, et al. Frequency and phenotype of SPG11 and SPG15 in complicated hereditary spastic paraplegia. J Neurol Neurosurg Psychiatry 2009; 80: 1402–4. [DOI] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332: 1429–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacka JJ, Klocke BJ, Shibata M, Uchiyama Y, Datta G, Schmidt RE, et al. Bafilomycin A1 inhibits chloroquine-induced death of cerebellar granule neurons. Mol Pharmacol 2006; 69: 1125–36. [DOI] [PubMed] [Google Scholar]
- Shibasaki Y, Tanaka H, Iwabuchi K, Kawasaki S, Kondo H, Uekawa K, et al. Linkage of autosomal recessive hereditary spastic paraplegia with mental impairment and thin corpus callosum to chromosome 15A13-15. Ann Neurol 2000; 48: 108–12. [PubMed] [Google Scholar]
- Shimada K, Motoi Y, Ishiguro K, Kambe T, Matsumoto SE, Itaya M, et al. Long-term oral lithium treatment attenuates motor disturbance in tauopathy model mice: implications of autophagy promotion. Neurobiol Dis 2012; 46: 101–8. [DOI] [PubMed] [Google Scholar]
- Shu XH, Wang LL, Li H, Song X, Shi S, Gu JY, et al. Diffusion efficiency and bioavailability of resveratrol administered to rat brain by different routes: therapeutic implications. Neurotherapeutics 2015; 12: 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slabicki M, Theis M, Krastev DB, Samsonov S, Mundwiller E, Junqueira M, et al. A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol 2010; 8: e1000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa SB, Ramos F, Garcia P, Pais RP, Paiva C, Beales PL, et al. Intellectual disability, coarse face, relative macrocephaly, and cerebellar hypotrophy in two sisters. Am J Med Genet A 2014; 164A: 10–4. [DOI] [PubMed] [Google Scholar]
- Spampanato C, Feeney E, Li L, Cardone M, Lim JA, Annunziata F, et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med 2013; 5: 691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadel D, Millarte V, Tillmann KD, Huber J, Tamin-Yecheskel BC, Akutsu M, et al. TECPR2 cooperates with LC3C to regulate COPII-dependent ER export. Mol Cell 2015; 60: 89–104. [DOI] [PubMed] [Google Scholar]
- Steele JW, Ju S, Lachenmayer ML, Liken J, Stock A, Kim SH, et al. Latrepirdine stimulates autophagy and reduces accumulation of alpha-synuclein in cells and in mouse brain. Mol Psychiatry 2013a; 18: 882–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele JW, Lachenmayer ML, Ju S, Stock A, Liken J, Kim SH, et al. Latrepirdine improves cognition and arrests progression of neuropathology in an Alzheimer's mouse model. Mol Psychiatry 2013b; 18: 889–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevanin G, Azzedine H, Denora P, Boukhris A, Tazir M, Lossos A, et al. Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain 2008; 131(Pt 3): 772–84. [DOI] [PubMed] [Google Scholar]
- Stevanin G, Santorelli FM, Azzedine H, Coutinho P, Chomilier J, Denora PS, et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet 2007; 39: 366–72. [DOI] [PubMed] [Google Scholar]
- Stirnimann CU, Petsalaki E, Russell RB, Muller CW. WD40 proteins propel cellular networks. Trends Biochem Sci 2010; 35: 565–74. [DOI] [PubMed] [Google Scholar]
- Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 2014; 16: 495–501. [DOI] [PubMed] [Google Scholar]
- Suzuki K. Selective autophagy in budding yeast. Cell Death Differ 2013; 20: 43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014; 83: 1131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdemir S, Sahin I, Cayir A, Yuce I, Ceylaner S, Tatar A. Vici syndrome in siblings born to consanguineous parents. Am J Med Genet A 2015. [DOI] [PubMed] [Google Scholar]
- Thomas AC, Williams H, Seto-Salvia N, Bacchelli C, Jenkins D, O'Sullivan M, et al. Mutations in SNX14 cause a distinctive autosomal-recessive cerebellar ataxia and intellectual disability syndrome. Am J Hum Genet 2014; 95: 611–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Bustos V, Flajolet M, Greengard P. A small-molecule enhancer of autophagy decreases levels of Abeta and APP-CTF via Atg5-dependent autophagy pathway. FASEB J 2011; 25: 1934–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 2010; 141: 1042–55. [DOI] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012; 488: 647–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, et al. PGC-1alpha rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med 2012; 4: 142ra97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goethem G, Livingston JH, Warren D, Oojageer AJ, Rice GI, Crow YJ. Basal ganglia calcification in a patient with beta-propeller protein-associated neurodegeneration. Pediatr Neurol 2014; 51: 843–5. [DOI] [PubMed] [Google Scholar]
- Vantaggiato C, Clementi E, Bassi MT. ZFYVE26/SPASTIZIN: a close link between complicated hereditary spastic paraparesis and autophagy. Autophagy 2014; 10: 374–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vantaggiato C, Crimella C, Airoldi G, Polishchuk R, Bonato S, Brighina E, et al. Defective autophagy in spastizin mutated patients with hereditary spastic paraparesis type 15. Brain 2013; 136(Pt 10): 3119–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga RE, Khundadze M, Damme M, Nietzsche S, Hoffmann B, Stauber T, et al. In Vivo evidence for lysosome depletion and impaired autophagic clearance in hereditary spastic paraplegia type SPG11. PLoS Genet 2015; 11: e1005454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven WM, Egger JI, Koolen DA, Yntema H, Olgiati S, Breedveld GJ, et al. Beta-propeller protein-associated neurodegeneration (BPAN), a rare form of NBIA: novel mutations and neuropsychiatric phenotype in three adult patients. Parkinsonism Relat Disord 2014; 20: 332–6. [DOI] [PubMed] [Google Scholar]
- Vicinanza M, Korolchuk VI, Ashkenazi A, Puri C, Menzies FM, Clarke JH, et al. PI(5)P regulates autophagosome biogenesis. Mol Cell 2015; 57: 219–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem 2010; 285: 9100–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscomi MT, D'Amelio M, Cavallucci V, Latini L, Bisicchia E, Nazio F, et al. Stimulation of autophagy by rapamycin protects neurons from remote degeneration after acute focal brain damage. Autophagy 2012; 8: 222–35. [DOI] [PubMed] [Google Scholar]
- Vite CH, Bagel JH, Swain GP, Prociuk M, Sikora TU, Stein VM, et al. Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease. Sci Transl Med. 2015;7:276ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodicka P, Lim J, Williams DT, Kegel KB, Chase K, Park H, et al. Assessment of chloroquine treatment for modulating autophagy flux in brain of WT and HD mice. J Huntingtons Dis 2014; 3: 159–74. [DOI] [PubMed] [Google Scholar]
- Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ, et al. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci USA 2012a; 109: 15024–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012b; 338: 956–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 2003; 278: 25009–13. [DOI] [PubMed] [Google Scholar]
- Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 2008; 4: 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PM, Puente C, Ganley IG, Jiang X. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 2013; 9: 124–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurzer B, Zaffagnini G, Fracchiolla D, Turco E, Abert C, Romanov J, et al. Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. Elife 2015; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong N, Jia M, Chen C, Xiong J, Zhang Z, Huang J, et al. Potential autophagy enhancers attenuate rotenone-induced toxicity in SH-SY5Y. Neuroscience 2011; 199: 292–302. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Simonsen A. The elimination of accumulated and aggregated proteins: a role for aggrephagy in neurodegeneration. Neurobiol Dis 2011; 43: 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, et al. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol 2012; 198: 219–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasin SA, Ali AM, Tata M, Picker SR, Anderson GW, Latimer-Bowman E, et al. mTOR-dependent abnormalities in autophagy characterize human malformations of cortical development: evidence from focal cortical dysplasia and tuberous sclerosis. Acta Neuropathol 2013; 126: 207–18. [DOI] [PubMed] [Google Scholar]
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010; 465: 942–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen S, Song L, Tang Y, Shen Y, Jia L, et al. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 2014; 10: 588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Zhao YG, Wang X, Xu L, Miao L, Feng D, et al. Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J Cell Biol 2013a; 200: 731–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YG, Sun L, Miao G, Ji C, Zhao H, Sun H, et al. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy 2015; 11: 881–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YG, Zhao H, Sun H, Zhang H. Role of Epg5 in selective neurodegeneration and Vici syndrome. Autophagy 2013b; 9: 1258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 2009; 11: 468–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.