

Abstract
Epidemiological investigations were conducted on recently emerging porcine reproductive and respiratory syndrome virus (PRRSV) strains in Shandong province in 2014–2015. The proportion of the NADC30 strain identified by ORF7 sequence alignment has been gradually increasing. Three emerging PRRSV strains were successfully isolated, and the complete genomic sequences were determined. Our results indicate the importance of recombinant strains in Shandong province, China. There was a varied degree of recombination of two or three strains (classical, HP-PRRSV and/or NADC30). Moreover, the recombination strains affected the pathogenicity of newly emerged strains.
Introduction
Porcine reproductive and respiratory syndrome (PRRS), a disease that is typically manifested by reproductive failures (e.g., late-term abortion, stillbirth and mummification) in pregnant sows and respiratory distress (e.g., interstitial pneumonia) in growing pigs, remains a substantial economic issue in the global swine industry1–3. Porcine reproductive and respiratory syndrome virus (PRRSV) is an enveloped, positive-sense, single-stranded RNA virus that belongs to the Arteriviridae family4. Its genome varies from 14.9 kb to 15.5 kb in length and possesses nine open reading frames (ORFs) that code seven structural proteins and 14 non-structural proteins5. The first two ORFs (ORF1a and ORF1b) encode an ORF1ab replicase polyprotein, and the remaining seven ORFs (ORF2a, 2b, and 3–7) code for minor structural proteins (GP2, GP3, and GP4) and major structural proteins (GP5, M, and N)3.
The initial characterization of circulating European genotype (genotype I, prototype virus Lelystad) and North American genotype (genotype II, prototype virus VR2332) isolates was found to be surprisingly genetically divergent. Although the overall disease phenotype, gross clinical symptoms, genomic organization and temporal emergence were all similar, these strains shared almost 60% homology at the nucleotide level. Genotype I was reported to be more virulent than genotype II in previous studies6–8.
PRRSV was first reported in North America in 19879 and in Western Europe in 199010. In China, PRRSV, belonging to genotype II, was first isolated in 199511,12. In 2006, HP-PRRSV (highly pathogenic) first emerged from a less-pathogenic variant circulating in southern China and quickly spread throughout the country, causing considerable damage to the swine industry13. A second major HP-PRRSV outbreak occurred in 2009–2010, resulting in increased mortality and affecting a wider geographical area including several Southeast Asian countries14.
The most recent evolving episode was the appearance of the virulent PRRSV NADC30 strains in the United States in 2008; additionally, several NADC30-like strains were isolated in China, which showed the highest nucleotide similarity to a group of PRRSVs represented by NADC3015–17. These NADC30-like PRRSV strains included Henan-XINX (accession no. KF611905), HNyc15 (accession no. KT945018), and JL580 (accession no. KR706343). Moreover, several reports showed that the NADC30-like strains were now beginning to recombine with the Chinese HP-PRRSV in the field18–20, increasing the difficulty of the prevention and control of PRRS.
In our study, the detection of different PRRSV strains in 13 different regions of Shandong province since June 2014 was analyzed, and the positive samples of classical PRRSV, HP-PRRSV and NADC30 were counted by sequence analysis of ORF7. To fully understand the evolutionary patterns and dynamics of PRRSV and to aid prevention and control policies against the disease, a genomic scale analysis was necessary. Among all the PRRSV-positive samples, three recombination strains (SDhz1512, SDlz1601 and SDYG1606) were isolated, and their complete genomic sequences were determined. Moreover, the protein structure predictions and pathogenicity experiments of the three recombinant strains were compared. The recombination of the three strains was quite complex and demonstrated different degrees of recombination formed by two or three strains (NADC30, HP-PRRSV and/or classical PRRSV). The temporal changes in the relative genetic diversity of PRRSV were illustrated by Bayesian Skyline plots (BSP). Hence, our study offers a unique opportunity to investigate the evolution and spread of new genotype II PRRSV strains in Shandong province, China. Moreover, the findings play an important role in preventing and controlling the occurrence and spread of PRRSV.
Results
Epidemiology of PRRSV and variability of ORF7
PRRSVs of recent outbreaks (since 2014) are characterized by severe sow abortion and respiratory symptoms of conservation piglets. PRRSV strains were present in 13 different regions of Shandong province. Within a total of 347 clinical samples of diseased pigs (182 lung and lymph node samples and 165 serum samples), 243 samples were PRRSV-positive in RT-PCR using primers that targeted the PRRSV ORF7 (N) gene. The percentage of PRRSV positive samples was 40%-50% from June 2014 to April 2017 (Supplementary Fig. 1). Comparative analyses of ORF7 sequences showed that ORF7 primers were able to distinguish classical PRRSV (represented by the virus VR2332), HP-PRRSV (represented by the virus JXA1) and NADC30. However, the proportion of NADC30 strains in the PRRSVs of recent outbreaks (since 2014) was 50.62%, which was higher than that of classical PRRSV (6.67%) or HP-PRRSV (42.80%) (Table 1).
Table 1.
Descriptive statistics for predictors tested for PRRS positive status for 13 different regions of Shandong province.
| Origin | No. of Clinical samples | No. of positive samples | Percent positive | Classical | HP-PRRSV | NADC30 |
|---|---|---|---|---|---|---|
| Linyi | 115 | 94 | 81.74% | 7 | 43 | 44 |
| Jining | 29 | 25 | 86.21% | 1 | 13 | 11 |
| Weifang | 32 | 19 | 59.38% | 0 | 8 | 11 |
| Laiwu | 19 | 11 | 57.89% | 2 | 3 | 6 |
| Yantai | 17 | 8 | 47.06% | 0 | 4 | 4 |
| Zibo | 14 | 9 | 64.29% | 1 | 3 | 5 |
| Binzhou | 25 | 19 | 76.00% | 1 | 7 | 11 |
| Jinan | 18 | 10 | 55.56% | 1 | 5 | 4 |
| Heze | 13 | 13 | 100.00% | 0 | 5 | 8 |
| Dongying | 17 | 9 | 52.94% | 1 | 2 | 6 |
| Taian | 15 | 6 | 40.00% | 0 | 4 | 2 |
| Qingdao | 12 | 7 | 58.33% | 1 | 3 | 3 |
| Liaocheng | 21 | 13 | 61.90% | 1 | 4 | 8 |
| Total | 347 | 243 | 70.03% | 16/6.67% | 104/42.80% | 123/50.62% |
To better understand the genetic relationship and evolution of PRRSV in Shandong province, China, an ORF7-based phylogenetic tree of 243 positive PRRSVs and 54 reference viruses was constructed. According to the phylogenetic tree (Fig. 1), these PRRSVs were divided into two distinct genotypes: genotype I and genotype II. All the PRRSVs of Shandong province belonged to genotype II, which was divided into three monophyletic lineages: the first sub-group contained 16 isolates belonging to classical PRRSV (represented by the virus VR2332), the second sub-group contained 104 isolates related to HP-PRRSV (represented by the virus JXA1), and the third sub-group contained 123 isolates (76 isolates from 2017) that were closely related to a group represented by NADC30.
Figure 1.

Phylogenetic tree based on the ORF7 gene of the 243 positive PRRSV and 54 reference viruses. Phylogenetic trees were constructed using the NJ method in MEGA7.0 with bootstrap values (1000 replicates), using the Kimura2-parameter model. The three isolates are marked with the red diamond.
Identification of PRRSV isolation
Three PRRSV strains (SDhz1512, SDlz1601 and SDYG1606) that caused piglets to have clinical respiratory distress and interstitial pneumonia were recovered by PAMs. PAMs inoculated with four PRRSV strains (SDhz1512, SDlz1601, SDYG1606 and SX-1) showed green fluorescence, and the positive control cells infected with SX-1 had died. No fluorescence signal was observed in the control cells (Fig. 2).
Figure 2.

The results of the immunofluorescence assay. (A) PAMs inoculated with SDlz1601. (B) PAMs inoculated with SDYG1606. (C) PAMs inoculated with SDhz1512. (D) Positive control cells inoculated with SX-1. (E) PAM control cells.
Genomic characterization of 3 PRRSV isolates
The complete genome sequences from three PRRSV isolates were determined and deposited in GenBank (accession no. KX980392, KX980393 and KY053458). They were classified as genotype II isolates but differed in length: 15069 nt for SDlz1601, 15367 nt for SDhz1512, and 15074 nt for SDYG1606. Full-length sequence analyses of SDlz1601, SDhz1512 and SDYG1606 were performed together with reference PRRSV strains including two North American isolates (VR2332 and NADC30) and one Chinese isolate (HP-PRRSV JXA1). The analysis showed that SDlz1601 shared 88.9% (JXA1), 84.5% (VR2332), 88.7% (NADC30) and 90.9% (JL580) nucleotide homology with the respective strains (data not shown); SDhz1512 shared 92.5% (JXA1), 89.8% (VR2332), 84.4% (NADC30), and 92.3% (HUN4) nucleotide homology with the respective strains (data not shown); and SDYG1606 shared 85.9% (JXA1), 89.8% (VR2332), and 91.5% (NADC30) nucleotide homology with the respective strains (Table 2). The nucleotide and amino acid homology of the 5′UTR, 3′UTR, ORF1a, ORF1b, and ORF2~7 genes in the three strains and the representative strains, including NADC30, VR2332 and JXA1, were compared. The results showed that the 5′UTR, ORF2~7 and 3′UTR of SDhz1512 shared 92.6~99.4% nucleotide (90.9~97.7% amino acid) homology with NADC30, which was higher than the homology shared with VR2332 and JXA1, whereas ORF1a of the SDhz1512 isolate shared 95.5% nucleotide (95.7% amino acid) identity with JXA1, which was higher than the others, and ORF1b of the SDhz1512 isolate shared 97.0% nucleotide (98.7% amino acid) identity with VR2332, which was also higher than the others. The SDlz1601 strain′s ORF1b, ORF2, ORF4, ORF6-7 and 3′UTR shared 91.1~97.5% nucleotide (90.2~97.7% amino acid) identity with NADC30, which was higher than the others, whereas 5′UTR, ORF1a, ORF3 and ORF5 shared 93.0~96.8% nucleotide (89.3~93.0% amino acid) identity with JXA1, which was higher than the other two strains. 3′UTR and ORF1b~ORF7 of SDYG1606 shared 87.0~97.3% nucleotide (90.9~98.0% amino acid) homology with NADC30; in addition, the 5′UTR region shared 97% nucleotide homology with the JXA1 strain (Table 2).
Table 2.
Genome positions, protein sizes, and percentages of nucleotide and amino acid (in parenthesis) identity of SDlz 1601, SDhz 1512 and SDYG 1606 to other representative isolates (%).
| Region | Length | Identities (%) nucleotide (amino acid) sequence identity | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| JXA1 | VR-2332 | NADC30 | SDlz1601 | SDhz1512 | SDYG1606 | SDlz1601 to JXA1 | SDhz1512 to JXA1 | SDYG1606 to JXA1 | SDlz1601 to VR2332 | SDhz1512 to VR-2332 | SDYG1606 to VR2332 | SDlz1601 to NADC30 | SDhz1512 to NADC30 | SDYG1606 to NADC30 | |
| Complete | 15319 | 15451 | 15047 | 15069 | 15367 | 15074 | 88.9 | 92.5 | 85.9 | 84.5 | 89.8 | 83.6 | 88.7 | 84.4 | 91.5 |
| 5′UTR | 188 | 190 | 191 | 189 | 189 | 189 | 96.8 | 92.6 | 97.3 | 89.5 | 93.1 | 90.5 | 89.5 | 99.4 | 91.0 |
| ORF1a | 7422(2475) | 7512(2502) | 7119(2375) | 7119(2375) | 7422(2475) | 7119(2375) | 87.3(87.6) | 95.5(95.7) | 82.0(82.9) | 81.4(80.7) | 87.0(85.8) | 79.6(80.7) | 83.3(85.1) | 76.2(77.1) | 87.9(89.3) |
| ORF1b | 4383(1462) | 4383(1462) | 4383(1462) | 4383(1462) | 4383(1462) | 4383(1462) | 87.7(96.3) | 92.5(96.5) | 89.9(96.6) | 87.4(95.5) | 97.0(98.7) | 88.6(96.2) | 95.2(97.8) | 89.0(96.4) | 94.6(98.0) |
| ORF2 | 768(256) | 768(256) | 768(256) | 768(256) | 768(256) | 768(256) | 90.3(90.6) | 86.5(87.5) | 85.9(83.9) | 89.5(88.6) | 89.2(91.4) | 89.4(86.7) | 91.1(90.2) | 95.4(94.9) | 93.6(92.1) |
| ORF3 | 770(254) | 770(254) | 770(254) | 770(254) | 770(254) | 770(254) | 93.0(89.3) | 82.0(78.7) | 82.0(81.1) | 88.0(87.4) | 82.0(80.7) | 84.0(83.4) | 87.5(87.7) | 95.0(94.0) | 92.6(90.9) |
| ORF4 | 534(178) | 534(178) | 534(178) | 534(178) | 534(178) | 534(178) | 88.0(85.9) | 87.0(84.8) | 85.0(87.0) | 86.8(85.9) | 86.0(87.6) | 87.2(87) | 94.0(96.6) | 96.6(97.1) | 96.2(96.0) |
| ORF5 | 600(200) | 600(200) | 600(200) | 600(200) | 600(200) | 600(200) | 94.8(93.0) | 83.8(84.5) | 85.5(84.5) | 87.3(85.5) | 84.1(82.5) | 85.5(83.0) | 87.1(87.5) | 92.5(92.5) | 95.5(93.0) |
| ORF6 | 522(174) | 522(174) | 522(174) | 522(174) | 522(174) | 522(174) | 89.2(94.2) | 89.2(94.2) | 88.5(94.2) | 90.6(94.2) | 90.2(94.2) | 89.4(94.2) | 97.5(97.7) | 97.5(97.7) | 97.3(98.8) |
| ORF7 | 369(123) | 369(123) | 369(123) | 369(123) | 369(123) | 369(123) | 90.5(90.2) | 90.5(91.8) | 88.8(89.4) | 91.0(91.8) | 91.7(93.4) | 91.0(92.6) | 96.7(97.5) | 97.2(97.5) | 96.4(95.9) |
| 3′UTR | 150 | 151 | 151 | 149 | 151 | 152 | 86.7 | 87.4 | 88.8 | 90 | 90.7 | 90.7 | 97.3 | 97.3 | 97.3 |
Amino acid analysis of NSP2, secondary structure and 3D-structure prediction
Non-structural protein 2 (NSP2) had the highest genetic diversity in the PRRSV genome. To further characterize the deletion regions, the NSP2 of the three isolated strains (SDhz1512, SDlz1601 and SDYG1606) was compared with 8 representative genotype II isolates. Multiple sequence alignment revealed that SDlz1601 and SDYG1606 had three discontinuous deletions (a total of 131 amino acids) in NSP2 that resembled previous NADC30-like PRRSVs, which can be used as molecular markers to distinguish them from other PRRSVs15. These deletions were not present in the Chinese HP-PRRSV strains (Fig. 3). Furthermore, the SDYG1606 isolate had the same 9 amino acid deletions (793–801) in NSP2 and was closely related to NADC30 (Fig. 3), but SDhz1512 had 30 amino acids deletion and was more similar to the HP-PRRSV strains (HUN4 and JXA1). Surprisingly, the 9 amino acid deletions (814~822) of NSP2 in SDlz1601 differed from those of previously sequenced NADC30-like isolates from China.
Figure 3.

The sequences alignment of NSP2. Three discontinuous amino acid deletions (323–433, 482, and 506–524) in NSP2 of SDlz1601 and SDYG1606 and a 793–801 aa deletion in SDYG1606. Two NADC30-like PRRSVs (JL580 and HNyc15) and other representative PRRSVs including NADC30 (JN654459), JXA1 (EF112445), HUN4 (EF635006), VR2332 (U87392), and CH-1a (AY032626) were included in the analysis.
NSP2 secondary structures of the three isolates (SDhz1512, SDlz1601 and SDYG1606) and three reference strains (VR2332, JXA1 and NADC30) were predicted using protein software of DNAStar version 7.1, Madison WI (Supplementary Fig. 2). SDhz1512 was consistent with the highly pathogenic strain JXA1, in which the α-helix was deleted and the β-strand was increased. Compared with the classical strain VR2332, SDlz1601 and SDYG1606 were more consistent with the new NADC30-like strain, and the deletion of a total of 111 amino acids (323~433) resulted in the lack of a large α-helix, β-strand and random coil. In addition, the Antigenic Index in this region (320–360) was reduced to a low level using Protein software (DNAStar, version 7.1, Madison WI). In conclusion, the absence of amino acid groups in the discontinuous large fragments in NSP2 of NADC30-like strains resulted in significant changes in the secondary structure of the protein.
The NSP2 amino acid sequences of the three isolated strains (SDhz1512, SDlz1601 and SDYG1606) and the three reference strains (VR2332, JXA1 and NADC30) were submitted to homology modeling by the I-TASSER Server21,22 for 3D-structure prediction. After the structure assembly simulation, I-TASSER uses the TM-align structural alignment program to match the first I-TASSER model to all structures in the PDB library. As a result, 5 models were established in all 5 strains, and the parameters of the first prediction model were the best. The parameters of VR2332 were as follows: TM-score 0.75, RMSD 10.8 and C-score −0.81. The parameters of the SDlz1601 strain were as follows: TM-score 0.65, RMSD 9.7 and C-score −0.50. The parameters of the SDYG1606 strain were as follows: TM-score 0.58, RMSD 11.0 and C-score −1.04. The parameters of the SDhz1512 strain were as follows: TM-score 0.65, RMSD 10.0 and C-score −0.51. The parameters of the JXA1 strain were as follows: TM-score 0.61, RMSD 10.0 and C-score −0.50. The parameters of the NADC30 strain were as follows: TM-score 0.65, RMSD 10.8, and C-score −0.81. The 3D spatial structure of the 6 virus strains was essentially the same, but the ligand binding sites differed (Fig. 4). The major amino acid residues are composed as follows: VR2332: R349, L352, T353, L356, S357, R364, E365, E366, D385, E386, D389, and Q390; JXA1: S371, L374, T404, M407, M408, V436, R439, K442, and S443; SDhz1512: S405, M408, A409, E413, P448, K451, P454, A455, G519, and V523; SDlz1601: T429, L433, A434, and D441; SDYG1606: P323, Q326, P327, A330, E331, S358, D362, and S366.
Figure 4.

Tertiary structure models for NSP2 of VR2332, SDlz1601, SDYG1606, SDhz1512, JXA1 and NADC30 using I-TASSER Server. TM-score values range from [0, 1]. A higher score indicates a better structural match. Statistically, a TM-score <0.17 means a randomly selected protein pair with the gapless alignment taken from PDB; a TM-score >0.5 corresponds approximately to two structures of similar topology. C-score values are in the confidence interval [−5, 2], with higher values indicating better models. The TM-score and RMSD are the indexes for evaluating the molecular structure. The correlation of the C-score with RMSD and the TM-score is extra high.
Recombination analyses in SDhz1512, SDlz1601 and SDYG1606
In this study, a comprehensive and novel approach was employed to characterize recombination in the complete genome of PRRSV. Using seven algorithms, RDP, GENECONV, MaxChi, Chimaera, Bootscan, SiScanand, and 3Seq methods in RDP4.70 software provided strong evidence of recombination events in most of the sequences analyzed. Seventeen recombination events were detected in the three isolates (Table 3). SDYG1606 had 3 potential recombination signals, whereas SDhz1512 had 8 potential recombination signals and SDlz1601 had 6 potential recombination signals (Table 3). Of all 16 potential recombination events, 14 recombination events were detected in SDYG1606. SDlz1601 had a remarkably high degree of certainty, with a P-value in at least four algorithms of <1 × 10−6 (Table 3). The other 14 recombination events with a high degree of certainty due to a recombinant score >0.6 were located at 1–1818, 4823–8190, and 12098–12358 of SDYG1606 (events 1–3); 56–365, 1944–2021, 5732–6302, 7303–8111, 9088–11240, 12098–12358, and 11241–14635 of SDhz1512 (events 4–10); and 3004–7473, 12986–13638, 12057–12662, and 1944–2021 of SDlz1601 (events 13–15 and 17) (Table 3). In addition, the remaining 3 recombination events had a fair likelihood since their recombinant score was between 0.429 and 0.549 (Table 3). Furthermore, there were similar recombinant events with the same locations and parents in different isolates including a recombination located at 1944–2021 with parents NADC30/JXA1 in SDhz1512 (event 7) and SDlz1601 (event 17) (Table 3).
Table 3.
Summary of possible recombination events in SDlz1601, SDhz1512 and SDYG1606 isolates identified by RDP4.
| Event number | Recombinant Sequence(s) | Breakpoint position(s) | Parental sequence | Recombinant score | P-Value for the six detection methods in RDP4 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| In Recombinant Sequence | Begin/End | Minor/Major | RDP | GENECONV | Bootscan | Maxchi | Chimaera | SiSscan | 3Seq | ||
| 1 | SDYG1606 | 4823/8190 | JXA1/NADC30 | 0.697 | 7.94E-113 | 8.32E-116 | 5.58E-120 | 1.54E-42 | 4.35E-09 | 1.46E-50 | 2.16E-11 |
| 2 | SDYG1606 | 1/1818 | SX1/JL580 | 0.685 | 1.88E-152 | 4.56E-131 | 1.21E-136 | 2.56E-35 | 4.50E-34 | NS | 2.16E-11 |
| 3 | SDYG1606 | 12098/12358 | HNyc15/NADC30 | 0.65 | 7.37E-06 | 1.63E-04 | NS | 2.72E-04 | 2.10E-04 | NS | 2.42E-05 |
| 4 | SDhz1512 | 9088/11240 | HN1/SX09 | 0.673 | NS | 1.67E-76 | 8.97E-80 | 8.10E-15 | 1.70E-14 | 4.21E-32 | NS |
| 5 | SDhz1512 | 5732/6302 | SD1100/SX1 | 0.661 | NS | 5.35E-32 | 2.59E-34 | 7.42E-09 | 1.82E-13 | 7.25E-13 | 2.16E-11 |
| 6 | SDhz1512 | 11241/14635 | NADC30/JXA1 | 0.741 | 5.53E-110 | NS | 5.07E-90 | 9.09E-32 | 2.47E-09 | 4.85E-33 | 2.88E-11 |
| 7 | SDhz1512 | 7303/8111 | SD1100/JXA1 | 0.67 | NS | 1.85E-31 | 7.03E-35 | 2.17E-11 | 6.16E-12 | 9.15E-13 | 2.16E-11 |
| 8 | SDhz1512 | 4702/14982 | WUH5/SX1 | 0.624 | 2.02E-14 | 8.23E-08 | 2.46E-18 | NS | NS | NS | 1.44E-11 |
| 9 | SDhz1512 | 56/365 | VR2332/JXA1 | 0.617 | 2.27E-17 | 2.61E-12 | 2.18E-11 | 7.33E-04 | 2.05E-04 | NS | 6.48E-11 |
| SDhz1512 | 1944/2021 | HNjz15/HLJB1 | 0.628 | 7.966E-10 | 3.40E-04 | 5.26E-03 | NS | NS | NS | 0.000249 | |
| 10 | SDhz1512 | 2062/3163 | SX1/Unknown | 0.429 | 2.03E-12 | 1.48E-20 | 1.09E-08 | 8.42E-07 | 7.58E-07 | 3.29E-13 | 7.20E-12 |
| 11 | SDlz1601 | 360/2020 | SX1/NADC30 | 0.549 | 4.12E-108 | 3.29E-113 | 1.53E-98 | 7.93E-38 | 1.58E-38 | 1.40E-39 | NS |
| 12 | SDlz1601 | 3004/7473 | JXA1/NADC30 | 0.629 | 1.36E-75 | 3.48E-87 | 2.41E-83 | 1.48E-44 | 1.77E-10 | 1.90E-64 | 3.78E-08 |
| 13 | SDlz1601 | 12986/13638 | JXA1/NADC30 | 0.674 | 4.37E-48 | 1.61E-40 | 1.28E-38 | 8.34E-14 | 3.55E-15 | 5.33E-16 | 1.44E-11 |
| 14 | SDlz1601 | 12057/12662 | TJbd141/NADC30 | 0.663 | NS | 5.92E-37 | 2.25E-44 | 1.01E-14 | 1.72E-12 | 6.58E-18 | 1.44E-11 |
| 15 | SDlz1601 | 1/354 | NVDCSDXX2013/JL580 | 0.549 | 7.19E-18 | 9.17E-12 | 1.61E-17 | 1.93E-02 | 3.91E-03 | 5.06E-04 | NS |
| 16 | SDlz1601 | 1944/ 2021 | NADC30/JXA1 | 0.628 | 7.966E-10 | 0.0003397 | 0.0052645 | NS | NS | NS | 0.000249 |
To further validate and confirm the putative recombination events and their breakpoint positions in isolates detected with RDP, the SimPlot method in the SimPlot v3.5.1 program was employed. This method records the coherence of the sequence relationship over the entire length of the isolate and its potential parents23. This study revealed that SDhz1512, SDlz1601 and SDYG1606 were the result of recombination between the NADC30-like viruses and the classical strains and HP-PRRSV strains present in outbreaks in China. From the similarity plot, we identified seven recombination breakpoints in SDhz1512: one was located in NSP1α (nucleotides [nt] 352), two points were located in NSP7 (nt6349 and 6938), two points were located in NSP9 (nt8009 and 8830), one point was located in NSP10 (nt10000), and the last point was located in ORF2a (nt12125) (Fig. 5Aa). The analysis revealed that SDlz1601 was the result of recombination between the NADC30 viruses and HP-PRRSV strains circulating in China. There were seven recombination breakpoints detected in the SDlz1601 isolate: two points were located in NSP2 (nt 2082 and 3919), one was located in NSP9 (nt8774), two points were located in ORF4 (nt 13398 and 14050), one was located in ORF5 (nt14344), and one was located in ORF6 (nt15015) (Fig. 5Ba). The analysis revealed that SDYG1606 was the result of recombination between the NADC30 viruses and the classic HP-PRRSV strains circulating in China. From the similarity plot, we identified three recombination breakpoints: one was located in NSP2 (nt1942) and the others were located in NSP4 (nt5759) and NSP9 (nt9296) (Fig. 5Ca). The breakpoints of SDhz1512 separate the genome into eight regions: three are closely related to the HP-PRRSV strains in China (represented by JXA1) (Fig. 5Ab), four are closely related to classical PRRSV strains from China (represented by China1a) (Fig. 5Ac), and remaining region is closely related to the PRRSV strains from North America (represented by NADC30) (Fig. 5Ad). The breakpoints of SDlz1601 separate the genome into eight regions: four are closely related to the classic HP-PRRSV strains in China (represented by JXA1) (Fig. 5Bb) and the other four are closely related to PRRSV strains from North America (represented by NADC30) (Fig. 5Bc). The breakpoints of SDYG1606 separate the genome into four regions: two are closely related to the classic HP-PRRSV strains in China (represented by JXA1) (Fig. 5Cb) and the other two are closely related to the PRRSV strains from North America (represented by NADC30) (Fig. 5Cc).
Figure 5.

Recombination analysis of the three PRRSV strains. Recombination analysis of SDhz1512 (A) SDlz1601 (B) and SDYG1606 (C) using SimPlot with a sliding window of 500 nt, moving in 20 nt steps. Genome scale similarity comparisons of three PRRSV strains (query) with NADC30 (parental group A, red), JXA1 (parental group B green), and VR2332 (outgroup C, blue). Recombination breakpoints are shown as dotted lines, with the locations indicated at the bottom. The background color of parental region A (with reference to the NADC30 strain) is light green, whereas that of parental region B (HP-PRRSV JXA1) is shaded light purple and that of parental region C (VR2332) is shaded white. The phylogenies of parental region A(c), parental region B(b), and parental region C(d) are shown below the similarity plot.
Interestingly, eight of the above 6 reported and 3 isolated strains had different parents of recombination with other PRRSV strains such as classical strains (VR2332 and/or CH1a), HP-PRRSV (JXA1 and/or 09NEN1), and NADC30 (Table 4). The recombination breakpoints analysis of the above recombinants demonstrated that they were similar to the phenomenon of fracture recombination, and the recombinant positions mainly occurred in NSP2, NSP9, ORF2, ORF4 and ORF7 (Table 4). These proteins may have been the hot regions for recombination event breakpoints.
Table 4.
Recombination analysis of nine PRRSV strains Simplot v3.5.1 software was used to analyze recombination.
| Virus | Recombination with | Recombination sites | GenBank Access NO. | Isolation year | Isolation sites |
|---|---|---|---|---|---|
| SDYG1606 | JXA1 | NSP2, NSP5~NSP9 | KY053458 | 2016 | Shandong province |
| NADC30 | NSP2~NSP5, NSP9~ORF7 | ||||
| SDhz1512 | VR2332/CH1a | NSP1α, NSP6-7, NSP9, ORF1b, ORF2, | KX980392 | 2015 | Shandong province |
| NADC30 | ORF2-7 | ||||
| JXA1 | NSP1α-NSP6, NSP7-9, NSP9-ORF1b | ||||
| SDlz1601 | JXA1 | NSP2, NSP9, ORF3, ORF4, ORF5, ORF6 | KX980393 | 2016 | Shandong province |
| NADC30 | NSP2, NSP9~ORF3, ORF4~ORF5, ORF6~7 | ||||
| JL580 | NADC30 | NSP2, NSP2~NSP3, NSP7~ORF2, ORF4~ORF7 | KR706343.1 | 2014 | Jilin province |
| 09HEN1 | NSP2, NSP2~NSP7, ORF2~ORF4 | ||||
| HNjz15 | / | / | KT945017.1 | 2015 | Henan province |
| HNyc15 | VR2332/CH1a | ORF2-4 | KT945018.1 | 2015 | Henan province |
| FJXS15 | JXA1 | NSP2~ORF4 | KX758250 | 2015 | Fujian province |
| NADC30 | NSP2, ORF4~7 | ||||
| HENAN-HEB | JXA1 | NSP2 | KJ143621.1 | 2013 | Henan province |
| HENAN-XINX | VR2332 | NSP2-5 | KF611905.1 | 2013 | Henan province |
Phylogenetic analysis
To determine the genetic relationship of the HP-PRRSV strains that have recently emerged in China, a phylogenetic tree based on the full genome sequences of SDhz1512, SDlz1601, SDYG1606 and another 73 published PRRSV isolates that were identified between 2013 and 2016 was generated. The phylogenetic analysis of the entire PRRSV genome was performed using RaxML and the BI tree, and its confidence was evaluated with 1000 bootstraps. As shown in Fig. 6, the results showed that genotype II isolates in China could be divided into three sub-genotypes: those closely related to NADC30-like isolates, China classical isolates, and HP-PRRSV isolates. The phylogenetic tree further revealed that SDhz1512, SDlz1601, and SDYG1606 were recombinant strains and had lower identity to other isolates. The complete genome of the three isolates shared only 89–92% identify, which differed from the published NADC30-like strains. This suggests that the virus gained genetic diversity by recombining with local HP-PRRSV, classical strains and North America isolates (represented by NADC30) in China, and these recombinants are becoming more common. The MCC tree revealed, with high resolution, the evolutionary history of NADC30-like viruses and related viruses in China, from which we could infer that NADC30-like PRRSVS were more closely related to NADC30 and were clustered into a separate branch, thus distinguishing them from the HP-PRRSV cluster represented by JXA1 and HuN4. The Bayesian skyline plot revealed two periods in which there was a marked increase in relative genetic diversity (Fig. 7) and whose timing and scale matched well with the epidemiological record of the HP-PRRSV and NADC30 PRRSVS outbreaks. The first growth phase corresponded to the initial HP-PRRSV outbreak in 2005–2010 whereas the second was mainly associated with the new HP-PRRSV variant NADC30-like outbreak in 2013–2017. Recently, outbreaks of this new HP-PRRSV variant have spread all over China.
Figure 6.

Maximum likelihood tree of whole PRRSV genomes under RaxML (n = 75).
Figure 7.
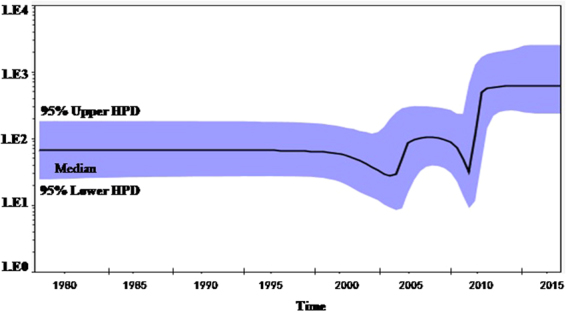
Bayesian skyline plot of global PRRSVs sampled between 2000 and 2016. The dark line in the Bayesian skyline plot shows the estimated effective population size over time. The gray area represents the 95% highest posterior density confidence intervals for this estimate.
PRRSV recombination strains exhibited pathogenicity for piglets
The pigs challenged by PRRSV SDhz1512, SDlz1601 and SDYG1606 strains developed similar clinical signs, such as fever (≥40 °C) and anorexia, and the clinical signs became more severe over the next few days. Three pigs in the SDlz1601 or SDYG1607 strain challenge group had respiratory distress, characterized by dyspnea, tachypnea and coughing, whereas two pigs in the SDhz1512 strain challenge group displayed similar symptoms. The piglets in the control group remained clinically normal throughout the study. The viral titers of the three isolates in sera reached peak levels at 14 days post-inoculation (dpi) (Supplementary Fig. 3), whereas all the pigs in the mock-inoculated control group were negative for PRRSV throughout the entire experimental period. The viruses were recovered from the sera of pigs in the three infected groups, and ORF7 genes were sequenced to confirm they were the original virus.
The major gross pathology finding was different levels of interstitial pneumonia in the PRRSV-challenged pigs (Fig. 8). Interstitial pneumonia was characterized by thickening of the alveolar septa and infiltration of infiltrating lymphocytes and macrophages. Necrotic bronchial epithelial cells could also be found in the trachea. No pathological lesion was identified in the control pigs. The morbidity was determined by clinical symptoms and pathological changes (Table 5).
Figure 8.

Microscopic lesions examination in lungs of the challenged piglets. (A) shows the thickening of the alveolar septa and a small amount of neutrophil infiltration. (B) shows thickening of the pulmonary alveolar wall, hyperplasia of alveolar epithelial cells, infiltration of monocytes and macrophages and exfoliated epithelial cells infiltrating in the bronchiole. (C) shows that the lung tissue alveolar structure disappeared, infiltration of monocytes, macrophages, neutrophils, and pulmonary parenchymal, and exfoliated epithelial cells infiltrating the bronchiole. (D) The lungs of a control pig. Original magnification.
Table 5.
The morbidity and mortality of pigs used for animal challenge.
| Group | Treatment | Number of pigs | Morbidity | Mortality |
|---|---|---|---|---|
| I | SDhz1512 | 5 | 60% (3/5) | 0 |
| II | SDlz1601 | 5 | 80% (4/5) | 20% (1/5) |
| III | SDYG1606 | 5 | 100% (4/5) | 20% (1/5) |
| IV | PAMs culture supernatant | 5 | 0 | 0 |
Discussion
Since 2013, the phenomenon of sow abortion has occurred frequently, resulting in significant losses to the global pig industry. The genetic diversity of HP-PRRSV has greatly increased by rapid evolution and recombination events14,19,24. Recently, several outbreaks of NADC30-like PRRSVs were reported in different provinces of China18. Strikingly, all NADC30-like strains carry the genetic marker of PRRSV MN184 strains, namely, the exact discontinuous deletions in the NSP2-coding region20. Some workers claimed that geographic separation is a factor influencing PRRSV evolution, based on ORF5 and genome sequences25,26. Yoon et al.27 have also reported that there was no immediate relationship between the date or place of collection and the topological distribution of ORF7 in the different PRRSVs. Conversely, Hao X et al.28 claimed that PRRSV evolution was also based on the ORF7 sequence. However, regarding the phylogenetic relationships among the PRRSVs, our results based on the whole genome sequence data of 73 clinical samples obtained between 2013 and 2016 using RaxML and a BI tree revealed that the Chinese and North American genotypes isolates could be divided into three sub-genotypes using ORF7 primers: those closely related to NADC30 isolates, classical isolates and HP-PRRSV isolates. Moreover, three PRRSV strains (SDlz1601, SDhz1512, and SDYG1606), which had more than 96% homology with NADC30 based on ORF7, were recombinant strains of two or three strains (HP-PRRSV, classical, and NADC30). The PRRSV variants have brought great challenges to the prevention and control of major diseases in the swine industry.
Recombination is not uncommon in PRRSV, and there are more and more reports about the recombination of PRRSVs5,19. Recently, Li Y et al.16 reported PRRSV HNyc15 isolation characterized by NADC30-like recombination with classical PRRSV strain VR2332 and CH-1a between ORF2 and ORF4. Zhao K et al.19 also indicated that the NADC30-like PRRSV strain from North America has spread to several provinces in China and recombined with local HP-PRRSV strains. Zhao H et al.24 reported that nine of 28 isolates and one isolate from another laboratory were potential complicated recombinants between the vaccine JXA1-R strains and predominant circulating strains, and the PRRSV recombination rate is increased under the current vaccination pressure. However, our study showed that SDlz1601 and SDYG1606 exhibited a 111 aa deletion at positions 323 to 433 and a 19 aa deletion at positions 506 to 524, which can be considered to be NADC30-like strains. Conversely, the SDhz1512 strain was a recombination strain of NADC30, classical and HP-PRRSV. A 30 aa sequence in the NSP2 gene was detected that was consistent with HP-PRRSV. Accordingly, not only NADC30-like strains but also recombinant strains of NADC30 with classical PRRSV and/or HP-PRRSV have become the current popular trend.
A recent publication indicated that piglets infected with NADC30-like viruses, such as HNjz15, CHsx1401, and FJ1402, showed fever and respiratory disorders, but no death29,30. Obviously, the virus NADC30-like pathogenicity is different from the Chinese HP-PRRSV, which is fatal for piglets31. In this study, animal challenge was used to determine the relationship between recombination and the virulence of three isolated PRRSV strains. The SDYG1606 strain led to more severe interstitial pneumonia than SDlz1601 although the sequence characteristics of it were similar to NADC30-like strains. The reason for the difference in virulence may result from varied recombination breakpoints. However, the piglet deaths were caused by secondary infection of bacteria. The new variant strain, SDhz1512, which was a triple recombination strain including NADC30, classical and HP-PRRSV, caused more minor clinical symptoms than the NADC30-like strains. Our present data indicated that the virulence of different PRRSV strains with different recombination breakpoints was varied, but the virulence of the recombinant strain with classical PRRSV was slightly weaker.
Bayesian phylodynamic models showed one remarkable improvement compared to traditional methods and could make use of associated epidemiological information to infer genetic relations. These inferences could be used to identify viral dispersion routes that correspond with transportation patterns involving high PRRSV risk32. In this study, 75 NSP2 sequences of genotype II PRRSVs from 2000 to 2016 of China obtained from GenBank were used to estimate the evolutionary history of PRRSV in China. The Bayesian skyline plot revealed two periods in which there was a marked increase in relative genetic diversity. The first growth phase corresponded to the initial HP-PRRSV outbreak in 2005~2010, whereas the second growth phase was mainly associated with the NADC30-like outbreak in 2013~2017. It matched well with the epidemiological record of the HP-PRRSV and NADC30 PRRSV outbreaks.
Currently, there are a variety of commercial PRRSV vaccines in the Chinese market. The current modified live vaccines do not provide complete cross-protection against heterologous PRRSV strains20. Recent studies suggest that several newly identified virulent PRRSV isolates have been introduced into swine populations through the inoculation of PRRSV-derived inactivated vaccines33,34. Recently, Shi M et al.13 indicated that recombinants appeared to be highly pathogenic, such that the recombination events that generated them either preserved or increased the pathogenicity of the parental strains. Zhao K et al.19 also confirmed the high pathogenicity of JL580 recombinants between NADC30-like and HP-PRRSV strains in piglet challenge. Several studies reported the outbreaks of NADC30-like PRRSVs in vaccinated pig herds with 30–50% fatality15. Thus, the current prevalence of recombinant strains may be the result of PRRSVs escaping vaccine immunization. However, whether the existing vaccine can provide protection for recombinant strains requires further study.
Material and Methods
Clinical samples
During the period from June 2014 to April 2017, suspected samples from stillborn piglets, serum samples from diseased sows and piglets, and lungs and lymph nodes of dead or diseased piglets were collected in 13 regions of Shandong Province, China. Clinical tissues were homogenized for RNA extraction and virus isolation, and the remaining samples or serums were kept at −80 °C until use. All animal experimental procedures were approved under the guidelines of the Shandong Province Animal Ethics Committee and conducted in accordance with the accepted policies of our institute and Chinese animal care authorities, in addition to the National Institute of Health Guide for the Care and Use of Laboratory Animals.
RNA extraction, RT-PCR amplification and sequencing
PRRSV RNAs were extracted using TRZOL reagent (Invitrogen, Carlsbad, CA) and dissolved in nuclease-free water. Reverse transcription PCR (RT-PCR) was performed with a PrimeScriptTM One Step RT-PCR Kit (Takara, Dalian, China) according to the manufacturer’s instructions. The primers specific for PRRSV ORF7 encode the nucleocapsid (N) protein. The primer sequences are F: 5′-ATGCCAAATAACAACGG-3′, R: 5′-TGCTGAGGGTGATGCTGT-3′. The PCR cycle parameters were as follows: 50 °C for 30 min, 95 °C for 1 min, 94 °C for 1 min, 52 °C for 1 min and 72 °C for 1 min. A 369 nt amplicon was obtained, analyzed by 1.2% agarose gel electrophoresis, stained with ethidium bromide and visualized under UV light. The PCR products were examined by gel electrophoresis and purified using an Agarose Gel DNA Extraction Kit (BioDev Co., Beijing, China) and then subjected to the Biotechnology Research Center Shandong academy of agricultural sciences (Jinan, China) for Sanger sequencing.
Isolation and identification of PRRSV
Three strains of PRRSV (SDhz1512, SDlz1601, and SDYG1606) were isolated from serum using porcine alveolar macrophages (PAMs) as previously described35. The inoculated cells were maintained at 37 °C in a 5% CO2 atmosphere for 72 h. The PRRSV strains were confirmed with an immunofluorescence assay using a monoclonal antibody directed against the PRRSV N protein, which was produced from hybridoma cells derived from Sp2/0 myeloma cells and spleen cells of BALB/c mice immunized with the N protein of PRRSV strain SX-1 (accession no. GQ857656.1).
Primers designed to determine the complete genome sequence
To determine the complete genomes of SDhz1512, SDlz1601 and SDYG1606 strains, 12 primers were designed on the basis of the genomic sequence of NADC30 available in GenBank (accession No. JN654459). Two Chinese isolates (JXA1 and SD1100) available from the National Center of Biotechnology Information (NCBI) were used as references. The primers used for full genome sequencing of the three isolates are shown in Table 6.
Table 6.
Primers used for sequencing the full-length genomes of PRRSV isolates.
| Fragment | Primer | Sequence (5′-3′) | Location |
|---|---|---|---|
| 1 | PRRSV-F1 | atgacgtataggtgttggct | 1–20 |
| PRRSV-R1 | atttaccatccggttggcgat | 1524–1544 | |
| 2 | PRRSV-F2 | acaagtggtatggtgctggga | 1327–1347 |
| PRRSV-R2 | gaagcacaacatcccaatcaaagg | 2194–2217 | |
| 3 | PRRSV-F3 | aaaattgaccagtacctccg | 2103–2122 |
| PRRSV-R3 | ggcggtgtctcgagaatcatctt | 3483–3505 | |
| 4 | PRRSV-F4 | tgatgcgtgaggcatgtgatg | 3328–3348 |
| PRRSV-R4 | ggaaagaaaggagctcggaatggaa | 4765–4789 | |
| 5 | PRRSV-F5 | aaacccatcgcgtatgccc | 4602–4620 |
| PRRSV-R5 | gtctttaattatgtggctgcc | 6180–6200 | |
| 6 | PRRSV-F6 | cattgttacgcgcccttcagg | 6071–6091 |
| PRRSV-R6 | gcggccacagcgggtcaagc | 7740–7759 | |
| 7 | PRRSV-F7 | tcggtatgatgaacgttgacg | 7621–7641 |
| PRRSV-R7 | ggtagtttggcatggagggag | 9268–9288 | |
| 8 | PRRSV-F8 | ttacttcaaaagcggtcaccc | 9146–9166 |
| PRRSV-R8 | ggtgaggacttgcccatatt | 10582–10601 | |
| 9 | PRRSV-F9 | ctgaagaccatctggagatt | 10464–10483 |
| PRRSV-R9 | gggcatctcaccatggtatg | 11834–11853 | |
| 10 | PRRSV-F10 | atttggacccctgcatggccct | 11714–11735 |
| PRRSV-R10 | gacaaaaacgccaaccaagc | 13051–13070 | |
| 11 | PRRSV-F11 | gctcatggtgaattatacggtgt | 12864–12886 |
| PRRSV-R11 | cgaaaaattgcatgtcctggcgc | 14201–14225 | |
| 12 | PRRSV-F12 | tttcctgtgttgactcatattg | 14026–14047 |
| PRRSV-R12 | ttttttttttaatttcggccgcatggttct | polyA |
Sequence assembly and alignment
Consensus sequences were assembled using sequence analysis software (DNAStar, version 7.1, Madison WI). Nucleotide and protein identities were searched with BLASTN and BLASTP programs implemented in the BLAST software package (http://www.ncbi.nlm.nih.gov/blast). The percentage of every ORF and most of the derived amino acids were calculated with other isolates using BioEdit software version 7.2.0 (TomHall, Carlsbad, CA, USA). To explore the evolution of the new PRRSV variant in more detail, we downloaded the full genome sequences (n = 73) from GenBank (Table 7). These genomes were aligned in MAFFT versions 7.263 with all other complete sequences from China.
Table 7.
A total of 76 PRRSV isolates were used in this study.
| No. | Virus strain | Accession no. | Country | Province | Time | No. | Virus strain | Accession no. | Country | Province | Time |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CH1a | AY032626 | China | Heilongjiang | 2000 | 39 | NVDCHBCZ2013 | KP771742 | China | Hubei | 2013 |
| 2 | HN1 | AY457635 | China | Hubei | 2003/11/4 | 40 | NVDCBJPG2013 | KP771743 | China | Beijing | 2013 |
| 3 | JXA1 | EF112445 | China | Jianxi | 2005 | 41 | NVDCHeB22013 | KP771744 | China | Hebei | 2013 |
| 4 | HuN4 | EF517962 | China | Hunan | 2007 | 42 | NVDCHeB12013 | KP771745 | China | Hebei | 2013 |
| 5 | VR2332 | EF536003 | USA | n/a | 2005/6/29 | 43 | NVDCMD22013 | KP771750 | China | n/a | 2013 |
| 6 | TJ | EU860248 | China | Tianjin | 2006/10/6 | 44 | NVDCMD12013 | KP771751 | China | n/a | 2013 |
| 7 | BJSY1 | FJ950744 | China | Beijing | 2007/10/7 | 45 | HEB2013000814 | KP771752 | China | Hebei | 2013 |
| 8 | BJSD | FJ950747 | China | Beijing | 2007/2/17 | 46 | HEB2013000813 | KP771753 | China | Hebei | 2013 |
| 9 | SX1 | GQ857656 | China | Shandong | 2008/12/31 | 47 | NVDC13SXJC2014 | KP771780 | China | Shanxi | 2014 |
| 10 | SD1100 | GQ914997 | China | Shandong | 2008 | 48 | NVDCHuNCS2014 | KP771781 | China | Hunan | 2014 |
| 11 | ZP1 | HM016159 | China | Shandong | 2009/11/24 | 49 | NVDCSD42014 | KP771784 | China | Shandong | 2014 |
| 12 | SD09 | HQ843180 | China | Shandong | 2009/4/9 | 50 | 14LY01FJ | KP780881 | China | Fujian | 2014/10/15 |
| 13 | SX09 | HQ843181 | China | Shanxi | 2009/1/1 | 51 | 14LY02FJ | KP780882 | China | Fujian | 2014/11/12 |
| 14 | ZCYZ | JF800911 | China | Shandong | 2009/12/25 | 52 | CHsx1401 | KP861625 | China | Shanxi | 2014/8/30 |
| 15 | NADC30 | JN654459 | USA | n/a | 2008 | 53 | FJE1 | KP998475 | China | Fujian | 2014 |
| 16 | GX1002 | JQ955658 | China | Guangxi | 2010 | 54 | JXja15 | KR149645 | China | Jiangxi | 2015 |
| 17 | YN2011 | JX857698 | China | Yunnan | 2011 | 55 | JL580 | KR706343 | China | Jilin | 2013 |
| 18 | HENANXINX | KF611905 | China | Henan | 2013/1/1 | 56 | HNxa14 | KT022071 | China | Hainan | 2014 |
| 19 | HenanA1 | KJ002451 | China | Henan | 2013/6/30 | 57 | HNyc13 | KT022072 | China | Hainan | 2013 |
| 20 | HeNanA9 | KJ546412 | China | Henan | 2013/7/4 | 58 | XF1129 | KT180169 | China | n/a | 2013/11/29 |
| 21 | HEB2013 | KJ591659 | China | Hebei | 2013/9/13 | 59 | HLJA1 | KT351739 | China | Henan | 2013/11/12 |
| 22 | MY486 | KJ609516 | China | Henan | 2013/7/23 | 60 | HLJB1 | KT351740 | China | Henan | 2013/1/12 |
| 23 | MY376 | KJ609517 | China | Henan | 2013/7/23 | 61 | GZgy151 | KT358728 | China | Guangdong | 2015 |
| 24 | HenanA12 | KJ819934 | China | Henan | 2014/4/13 | 62 | HNP5 | KT445876 | China | Henan | 2014/7/10 |
| 25 | NMG2014 | KM000066 | China | Neimeng | 2014/4/2 | 63 | HNjz15 | KT945017 | China | Henan | 2015 |
| 26 | HB2014001 | KM261784 | China | Hubei | 2014/3/14 | 64 | HNyc15 | KT945018 | China | Henan | 2015 |
| 27 | BB0907s34 | KM453698 | China | Guangxi | 2014/2/14 | 65 | 15LY01FJ | KU215416 | China | Fujian | 2015 |
| 28 | BB0907F44 | KM453699 | China | Guangxi | 2014/2/14 | 66 | 15LY02FJ | KU215417 | China | Fujian | 2015 |
| 29 | HUN2014 | KP330232 | China | Hunan | 2014/2/20 | 67 | WUH5 | KU523366 | China | Hubei | 2015/9/15 |
| 30 | TJbd141 | KP742986 | China | Tianjin | 2014 | 68 | HENPDS2 | KU950370 | China | Henan | 2015/4/1 |
| 31 | TJbd142 | KP742987 | China | Tianjin | 2014 | 69 | HENXX1 | KU950372 | China | Henan | 2014/12/14 |
| 32 | NVDCSHH022014 | KP771735 | China | Shanghai | 2014 | 70 | HENZK1 | KU950373 | China | Henan | 2014/3/14 |
| 33 | NVDCshh012014 | KP771736 | China | Shanghai | 2014 | 71 | HENZZ8 | KU950375 | China | Henan | 2015/11/15 |
| 34 | NVDCSD62014 | KP771737 | China | Shandong | 2014 | 72 | GDKP | KU978619 | China | Guangdong | 2015/10/12 |
| 35 | NVDCSD12014 | KP771738 | China | Shandong | 2014 | 73 | SDhz1512 | KX980392 | China | Shandong | 2015/12/1 |
| 36 | NVDCSC12014 | KP771739 | China | Sichuan | 2014 | 74 | SDlz1601 | KX980393 | China | Shandong | 2016/1/1 |
| 37 | NVDCSXJC2013 | KP771740 | China | Shanxi | 2013 | 75 | SDYG1606 | KY053458 | China | Shandong | 2016/6/1 |
| 38 | NVDCSDXX2013 | KP771741 | China | Shandong | 2013 | 76 | PRRSVLV421 | AY588319 | Netherlands | n/a | 38079 |
n/a: not a vailable.
NSP2 amino acid sequence alignment, secondary structure and 3D-structure prediction
The NSP2 amino acid sequences of the three PRRSV isolates (SDhz1512, SDlz1601 and SDYG1606) were analyzed by Clustal W software and compared with the standard reference strain sequences. The secondary structure was predicted by the matrix algorithm with the best structure or base pairing using Protein software (DNAStar, version 7.1, Madison WI). The amino acid sequences of the three PRRSV isolates (SDhz1512, SDlz1601 and SDYG1606) and the reference strains (VR2332, JXA1 and NADC30) were submitted to homology modeling of I-TASSER Server21,22 for 3D-structure prediction.
Recombination analysis
RDP4.70 software36 was used to test for potential recombination sequences, their probable parental sequences, and the positions of breakpoints. The methods included RDP37, GENECONV38, MaxChi39, Bootscan/Recscan39, SiScan40 and 3Seq41. Default settings were used and the threshold p-value set at 0.05 using the Bonferroni correction. To avoid false positive results, recombination events supported by at least six different methods were considered. The putative recombination events and their breakpoint positions were further validated and confirmed via the SimPlot program (version 3.5.1), which performs similarity comparisons within a 500-bp window sliding along the genome (10 bp step size) using the similarity score and Bootscan plot42 based on 26 representative genome sequences. The parental sequences detected were further checked for the presence of recombination. A cluster analysis maximizing the value of x2 was then used to select breakpoints among the clusters38. These breakpoints were used to divide the alignment into segments for phylogenetic tree construction.
Pathogenicity study of the three PRRSV isolated strains
To determine the pathogenicity differences among SDhz1512, SDlz1601 and SDYG1606, an animal experiment was designed. Twenty four-week-old piglets were randomly divided into four groups and maintained in individual rooms. All the piglets were negative for antibodies of porcine circovirus type 2 (PCV2) and PRRSV before the experiment. Each piglet in the PRRSV strain group (SDhz1512, SDlz1601 or SDYG1606) was intranasally administered 2 ml of the virus containing 2 × 105 TCID50. Each animal in the control group was given the same dosage of PAM culture supernatant. The viremia of PRRSV-inoculated animals was detected at 0, 3, 7, 14, and 21 dpi by an IFA-microtitration infectivity assay. The animals were euthanized and lung samples were collected for histopathology at 14 dpi.
Phylogenetic classification of PRRSV
The ORF7 nucleotide phylogenetic and molecular evolutionary analyses of 243 positive PRRSVs were conducted using MEGA version 7, along with 54 sequences of representative PRRSVs available in GenBank from various countries and areas.
Seventy-five complete genomes representing the main PRRSV strains were obtained from GenBank. Multiple sequence alignments were performed in MAFFT version 7.263, and the nucleotide substitution model was evaluated by j-ModelTest v2.1.10 using the Akaike Information Criterion (AIC)43. A general-time reversible (GTR) model of nucleotide substitution was used with a gamma-distributed (Γ) rate variation among sites, with a proportion of invariant sites (I). A maximum likelihood (ML) tree was reconstructed using RaxML v8.2.1044, and its confidence was evaluated by 1000 bootstraps.
Time dynamic evolution analysis of PRRSV
According to the description of Shi M et al.13 and Sook Hee Yoon26, 75 NSP2 gene sequences of genotype II PRRSVs from 2000 to 2016 of China obtained from GenBank were used to estimate the evolutionary history of PRRSV in China. Alignments with recombinant regions removed were utilized for phylogenetic analyses using the Bayesian Markov chain Monte Carlo (MCMC) approach available in BEAST v1.8.245. A Bayesian skyline non-parametric coalescent model and uncorrelated lognormal (UCLN) relaxed molecular clock were selected. Bayesian MCMC chains were run for 500 million generations, 10% of which were removed as burn-in, and sampled every 50,000 steps. Convergence and uncertainty in parameter estimates were evaluated by calculating the active sample size46 in Tracer v1.6.
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines, please flag it as inappropriate.
Electronic supplementary material
Acknowledgements
This study was supported by the National Natural Science Funds (31340047, 31572550, 31600106), the National Key Research and Development Program of China (2017YFD0500602), the Taishan Scholars Project, the Shandong Provincial Modern Agricultural Industry and Technology System (SDAIT), the Major Science and Technology Achievement Plan (2014CGPY04), the Shandong Province Major Application of Agricultural Technology Innovation Projects, and the Agricultural Science and Technology Innovation Project of Shandong Academy of Agricultural Sciences (CXGC2016B14).
Author Contributions
Y.L. performed the experiments and wrote the relative manuscript; J.L. was responsible for the animal challenge; J.Y. prepared the reagents and helped with virus separation; H.Z., L.G. and S.R. were responsible for clinical samples collection and RT-PCR amplification; W.S., Z.C. and X.C. were involved in data discussion; L.C. and J.S. prepared Table 4 and Figure. 2; Y.D. and J.L. corrected the manuscript; J.Y. guided the virus isolation, sequence analysis and wrote the manuscript; and J.W and J.W. initiated the study, designed the experiments and supplied the manuscript. All authors reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Yanyan Liu and Jianda Li contributed equally to this work.
A correction to this article is available online at https://doi.org/10.1038/s41598-018-25625-z.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-22494-4.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
5/23/2018
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has been fixed in the paper.
Contributor Information
Jinbao Wang, Email: wangjb@saas.ac.cn.
Jiaqiang Wu, Email: wujiaqiang2000@sina.com.
Jiang Yu, Email: yujiang_2213@163.com.
References
- 1.Dea S, Gagnon CA, Mardassi H, Pirzadeh B, Rogan D. Current knowledge on the structural proteins of porcine reproductive and respiratory syndrome (PRRS) virus: comparison of the North American and European isolates. Archives of virology. 2000;145:659–688. doi: 10.1007/s007050050662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou L, Yang H. Porcine reproductive and respiratory syndrome in China. Virus research. 2010;154:31–37. doi: 10.1016/j.virusres.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 3.Snijder EJ, Meulenberg JJ. The molecular biology of arteriviruses. The Journal of general virology. 1998;79(Pt 5):961–979. doi: 10.1099/0022-1317-79-5-961. [DOI] [PubMed] [Google Scholar]
- 4.Benfield DA, et al. Characterization of swine infertility and respiratory syndrome (SIRS) virus (isolate ATCC VR2332) Journal of veterinary diagnostic investigation: official publication of the American Association of Veterinary Laboratory Diagnosticians, Inc. 1992;4:127–133. doi: 10.1177/104063879200400202. [DOI] [PubMed] [Google Scholar]
- 5.Kappes MA, Faaberg KS. PRRSV structure, replication and recombination: Origin of phenotype and genotype diversity. Virology. 2015;479-480:475–486. doi: 10.1016/j.virol.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han K, et al. Comparison of the virulence of European and North American genotypes of porcine reproductive and respiratory syndrome virus in experimentally infected pigs. Veterinary journal. 2013;195:313–318. doi: 10.1016/j.tvjl.2012.06.035. [DOI] [PubMed] [Google Scholar]
- 7.Han K, et al. Comparative virulence of reproductive diseases caused by type 1 (European-like) and type 2 (North American-like) porcine reproductive and respiratory syndrome virus in experimentally infected pregnant gilts. Journal of comparative pathology. 2014;150:297–305. doi: 10.1016/j.jcpa.2013.11.205. [DOI] [PubMed] [Google Scholar]
- 8.Martinez-Lobo FJ, et al. Comparative pathogenicity of type 1 and type 2 isolates of porcine reproductive and respiratory syndrome virus (PRRSV) in a young pig infection model. Veterinary microbiology. 2011;154:58–68. doi: 10.1016/j.vetmic.2011.06.025. [DOI] [PubMed] [Google Scholar]
- 9.Russell P, Atkinson K, Krishnan L. Recurrent reproductive failure due to severe placental villitis of unknown etiology. The Journal of reproductive medicine. 1980;24:93–98. [PubMed] [Google Scholar]
- 10.Wensvoort G, et al. Mystery swine disease in The Netherlands: the isolation of Lelystad virus. The Veterinary quarterly. 1991;13:121–130. doi: 10.1080/01652176.1991.9694296. [DOI] [PubMed] [Google Scholar]
- 11.Valicek L, et al. Isolation and identification of porcine reproductive and respiratory syndrome virus in cell cultures. Veterinarni medicina. 1997;42:281–287. [PubMed] [Google Scholar]
- 12.Chen J, Liu T, Zhu CG, Jin YF, Zhang YZ. Genetic variation of Chinese PRRSV strains based on ORF5 sequence. Biochemical genetics. 2006;44:425–435. doi: 10.1007/s10528-006-9039-9. [DOI] [PubMed] [Google Scholar]
- 13.Shi M, Holmes EC, Brar MS, Leung FC. Recombination is associated with an outbreak of novel highly pathogenic porcine reproductive and respiratory syndrome viruses in China. Journal of virology. 2013;87:10904–10907. doi: 10.1128/JVI.01270-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu X, et al. New genomic characteristics of highly pathogenic porcine reproductive and respiratory syndrome viruses do not lead to significant changes in pathogenicity. Veterinary microbiology. 2012;158:291–299. doi: 10.1016/j.vetmic.2012.02.036. [DOI] [PubMed] [Google Scholar]
- 15.Zhou L, et al. NADC30-like Strain of Porcine Reproductive and Respiratory Syndrome Virus, China. Emerging infectious diseases. 2015;21:2256–2257. doi: 10.3201/eid2112.150360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, et al. Complete Genome Sequence of an NADC30-Like Porcine Reproductive and Respiratory Syndrome Virus Characterized by Recombination with Other Strains. Genome announcements. 2016;4(2):e00303–16. doi: 10.1128/genomeA.00330-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L, et al. The Chinese highly pathogenic porcine reproductive and respiratory syndrome virus infection suppresses Th17 cells response in vivo. Veterinary microbiology. 2016;189:75–85. doi: 10.1016/j.vetmic.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 18.Li C, et al. Outbreak Investigation of NADC30-Like PRRSV in South-East China. Transboundary and emerging diseases. 2016;63:474–479. doi: 10.1111/tbed.12530. [DOI] [PubMed] [Google Scholar]
- 19.Zhao K, et al. Importation and Recombination Are Responsible for the Latest Emergence of Highly Pathogenic Porcine Reproductive and Respiratory Syndrome Virus in China. Journal of virology. 2015;89:10712–10716. doi: 10.1128/JVI.01446-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han J, Zhou L, Ge X, Guo X, Yang H. Pathogenesis and control of the Chinese highly pathogenic porcine reproductive and respiratory syndrome virus. Veterinary microbiology. 2017;209:30–47. doi: 10.1016/j.vetmic.2017.02.020. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X, et al. Recombination and natural selection in hepatitis E virus genotypes. Journal of medical virology. 2012;84:1396–1407. doi: 10.1002/jmv.23237. [DOI] [PubMed] [Google Scholar]
- 24.Zhao H, et al. Emergence of mosaic recombinant strains potentially associated with vaccine JXA1-R and predominant circulating strains of porcine reproductive and respiratory syndrome virus in different provinces of China. Virology journal. 2017;14:67. doi: 10.1186/s12985-017-0735-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cha SH, et al. Molecular characterization of recent Korean porcine reproductive and respiratory syndrome (PRRS) viruses and comparison to other Asian PRRS viruses. Veterinary microbiology. 2006;117:248–257. doi: 10.1016/j.vetmic.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 26.Zhu L, et al. Complete genomic characterization of a Chinese isolate of porcine reproductive and respiratory syndrome virus. Veterinary microbiology. 2011;147:274–282. doi: 10.1016/j.vetmic.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Yoon SH, et al. Genetic characterization of the Korean porcine reproductive and respiratory syndrome viruses based on the nucleocapsid protein gene (ORF7) sequences. Archives of virology. 2008;153:627–635. doi: 10.1007/s00705-007-0027-0. [DOI] [PubMed] [Google Scholar]
- 28.Hao X, et al. Polymorphic genetic characterization of the ORF7 gene of porcine reproductive and respiratory syndrome virus (PRRSV) in China. Virology journal. 2011;8:73. doi: 10.1186/1743-422X-8-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou L, et al. Efficacy evaluation of three modified-live virus vaccines against a strain of porcine reproductive and respiratory syndrome virus NADC30-like. Veterinary microbiology. 2017;207:108–116. doi: 10.1016/j.vetmic.2017.05.031. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Q, et al. Pathogenicity and antigenicity of a novel NADC30-like strain of porcine reproductive and respiratory syndrome virus emerged in China. Veterinary microbiology. 2016;197:93–101. doi: 10.1016/j.vetmic.2016.11.010. [DOI] [PubMed] [Google Scholar]
- 31.Zhou L, et al. The 30-amino-acid deletion in the Nsp2 of highly pathogenic porcine reproductive and respiratory syndrome virus emerging in China is not related to its virulence. Journal of virology. 2009;83:5156–5167. doi: 10.1128/JVI.02678-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alkhamis MA, Perez AM, Murtaugh MP, Wang X, Morrison RB. Applications of Bayesian Phylodynamic Methods in a Recent U.S. Porcine Reproductive and Respiratory Syndrome Virus Outbreak. Frontiers in microbiology. 2016;7:67. doi: 10.3389/fmicb.2016.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nielsen J, Botner A, Bille-Hansen V, Oleksiewicz MB, Storgaard T. Experimental inoculation of late term pregnant sows with a field isolate of porcine reproductive and respiratory syndrome vaccine-derived virus. Veterinary microbiology. 2002;84:1–13. doi: 10.1016/S0378-1135(01)00450-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian K, et al. Emergence of fatal PRRSV variants: unparalleled outbreaks of atypical PRRS in China and molecular dissection of the unique hallmark. PloS one. 2007;2:e526. doi: 10.1371/journal.pone.0000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Labarque GG, Nauwynck HJ, Van Reeth K, Pensaert MB. Effect of cellular changes and onset of humoral immunity on the replication of porcine reproductive and respiratory syndrome virus in the lungs of pigs. The Journal of general virology. 2000;81:1327–1334. doi: 10.1099/0022-1317-81-5-1327. [DOI] [PubMed] [Google Scholar]
- 36.Martin DP, et al. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26:2462–2463. doi: 10.1093/bioinformatics/btq467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Padidam M, Sawyer S, Fauquet CM. Possible emergence of new geminiviruses by frequent recombination. Virology. 1999;265:218–225. doi: 10.1006/viro.1999.0056. [DOI] [PubMed] [Google Scholar]
- 38.Smith JM. Analyzing the mosaic structure of genes. Journal of molecular evolution. 1992;34:126–129. doi: 10.1007/BF00182389. [DOI] [PubMed] [Google Scholar]
- 39.Martin DP, Posada D, Crandall KA, Williamson C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS research and human retroviruses. 2005;21:98–102. doi: 10.1089/aid.2005.21.98. [DOI] [PubMed] [Google Scholar]
- 40.Gibbs MJ, Armstrong JS, Gibbs AJ. Sister-scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics. 2000;16:573–582. doi: 10.1093/bioinformatics/16.7.573. [DOI] [PubMed] [Google Scholar]
- 41.Boni MF, Posada D, Feldman MW. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics. 2007;176:1035–1047. doi: 10.1534/genetics.106.068874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lole KS, et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. Journal of virology. 1999;73:152–160. doi: 10.1128/jvi.73.1.152-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Posada D. jModelTest: phylogenetic model averaging. Molecular biology and evolution. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- 44.Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005;21:456–463. doi: 10.1093/bioinformatics/bti191. [DOI] [PubMed] [Google Scholar]
- 45.Bose ME, et al. Sequencing and analysis of globally obtained human respiratory syncytial virus A and B genomes. PloS one. 2015;10:e0120098. doi: 10.1371/journal.pone.0120098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Melander A, et al. 35th Annual Meeting of the European Association for the Study of Diabetes: Brussels, Belgium, 28 September-2 October 1999. Diabetologia. 1999;42:A1–A330. doi: 10.1007/BF03375458. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.