

Abstract
The three human monoamine transporters (MATs) i.e. the dopamine transporter (DAT), the serotonin transporter (SERT) and the norepinephrine transporter (NET) are primarily responsible for controlling the neurotransmission mediated by their respective monoamine substrates, dopamine (DA), serotonin (5-HT) and norepinephrine (NE). MATs terminate the action of the neurotransmitters by translocating them from the synaptic space back into the presynaptic neurons. These neurotransmitters are responsible for controlling a number of physiological, emotional and behavioral functions and their transporters are a target of many marketed drugs prescribed for treatment and relief of disorders such as depression, anxiety, ADHD, schizophrenia, and psychostimulant abuse. This unit provides an overview of the location and regulation of the MATs and the structural aspects that mediate the translocation process. A brief description of the evolution of ligands that target these transporters is provided as well as more current insights regarding pharmacological effects, mechanisms of synthetic substrates, inhibitors and allosteric modulators. This unit also summarizes known data and evidence that provide structural details on the existence and significance of allosteric binding sites other than the natural substrate binding site. The purpose of this unit is to introduce researchers new to this field to basic background information on MATs, and the significance and application of unique effects elicited by ligands that interact with these transporters.
Keywords: Transporters, Dopamine, Serotonin, Norepinephrine, Inhibitors, Allosteric modulators, substrates, releasers
Introduction
The monoamine neurotransmitter transporters (MATs) are part of the solute carrier 6 (SLC6) family of transporters (Kristensen et al., 2011; Rudnick et al., 2014). They are expressed in the central nervous system (CNS) among other places, where they play a critical al role in regulating monoamine neurotransmitter homeostasis. There are three types of MATs, namely dopamine transporter (DAT, SLC6A3), norepinephrine transporter (NET, SLC6A2), and serotonin transporter (SERT, SLC6A4) which mediate the uptake of monoamine neurotransmitters dopamine (DA), norepinephrine (NE), and serotonin (5-HT), respectively, from the extracellular space into the intracellular compartment (Figure 1). All three transporters are membrane-embedded proteins and in the CNS exclusively expressed in the presynaptic neuronal terminals of their respective pathways. They mediate a relatively rapid uptake of neurotransmitters (turnover rate ~1 molecule/second) from the synaptic cleft, into the pre-synaptic neuronal terminals, where the neurotransmitters are sequestered into synaptic vesicles (via vesicular monoamine transporters or VMAT) for recycling or are degraded by monoamine oxidase enzymes. The translocation of monoamine substrates involves co-transport of Na+ ions with one molecule of substrate through the transporters along with other ions. The transport of substrates by the transporters is favored by the energy gradient produced by the movement of Na+ ions inside the cell, and driven by the concentration gradient created by Na+/K+ ATPase. DAT and NET transport one dopamine or norepinephrine molecule along with two Na+ ions and one Cl− ion, whereas SERT co-transports one 5-HT molecule with one Na+ and one Cl− together with one K+ ion in the opposite direction. Thus, MATs are sometimes also referred to as Na+/ Cl−-symporters.
Figure 1.

Chemical structures of the three monoamine neurotransmitters.
The MATs play a pivotal role in controlling the signal amplitude and duration of monoaminergic neurotransmission by altering the concentration of monoamines in the extracellular space of the CNS. Therefore, direct or indirect modulation of the MATs can significantly affect the regulation of neuronal activity (Howell & Negus, 2014). There are a plethora of compounds that are used as therapeutic drugs or as pharmacological tools to modulate or control monoamine neurotransmission in the brain (Table 1). In addition, the MATs are also the primary targets of action of a number of psychostimulants and recreational drugs of abuse such as cocaine, methamphetamine, 3,4-methylenedioxy–methamphetamine (“ecstasy” or MDMA), cathinones (or “bath salts”) and many more which block or reverse the transport of neurotransmitters and increase the synaptic neurotransmission leading to stimulatory effects.
Table 1.
Commonly known ligands of monoamine transporters.
| Transporters | DAT | NET | SERT | |||
|---|---|---|---|---|---|---|
| Endogenous Substrates | dopamine (DA) | norepinephrine (NE), dopamine (DA) | 5-hydroxytryptamine (5-HT, serotonin) | |||
| Synthetic substrates | amphetamine, methamphetamine, 1-methyl-4-phenylpyridinium (MPP+) | amphetamine, methamphetamine, 1-methyl-4-phenylpyridinium (MPP+) | MDMA, 1-methyl-4-phenylpyridinium (MPP+) | |||
| Releasers or efflux promoters | amphetamine, MDMA, phenmetrazine | amphetamine, MDMA, phenmetrazine | amphetamine, MDMA, 1-(3-trifluoromethylphenyl) Piperazine (TPP) | |||
| Inhibitors | Compound | Function | Compound | Function | Compound | Function |
| Cocaine | Drug of abuse | Nisoxetine | Pharmacological tool | Paroxetine | Therapeutic drug | |
| Bupropion | Therapeutic drug | Talopram | Pharmacological tool | Sertraline | Therapeutic drug | |
| GBR12935 | Pharmacological tool | Reboxetine | Therapeutic drug | citalopram | Therapeutic drug | |
| Methylphenidate | Therapeutic drug | Nomifensin | Therapeutic drug | Fluoxetine | Therapeutic drug | |
| WIN35428 | Pharmacological tool | Mazindol | Therapeutic drug | Fluvoxamine | Therapeutic drug | |
| RTI-55 | Pharmacological tool | Amphetamine | Drug of abuse | amitriptyline | Therapeutic drug | |
| nomifensin | Therapeutic drug | MDMA | Drug of abuse | desipramine | Therapeutic drug | |
| benztropine | Pharmacological tool | methylphenidate | Therapeutic drug | trimipramine | Therapeutic drug | |
| JHW 007 | Pharmacological tool | Clomipramine | Therapeutic drug | Clomipramine | Therapeutic drug | |
| Commonly used radiolabelled inhibitors | [3H]-GBR12935, [3H]-WIN35428, [3H]-RTI-55 | [3H]-mazindol, [3H]-nisoxetine, [3H]-RTI-55 | [3H]-paroxetine, [3H]-citalopram, [3H]-RTI-55 | |||
As evident from Table 1, a number of drug discovery efforts have resulted in the availability of inhibitors for DAT, NET and SERT that have high affinity and selectivity. Tricyclic antidepressants (TCAs) (for e.g., clomipramine and amitriptyline) and the selective inhibitors of SERT (also known as selective serotonin-reuptake inhibitors or SSRIs) such as fluoxetine, sertraline, escitalopram and paroxetine are commonly prescribed for depression, anxiety, and panic disorders. Bupropion, a DAT inhibitor, is an anti-depressant and smoking cessation aid. Methylphenidate, another DAT-inhibitor is marketed for attention-deficit hyperactivity disorder (ADHD). The NET inhibitor reboxetine is approved and marketed for depression, panic disorders and ADHD outside of the U.S. Synthetic compounds that act as non-selective substrates of MATs and promote monoamine efflux have also found application for clinical use. For example, Adderall® (a racemic mixture of amphetamine isomers) is prescribed in low doses to treat ADHD, and is commonly used off-label to improve cognition and to help in narcolepsy by promoting wakefulness. However, such drugs along with many other cognition-enhancing drugs that interact with MATs are strictly controlled and regulated because of their rewarding properties and abuse liability.
Apart from a plethora of known MAT-inhibitors as therapeutic drugs, there are many highly potent and selective blockers of MATs that are commonly used as important pharmacological tools (some in the form of radioactive or fluorescence-based ligands) for in vitro and in vivo studies related to MAT’s biochemistry and molecular pharmacology. Despite several years of research in studying MATs structure and function, there is still information lacking about the exact mechanism of transport and inhibition of the MATs and the structure of their binding sites. The breakthrough discoveries such as co-crystal structures of the homologous bacterial (Aquifex aeolicus) leucine transporter (LeuT) (Singh et al., 2007; Yamashita et al., 2005; Zhou et al., 2007), Drosophila melanogaster dopamine transporter (dDAT) (Penmatsa et al., 2013, 2015; K. H. Wang et al., 2015), and human serotonin transporter (hSERT) (Coleman et al., 2016) have been pivotal in enhancing our understanding of the structural biology of the MATs and in guiding further studies to elucidate underlying molecular mechanisms of ligand transport and interaction.
The aim of this overview is to provide a brief background on DAT, SERT and NET to introduce the readers to the human monoamine transporters and their significance in CNS-related diseases. Information on a number of marketed CNS-drugs that target these transporters for their respective therapeutic applications is also provided. Special emphasis has been given to the recent elucidation of structural characteristics and transport mechanisms utilized by these transporters. This unit also covers details on what is currently known about the unique pharmacological effects of substrates, inhibitors as well as allosteric modulators of MATs.
Localization
All three monoamine transporters are distributed throughout the brain, but data shows that NET and SERT are also found in adrenal chromaffin cells, mast cells and blood platelets (Ramamoorthy et al., 2011). Due to their extensive distributions in the brain, they control a wide variety of physiological and behavioral functions. In the CNS, MATs are exclusively expressed in their corresponding monoaminergic neurons that have projections throughout the neocortex, basal ganglia and limbic forebrain areas (Lin et al., 2011). These monoaminergic projections interact with and innervate other neurons in the brain in the cortex, hippocampus, amygdala and hypothalamus. Dopaminergic neurons that express DAT project from the VTA (ventral tegmental area) and substantia nigra to pre-frontal cortex, nucleus accumbens and striatum. These projections control functions such as memory, planning, attention, motivation, reward and reinforcement processes, and movement. SERT is expressed in the serotonergic neurons that project from raphe nuclei of pons and upper brain stem to hypothalamus, thalamus, amygdala, striatum, cortical mantle and many other regions. These neurons regulate mood, emotion, learning, cognition, memory, sleep, and appetite. Other than its presence in the serotonergic neurons in the central nervous system, SERT is also known to be expressed in many peripheral sites such as the gut, lung, placenta, platelets, blood lymphocytes, and adrenal chromaffin cells (Ramamoorthy et al., 2011). NET is expressed in noradrenergic neurons present in the locus coeruleus and the lateral tegmental group that innervate regions such as striatum, amygdala, hypothalamus, thalamus, cerebellum, VTA, brain stem, spinal cord, and cortex. These neurons control processes such as modulation of arousal, sleep-wake cycle, and cognition. Noradrenergic neurons also mediate the stress and fear-related response by controlling the endocrine and the autonomic nervous system.
Structural Insights and Transport Mechanism
In the early 1990’s, the complementary DNAs (cDNAs) of NET, DAT and SERT were cloned for the first time from rat (Kilty et al., 1991) following the methods used successfully to clone cDNAs of rat and human brain GABA transporters. The availability of the primary amino acid sequences of the MATs thereafter increased the impetus to delineate the structural features of the transporters involved in ligand binding and transport through extensive biochemical and mutagenesis studies (Torres et al., 2003). Since the primary sequences of DAT, NET and SERT were found to have high amino acid similarity (~40%), all three transporters were proposed to possess a very similar general structure (Figure 2). A major breakthrough in the SLC6 transporter field occurred when, in 2005, the first x-ray crystal structure of the bacterial leucine transporter (LeuT) that shares 20–25 % overall homology with the MATs was published (Yamashita et al., 2005). This structure, for the first time, gave insights into the tertiary arrangement of the transporters and their functioning, especially of the core region which shares ~ 60% homology with the MATs. This discovery has since triggered several homology-based molecular modeling studies to reveal the structure and mechanisms of the MATs using the LeuT structure as the template (Manepalli et al., 2012). Subsequently, over the years, an arsenal of new crystal structures bound with various ligands have been published which have further provided much clearer insight into the ligand binding sites, conformational changes and transport mechanism of the MATs (Table 2) (Koldso et al., 2015). More recent structures are the co-crystal structures of Drosophila melanogaster DAT (dDAT bound to the substrate DA, D-amphetamine, methamphetamine, cocaine, and other ligands) which has 50–55% homology with the MATs (Penmatsa et al., 2013, 2015; K. H. Wang et al., 2015), and the human SERT in complex with paroxetine and escitalopram (Coleman et al., 2016).
Figure 2.

Alignment of primary sequences of hDAT, hSERT, hNET and bacterial LeuT.
Table 2.
A list of relevant x-ray co-crystal structures of transporters discovered to date bound to various ligands.
| Transporter | Ligand | Binding site/Conformation | Reference |
|---|---|---|---|
| LeuT | Leucine | Substrate binding site (S1), outward-occluded | (Yamashita et al., 2005) |
| LeuT | Tryptophan | 2 molecules bound, one at S1 and other at the external vestibule site, outward-open | (Singh et al., 2008) |
| LeuT | n-octyl-β-D-glucopyranoside detergent | External vestibule site, outward-open | (Quick et al., 2009) |
| LeuT | TCAs (desipramine, clomipramine, imipramine), SSRIs (sertraline, (R)/(S)-fluoxetine) | External vestibule site, outward-open | (Singh et al., 2007) |
| LeuBAT | Antidepressants (SSRIs, SNRIs, Clomipramine), Mazindol | S1, outward-open | (H. Wang et al., 2013) |
| dDAT | 3,4-dichlorophenethylamine (DCP, a dopamine analog) | S1, outward-partially occluded | (K. H. Wang et al., 2015) |
| dDAT | cocaine, RTI-55, SNRIs, NRIs (nisoxetine, reboxetine), SSRIs, nortriptyline, dopamine, D-amphetamine, (+)-methamphetamine | S1, outward-open | (Penmatsa et al., 2015; K. H. Wang et al., 2015) |
| hSERT | (S)-citalopram | 2 molecules bound, one at S1 and other at external vestibular site, outward-open | (Coleman et al., 2016) |
| hSERT | paroxetine | S1, outward-open | (Coleman et al., 2016) |
The structure of all MATs includes 12 alpha-helical transmembrane spanning domains connected with flexible intracellular and extracellular loops (Figure 3). The N- and C-termini lie in the intracellular region. The high affinity primary substrate binding site, the S1 site, lies at the core of the translocation pathway located between TM1 and TM6. The S1 site binds a substrate together with one or two Na+ ions. The S1 pocket is comprised of a hydrophobic region, which binds the aromatic substituents of the substrates, and a hydrophilic or a polar region that contains a conserved aspartate residue (SERT: D98, DAT: D79, NET: D75) to form ionic interactions with the amino group of the substrates.
Figure 3.

A general 2D representation of the 12-transmembrane domain structure of monoamine transporters showing the placement of N-terminal, C-terminal, extracellular- and intracellular loops.
Substrate translocation by these transporters is believed to follow a three-state “alternating access” mechanism (Figure 4) (Kristensen et al., 2011). This implies that the transporter is capable of adopting distinct conformations in which it can seal the access to the S1 binding site from either extracellular or intracellular side of the membrane in an alternating manner. The gating networks, formed by two pairs of charged residues along with a few hydrophobic amino acids, present above and below the S1 site, mediate these conformations. It has been proposed that the influx of substrates and ions is achieved through a series of sequential binding and conformational changes. This sequence is initiated by the binding of a Na+ ion on the extracellular side which is followed by the substrate binding within the central S1 site when the transporter adopts an outward-facing conformation exposed to the extracellular side (state 1). Substrate binding to S1 is the key for further triggering conformational changes in the transporter which closes the extracellular gate of the transporter to form an occluded-state in which the ions and the substrate lie in the middle of the channel and are protected from the either side of the membrane by gating networks (state 2). Next, the occluded state is followed by the opening of the intracellular gate, leading to an inward-facing conformation, which releases the ions and substrate into the cytoplasm via diffusion facilitated by hydration of the site (state 3).
Figure 4.

Cartoon representation of the proposed alternating access mechanism of translocation of substrates and ions through monoamine transporters.
The MATs are proposed to have another low affinity binding site in the extracellular vestibular region that lies in the solvent-accessible pathway which connects the extracellular milieu of the transporter with the S1 site (Shi et al., 2008). Though the exact molecular and structural determinants of ligand binding to this region of the transporter are unknown, there is evidence that several inhibitors can bind in this extracellular vestibular region. For example, this putative secondary binding site region (which we will refer to as LeuT/TCA site in this review) is shown to be occupied by several TCAs and SSRIs in molecular modeling and x-ray crystal structures of bacterial LeuT (Loland, 2015; Singh et al., 2007; Zhou et al., 2007). This LeuT/TCA site is formed by LeuT residues R30, Q34, F253, A319, F320, L400, D401, and D404. However, co-crystal structures of dDAT in complex with DA, amphetamine, cocaine, tricyclic antidepressants and several other inhibitors only show the occupancy of the S1 site (K. H. Wang et al., 2015) which is in contrast to the LeuT structures which are co-crystallized with antidepressants in the secondary LeuT/TCA-site located in the extracellular vestibule. It is important to note that the affinity of certain inhibitors to LeuT is much lower compared to its mammalian counterparts. Thus, it is very unlikely that the LeuT/TCA-site of LeuT for which the ligands have low-affinity will correspond to a similar secondary binding site in human MATs for which they will have high affinity. Though the functional relevance of the binding sites present in this extracellular vestibule region is still being explored in human MATs, evidence shown by the recent hSERT crystal structure with two (S)-citalopram molecules simultaneously bound (one in the S1 site and the other in the extracellular vestibule region) strongly indicates the presence of distinct low-affinity ligand binding sites (similar to the LeuT/TCA-site) in the extracellular region within MATs other than the high-affinity primary binding site (i.e. S1). The drastic increase in affinity of some bivalent ligands for SERT and DAT (Andersen et al., 2016) compared to the common monovalent ligands also supports the presence of distinct low-affinity ligand binding sites. These results add to the earlier data on antagonist dissociation experiments of SERT that showed that there is a low-affinity allosteric site in SERT that slows the dissociation of inhibitors from a separate high-affinity site (Plenge et al., 2012).
Though the role of S1 in driving the alternating access mechanism of translocation is proven, it is yet to be determined if binding of substrates to secondary binding sites is also necessary to trigger substrate translocation. Molecular dynamics studies using LeuT crystal structure revealed that along with a leucine molecule present in the primary binding site, another leucine simultaneous can occupy a secondary substrate binding site (designated as LeuT/S2) lined by hydrophobic residues i.e. Leu29, Tyr107, Ile111, Trp114, Ala319, Phe320, Phe324, and Leu400, and hydrophilic resides i.e., Asp404 and Arg30 (the residues that form the extracellular gate in LeuT) (Loland, 2015; Shi et al., 2008). This second leucine binding site (LeuT/S2) in the extracellular vestibule, that lies on top of the extracellular gate, is proposed to “allosterically” trigger conformational change in the LeuT transporter from substrate-bound occluded state to inward-open state which facilitates the release of the substrate and Na+ ions through hydration of the primary site from the intracellular side. The binding of a Na+ ion from the extracellular side then reorganizes the transporter to the extracellular-open conformation to begin another transport cycle. Interestingly, this proposed leucine-bound LeuT/S2 site lies adjacent to the LeuT/TCA site and both have some structural features in common. However, the binding mode of TCAs, such as clomipramine, to the LeuT/TCA-site is proposed to be different from that of leucine. Thus, TCAs, when bound to the LeuT/TCA-site, functionally act as inhibitors of the substrate transport probably by inhibiting the conformational change that facilitates the substrate and ion release into the intracellular side (Loland, 2015). In addition to the studies performed in LeuT to elucidate the functional relevance of secondary sites, combined computational and mutagenesis approaches have shown that SERT also contains a secondary binding site (which we will refer to as the SERT/A1 site in this review) that is located in the extracellular vestibule which is analogous to the LeuT/S2 site (Plenge et al., 2012). SERT ligands such as 5-HT, and some TCAs and SSRIs, that exert an allosteric effect by functionally modulating the dissociation of ligands bound to the high affinity primary site, were proposed to bind to this secondary site SERT/A1 (Plenge et al., 2012).
Mutational and computational studies identified some key residues that constitute SERT/A1 site such as L99, W103, I179, A486, V489, K490 and G402 along with R104 and E493, which are a part of the extracellular gate (Figure 5). This SERT/A1 site closely resembles the earlier proposed LeuT/S2 site. Mutation at these residues tend to affect substrate transport, decrease allosteric potency of ligands bound to the secondary site as well as reduce dissociation rates of ligands bound to the primary site but do not affect the binding affinity of ligands towards the primary site (Plenge et al., 2012). However, direct or indirect effects of such mutations through long range conformational changes still cannot be proven. Interestingly, in another study, molecular dynamics simulations and comparative genomics techniques identified one more allosteric pocket (Kortagere et al., 2013; Mortensen & Kortagere, 2015) in the extracellular vestibule of SERT (we will refer to this site as SERT/A2). This SERT/A2 pocket, which is conserved in all MATs, is lined by residues Q111, N112, I327, D328, A331, Q332, K490, and E494 (Figure 5). Virtual screening identified a few low potency ligands, which engage with this SERT/A2 site to specifically modulate the function of SERT ligands known to bind to the high-affinity S1 site.
Figure 5.

Side (left) and top (right) views of the human SERT crystal structure bound with two escitalopram molecules (PDB ID 5I73), with a few protein backbone structures truncated for the ease of visibility. One escitalopram molecule (purple) is bound to the primary, high-affinity S1 binding site with residues shown in yellow. The residues of the secondary allosteric site SERT/A1 in the extracellular vestibule are depicted in blue. The SERT/A2 site residues are shown in orange. The second escitalopram molecule of the crystal structure bound in the extracellular vestibule is shown in green.
The recently resolved x-ray crystal structure of human SERT co-crystallized with (S)-citalopram adopts an outward-open conformation bound with two molecules of (S)-citalopram (Coleman et al., 2016) (Figure 5). One molecule of (S)-citalopram occupies the primary substrate binding site S1 which is analogous to the central binding sites seen in LeuT and dDAT structures. The second (S)-citalopram molecule is present in the extracellular vestibule and occupies a secondary site (we will refer to this site as SERT/A3). According to our observation, the SERT/A3 site more closely overlaps with the LeuT/TCA and the proposed SERT/A2 sites. However, the SERT/A3 site is distinct from the LeuT/S2 site (Shi et al., 2008) and also from the SERT/A1 site that was proposed in the previous mutagenesis and computational studies of SERT (Plenge et al., 2012), with a few overlapping residues common to all sites. The amino acids constituting this SERT/A3 site that interact with (S)-citalopram, as seen in the crystal structure, are R104, D328, A331, Q332, F335, E494, F556, and P561. Overall, the predictions based on computational and biochemical studies, along with further corroborating evidences from co-crystal structures of LeuT and SERT suggest a physiological role of putative secondary “allosteric” binding sites present in the extracellular vestibule in substrate translocation. Thus, in addition to the primary binding site S1 observed in the crystal structures of LeuT and SERT, there is a possibility that multiple low affinity binding sites are present in regions distinct from the direct translocation pathway that could serve as allosteric sites for modulating conformational changes in the transporter and affect its function. The relevance of these allosteric sites as functional binding sites for monoamine substrates during translocation and for inhibitors is still strongly debated by many as, to date, there is no known crystal structure of any MATs or their homologs that have a substrate molecule simultaneously bound to any site other than the primary S1 site. Moreover, a large number of structures of dDAT have been co-crystallized with the substrate DA, NET-specific reuptake inhibitors (NRIs), SSRIs, TCAs, serotonin-norepinephrine reuptake inhibitors (SNRIs), cocaine and its analog RTI-55, and amphetamines bound only at the conserved primary binding site, S1 (Penmatsa et al., 2015; K. H. Wang et al., 2015). In addition, several crystal structures of LeuBAT (LeuT with key residues in the central binding site mutated to corresponding amino acids of human SERT) have been published with mazindol, TCAs, SSRIs, SNRIs, etc., all occupying the S1 site (H. Wang et al., 2013). Hence, there is still a further need to establish the presence and relevance of allosteric binding sites, similar to those in LeuT and SERT, in the extracellular vestibule of DAT and NET via crystallographic studies. Several computational, pharmacological and structure-function studies have discovered a few ligands that act as allosteric modulators of DAT function (Rothman et al., 2015) and NET (Paczkowski et al., 2007) confirming the possibility that DAT and NET also might contain functionally relevant allosteric binding sites outside of the S1 site. However, additional studies related to elucidation of the location and molecular determinants of the binding pockets of these allosteric ligands will be highly desirable.
Pharmacology
A wide range of MAT-interacting compounds have been developed to date as pharmacological and therapeutic tools to modulate and regulate neurotransmission (Table 1). Different classes of non-selective or selective drugs that can inhibit, modulate or promote the activity of the three transporters have been extensively developed via design and synthesis efforts and evaluated for potential efficacy in the treatment of many CNS-related diseases such as depression and the abuse of psychostimulants like cocaine and amphetamines.
Inhibitors
DAT: Cocaine is one of the most widely abused psychostimulants and while a non-selective inhibitor of DAT, NET and SERT, its reinforcing and rewarding effects lead to abuse and addiction that are primarily mediated by inhibiting DA uptake by blocking the DA binding site of DAT increasing perisynaptic DA levels. Efforts are ongoing to develop cocaine antagonists that can competitively or non-competitively prevent cocaine binding to DAT without the associated stimulatory effects exhibited by cocaine. These efforts have led DAT inhibitors like WIN-35,428 (β-CFT), benztropine (and its analog JHW-007), GBR-12909, methylphenidate, RTI-55, and more (Figure 6) (Reith et al., 2015; Schmitt et al., 2013). WIN-35,428 and RTI-55 are more structurally rigid, metabolically stable and higher affinity cocaine analogs with pharmacological effects similar to cocaine. Due to their “cocaine-like” psychostimulant effects and addictive liability, they lack therapeutic utility but as radioactive ligands are used as pharmacological tools instead of cocaine in DAT-related assays. Benztropines are another class of high affinity DAT inhibitors that are structurally analogous to cocaine. Despite their higher affinity and selectivity for DAT, benztropine itself and its analog, JHW-007, do not demonstrate cocaine-like stimulatory behavioral effects in animal models of addiction (Schmitt et al., 2013, Velázquez-Sánchez, C. et al., 2010). Such ‘atypical’ non-addictive behavioral effects of these compounds have resulted in their exhaustive exploration for the treatment of cocaine dependence. GBR-12909, with a distinct structure from that of cocaine, is a potent and selective DAT inhibitor with non-addicting properties. Despite an ideal therapeutic profile for psychostimulant abuse treatment, GBR-12909 prolonged the QTc interval causing ventricular arrhythmias that led to its clinical failure. Methylphenidate, another potent DAT inhibitor marketed as Ritalin® in low doses to treat attention-deficit hyperactivity disorder (ADHD), also has psychostimulant activity. Bupropion is approved as a smoking cessation aid and for the treatment of major depressive disorder and seasonal affective disorder. The pharmacological properties of buproprion have been attributed to weak DAT inhibitory potency, inhibition of NE uptake and non-competitive inhibition of select nicotinic acetylcholine receptors (nAChRs) (Carroll, et al., 2014). At low doses, it lacks psychostimulant activity in humans and is of interest for development as a treatment for cocaine abuse (Carroll, et al., 2010). Modafinil continues to be one of the most sort after DAT ligands for development into a potential treatment for cocaine and other psychostimulant dependence (Mereu et al., 2017). It is a schedule IV controlled drug approved for promoting wakefulness, it is commonly used “off-label” as a cognition-enhancer. Preclinical and clinical studies on modafinil have produced mixed results while evaluating its ability for abuse potential. Though evidence from animal studies indicate that modafinil does have some level of abuse liability, it is relatively lower than cocaine which is attributed to its weak DAT inhibitory efficacy (Schmitt et al., 2011). Due to modafinil’s unique in vivo pharmacological profile as an “atypical” DAT ligand, structure-activity relationship studies continue to explore its mechanism of action (Mereu et al., 2017, Zhang et al., 2017).
Figure 6.

Chemical structures of a few DAT ligands.
The discovery of several potent and selective DAT inhibitors that lack “cocaine-like” abuse liability led to an increase in elucidation of their mechanism of interaction with DAT. The findings that “atypical” DAT-inhibitors such as GBR-12909, benztropine, JHW-007, bupropion and modafinil, despite their ability to inhibit DA uptake, lack the same psychostimulant and abuse liability profile of cocaine has piqued further interest in determining their molecular interactions with DAT. It is suggested that these ligands bind and stabilize a DAT conformation which is disparate from that of cocaine-bound DAT conformation resulting in contrasting psychostimulant effects (Schmitt et al., 2013). Experimental evidence suggests that cocaine and methylphenidate stabilize outward-open DAT conformation whereas JHW-007, GBR-12909, modafinil and bupropion tend to be less affected by conformational changes and most likely favor a more inward-facing occluded conformation (Schmitt et al., 2013). This conformational “preference” could be responsible for the lack of addiction potential of atypical inhibitors (Reith et al., 2015). Another reason for reduced addiction liability of atypical DAT inhibitors could be their slow rate of onset of action in the brain or their off-target effects that might also contribute to their reduced behavioral reinforcing effects (Reith et al., 2015). Hence, further studies related to specific structural basis of interaction of DAT ligands are needed to promote the discovery and development of improved medications for psychostimulant addiction.
SERT
TCAs that include imipramine, clomipramine, desipramine, and amitriptyline were among the first-generation MAT inhibitors marketed as antidepressants (Stahl et al., 2013). They mediated their antidepressant properties by increasing synaptic levels of 5-HT and NE neurotransmitters via inhibition of SERT and NET. Though they do not bind to DAT, they displayed activity across many other receptors which is associated with many adverse effects. Since their antidepressant effect was mainly attributed to the inhibition of SERT, this led to the development of more selective SERT inhibitors (Stahl et al., 2013). The selective serotonin reuptake inhibitors (SSRIs) fluoxetine, citalopram, paroxetine (and sertraline or Zoloft®) were categorized among the second-generation antidepressants (Figure 7) (Stahl et al., 2013). SSRIs are selective and potent SERT inhibitors with relatively fewer side-effects as compared to TCAs. In particular, citalopram, in its racemic mixture, is a potent and selective SERT inhibitor devoid of any affinity for DAT and NET. Upon resolution of citalopram enantiomers, S-citalopram (or escitalopram) was found to be the active, higher efficacy enantiomer as compared to the R-enantiomer (Jacobsen et al., 2014). In fact, studies showed that R-citalopram can also weakly attenuate the activity of escitalopram.
Figure 7.

Chemical structures of a few SERT ligands.
Though LeuT was co-crystallized with TCAs and SSRIs bound in the secondary LeuT/TCA site in the extracellular vestibule region, compelling evidence has supported the observation that these antidepressants bind to the primary site S1 in mammalian transporters (Penmatsa et al., 2013). Several crystal structures of dDAT are now available which are in complex with various TCAs and SSRIs bound to the S1 site (see Table 2 for the list of references). However, citalopram, a SERT inhibitor, displayed unique in vitro kinetic behavior where it slowed down its own dissociation-rate from SERT and possessed allosteric modulatory effects (Plenge et al., 2012). The recently published x-ray co-crystal structure of escitalopram-bound hSERT confirmed the binding of escitalopram simultaneously to both high affinity S1 site and low affinity SERT/A3 allosteric site in the extracellular vestibule (Coleman et al., 2016). This unambiguously proved that escitalopram allosterically modulates its affinity to the primary site by simultaneously binding to the extracellular region. Interestingly, in addition to R-citalopram’s weaker affinity for the S1 site than escitalopram, it is hypothesized to weakly interact with the allosteric SERT/A3 site also and compete with escitalopram binding to this site. Many studies continue to explore the in vitro allosteric effects of the two enantiomers of citalopram and other antidepressants, and how their interaction with SERT translates to their behavioral effects in animal models.
NET
Discovery and development of the later generation of antidepressants has seen a paradigm shift where additional inhibition of norepinephrine reuptake activity has been combined with the SSRIs. These SERT/NET inhibitors have been suggested to have additional therapeutic benefits compared to SSRIs such as improved antidepressant efficacy and faster onset of action. SNRIs such as duloxetine and venlafaxine increase DA levels specifically in the prefrontal cortex area of the brain along with increasing in 5HT and NE. This effect is thought to improve cognition and motivation (Stahl et al., 2013), and adds to the anti-depressant efficacy of these inhibitors. Selective norepinephrine reuptake inhibitors (NRIs) such as reboxetine and nisoxetine (Figure 8) are also in use for depression and ADHD (Stahl et al., 2013). Dual NET/DAT inhibitors (eg, nomifensin) and triple reuptake inhibitors (SERT/DAT/NET inhibitors eg, indatraline and mazindol) are also being explored for their efficacy in depression and other CNS-disorders (Stahl et al., 2013).
Figure 8.
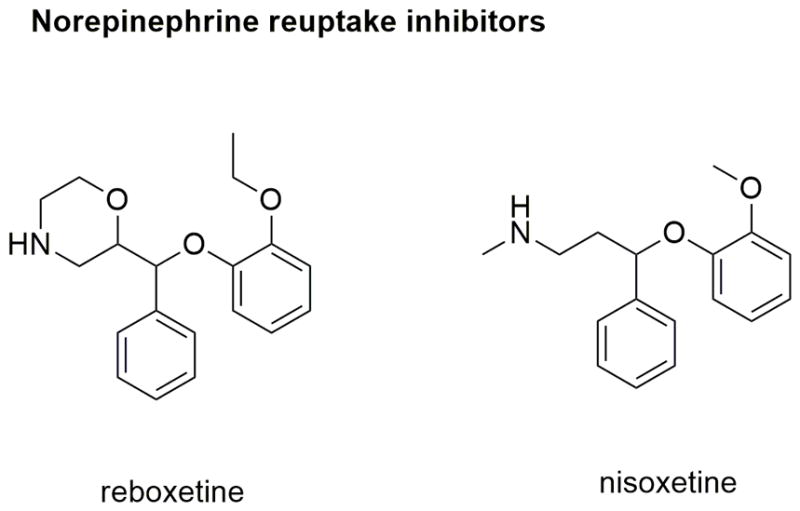
Chemical structures of selective NET inhibitors.
Human NET and DAT share 80% overall sequence homology. Even though structural variations in the non-conserved regions of NET versus DAT imparts transporter selectivity to their respective ligands, their pharmacological profiles resemble one another as both transporters can translocate DA and NE across the membrane (Andersen et al., 2015). For these reasons, the dDAT crystal structure bound with the NRIs reboxetine and nisoxetine in the S1 site is suggested to be the closest representative of inhibitor binding to hNET (Andersen et al., 2015).
Synthetic substrates/Releasers/Substrate efflux inducers
Amphetamine, and its derivatives e.g., methamphetamine, are recognized as substrates and translocated by the MATs through the plasma membrane and compete with the uptake of physiological monoamines. In addition, these synthetic substrates, once translocated, elicit the efflux of monoamines through the transporters into the synaptic cleft (Sitte & Freissmuth, 2015). Transported amphetamines are accumulated in the synaptic vesicles through the VMATs (vesicular monoamine transporters) and displace the monoamines out from the vesicles (Sitte & Freissmuth, 2015). The combined effects of amphetamine-induced natural substrate reuptake inhibition and efflux results in an increased synaptic concentration of the monoamines leading to psychostimulant effects. Other amphetamine-like compounds with a similar mode of action are MDMA (methylenedioxymethamphetamine), cathinone, methcathinone, mephedrone, methylone, phenmetrazine and others (Figure 9) (Baumann & Volkow, 2016). Although the mechanism of reverse transport is not well understood, evidence indicates that amphetamines could facilitate reverse monoamine transport by altering ion conductance across the plasma membrane that couples the monoamine transport process (Sitte & Freissmuth, 2015). Since the increase in monoamine concentrations could either result from their reuptake inhibition or reverse transport or both, the activity of any MAT ligand should be experimentally evaluated in both in vitro uptake inhibition assays as well as efflux assays.
Figure 9.

Chemical structures of monoamine substrate releasers or efflux inducers.
Amphetamine and its analogs are currently being explored as “agonist medications” for stimulant addiction (Howell & Negus, 2014; Negus & Henningfield, 2015). The antagonist approach using MAT inhibitors generally suffers from their own abuse liability and low compliance due to extreme withdrawal symptoms. On the other hand, ideal agonist-based medications will likely compete with the psychostimulants for the transporters and attenuate their action, and will be more compliant as they will provide a mild reinforcing effect of their own to provide relief in withdrawal symptoms. In addition, their pharmacokinetic profile (such as bioavailability, slow onset of action, long duration of action) could be modified to make them less likely to be abused and increase their safety and efficacy. Interestingly, a set of compounds characterized as “partial releasers” could be ideal candidates for the agonist-based therapy (Reith et al., 2015; Rothman et al., 2012). Structural analogs of amphetamines, e.g., MDEA (methylenedioxy-ethylamphetamine) do not elicit the maximum efflux of intracellular substrates even at highest concentrations as compared to “full” substrates. Further studies are required to evaluate the in vivo efficacy of such ligands in attenuating the effects of psychostimulants in animal models of addiction. Many recent studies have suggested that the mechanism of substrate uptake and efflux are distinct from each other and can be differentially regulated. Experimental evidence indicates that there are certain mutations as well as MAT-ligands that modulate monoamine uptake without having any effect on efflux. The functional significance of such a mechanism, as well as exact molecular determinants and binding site location of these ligands will require further evaluation.
Allosteric modulators
The X-ray crystal structure of LeuT bound with TCAs and SSRIs in the secondary LeuT/TCA site present in the extracellular vestibule of the translocation pathway provided a possibility for the presence of corresponding secondary or allosteric sites in mammalian transporters as well. This notion was strengthened by previous observations where some antidepressants could slow the dissociation rate of other SERT-bound ligands. It was demonstrated that mutation of some residues in the extracellular region of SERT reduced the allosteric potency of antidepressants in modulating the dissociation of ligands bound to the high affinity primary S1 site (Plenge et al., 2012). This further indicated that the S1 site in SERT is allosterically associated with a functionally-relevant secondary site in SERT. Biochemical, pharmacological, and computational approaches predicted the location and molecular determinants of this second site (SERT/A1) (Plenge et al., 2012) which, according to our observations, is slightly distinct from the location of the corresponding LeuT/TCA site (Singh et al., 2007) but more closely overlaps with the LeuT/S2 site (Shi et al., 2008). Another allosteric site was identified in SERT (SERT/A2) through molecular dynamics and comparative genomics techniques which is distinct from the SERT/A1 site reported earlier. A hybrid structure-based method was utilized to screen for compounds that bind to the SERT/A2 site. ATM7 was one of the ligands identified using this approach which allosterically modulated the 5-HT uptake as well as MDMA-elicited reverse transport (Kortagere et al., 2013; Mortensen & Kortagere, 2015). The recently published x-ray crystal structure of hSERT with a molecule of escitalopram bound to the S1 site and another escitalopram molecule bound in the extracellular vestibule region finally provided a direct evidence of the presence of an allosteric site in SERT (Coleman et al., 2016). This second site (SERT/A3) had low affinity for escitalopram which upon interaction could functionally modulate the dissociation rates of ligands bound to the high affinity S1 site. We observed that the predicted location of the SERT/A2 site is close to the SERT/A3 site and both have many overlapping residues (Figure 5). These results together suggest the presence of one or more functionally-relevant allosteric sites within SERT, and that continued computational approaches along with biochemical and structure-function studies could lead to the discovery of more efficacious allosteric modulators for use in the treatment of many CNS-related diseases involving monoamines.
There is still inadequate experimental evidence for the presence of functionally-relevant allosteric sites, and their location and molecular determinants, in DAT and NET. The availability of the dDAT crystal structure with a variety of ligands seen to be bound only to the S1 site with nothing in the extracellular region adds to the uncertainty of whether a secondary site is present in hDAT and hNET or not. Interestingly, a study showed that a peptide called Chi-conotoxin, isolated from a marine Conus marmoreus snail, noncompetitively inhibited NET function by interacting with the extracellular vestibule region of NET (Paczkowski et al., 2007). Another chemical library screening effort identified a series of ligands (known as the SoRI ligands) with moderate DAT affinity and exhibited partial allosteric modulation of DAT function (Rothman et al., 2015; Rothman et al., 2012). None of the SoRI ligands displayed dose-dependent complete inhibition of a DAT radioligand (known to occupy S1) which is a typical characteristic of noncompetitive binding. Further evaluation using uptake and kinetic assays suggested that the SoRI ligands probably bind to a site distinct from that of the primary site S1. Remarkably, one of the SoRI compounds (SRI-20041, Rothman et al., 2009) inhibited DA uptake like the others in the series, but had no effect on the amphetamine-induced efflux of the substrate. This observation led to the conclusion that the two processes of substrate translocation in and out of the MATs are independent of each other and allosteric modulators could be designed to modulate only one single cycle of substrate transport or the other (Schmitt et al., 2013). The molecular determinants of the binding site of SoRI compounds and the mechanism of action of these compounds at molecular level will be an interesting addition to aid in understanding of the function of DATs at the molecular level.
Regulation and trafficking
Though MATs have high similarity in their core TM region, their extracellular loops, N-, and C-termini differ to a great extent in length and sequence of the amino acids (Kristensen et al., 2011) (Figure 2). The sites present in these regions are involved in various post-translational modifications and protein-protein interactions with several signaling proteins that control transporter localization, stability, and activity of each MAT. For example, the long extracellular loop, EL2, between TM 3 and 4 of the MATs contains several sites for N-glycosylation that are required for the stability of DAT and NET embedded in the plasma membrane (Kristensen et al., 2011). However differences in the N-terminus of DAT and NET could explain differences observed in the role of posttranslational ubiquitinylation. The DAT is ubiquitylated following activation of PKC triggering the internalization of DAT (Miranda et al., 2007; Miranda et al., 2005; Mortensen, 2003). On the other hand, NET does not appear to be ubiquitylated (Mortensen et al., 2017). This difference is perhaps not surprising as NET lacks all the N-terminal lysines that are ubiquitylated in DAT (Figure 2). The physiological relevance of such differences in regulation between DAT and NET is highlighted by studies on the ultrastructural localization of DAT and NET. These studies have found the expression of NET in some areas of the prefrontal cortex is predominantly intracellular (Miner et al., 2003). This is in contrast to the DAT that was found to be mainly expressed on the plasma membrane (Hersch et al., 1997). These results implies that there must be differences in how trafficking and cell surface expression of these two related transporter is regulated. It could be hypothesized that differences in the primary sequences of intracellular domains of DAT and NET recruits different regulatory proteins.
MATs also undergo regulation directly or indirectly by a number of kinases e.g., protein kinase C (PKC), cAMP-dependent protein kinase (protein kinase A, PKA), cGMP-dependent protein kinase (protein kinase G), Ca2+/calmodulin-dependent protein kinase II, ERK1/2, p38 mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K)/Akt and tyrosine kinases. Details regarding the effects of every signaling pathway on MAT structure and function is beyond the scope of this overview and the readers are directed to review articles on this topic (Bermingham & Blakely, 2016; Kristensen et al., 2011).
Conclusions
DAT, NET and SERT represent important therapeutic targets for a number of CNS-related pathophysiological conditions. The discovery of crystal structures of transporter homologs bound with substrate and inhibitors have substantially advanced our understanding of structure-function profile of MATs and has aided in several medicinal chemistry-based drug discovery efforts. However, knowledge of specific ligand-directed conformational changes at the molecular level and the subsequent behavioral effects elicited through the transporters is still lacking. The exact regulatory and functional significance of the extracellular vestibular region of the human MATs at a physiological level also remains unresolved. The complex mechanisms that dictate the binding interactions and effects of substrates, inhibitors and releasers on the transporters also remain a critical topic of research. Careful design and interpretation of biochemical and behavioral assays to evaluate the function and pharmacology of MAT ligands is necessary to delineate them as competitive inhibitors, allosteric modulators, substrates or releasers.
Acknowledgments
The authors acknowledge support from NIMH grant MH106912.
References
- Andersen J, Ladefoged LK, Kristensen TN, Munro L, Grouleff J, Stuhr-Hansen N, Kristensen AS, Schiott B, Stromgaard K. Interrogating the Molecular Basis for Substrate Recognition in Serotonin and Dopamine Transporters with High-Affinity Substrate-Based Bivalent Ligands. ACS Chem Neurosci. 2016;7(10):1406–1417. doi: 10.1021/acschemneuro.6b00164. [DOI] [PubMed] [Google Scholar]
- Andersen J, Ringsted KB, Bang-Andersen B, Stromgaard K, Kristensen AS. Binding site residues control inhibitor selectivity in the human norepinephrine transporter but not in the human dopamine transporter. Sci Rep. 2015;5:15650. doi: 10.1038/srep15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann MH, Volkow ND. Abuse of New Psychoactive Substances: Threats and Solutions. Neuropsychopharmacology. 2016;41(3):663–665. doi: 10.1038/npp.2015.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermingham DP, Blakely RD. Kinase-dependent Regulation of Monoamine Neurotransmitter Transporters. Pharmacol Rev. 2016;68(4):888–953. doi: 10.1124/pr.115.012260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll FI, Blough BE, Mascarella SW, Navarro HA, Eaton JB, Lukas RJ, Damaj MI. Synthesis and biological evaluation of bupropion analogues as potential pharmacotherapies for smoking cessation. J Med Chem. 2010;53(5):2204–2214. doi: 10.1021/jm9017465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll FI, Blough BE, Mascarella SW, Navarro HA, Lukas RJ, Damaj MI. Bupropion and bupropion analogs as treatments for CNS disorders. Adv Pharmacol. 2014;69:177–216. doi: 10.1016/B978-0-12-420118-7.00005-6. [DOI] [PubMed] [Google Scholar]
- Coleman JA, Green EM, Gouaux E. X-ray structures and mechanism of the human serotonin transporter. Nature. 2016;532(7599):334–339. doi: 10.1038/nature17629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersch SM, Yi H, Heilman CJ, Edwards RH, Levey AI. Subcellular localization and molecular topology of the dopamine transporter in the striatum and substantia nigra. J Comp Neurol. 1997;388(2):211–227. doi: 10.1002/(SICI)1096-9861(19971117)388:2<211::AID-CNE3>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Howell LL, Negus SS. Monoamine transporter inhibitors and substrates as treatments for stimulant abuse. Adv Pharmacol. 2014;69:129–176. doi: 10.1016/B978-0-12-420118-7.00004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen JP, Plenge P, Sachs BD, Pehrson AL, Cajina M, Du Y, Roberts W, Rudder ML, Dalvi P, Robinson TJ, O’Neill SP, Khoo KS, Morillo CS, Zhang X, Caron MG. The interaction of escitalopram and R-citalopram at the human serotonin transporter investigated in the mouse. Psychopharmacology (Berl) 2014;231(23):4527–4540. doi: 10.1007/s00213-014-3595-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilty JE, Lorang D, Amara SG. Cloning and expression of a cocaine-sensitive rat dopamine transporter. Science. 1991;254(5031):578–579. doi: 10.1126/science.1948035. [DOI] [PubMed] [Google Scholar]
- Koldso H, Grouleff J, Schiott B. Insights to ligand binding to the monoamine transporters-from homology modeling to LeuBAT and dDAT. Front Pharmacol. 2015;6:208. doi: 10.3389/fphar.2015.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortagere S, Fontana AC, Rose DR, Mortensen OV. Identification of an allosteric modulator of the serotonin transporter with novel mechanism of action. Neuropharmacology. 2013;72:282–290. doi: 10.1016/j.neuropharm.2013.04.026. [DOI] [PubMed] [Google Scholar]
- Kristensen AS, Andersen J, Jorgensen TN, Sorensen L, Eriksen J, Loland CJ, Stromgaard K, Gether U. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol Rev. 2011;63(3):585–640. doi: 10.1124/pr.108.000869. [DOI] [PubMed] [Google Scholar]
- Lin Z, Canales JJ, Bjorgvinsson T, Thomsen M, Qu H, Liu QR, Torres GE, Caine SB. Monoamine transporters: vulnerable and vital doorkeepers. Prog Mol Biol Transl Sci. 2011;98:1–46. doi: 10.1016/B978-0-12-385506-0.00001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loland CJ. The use of LeuT as a model in elucidating binding sites for substrates and inhibitors in neurotransmitter transporters. Biochim Biophys Acta. 2015;1850(3):500–510. doi: 10.1016/j.bbagen.2014.04.011. [DOI] [PubMed] [Google Scholar]
- Manepalli S, Surratt CK, Madura JD, Nolan TL. Monoamine transporter structure, function, dynamics, and drug discovery: a computational perspective. AAPS J. 2012;14(4):820–831. doi: 10.1208/s12248-012-9391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereu M, Chun LE, Prisinzano TE, Newman AH, Katz JL, Tanda G. The unique psychostimulant profile of (+/−)-modafinil: investigation of behavioral and neurochemical effects in mice. Eur J Neurosci. 2017;45(1):167–174. doi: 10.1111/ejn.13376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner LH, Schroeter S, Blakely RD, Sesack SR. Ultrastructural localization of the norepinephrine transporter in superficial and deep layers of the rat prelimbic prefrontal cortex and its spatial relationship to probable dopamine terminals. J Comp Neurol. 2003;466(4):478–494. doi: 10.1002/cne.10898. [DOI] [PubMed] [Google Scholar]
- Miranda M, Dionne KR, Sorkina T, Sorkin A. Three ubiquitin conjugation sites in the amino terminus of the dopamine transporter mediate protein kinase C–dependent endocytosis of the transporter. Mol Bio Cell. 2007;18(1):313–323. doi: 10.1091/mbc.E06-08-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda M, Wu CC, Sorkina T, Korstjens DR, Sorkin A. Enhanced ubiquitylation and accelerated degradation of the dopamine transporter mediated by protein kinase C. J Bio Chem. 2005;280(42):35617–35624. doi: 10.1074/jbc.M506618200. [DOI] [PubMed] [Google Scholar]
- Mortensen OV, Larsen MB, Amara S. MAP Kinase Phosphatase 3 (MKP3) Preserves Norepinephrine Transporter Activity by Modulating ERK1/2 Kinase-Mediated Gene Expression. Front Cell Neurosci. 2017;11:253. doi: 10.3389/fncel.2017.00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen OV, Kortagere S. Designing modulators of monoamine transporters using virtual screening techniques. Front Pharmacol. 2015;6:223. doi: 10.3389/fphar.2015.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen OV. MKP3 eliminates depolarization-dependent neurotransmitter release through downregulation of L-type calcium channel Cav1. 2 expression. Cell Calcium. 2013;53(3):224–230. doi: 10.1016/j.ceca.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Negus SS, Henningfield J. Agonist Medications for the Treatment of Cocaine Use Disorder. Neuropsychopharmacology. 2015;40(8):1815–1825. doi: 10.1038/npp.2014.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paczkowski FA, Sharpe IA, Dutertre S, Lewis RJ. chi-Conotoxin and tricyclic antidepressant interactions at the norepinephrine transporter define a new transporter model. J Biol Chem. 2007;282(24):17837–17844. doi: 10.1074/jbc.M610813200. [DOI] [PubMed] [Google Scholar]
- Penmatsa A, Wang KH, Gouaux E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature. 2013;503(7474):85–90. doi: 10.1038/nature12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa A, Wang KH, Gouaux E. X-ray structures of Drosophila dopamine transporter in complex with nisoxetine and reboxetine. Nat Struct Mol Biol. 2015;22(6):506–508. doi: 10.1038/nsmb.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenge P, Shi L, Beuming T, Te J, Newman AH, Weinstein H, Gether U, Loland CJ. Steric hindrance mutagenesis in the conserved extracellular vestibule impedes allosteric binding of antidepressants to the serotonin transporter. J Biol Chem. 2012;287(47):39316–39326. doi: 10.1074/jbc.M112.371765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick M, Winther AM, Shi L, Nissen P, Weinstein H, Javitch JA. Binding of an octylglucoside detergent molecule in the second substrate (S2) site of LeuT establishes an inhibitor-bound conformation. Proc Natl Acad Sci U S A. 2009;106(14):5563–5568. doi: 10.1073/pnas.0811322106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamoorthy S, Shippenberg TS, Jayanthi LD. Regulation of monoamine transporters: Role of transporter phosphorylation. Pharmacol Ther. 2011;129(2):220–238. doi: 10.1016/j.pharmthera.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith ME, Blough BE, Hong WC, Jones KT, Schmitt KC, Baumann MH, Partilla JS, Rothman RB, Katz JL. Behavioral, biological, and chemical perspectives on atypical agents targeting the dopamine transporter. Drug Alcohol Depend. 2015;147:1–19. doi: 10.1016/j.drugalcdep.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Ananthan S, Partilla JS, Saini SK, Moukha-Chafiq O, Pathak V, Baumann MH. Studies of the biogenic amine transporters 15. Identification of novel allosteric dopamine transporter ligands with nanomolar potency. J Pharmacol Exp Ther. 2015;353(3):529–538. doi: 10.1124/jpet.114.222299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Partilla JS, Baumann MH, Lightfoot-Siordia C, Blough BE. Studies of the biogenic amine transporters. 14. Identification of low-efficacy “partial” substrates for the biogenic amine transporters. J Pharmacol Exp Ther. 2012;341(1):251–262. doi: 10.1124/jpet.111.188946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Dersch CM, Ananthan S, Partilla JS. Studies of the biogenic amine transporters. 13. Identification of “agonist” and “antagonist” allosteric modulators of amphetamine-induced dopamine release. J Pharmacol Exp Therap. 2009;329(2):718–728. doi: 10.1124/jpet.108.149088. doi: https://doi.org/10.1124/jpet.108.149088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnick G, Kramer R, Blakely RD, Murphy DL, Verrey F. The SLC6 transporters: perspectives on structure, functions, regulation, and models for transporter dysfunction. Pflugers Arch. 2014;466(1):25–42. doi: 10.1007/s00424-013-1410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt KC, Reith ME. The atypical stimulant and nootropic modafinil interacts with the dopamine transporter in a different manner than classical cocaine-like inhibitors. PloS one. 2011;6(10):e25790. doi: 10.1371/journal.pone.0025790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt KC, Rothman RB, Reith ME. Nonclassical pharmacology of the dopamine transporter: atypical inhibitors, allosteric modulators, and partial substrates. J Pharmacol Exp Ther. 2013;346(1):2–10. doi: 10.1124/jpet.111.191056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. The mechanism of a neurotransmitter:sodium symporter--inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol Cell. 2008;30(6):667–677. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322(5908):1655–1661. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Yamashita A, Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448(7156):952–956. doi: 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- Sitte HH, Freissmuth M. Amphetamines, new psychoactive drugs and the monoamine transporter cycle. Trends Pharmacol Sci. 2015;36(1):41–50. doi: 10.1016/j.tips.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl SM, Lee-Zimmerman C, Cartwright S, Morrissette DA. Serotonergic drugs for depression and beyond. Curr Drug Targets. 2013;14(5):578–585. doi: 10.2174/1389450111314050007. [DOI] [PubMed] [Google Scholar]
- Torres GE, Gainetdnov RR, Caron MG. Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neuroscience. 2003;4(1):13–25. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- Velázquez-Sánchez C, Ferragud A, Murga J, Cardá M, Canales JJ. The high affinity dopamine uptake inhibitor, JHW 007, blocks cocaine-induced reward, locomotor stimulation and sensitization. Eur Neuropsychopharmacol. 2010;20(7):501–508. doi: 10.1016/j.euroneuro.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Wang H, Goehring A, Wang KH, Penmatsa A, Ressler R, Gouaux E. Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature. 2013;503(7474):141–145. doi: 10.1038/nature12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KH, Penmatsa A, Gouaux E. Neurotransmitter and psychostimulant recognition by the dopamine transporter. Nature. 2015;521(7552):322–327. doi: 10.1038/nature14431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature. 2005;437(7056):215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Zhang HY, Bi GH, Yang HJ, He Y, Xue G, Cao J, Tanda G, Gardner EL, Newman AH, Xi ZX. The Novel Modafinil Analog, JJC8–016, as a Potential Cocaine Abuse Pharmacotherapeutic. Neuropsychopharmacology. 2017;42(9):1871–1883. doi: 10.1038/npp.2017.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith ME, Wang DN. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science. 2007;317(5843):1390–1393. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]