

Abstract
Lytic transglycosylases (LTs) catalyze the non-hydrolytic cleavage of the bacterial cell wall by an intramolecular transacetalization reaction. This reaction is critically and broadly important in modifications of the bacterial cell wall in the course of its biosynthesis, recycling, manifestation of virulence, insertion of structural entities such as the flagellum and the pili, among others. The first QM/MM analysis of the mechanism of reaction of an LT, that for the Escherichia coli MltE, is undertaken. The study reveals a conformational itinerary consistent with an oxocarbenium-like transition state, characterized by a pivotal role for the active-site glutamic acid in proton transfer. Notably, an oxazolinium intermediate, as a potential intermediate, is absent. Rather, substrate-assisted catalysis is observed through a favorable dipole provided by the N-acetyl carbonyl group of MurNAc saccharide. This interaction stabilizes the incipient positive charge development in the transition state. This mechanism coincides with near-synchronous acetal cleavage and acetal formation.
Introduction
The lysozyme family of the glycoside hydrolases (GHs) catalyzes the cleavage of the β-1 → 4-glycosidic linkage connecting the N-acetylmuramic acid (MurNAc) and N-acetyl-d-glucosamine (GlcNAc) saccharides of the (MurNAc-GlcNAc)n polymer (the peptidoglycan) of the cell wall of bacteria. While the non-bacterial lysozymes themselves are hydrolytic catalysts, the lytic transglycosylase (LT) sub-families of the GHs are not1,2. The LTs act on the same MurNAc-β-1 → 4-GlcNAc glycosidic linkage of the peptidoglycan to accomplish a non-hydrolytic scission so as to create two daughter strands having (respectively) 1,6-anhydroMurNAc and GlcNAc termini (Fig. 1)3–5. LT catalysis is used by bacteria for a host of functional transformations, including peptidoglycan biosynthesis, remodeling, recycling, and excavation for insertion of secretion systems and of flagella and pili. Evidence also correlates LT dysregulation to the bactericidal mechanism of the β-lactam antibiotics6. The range of cell-wall processes that the LT reactions enable is impressive. This first QM/MM analysis of the LT reaction was undertaken to shed light on this unique transformation, critical for homeostasis of the bacterial cell wall.
Figure 1.

Proposed mechanism for lytic transglycosylases. The catalytic Glu/Asp acts initially as a general acid, donating its proton to the glycosidic oxygen of the scissile bond. The developing positive charge in the oxocarbenium transition state (Ts I) may be stabilized through the formation of a putative oxazolinium intermediate involving the N-acetyl group of −1 MurNAc. The deprotonated glutamate/aspartate then acts as a general base to activate the C6-OH for intramolecular attack at the anomeric carbon. This process collapses the oxazolinium intermediate with the concomitant formation of the 1,6-anhydroMurNAc reaction product, having all of its substituents in an axial orientation.
The stereochemistry of LT catalysis is overall retention with respect to the anomeric carbon of the MurNAc saccharide. While the origin of the anhydroMurNAc product is that of an intramolecular interception of an oxocarbenium entity, the steps leading to this event are uncertain. One proposed mechanism uses substrate-assistance by forming an oxazolinium intermediate. In this mechanism, the MurNAc amide functional group acts first as a nucleophile and then as a nucleofuge (Fig. 1)7. Oxazolinium intermediates in GlcNAc glycosyl transfer are well recognized8, and invoking this intermediate would account for the retention of configuration by the LTs through a sequence of two half-reactions, each requiring inversion9. This proposed intermediate was also suggested to account for dispersing the charge developed at the anomeric carbon during bond breaking10.
Within the LT sub-family, a glutamic (or aspartic) acid has the pivotal catalytic role. We elected to use QM/MM analysis to elucidate the LT reaction mechanism, using the membrane-bound lytic transglycosylase E (MltE) enzyme of Escherichia coli as our example. The small mass of this protein (approximately 21 kDa), the absence of peripheral domains that may impart influence, and the availability of quality crystal structures for MltE made it the appropriate choice for this study. MltE is a lipoprotein catalyst involved in the late stages of type VI secretion system assembly11. MltE is also the primary endolytic LT (i.e., cleavage in the middle of a peptidoglycan strand) of E. coli12,13. Its QM/MM study was anticipated to provide the first insights into the enigmatic mechanism of the LT enzymes.
We analyzed the molecular-dynamics production-phase trajectory of the complex and selected several snapshots that had suitable distances (d3, d4, and d5) for the proton transfer events (Fig. 2 and Supplementary Fig. 1). One of the snapshots with appropriate distance parameters after MM optimizations was selected for the QM/MM calculations (see Supplementary Computational Methods for details). The selection of residues for the QM layer (Fig. 2) was made with attention to a comparative sequence analysis of the LT family and available X-ray structures (Supplementary Figs 2–5). Residue E64 (the catalytic glutamic acid); the side chains of S73, S75, and Y192; and the two active-site water molecules (Wat1 and Wat2) were carefully selected. The Michaelis complex (Fig. 3a and Supplementary Fig. 6) was obtained following two-layer ONIOM14 QM/MM energy minimization method. The dominant features of the Michaelis complex are the hydrogen-bonding pattern of E64 and the E1 conformation of the −1 MurNAc saccharide (see Supplementary Information for details: the hexose conformers are described as boat (B), chair (C), envelope (E, previously “sofa”), half-chair (H, sometimes called half-boat or twist), and skew (S, sometimes called twist-boat) conformations (Fig. 4), according to the Cremer and Pople nomenclature)15–17. The competence of this Michaelis complex was tested for its ability to traverse the full reaction coordinate across a 1D potential-energy surface (PES) scan (see Supplementary Information for details). Coordinates obtained from the 1D PES provided the requisite starting points for two subsequent 2D-PES calculations of the key reaction steps. The first 2D-PES scan corresponds to the formation of the local energy minimum (II2D), and the second scan to the formation of 1,6-anhydroMurNAc in a B3,O conformation (III2D) (Fig. 3).
Figure 2.
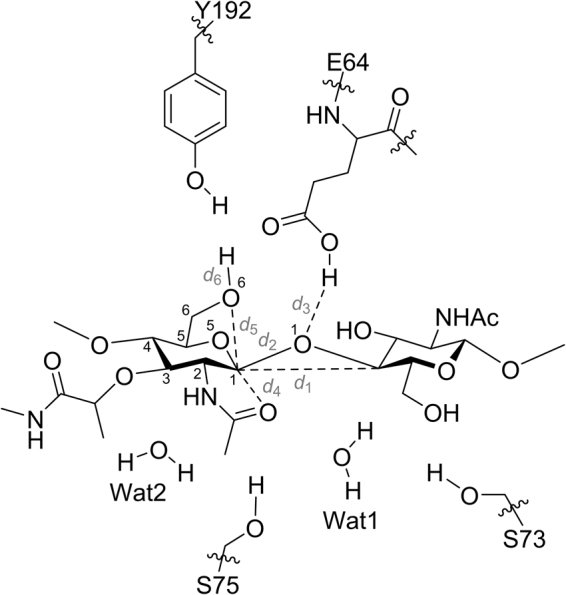
Atoms included in the QM layer and the reaction parameters. The QM layer includes 123 atoms: the MurNAc-GlcNAc substrate; residue E64; the side chains of S73, S75, and Y192; and the two active-site water molecules (Wat1 and Wat2).
Figure 3.

MltE transformation. (a) Stereo representation of the Michaelis complex I. (b) The potential-energy surface with respect to the d2 and d3 reaction coordinates. (c) Intermediate II2D. (d) Transition species Ts22D between II2D and III2D. (e) The reaction path (orange arrows) from II2D through Ts22D to III2D. (f,g) 1,6-anhydroMurNAc in the B3,O conformation (III2D and IIIQM). (h) The QM potential-energy surface for B3,O to 1C4 conformational change. (i) 1,6-anhydroMurNAc in the 1C4 conformation (IVQM). Hydrogen bonds and the d2 are shown as blue and red dashed lines, respectively.
Figure 4.

Conformational itinerary of −1 MurNAc along MltE transformation. I denotes the Michaelis complex; Ts12D, the transition species between I and II2D; II2D, the intermediate; Ts22D, the transition species between II2D and III2D; III2D and IIIQM, the 1,6-anhydroMurNAc in the B3,O conformation; Ts3QM, the transition species between IIIQM and IVQM; IVQM, the 1,6-anhydroMurNAc in the 1C4 conformation. A −1 MurNAc adopts boat (B), chair (C), envelope (E), and skew (S) conformations. The B3,O to 1C4 transformation is off-enzyme reaction.
The first 2D-PES scan starts from the Michaelis complex (I) and uses the glycosidic bond (d2, scanned at 0.10 Å intervals from 1.40 to 2.20 Å) and the distance between the glycosidic oxygen (O1) and the Oε2 hydrogen (Hε2) of E64 (d3, scanned at –0.10 Å intervals from 2.50 to 1.00 Å) as the reaction coordinates. The lengthening of the C–O glycosidic bond is accompanied by the approach of hydrogen atom from E64 to O1. In this progression, the MurNAc undergoes an E1 → [E1]⧧ → 2,5B conformational path. The locations of the Michaelis complex I (Fig. 3a and Supplementary Fig. 6: d2 = 1.40 Å, d3 = 2.50 Å) and the local minimum II2D (Fig. 3c and Supplementary Fig. 7: d2 = 1.80 Å, d3 = 1.50 Å, see Supplementary Information for details) are indicated on the 2D PES (Fig. 3b). The arrows show the progression from I to II2D. The energy of II2D is 6.55 kcal·mol−1 higher than that of I. Transition point 1 (Supplementary Fig. 8: species Ts12D at d2 = 1.70 Å and d3 = 2.40 Å) retains the E1 conformation and is 17.09 kcal·mol−1 higher in energy than I.
Species II2D progresses toward the transition species Ts22D, having the fully broken (2.90 Å) glycosidic bond in our second 2D-PES calculation. Continued progression results in the formation of 1,6-anhydroMurNAc in a B3,O conformation (species III2D), as shown in Fig. 3. Proton transfer mediated by the E64 carboxylate acting as a general base directs intramolecular bond formation between the O6 and the anomeric C1 of MurNAc. The reaction coordinates d5 (distance between O6 and C1 of MurNAc, corresponding to bond formation) and d6 (O6–H bond of MurNAc, corresponding to proton transfer) define the path to formation of species III2D (Fig. 2). These distances were scanned at 0.10 Å intervals (d5 from 3.29 to 1.39 Å, and d6 from 0.90 to 2.00 Å). The resulting PES (Fig. 3e) shows the second transition species in a 2SO skew conformation (Fig. 3d: species Ts22D at d5 = 2.29 Å and d6 = 1.00 Å) and the 1,6-anhydroMurNAc in a B3,O conformation (Fig. 3f: species III2D at d5 = 1.49 Å and d6 = 1.80 Å). Progression along this path (arrows) to species III2D at −3.91 kcal·mol−1 is exothermic with respect to the Michaelis complex. The transition species Ts22D, between species II2D and III2D, is 10.39 kcal·mol−1 higher in potential energy than II2D.
The progress from II2D to Ts22D coincides with an increased positive charge on C1 (from 0.451 to 0.522 e), as calculated by natural population atomic charges (Supplementary Table 1). During this progress, the distance between the N-acetyl carbonyl oxygen of MurNAc (ONAc) and the C1 shortens in order to stabilize the incipient positive charge on C1. As d2 increases from 1.80 to 2.90 Å, d4 (the distance between C1 and ONAc) decreases from 3.14 to 2.83 Å. The increase in d2 provides the necessary space between C1 and O1 for the H1 (the hydrogen on the C1) to assume the planar arrangement for the oxocarbenium species in Ts22D. The MurNAc of Ts22D shows a dihedral angle of C5–O5–C1–C2 (37.4°) and an out-of-plane angle of θH1 (10.9°). Additionally, the distance between C1–O5 shortens from 1.35 to 1.28 Å. These changes reflect the oxocarbenium character of species Ts22D.
Species Ts22D has a 2SO conformation, indicating a 2,5B → [2SO]⧧ → B3,O conformational path. The 2SO conformation provides a favorable orientation for the in-line approach of O6 to C1. The O6 of MurNAc has more negative charge than the ONAc (–0.700 e and –0.619 e, respectively). At the same time, the distance of C1 to O6 (2.29 Å) is less than that of C1 to ONAc (2.83 Å). The suitably positioned (now serving as a general base) E64 Oε1 activates the C6 hydroxyl by proton abstraction. Interception of the oxocarbenium by O6 transforms Ts22D to the 1,6-anhydroMurNAc product in a B3,O conformation (III2D).
We note the importance of solvation within the active site for catalysis. In the Ts22D species, Wat1 bridges between the Hγ of S75 and the ONAc via hydrogen bonds (1.83 Å for both, Fig. 3d). In addition, Wat1 forms another hydrogen bond (1.89 Å) with the oxygen of C3 hydroxyl group of GlcNAc. These hydrogen bonds are maintained throughout the transition from species II2D to Ts22D. The location of Wat1 prevents oxazolinium formation. Interestingly, a water molecule poised in a similar location is observed in the X-ray co-crystal structures (PDB IDs: 4HJZ, 1QTE, 1QUT, 1D0K, 5AO7, 3D3D, and 1D9U) of LT enzymes MltE, Slt70, Slt35, SltB3, and bacteriophage endolysin lambda18–23. The failure to observe formation of an oxazolinium intermediate is attributable in part to the proximity of C1 to O1 (a short distance of 1.80 Å). This proximity prevents the H1 from achieving planarity. In addition, PES calculated after computational deletion of Wat1 from the QM layer gave Ts1 at a value of 6.8 kcal·mol−1 higher than that with Wat1 present (23.9 kcal·mol−1 vs 17.1 kcal·mol−1 respectively; Supplementary Fig. 9). Hence, conservation of this active-site water molecule is not merely structural, but also contributes to transition-state stabilization.
The absence of the oxazolinium species from the two PESs likely is linked with the lack of a second carboxylic acid in the MltE active site. In retaining GH enzymes (GH18, 20, 25, 56, 84, 85, and 123) where this second carboxylate is present, the energetic demand for formation of an oxocarbenium intermediate is mitigated by its interception by the proximal acetamide, to form an oxazolinium intermediate8,24–31. In our computational study, however, the oxazolinium is not a local energy minimum. A 2D PES (Supplementary Fig. 10) generated by scanning d4 (the distance between C1 and ONAc) and d5 (the distance between O6 and C1 of MurNAc) as the coordinates further interrogated its structure. We examined the PES to verify that the dipole interaction with ONAc contributes greater stability to the charge development on C1 compared to the bonding represented by an oxazolinium cation (defined by a d4 value of 1.58 Å). None of the species with d4 = 1.58 Å was more stable than species with d4 > 1.58 Å. Formation of the oxazolinium in the absence of energetic compensation by the second catalytic residue is unfavorable due to the charge development on N-acetyl nitrogen of MurNAc. The water molecules, including Wat2, cannot provide sufficient stabilizing effect for the charge development on the nitrogen of the 2-acetamido group of the oxazolinium. We followed up the observation from the calculation with experiments. The oxazolinium moiety is relatively unstable in solution, however the thiazoline (thiazolinium) analog is stable, and its inhibitory activity against GH enzymes that form an oxazolinium intermediate is regarded as a diagnostic of this mechanistic pathway32. We synthesized two MurNAc-based thiazoline derivatives (compounds 1 and 2, Fig. 5). If the oxazolinium species were an enzyme intermediate, one would expect that thiazolines 1 or 2 would provide some degree of inhibition of MltE. At concentrations as high as 1 mM, we observed no inhibition of MltE by either compound (Supplementary Fig. 11).
Figure 5.
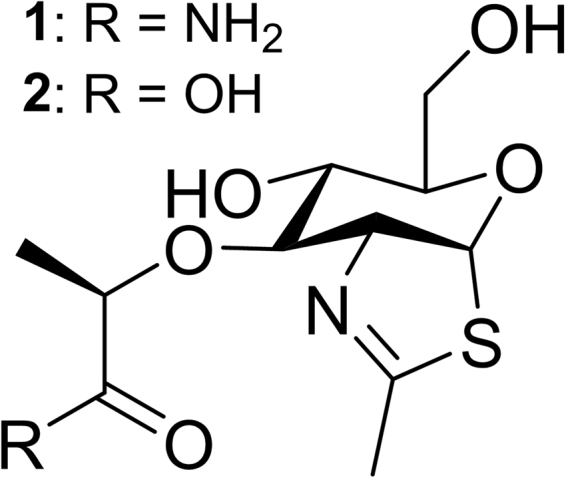
Chemical structures of compounds 1 and 2. The compounds 1 and 2 are MurNAc-based thiazoline derivatives, analogs of the putative oxazolinium intermediate (see Fig. 1).
The final step of the reaction is a boat-to-chair transformation. The ground-state conformation of 1,6-anhydroMurNAc is a 1C4 chair. Before 2D-PES calculations, the relaxation of the B3,O conformation of III2D to the 1C4 conformation first was assessed by a 1D-PES scan of the MurNAc C2–C3–C4–C5 dihedral angle of species III2D. During this QM/MM scan, the energy of the system increased continuously and the potential-energy surface failed to give a local energy minimum. This progressive increase in energy results from a steric clash between the C3 lactyl moiety and the protein surface. The B3,O-to-1C4 transformation cannot occur within the active site, but must occur during the course of (or subsequent to) release of the 1,6-anhydroMurNAc product. Indeed, the favorable energy change of this transformation might be a critical driving force for product release. To gain insight into the energy barrier for this boat-to-chair transformation, a QM 2D-PES scan was conducted for the 1,6-anhydroMurNAc outside of the active site. Two dihedral angles were scanned in this calculation; dihedral C2–C3–C4–C5 (from 63.9 to −46.1° with −5.0° intervals) and dihedral H2–C2–N′–C′ (from −180.0 to 150.0° with 30.0° intervals). The 2D PES shows a potential energy for the B3,O boat that is +2.00 kcal·mol−1 above the 1C4 chair (Fig. 3h). The transition point (Supplementary Fig. 12: species Ts3QM at C2–C3–C4–C5 = −31.1° and H2–C2–N′–C′ = −150.0°) adopts an EO conformation that is +5.50 kcal·mol−1 higher than the B3,O conformation. Interestingly, concomitant rotation occurs about the C2–N′ single bond, with the boat-to-chair transition. This rotation can be attributed to a relieving of the electrostatic repulsions in the 1C4 conformation among the O5, C4 oxygen, and N-acetyl carbonyl oxygen.
GHs bind their carbohydrate substrates in the non-ground-state conformation that optimally positions the exocyclic moiety at the anomeric carbon for departure as a leaving group. Understanding this conformational distortion is recognized as having widespread value for the development of GH inhibitors as antibiotics or potentiators of clinical antibiotics33. Our calculations support existence of such a substrate distortion in MltE. The conformation of the MurNAc in the peptidoglycan in solution is 1C4. On binding to MltE, overall turnover chemistry uses an E1 → [E1]⧧ → 2,5B → [2SO]⧧ → B3,O → [EO]⧧ → 1C4 conformational itinerary (Figs 4 and 6). The initial E1 conformation imposed by MltE on its MurNAc substrate enables access to the transition point 1 (Ts12D) through least motion of the nuclei33. Formation of the intermediate II2D, by lengthening of the C–O glycosidic bond in response to hydrogen bonding by E64 (activation barrier of 17.09 kcal·mol−1) is overall endothermic by 6.55 kcal·mol−1. Subsequent further glycosidic-bond lengthening and proton transfer gives a free-base E64 poised to activate the C6 hydroxyl for interception of the oxocarbenium (Ts22D). The 2SO conformation for Ts22D provides a favorable in-line approach of the nucleophilic O6 oxygen. Intramolecular bond formation between O6 and C1 gives the 1,6-anhydroMurNAc product in a B3,O conformation (species III2D). The transition species Ts22D is 10.39 kcal·mol−1 higher in potential energy relative to II2D, and formation of III2D is exothermic by −3.91 kcal·mol−1. There does not appear to be a unique rate-limiting step, as the two transition-step species are essentially of equal energy (Fig. 6). Relaxation of the B3,O boat to the 1C4 1,6-anhydroMurNAc chair is concurrent with, or subsequent to, product release. Our calculations are consistent with a mechanism of near-synchronous bond formation and bond cleavage, enabled by complementary conformational and electrostatic stabilization. In this particular aspect, the reaction of MltE is an example of the mechanistically challenging front-face retaining glycoside-transferase enzymes9, exhibiting a near-synchronous pathway involving a short-lived oxocarbenium-like species34.
Figure 6.
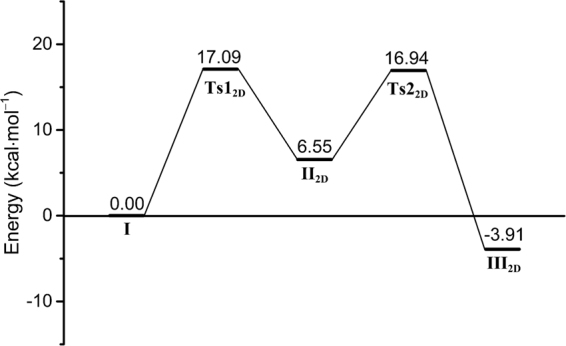
Potential-energy profiles for MltE transformations. I denotes the complex of the substrate bound into the MltE active site; Ts12D, the transition point between species I and II2D; II2D, the intermediate; Ts22D, the transition point between species II2D and III2D; III2D, the 1,6-anhydroMurNAc in the B3,O conformation.
The LT family is implicated in a host of transformations preserving the function and integrity of the bacterial cell wall. This study establishes a mechanistic framework for further interrogation of the critical roles this family has in the biosynthesis, maturation and turnover of this important biopolymer.
Methods
Calculations
Molecular dynamics (MD) simulations used the AMBER 11 suite35. AMBER FF99 and GAFF provided simulation parameters. A snapshot was selected as the starting point, chosen by monitoring the distances and angles from the production phase trajectory, for the design of the QM/MM calculation. The two-layer version of the ONIOM14 method implemented in Gaussian 0936 was used. In a two-layer ONIOM method, the total energy of the system is obtained from three independent calculations: EONIOM = Ereal,MM + Emodel,QM – Emodel,MM, where ‘real’ refer to the whole system and ‘model’ refers to the chemically important part of the system (QM layer). The real system is calculated at MM level. MM method cannot describe bond breaking or formation. The model system is treated with more accurate, but considerably expensive QM method. The QM layer used the B3LYP/6-311++G(d,p)//B3LYP/6-31 G(d) level of theory while the MM layer used the AMBER FF99 force field. The QM layer included 123 atoms: the MurNAc-GlcNAc substrate; the catalytic residue E64; the side chains of S73, S75, and Y192; and the two active-site water molecules (Wat1 and Wat2). Potential-energy points in the QM/MM calculations were generated over a two-dimensional grid of two direct coordinates. All the stationary points (i.e. species I, Ts12D, II2D, Ts22D, III2D, IIIQM, Ts3QM, and IVQM) were fully optimized with no reaction coordinate constraints before characterization by frequency calculations. Frequency calculations were performed with scale factors of 0.873 and 0.944 at B3LYP and M06-2X levels of theory, respectively, at 25 °C and 1 atm. For more details, see Supplementary Computational Methods.
(2R)-2-[[(3aR,5R,6S,7R,7aR)-3a,6,7,7a-tetrahydro-6-hydroxy-5-(hydroxymethyl)-2-methyl-5H-pyrano[3,2-d]thiazol-7-yl]oxy]propanamide (1)
Compound 6 (0.10 g, 0.32 mmol, see Supplementary Information) was dissolved in methanol (4 mL) and 7 N ammonia in methanol was added (3 mL, 21 mmol). The reaction mixture was stirred overnight at RT, similar to a previously reported method)37. The reaction mixture was filtered through a cotton plug and concentrated by rotary evaporation. The titled compound was obtained as a white solid (97 mg, 99%) after vacuum drying: TLC (1:9 MeOH:CH2Cl2): Rf = 0.17. 1H NMR (400 MHz, CD3OD) δ 1.43 (d, J = 6.9 Hz, 3 H), 2.27 (d, J = 2.0 Hz, 3 H) 3.37 (ddd, J = 11.3, 5.6, 2.7 Hz, 1 H), 3.63–3.69 (m, 1 H), 3.69–3.73 (m, 1 H), 3.73–3.80 (m, 1 H), 3.91 (t, J = 4.8 Hz, 1 H), 4.31 (q, J = 6.9 Hz, 1 H), 4.48 (dddq, J approx 7.1, 5.1, 1.0, 2.0 Hz, 1 H), 6.38 (d, J = 7.1 Hz, 1 H); 13C NMR (101 MHz, CD3OD) δ 18.16, 19.38, 61.51, 68.45, 74.95, 75.76, 77.34, 80.44, 89.48, 169.55, 177.90. MS (m/z): [M + H]+, calcd for C11H19N2O5S, 291.1009; found, 291.1023.
(2R)-2-[[(3aR,5R,6S,7R,7aR)-3a,6,7,7a-tetrahydro-6-hydroxy-5-(hydroxymethyl)-2-methyl-5H-pyrano[3,2-d]thiazol-7-yl]oxy]propanoic acid (2)
Compound 6 (0.10 g, 0.32 mmol) was dissolved in 1:1 THF:water (2 mL). Solid LiOH monohydrate (15 mg, 0.36 mmol) was added. The mixture was stirred for 2 h at RT. The solution was filtered using a cotton plug and concentrated. The solid was vacuum dried to give (100 mg, 99%) of an off-white solid: 1H NMR (400 MHz, CD3OD) δ 1.35 (d, J = 6.9 Hz, 3 H), 2.25 (d, J = 2.5 Hz, 3 H), 3.08–3.21 (m, 1 H), 3.54 (dd, J = 12.0, 6.4 Hz, 1 H), 3.63–3.76 (m, 2 H), 4.04–4.19 (m, 2 H), 4.67 (dddq, J approx. 7.1, 5.1, 1.0, 2.0 Hz, 1 H), 6.34 (d, J = 7.1 Hz, 1 H); 13C NMR (101 MHz, CD3OD) δ 18.61, 18.80, 62.49, 68.16, 73.98, 76.12, 77.76, 78.80, 88.84, 168.73, 180.25. MS (m/z): [M + H]+, calcd for C11H17LiNO6S, 298.0931; found, 298.0915.
Cloning and purification of MltE wild-type
The cloning and purification of MltE from E. coli K12 substrain MG1655 was previously reported by our lab38. MltE wild-type was cloned into pET-24a(+) vector (Novagen) using restriction enzyme NdeI to XhoI. The gene encodes for residues 19–203 of MltE, an N-terminal methionine, and a non-cleavable C-terminal LEHHHHHH (membrane anchor and signal peptide removed; residues 1–18 of MltE). The wild-type MltE was expressed and purified as previously reported. The final concentration of the MltE wild-type was determined by a BCA (Bicinchoninic Acid) Protein Assay kit (Pierce). The final yield of the purification was approximately 56 mg of protein per 0.5 L of liquid culture. The proteins were stored at –80 °C and after thawing on ice, no precipitate formed.
Lytic transglycosylase activity assay
The E. coli MltE fluorescence activity assays were conducted using a EnzChek® Lysozyme Assay Kit (Invitrogen). The kit includes fluorescein-labeled sacculus (cell wall) from the Gram-positive bacteria Micrococcus lysodeikticus. Sacculus of Micrococcus species is commonly used in the analysis of LT activity, as it is commercially available and provides a high-level of reactivity with LTs18. MltE reactions (100 μL) were prepared by incubation of 50 μL of succulus (substrate at a 1X dilution in 100 μL, as described in the kit) and 50 μL of MltE (final protein concentration 8 μM). Immediately after mixing, the change in fluorescent intensity was monitored for 30 minutes at room temperature on a Cary Eclipse Fluorescence Spectrophotometer (Agilent). Prior to the experiment, the protein was buffer exchanged into 100 mM NaPO4, pH 7.5 supplemented with 100 mM NaCl using a Zeba Desalting Column (Thermo Fisher Scientific). Reactions containing compound 1 or 2 were incubated on ice for 20 min in the presence of MltE, prior to incubation with the sacculus at the start of the reaction. Fluorescence readings were obtained at an excitation wavelength of 485 nm and an emission wavelength of 516 nm. The results are displayed in Supplementary Fig. 11.
Electronic supplementary material
Acknowledgements
This work was supported by grant GM61629 from the USA National Institutes of Health (to SM). DAD was supported by NIH Training Grant T32GM075762 and by the ECK Institute for Global Health. ES is the recipient of a Berry Family Foundation Graduate Fellowship in Advanced Diagnostics & Therapeutics.
Author Contributions
B.B., K.V.M. and M.K. performed the calculations. D.R.M. and E.S. synthesized the compounds 1 and 2 and carried out activity assay. D.A.D., J.F.F. and J.A.H. participated in mechanistic discussions. B.B., J.F.F. and S.M. wrote the paper. All authors edited the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-22527-y.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Grütter MG, Weaver LH, Matthews BW. Goose lysozyme structure: an evolutionary link between hen and bacteriophage lysozymes? Nature. 1983;303:828–831. doi: 10.1038/303828a0. [DOI] [PubMed] [Google Scholar]
- 2.Wohlkönig A, Huet J, Looze Y, Wintjens R. Structural relationships in the lysozyme superfamily: significant evidence for glycoside hydrolase signature motifs. PLoS ONE. 2010;5:e15388. doi: 10.1371/journal.pone.0015388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Höltje J-V, Mirelman D, Sharon N, Schwarz U. Novel type of murein transglycosylase in. Escherichia coli. J. Bacteriol. 1975;124:1067–1076. doi: 10.1128/jb.124.3.1067-1076.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thunnissen A-MWH, Isaacs NW, Dijkstra BW. The catalytic domain of a bacterial lytic transglycosylase defines a novel class of lysozymes. Proteins. 1995;22:245–258. doi: 10.1002/prot.340220305. [DOI] [PubMed] [Google Scholar]
- 5.Scheurwater E, Reid CW, Clarke AJ. Lytic transglycosylases: bacterial space-making autolysins. Int. J. Biochem. Cell Biol. 2008;40:586–591. doi: 10.1016/j.biocel.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 6.Cho H, Uehara T, Bernhardt TG. Beta-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell. 2014;159:1300–1311. doi: 10.1016/j.cell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reid CW, Legaree BA, Clarke AJ. Role of Ser216 in the mechanism of action of membrane-bound lytic transglycosylase B: further evidence for substrate-assisted catalysis. FEBS Lett. 2007;581:4988–4992. doi: 10.1016/j.febslet.2007.09.037. [DOI] [PubMed] [Google Scholar]
- 8.Jitonnom J, Limb MAL, Mulholland AJ. QM/MM free-energy simulations of reaction in Serratia marcescens chitinase B reveal the protonation state of Asp142 and the critical role of Tyr214. J. Phys. Chem. B. 2014;118:4771–4783. doi: 10.1021/jp500652x. [DOI] [PubMed] [Google Scholar]
- 9.Ardèvol A, Iglesias-Fernández J, Rojas-Cervellera V, Rovira C. The reaction mechanism of retaining glycosyltransferases. Biochem. Soc. Trans. 2016;44:51–60. doi: 10.1042/BST20150177. [DOI] [PubMed] [Google Scholar]
- 10.Mark BL, et al. Crystallographic evidence for substrate-assisted catalysis in a bacterial β-hexosaminidase. J. Biol. Chem. 2001;276:10330–10337. doi: 10.1074/jbc.M011067200. [DOI] [PubMed] [Google Scholar]
- 11.Santin YG, Cascales E. Domestication of a housekeeping transglycosylase for assembly of a Type VI secretion system. EMBO Rep. 2017;18:138–149. doi: 10.15252/embr.201643206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kraft AR, Templin MF, Höltje J-V. Membrane-bound lytic endotransglycosylase in. Escherichia coli. J. Bacteriol. 1998;180:3441–3447. doi: 10.1128/jb.180.13.3441-3447.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee M, et al. Reactions of all Escherichia coli lytic transglycosylases with bacterial cell wall. J. Am. Chem. Soc. 2013;135:3311–3314. doi: 10.1021/ja309036q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Svensson M, et al. ONIOM: a multilayered integrated MO + MM method for geometry optimizations and single point energy predictions. A test for Diels-Alder reactions and Pt(P(t-Bu)3)2 + H2 oxidative addition. J. Phys. Chem. 1996;100:19357–19363. doi: 10.1021/jp962071j. [DOI] [Google Scholar]
- 15.Schwarz, J. C. P. Rules for conformation nomenclature for five- and six-membered rings in monosaccharides and their derivatives. J. Chem. Soc., Chem. Commun. 505–508 (1973).
- 16.Cremer D, Pople JA. A general definition of ring puckering coordinates. J. Am. Chem. Soc. 1975;97:1354–1358. doi: 10.1021/ja00839a011. [DOI] [Google Scholar]
- 17.IUPAC-IUB Joint Commission on Biochemical Nomenclature Conformational nomenclature for five and six-membered ring forms of monosaccharides and their derivatives. Eur. J. Biochem. 1980;111:295–298. doi: 10.1111/j.1432-1033.1980.tb04941.x. [DOI] [PubMed] [Google Scholar]
- 18.Fibriansah G, Gliubich FI, Thunnissen A-MWH. On the mechanism of peptidoglycan binding and cleavage by the endo-specific lytic transglycosylase MltE from Escherichia coli. Biochemistry. 2012;51:9164–9177. doi: 10.1021/bi300900t. [DOI] [PubMed] [Google Scholar]
- 19.van Asselt EJ, Thunnissen A-MWH, Dijkstra BW. High resolution crystal structures of the Escherichia coli lytic transglycosylase Slt70 and its complex with a peptidoglycan fragment. J. Mol. Biol. 1999;291:877–898. doi: 10.1006/jmbi.1999.3013. [DOI] [PubMed] [Google Scholar]
- 20.van Asselt EJ, et al. Crystal structure of Escherichia coli lytic transglycosylase Slt35 reveals a lysozyme-like catalytic domain with an EF-hand. Structure. 1999;7:1167–1180. doi: 10.1016/S0969-2126(00)80051-9. [DOI] [PubMed] [Google Scholar]
- 21.van Asselt EJ, Kalk KH, Dijkstra BW. Crystallographic studies of the interactions of Escherichia coli lytic transglycosylase Slt35 with peptidoglycan. Biochemistry. 2000;39:1924–1934. doi: 10.1021/bi992161p. [DOI] [PubMed] [Google Scholar]
- 22.Lee M, et al. Turnover of bacterial cell wall by SltB3, a multidomain lytic transglycosylase of Pseudomonas aeruginosa. ACS Chem. Biol. 2016;11:1525–1531. doi: 10.1021/acschembio.6b00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leung AK-W, Duewel HS, Honek JF, Berghuis AM. Crystal structure of the lytic transglycosylase from bacteriophage lambda in complex with hexa-N-acetylchitohexaose. Biochemistry. 2001;40:5665–5673. doi: 10.1021/bi0028035. [DOI] [PubMed] [Google Scholar]
- 24.van Aalten DMF, et al. Structural insights into the catalytic mechanism of a family 18 exo-chitinase. Proc. Natl. Acad. Sci. USA. 2001;98:8979–8984. doi: 10.1073/pnas.151103798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams SJ, Mark BL, Vocadlo DJ, James MNG, Withers SG. Aspartate 313 in the Streptomyces plicatus hexosaminidase plays a critical role in substrate-assisted catalysis by orienting the 2-acetamido group and stabilizing the transition state. J. Biol. Chem. 2002;277:40055–40065. doi: 10.1074/jbc.M206481200. [DOI] [PubMed] [Google Scholar]
- 26.Greig IR, Zahariev F, Withers SG. Elucidating the nature of the Streptomyces plicatus β-hexosaminidase-bound intermediate using ab initio molecular dynamics simulations. J. Am. Chem. Soc. 2008;130:17620–17628. doi: 10.1021/ja805640c. [DOI] [PubMed] [Google Scholar]
- 27.Martinez-Fleites C, et al. The crystal structure of a family GH25 lysozyme from Bacillus anthracis implies a neighboring-group catalytic mechanism with retention of anomeric configuration. Carbohydr. Res. 2009;344:1753–1757. doi: 10.1016/j.carres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 28.Marković-Housley Z, et al. Crystal structure of hyaluronidase, a major allergen of bee venom. Structure. 2000;8:1025–1035. doi: 10.1016/S0969-2126(00)00511-6. [DOI] [PubMed] [Google Scholar]
- 29.Çetinbaş N, Macauley MS, Stubbs KA, Drapala R, Vocadlo DJ. Identification of Asp174 and Asp175 as the key catalytic residues of human O-GlcNAcase by functional analysis of site-directed mutants. Biochemistry. 2006;45:3835–3844. doi: 10.1021/bi052370b. [DOI] [PubMed] [Google Scholar]
- 30.Abbott DW, Macauley MS, Vocadlo DJ, Boraston AB. Streptococcus pneumoniae endohexosaminidase D, structural and mechanistic insight into substrate-assisted catalysis in family 85 glycoside hydrolases. J. Biol. Chem. 2009;284:11676–11689. doi: 10.1074/jbc.M809663200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roth C, et al. Structural and mechanistic insights into a Bacteroides vulgatus retaining N-acetyl-β-galactosaminidase that uses neighbouring group participation. Chem. Commun. 2016;52:11096–11099. doi: 10.1039/C6CC04649E. [DOI] [PubMed] [Google Scholar]
- 32.Knapp S, et al. NAG-thiazoline, an N-acetyl-β-hexosaminidase inhibitor that implicates acetamido participation. J. Am. Chem. Soc. 1996;118:6804–6805. doi: 10.1021/ja960826u. [DOI] [Google Scholar]
- 33.Speciale G, Thompson AJ, Davies GJ, Williams SJ. Dissecting conformational contributions to glycosidase catalysis and inhibition. Curr. Opin. Struct. Biol. 2014;28:1–13. doi: 10.1016/j.sbi.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ardèvol A, Rovira C. The molecular mechanism of enzymatic glycosyl transfer with retention of configuration: evidence for a short-lived oxocarbenium-like species. Angew. Chem. Int. Ed. 2011;50:10897–10901. doi: 10.1002/anie.201104623. [DOI] [PubMed] [Google Scholar]
- 35.Case, D. A. et al. AMBER 11, University of California, San Francisco, (2010).
- 36.Frisch, M. J. et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, (2013)
- 37.Bacik J-P, Whitworth GE, Stubbs KA, Vocadlo DJ, Mark BL. Active site plasticity within the glycoside hydrolase NagZ underlies a dynamic mechanism of substrate distortion. Chem. Biol. 2012;19:1471–1482. doi: 10.1016/j.chembiol.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 38.Artola-Recolons C, Llarrull LI, Lastochkin E, Mobashery S, Hermoso JA. Crystallization and preliminary X-ray diffraction analysis of the lytic transglycosylase MltE from Escherichia coli. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011;67:161–163. doi: 10.1107/S1744309110049171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.