

Abstract
Bivalent compounds anchoring in different manners to the membrane were designed and biologically characterized to understand the contribution of the anchor moiety to their biological activity as neuroprotectants for Alzheimer’s disease. Our results established that the anchor moiety is essential and we identified a preference for diosgenin, as evidenced by 17MD. Studies in primary neurons and mouse brain mitochondria also identified 17MD as exhibiting activity on neuritic outgrowth and the state 3 oxidative rate of glutamate while preserving the coupling capacity of the mitochondria. Significantly, our studies demonstrated that the integrated bivalent structure is essential to the observed biological activities. Further studies employing bivalent compounds as probes in a model membrane also revealed the influence of the anchor moiety on how they interact with the membrane. Collectively, our results suggest diosgenin to be an optimal anchor moiety, providing bivalent compounds with promising pharmacology that have potential applications for Alzheimer’s disease.
Graphical Abstract
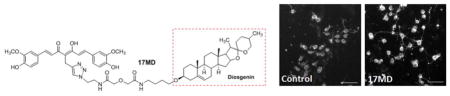
Introduction
Alzheimer’s disease (AD) is a devastating neurodegenerative disease and the most common cause of dementia.1 Multiple pathogenic factors have been suggested to play essential roles in the development of AD, including aggregation of beta-amyloid (Aβ),2, 3 tau protein,4 oxidative stress,5 neuroinflammation,6, 7 and mitochondrial dysfunction,8 among others. As a result, drug development to provide effective disease-modifying agents for AD still remains a challenging and unmet task. To address this challenge, a multifunctional strategy that tackles multiple risk factors by small molecules has recently emerged as an alternative and innovative approach to surmount the paucity of effective disease-modifying agents in the AD therapeutics development pipeline.9–12 Note that applying a combination therapy strategy with compounds possessing different mechanisms of action in AD clinical trials presages the idea of developing multifunctional compounds.
Recent studies have established that neuronal membrane/lipid rafts play important roles in regulating the production, aggregation, and consequent cytotoxicity of Aβ,13–16 thus strongly suggesting the membrane/lipid rafts domain as an important platform involving multiple AD risk factors. Cell membrane targeting has long been recognized as an efficient means for modifying the pharmacokinetic properties and efficacy of drug molecules.17–20 Peterson’s group proposed and developed a synthetic receptors/antigen strategy by incorporating cholesterol or cholesterylamine as membrane anchors to improve transmembrane uptake.21–23 A tripartite strategy has also been successfully applied to design compounds that include a β-secretase inhibitor, a spacer and a raftophile; subsequent studies demonstrated theirs significantly improved efficacy both in vitro and in vivo for β-secretase inhibition,18, 24–27 thus providing a critical proof of principle for development of such inhibitors. Recently, our group embarked on development of novel bivalent compounds as potential neuroprotectants for AD by linking a multifunctional “warhead” with a membrane anchor moiety via a spacer.28–33 The hypothesis behind this design was that by incorporating a membrane-anchoring moiety into molecular design, the resulting bivalent compounds would exhibit improved accessibility of the anchored warhead to the multiple risk factors of AD, consequently leading to improved efficacy. Conceptually, the bivalent strategy originated from the development of chemical probes to study the dimerization/oligomerization of opioid receptors.34 Recently, this concept has been extended to neurodegenerative diseases by the development of multi-targeted ligands for AD and other diseases.35 Considering the multifactorial nature of AD, we employed curcumin, an important phytochemical with known antioxidant, anti-inflammatory and anti-Aβ activities, as the warhead moiety in our bivalent compounds.36, 37 On the other end, we employed a steroid, i.e., cholesterol, cholesterylamine or diosgenin as the membrane-anchoring pharmacophore. Sterol and analogs such as cholesterol, dihydrocholesterol and cholesterylamine have demonstrated utility as membrane anchors by many research groups including ours.23–26, 28, 29, 31, 33 Diosgenin and its analogs have shown neuroprotective activities in various AD models with a number of mechanisms suggested for their observed pharmacological activities.38, 39 Therefore, incorporation of diosgenin into our molecular design may offer a double role: anchoring to the membrane and neuroprotective activity, which was clearly supported by the results of our recent studies.29
Our previous studies, as summarized in Figure 1, established that bivalent compounds we designed exhibit significantly improved protection compared to the curcumin warhead or the membrane anchor alone, or simultaneous treatment with these two molecules in cellular AD models. The optimal attachment position on curcumin resides on the methylene carbon between the diketone moieties.33 Our studies also reported that the spacer length between the warhead and the anchor is crucial to the observed protective activities, consistent with the results of the tripartite compounds.24, 26 Specifically, the optimal spacer length ranges from 17 to 21 atoms in our models.29, 31, 33 Notably, our studies suggested that the spacer length and the anchor moiety of the bivalent compounds may determine the sub-cellular localization of these bivalent compounds with different, yet overlapping, biological activities.28, 30 In addition, bivalent compounds with different spacers showed different profiles with respect to their biometal chelating properties.29 Although our bivalent compounds exhibited promising activities as potential neuroprotectants for AD, these results were derived from analogs with structural modifications on both the spacer composition/length and the anchor moiety at the same time. In this work we dissect the contributions of these individual components within the bivalent structure scaffold, as we believe that the results of such should effectively guide the design and development of new and more potent analogs. Herein, we report the synthesis and biological characterization of a series of bivalent compounds with varying membrane anchor moieties. With these results, we believe that the impact of the anchor moiety on their biological activities can be understood.
Figure 1.

Summary of previous studies on the bivalent compounds.
Design and Chemistry
Our previous work established that bivalent compounds with a 17-atom spacer exhibited the most potent protection in our cellular models as demonstrated by compound 17MN (Figure 2); our naming convention is as follows: 17 denotes the number of atoms in the spacer, M indicates the “middle” position between the diketone moiety of curcumin, and N indicates attachment to the steroid moiety (cholesterylamine) via a nitrogen atom.29, 31 To understand the impact of the membrane anchor on the biology, we retained the spacer composition of 17MN and varied the anchor moiety within the set of structures. As shown in Figure 2, three of our previously studied membrane-anchoring molecules were employed for this study: cholesterol, cholesterylamine and diosgenin. For each anchor moiety, two compounds, with either a 17-atom spacer or a 21-atom spacer, were designed. To further confirm the essential role of the bivalent structure for the biological activity, two monovalent control compounds, 17SC (no steroid) and 17SD (D indicates the diosgenin moiety, no curcumin), were also designed.
Figure 2.

Structures of the designed bivalent compounds and monovalent controls.
The chemical syntheses of 17MN and 21MN were achieved by following previously reported procedures from our laboratory.31 The synthesis of 17MO and 21MO began with the alkylation of cholesterol (1) with 1,4-dibromobutane, and then followed by reaction with phathalimide to give intermediate 3 (Scheme 1). Refluxing of 3 with hydrazine in ethanol was followed by a coupling reaction with azido carboxylic acid 5 or 6, which were synthesized from 2-azido-ethylamine or 4-azido-1-butylamine and diglycolic anhydride, and afforded intermediate 7 or 8, respectively. Finally, the click reaction of 7 or 8 with alkyne 11, which was obtained as previously reported via the well-established Pabon reaction,33 yielded bivalent compounds 17MO or 21MO (O indicates attachment to the steroid moiety (cholesterol) via an oxygen atom), respectively. Similarly, the syntheses of 17MD and 21MD were achieved by following the same conditions with diosgenin (2) as the starting material. The syntheses of the monovalent control compounds 17SC and 17SD were as outlined in Scheme 2. Briefly, the coupling reaction of carboxylic acid 12, synthesized from 2-azido-1-ethylamine and diglycolic anhydride, with 1-aminobutane gave intermediate 13. The click reaction of 13 with alkyne 11 afforded 17SC. The click reaction of 9 with 4-methyl-1-pentyne gave monovalent compound 17SD.
Scheme 1a.

a Reagents and conditions: a) i. NaH, 1,4-dibromobutane, THF, reflux; ii. Phthalimide, K2CO3, DMF. b) i. NH2NH2, EtOH, reflux; ii. 5 or 6, EDC, dichloromethane. c) CuSO4, sodium ascorbate, THF/H2O (1:1). d) i. 5, 1-aminopetane, EDC, dichloromethane; ii. CuSO4, sodium ascorbate, THF/H2O (1:1).
Scheme 2a.

a Reagents and conditions: a) 1-butylamine, EDC, dichloromethane, b) 11 or 4-methyl-1-pentyne, CuSO4, sodium ascorbate, THF/H2O (1:1).
Results
Protective effects in MC65 cells
MC65 is a neuronal cell line that conditionally expresses C99, the C-terminus fragment of amyloid precursor protein (APP) using tetracycline (TC) as transgene suppressor.40, 41 Upon removal of TC, MC65 cells produce intracellular Aβ aggregates including small Aβ oligomers (AβOs). More importantly, the induced cytotoxicity in these cells by TC removal has been associated with the accumulation of AβOs and oxidative stress, two of the well studied risk factors in AD development. Studies from our group and others have demonstrated that this cellular assay is a suitable screening model to test small molecule compounds with potential protective activities.31, 33, 42–45 Thus, we first tested the six bivalent compounds in MC65 cells for their potency to rescue these cells from cytotoxicity induced by TC removal. As shown in Table 1, overall, the results of the six bivalent compounds are in agreement with our previous studies and show that compounds with 17-atom spacers are more potent than those with a 21-atom spacer (~ 3–6 times more potent).29, 31 Specifically, 17MN and 21MN rescued MC65 cells with an EC50 being 56.50 ± 9.08 and 212.28 ± 54.87 nM, respectively, consistent with our previously reported results.31 Surprisingly, 21MO showed a significantly reduced potency (1848.67 ± 325.13 nM) compared our previously reported compound with a 21-atom spacer but different spacer composition.33 This suggests that the nature of the spacer does play an essential role in the observed biological activities of these bivalent compounds. Notably, 17MD exhibited the most potent protection under the current experimental conditions with an EC50 of 8.30 ± 0.52 nM, which represents a 14-fold increase compared to the bivalent compound we previously reported with diosgenin as the anchor moiety but a different 17-atom spacer.29 This supports our contention that the nature and composition of the spacer within bivalent compounds is a key factor in optimizing potency.
Table 1.
Potency of bivalent compounds in rescuing MC65 cells from TC removal induced cytotoxicity (Mean ± SEM).
| 17MN | 21MN | 17MO | 21MO | 17MD | 21MD | |
|---|---|---|---|---|---|---|
| EC50 (nM) | 56.50 ± 9.08 | 212.28 ± 54.87 | 296.30 ± 11.62 | 1848.66 ± 325.13 | 8.30 ± 0.52 | 30.33 ± 1.42 |
To confirm that the observed rescue activity is by our bivalent compound, we tested 17SC itself and the combination of 17SC and 17SD. We also tested the combination of curcumin and diosgenin as another control. No significant rescue was observed for any of these controls (Figure 3A). These results provide strong evidence that the entire skeleton of 17MD is essential for the observed biological activity. Curcumin itself has been suggested as one of the promiscuous Pan-Assay Interference Compounds (PAINS) due to the presence of a Michael acceptor moiety within the structure.46 The distinct results of the control compounds that contain curcumin or curcumin itself when compared to that of 17MD in our experiments clearly indicate that the observed biological activity is solely due to the presence of the bivalent structure, and not by the promiscuity of the curcumin moiety. Our bivalent compounds thus should not be considered as PAINS. When the protective potencies of 17MN, 17MO, 17MD were compared, 17MD is the most potent, followed by 17MN, then 17MO. The same ranking was observed for the 21-atom spacer analogs. These identical rankings strongly suggest that the identity of the anchor moiety is essential to the observed protective activity and diosgenin is preferred among this series, followed by cholesterylamine, then cholesterol.
Figure 3. Protective activities in MC65 cells and neuritic outgrowth stimulating effects in N2a cells by tested compounds.

(A) MC65 cells were treated with indicated compounds or the combination of indicated compounds (0.3 μM) under –TC conditions for 72 h. Then cell viability was measured by MTT assay. Data were presented as mean (n=3) ± SEM (**p < 0.01 compared to -TC). (B) Neuronal N2a cells were seeded in 24-well plates and treated with indicated compounds (0.3 μM) for 48 h. Cells were stained with the Neurite Outgrowth Staining kit (Life Technologies) and images were recorded by Zeiss Axiovert 200M fluorescence microscopy using 10× objective and average neurite length was analyzed using Image J program. Data were presented as mean (n=3) ± SEM (**p < 0.01 compared to control). (C) Representative images from three independent experiments. Scale bar: 100 μm.
Neurotrophic effects of the designed bivalent compounds
Neuritic atrophy and disruption of neuronal networks are common features of neurodegenerative disorders including AD.47–52 In addition to efforts targeting the well characterized protein aggregation process or other pathological hallmarks, re-establishment of damaged neuronal and synaptic network by neurotrophic agents has attracted extensive attention as an alternative and efficient way to treat neurodegenerative disorders and neuronal injuries.53–56 Polyphenols and steroid analogs have been shown to induce neuronal differentiation and neurite outgrowth.39, 57, 58 We thus examined whether our bivalent compounds provide neurotrophic activity to stimulate neurite outgrowth, which would add an additional layer of benefits to the scaffold. We first tested the ability of these six compounds to stimulate the differentiation and neuritic outgrowth of neuronal N2a cells, a cell line that has been widely used for neuritic outgrowth studies. As shown in Figure 3B, at 0.3 μM concentration, only 17MD and 21MD exhibited significant activity on stimulating neuritic outgrowth of N2a cells. On the other hand, 17SC or the combination of 17SC and 17SD, did not show activity under the same experimental conditions, confirming that the observed activity was caused by the entire structure of 17MD. This is consistent with the results from the MC65 cell rescue experiments, and further eliminates PAINS concerns for our bivalent compounds. The 17MD and 21MD stimulation activity on neuritic outgrowth can also be visually observed (Figure 3C). To further validate these effects on neuritic outgrowth, we also tested the compounds in rat primary cortical neurons, a model that is more relevant to the physiological system. Because 17MN was seen to be toxic to the primary neurons in preliminary studies, this compound was not included in this experiment. Consistent with the N2a cell studies, only 17MD and 21MD exhibited significant stimulating activity on neuritic outgrowth (Figures 4A and 4B). The observed neuritic outgrowth activity could be due to the presence of the diosgenin moiety since it has been recently reported that diosgenin can improve cognition function in AD animal models by targeting the 1,25D3-membrane-associated, rapid response steroid-binding protein (1,25D3-MARRS).39, 58 However, the lack of stimulating activity by the combination of 17SC and 17SD may rule out this possibility as the diosgenin pharmacophore is also present in the combination. Further studies are needed to investigate the roles of 1,25D3-MARRS and to elucidate the mechanisms of action for the observed activity of 17MD and 21MD.
Figure 4. Effects on neuritic outgrowth of mouse primary cortical neurons and the enzymatic activity of ETC complexes of mouse brain mitochondria.

(A) Cortical neurons from E17 embryos were treated with DMSO or compounds for 4 days. Neurons were immunostained with the neuronal marker beta III tubulin (red) and nuclei were detected by use of DAPI (blue). Quantification of the average length of the total neurites was quantified by Image J analysis. Data were presented as mean ± SEM) (** P < 0.01, *** P < 0.001). (B) Representative images of neurons for the corresponding groups to illustrate the length of neurite are shown in (A). Scale bar: 20 μm. (C) Detergent solubilized mouse brain mitochondria were treated with 17MD (3.3 μM) and the respiratory complex activities were determined using spectrophotometer. No difference in complex I, II, III, and IV was observed between control and 17MD treated mitochondria. Data were presented as mean ± SEM (n=4).
Effects on the oxidative phosphorylation (OXPHOS) of isolated brain mitochondria by the bivalent compounds
Our previous studies suggested potential interactions between bivalent compounds and mitochondria.28, 30 To further examine how the anchor moiety could impact the interactions with, and the functions of, mitochondria, we employed 17MN, 17MO, and 17MD as probes on isolated mouse brain mitochondria for the effects on OXPHOS. As shown in Table 2, among the tested bivalent compounds, only treatment of mitochondria with 17MD decreased the state 3 respiratory rate of glutamate, and the difference reached statistical significance when compared to the control by SigmaStat t-test analysis (p = 0.024). A decreased respiratory rate was also shown for maximal rates of ADP stimulated respiration using 2 mM ADP (p = 0.04). The respiratory control ratio (RCR, state 3/state 4) and ADP/O ratio were not changed in the presence of 17MD (3.9 ± 0.5 and 2.3 ± 0.3, respectively) compared to control (3.8 ± 0.5 and 2.3 ± 0.3, respectively). A change in RCR, the ratio of state 3 (ADP stimulated) to state 4 (ADP initiated) respiration, which is an index of mitochondria respiration coupling, indicates uncoupled respiration due to a change of the electrochemical gradient across the inner mitochondria membrane. The ratio ADP/O is the number of ATP molecules generated by OXPHOS upon traverse of a pair of electrons derived from NADH along the electron transport chain (ETC) to oxygen, and is a key indicator of the efficiency of ATP generation. Despite the decline in state 3 oxidation of glutamate, our results suggest that the coupling ability of mitochondria is preserved under treatment with 17MD.
Table 2.
Effects of bivalent compounds (3.3 μM) on the OXPHOS of glutamate (succinate) in mouse brain mitochondria (n=4, Mean ± SD, *p<0.05, **p<0.01, ***p<0.001).
| Control | 17MN | 17MO | 17MD | 17M/17D | |
|---|---|---|---|---|---|
| State 3 | 168.3 ± 9.5 (170.8 ± 4.2) | 145.0 ± 17.0(165.0 ± 15.0) | 160.0 ± 7.5 (171.8 ± 9.5) | 149.0 ± 9.0* (161.0 ± 21.0) | 125.0 ± 13.0** (113.0 ± 12.0)*** |
| State 4 | 44.5 ± 4.4 (73.3 ± 5.2) | 37.0 ± 3.0 (69.0 ± 17.0) | 37.3 ± 6.1 (73.3 ± 3.5) | 39.0 ± 3.0 (79.0 ± 9.0) | 49.0 ± 7.0 (99.0 ± 8.0)** |
| RCR | 3.8 ± 0.5 (2.3 ± 0.2) | 3.9 ± 0.2 (2.6 ± 1.0) | 4.4 ± 0.7 (2.4 ± 0.2) | 3.9 ± 0.5 (2.1 ± 0.2) | 2.6 ± 0.5** (1.2 ± 0.10)*** |
| ADP/O | 2.3 ± 0.3 (1.3 ± 0.2) | 2.4 ± 0.3 (1.1 ± 0.2) | 2.4 ± 0.3 (1.2 ± 0.2) | 2.3 ± 0.3 (1.1 ± 0.2) | 1.9 ± 0.3 (0.90 ± 0.20)* |
| 2 mM ADP | 177.5 ± 13.0 (163.8 ± 20.8) | 151.0 ± 21.0 (149.0 ± 25.0) | 167.3 ± 14.4 (169.3 ± 27.7) | 154.0 ± 13.0* (146.0 ± 26.0) | 120.0 ± 16.0*** (106.0 ± 13.0)** |
A normal oxidation rate was observed for succinate, a complex II substrate, under treatment with 17MD (numbers in parentheses in Table 2). An unaltered rate of succinate oxidation indicates that the ETC distal to complex I remains intact. When the effects of the combination of 17SC and 17SD were examined, a significant decrease was noted for both state 3 respiratory rate and RCR when using glutamate as a complex I substrate. When succinate oxidation was examined, treatment with the combination of 17SC and 17SD led to a significant decrease of state 3 and state 4 respiratory rates, RCR, and ADP/O (Table 2), indicating impairment of both electron transport and the coupling capacity of mitochondria. The results of 17SC/17SD may suggest potential toxicity on mitochondria by physically combining individual compounds together, while 17MD, as an integrated bivalent compound, does not show such toxic potential. The results also suggest that 17MD provided mild modulating activities on state 3 oxidation of glutamate (~ 6% change compared to control) and this may suggest a potential contribution to the overall protective activity of this compound.
Taken as a whole, our results highlight the important roles of the anchor moiety in the bivalent compounds to guide the interactions with different components of the mitochondria OXPHOS process. Since glutamate and succinate represent the substrates for complex I and complex II, respectively, we next examined the effects of 17MD treatment on the activity of ETC complexes. Interestingly, no significant effect was observed under treatment with 17MD for individual activities of the ETC enzymes (Figure 4C). Glutamate oxidation reflects a multi-step process, including glutamate uptake via transporter, glutamate dehydrogenase, the activity of ETC, formation supercomplexes, and the phosphorylation process.59 Since 17MD did not directly inhibit complex I activity, the decreased OXPHOS by 17MD may be due to its effect on integrated respiration. The results cannot exclude the possibility that 17MD treatment may interfere with glutamate uptake or its redox reaction. Further studies are warranted to help elucidate the mechanism underlying the mitochondrial modulating activity of 17MD.
Interaction of bivalent compounds with model membrane system
The bivalent compounds are lipophilic in nature and curcumin has been reported to modulate membrane structure.60, 61 Furthermore, our studies above on isolated mitochondria suggest membrane interaction effects of the anchor moiety in the bivalent compounds. We next studied 17MN, 17MO, and 17MD as probes to examine whether and how the anchor moieties influence their localization and thus impact membrane structure in 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), a widely used membrane model. A combination of two physical techniques, differential scanning calorimetry (DSC) and X-ray diffraction, were employed.
The pre-transition of pure DPPC (transition from Lβ′ to Pβ′- from gel phase to a rippled gel phase) started at 33 °C and the onset of the main transition (from Pβ′ to Lα, the fluid phase) started at 41 °C (Tc), consistent with previous reports (Figures 5A-5C).62, 63 The addition of our bivalent compounds to DPPC, even at a low molar ratio of 40:1, led to disappearance of the pre-transition. Increasing concentrations of these compounds resulted in a progressive broadening and reduction in amplitude of the main transition peak. The Tc transition temperature (onset of the transition) was also shifted to lower temperatures with increasing concentrations of added compounds (Figure 5D). This behavior suggests a reduction in the order of the membrane induced by the interposition of the bivalent compound between the acyl chains of DPPC, which disrupts their regular packing in the Lβ′ and Pβ′ phases. The behavior observed herein is similar to that of curcumin61 except that disappearance of the pre-transition only occurred in the presence of higher concentrations of curcumin.61 This may suggest that both curcumin and our bivalent compounds can insert themselves between the acyl chains of DPPC. When the shift of the phase transition completion temperatures were examined, only a slight change was observed, i.e., from 43.1 °C in pure DPPC to 42.4 °C, even in the presence of the maximum concentration of 17MN or 17MD (5:1 molar ratio) (Figure 5E). Such small changes suggest fluid-fluid immiscibility, i.e., the bivalent compounds segregate from the bulk DPPC and accumulate in a laterally separated and compound-enriched domain. In contrast, addition of curcumin alone to DPPC membrane systems has been reported to slightly increase the completion temperature of the DPPC phase transition and reach a plateau until a 10:1 molar ratio is reached.61 The conclusion is that, although both curcumin and our bivalent compounds can interposition themselves into the hydrophobic palisade of the membrane bilayer, they may interact differently with the acyl chain of the DPPC.
Figure 5. Heating thermograms of DPPC/compounds by DSC.

DSC measurements of mixtures of DPPC with 17MN (A), 17MO (B); 17MD (C) under various DPPC/intrinsic molecule molar ratios. Onset temperatures (D) and completion temperatures (E) of the heating thermograms as a function of the molar ratios of intrinsic molecules/DPPC. (F) Effects on the ΔH change with increasing concentrations of the intrinsic molecules.
The effects of these bivalent compounds on DPPC phase transitions were also analysed calorimetrically with ΔH analysis (Figure 5F). In general, ΔH decreased from 8.7 kcal/mol for pure DPPC to ~3–5 kcal/mol in the presence of compounds. Notably, the most significant decrease was observed after addition of the minimum compound concentrations, and subsequent increases in concentration did not significantly impact ΔH. This can be interpreted in terms of limited miscibility of the bivalent compounds within the DPPC membrane system. To further understand the impact of the bivalent compounds’ anchor moiety on the structure of DPPC, studies of pure DPPC and DPPC/compound were performed using small angle X-ray diffraction (SAXD) and wide angle X-ray diffraction (WAXD). These techniques define not only macroscopic structure but also the interlamellar repeat distance, which is reflected by the largest first order reflection component or d value. Both the bilayer thickness and the thickness of the water layer between bilayers compose the d value.64 Typically, when phospholipids form a multilamellar bilayer membrane, the d-spacing observed with SAXD manifests as 1:1/2:1/3:1/4:1/5.65 As shown in Figure 6A, the SAXD and WAXD patterns of pure DPPC demonstrated the existence of multilamellar vesicles, the presence of an Lβ′ gel phase with hydrocarbon side-chains tilted relative to the normal bilayer phase and pseudohexagonal chain packing at 25 °C, and a bilayer structure in the La phase at 50 °C, results that are all consistent with those previously reported for DPPC.66
Figure 6. Small angle and wide angle diffraction profiles of pure DPPC and DPPC/compounds.

(A) Pure DPPC. (B) DPPC/17MN. (C) DPPC/17MO. (D) DPPC/17MD. Molar ratio is 10:1 in all of these experiments. Temperatures are shown next to the traces.
The incorporation of 17MN to DPPC at 25 °C (Figure 6B) led to a disordered membrane structure as evidenced by the diffused d-spacing. However, the presence of two reflections at 23.2 and 16.0 Å may suggest observation of the third and fourth order reflections of a bilayer, and the resulting broad peak may hide the first order Bragg reflection. The WAXD diffractogram at the same temperature exhibited a peak at 4.14 Å, characteristic of a membrane in which lipids exhibit hexagonal packing in a Lβ structure in which the acyl chains are perpendicular to the plane of the bilayer rather than the tilted Lβ′ structure present in pure DPPC.66 At 50 °C, the SAXD diffractogram showed a first spacing at 81.0 Å that may suggest a structure of increased membrane thickness. However, we cannot exclude the possibility that this could be due, at least partially, to an increase in the thickness of the water layer between the bilayers. The second and third order spacing appeared at 40.7 and 20.6 Å, respectively, confirming a bilayer structure (Figure 6B). No change was observed for the WAXD at this temperature when compared to pure DPPC. Overall, the SAXD and WAXD patterns observed for DPPC/17MN above the temperature of phase transition are very similar to those reported previously for curcumin at a similar concentration.61
Notably, the DPPC/17MO and DPPC/17MD gave a very different SAXD profile (Figures 6C and 6D) relative to DPPC/17MN. At 25 °C a broad and diffuse Bragg reflection peak was observed indicating a disordered membrane structure. However, a first order d-spacing appeared (at 55.9 Ǻ for 17MO and 55.8 Å for 17MD) out of the broad peak. At 50 °C two first order reflections were detected (80.9/55.6 Ǻ for 17MO; 81.1/55.6 Å for 17MD, respectively). Finally, a second order d-spacing was observed (40.1 Ǻ for 17MO and 40.8 Å for 17MD). These results clearly indicate a phase separation in the membrane structure, with one phase being an expanded interlamellar repeat spacing (80.9 or 81.1 Å) containing a low concentration of 17MO or 17MD and another one being enriched with them (55.6 Å). The first order d-spacing observed (55.9 and 55.8 Ǻ) at 25 °C may also reflect the existence of this phase separation and compound-enriched phase. The WAXD diffractograms for 17MO and 17MD are essentially the same as those of 17MN, suggesting a Lβ structure at 25 °C and a fluid state at 50 °C.
Discussion and Conclusions
Multiple pathogenic factors have been implicated in the development of AD;2, 6, 7, 67–69 consequently, design of compounds that can tackle multiple risk factors simultaneously has attracted much interest as routes to develop more effective treatment agents. Our recent research has led to the design and development of bivalent compounds that contain a multifunctional warhead with antioxidant, anti-inflammatory and anti-Aβ activities while containing a membrane-anchoring moiety linked to the warhead by a spacer.28–33 We clearly established the superior activity of such bivalent compounds in rescuing MC65 cells from TC withdrawal-induced cytotoxicity compared to the physical combination of the two parent (pharmacophore) compounds. To understand the specific contribution of the anchor moiety to the observed biological activity, bivalent compounds with different anchoring pharmacophores were designed and synthesized. Our results here identified diosgenin being the preferred anchor to give the most potent bivalent compound for protecting MC65 cells from TC removal-induced cytotoxicity. The signifcantly increased potency of 17MD may be due to the fact that diosgenin and related analogs, such as caprospinol, have been demonstrated to have neuroprotective activities in various AD models.38, 70, 71 However, the combination of 17SC and 17SD, two monovalent control compounds of 17MD that contain curcumin and diosgenin moiety, respectively, did not provide rescue effects under the same experimental conditions. This clearly indicates that the nature of the bivalent structure is essential to the observed biological activity and, in particular, the incorporation of diosgenin may lead to a distinct mechanism of action compared to the parent structures.
In neuritic outgrowth studies, only bivalent compounds with diosgenin as the anchor moiety exhibited neurotrophic effects. Diosgenin and its analogs have been shown to exhibit axonic outgrowth and cognitive improvement activities by targeting the 1,25D3-MARRS.39, 58 Because the combination of 17SC and 17SD did not exhibit neurotrophic effects, 1,25D3-MARRS might not be a significant biological target for the neuritic outgrowth activity of 17MD. This compound also exhibited modulating activities on the state 3 oxidative rate of glutamate while maintaining coupling capacity of the mitochondria in mouse brain, whereras the 17SC and 17SD combination led to potential mitochondrial toxicity with significant effects on the oxidation of both glutamate and succinate during the OXPHOS process. From our results, diosgenin is the essential moiety within our unique bivalent compound set for neuritic outgrowth and mitochondria activity. The conclusion is that incorporation of diosgenin into bivalent compounds results in unqiue membrane interactions and/or destinations that give superior biological activities. This is certainly due in part to the unique structure of diosgenin, where the cyclized side chain contains a ketal moiety different from the other steroid anchor moieties we studied.
The model membrane DPPC, when treated with 17MN, 17MO and 17MD as the representative bivalent probes, also indicated that the anchor moiety plays a role in its interactions with the membrane system. DSC experiments of completion temperature as a function of compound concentration suggest that bivalent compounds are essentially immiscible with fluid DPPC, as previously shown with curcumin.61 But, judging from the analysis of changes in the transition state onset temperature, some miscibility was implicated in the DPPC gel state with compounds 17MN, 17MO and 17MD. Furthermore, analyses of changes in ΔH also support the notion that these bivalent compounds influence the ordering of the DPPC membrane, but remain immiscible in fluid DPPC, which was confirmed by SAXD diffraction results.
However, a few notable features were observed from the SAXD analysis: firstly, addition of all three compounds to the DPPC led to a high d-spacing in the fluid phase, which suggests the formation of an expanded interlamellar repeat spacing; secondly, the clear observation of a second phase in fluidic DPPC for compounds 17MO and 17MD possibly represents the formation of a compound-enriched phase separated from a phase with an increased interlamellar repeat spacing. A similar result was previously observed for DPPC/curcumin.64 The increase at 50 °C in the interlamellar spacing could be due to an increase in the membrane thickness induced by interacting with the bivalent compounds or to an increase in thickness of the water layer between the two bilayers. But, given the complexity of this system and the resulting diffractograms, it is difficult to discern between these two possibilities, since calculation of the electronic density of this membrane would be very challenging. Nevertheless, the results strongly indicate that the anchor moiety does influence how the bivalent compounds interact with the DPPC model membrane, and in particular, bivalent compounds with a O-linkage to the anchor moiety, i.e., cholesterol or diosgenin, can potentially lead to phase separation of the DPPC model membrane.
In summary, the studies of bivalent compounds we reported here identified diosgenin as the optimal anchor moiety within this series that provides compounds with superior protective activity, mitochondria modulating activity, and neuritic outgrowth stimulating activity. Comparison with the activities of the combination of two monovalent control compounds in all of these assays indicated that the observed biological activities of the diosgenin-containing compounds are due to the explicit nature of the bivalent compounds. Studies in model membrane system also illuminate the differential roles of anchor moiety in the interactions, especially when comparing the steroid skeleton with an oxygen atom at position-3 with one possessing a nitrogen atom at the same position. Collectively, the results suggest that diosgenin may represent a novel anchor moiety that truly gives bivalent compounds with multifunctional properties and strongly encourages further development of such bivalent compounds as potential treatments for AD.
Experimental Section
Chemistry
Reagents and solvents were obtained from commercial suppliers and used as received unless otherwise indicated. All reactions were carried out under inert atmosphere (N2) unless otherwise noted. Reactions were monitored by thin-layer chromatography (TLC) (pre-coated silica gel 60 F254 plates, EMD Chemicals) and visualized with UV light or by treatment with Phosphomolybdic acid (PMA). Flash chromatography was performed on silica gel (200–300 mesh, Fisher Scientific) using solvents as indicated. 1HNMR and 13CNMR spectra were routinely recorded on Bruker ARX 400 spectrometer. The NMR solvent used was CDCl3 or DMSO-d6 as indicated. Tetramethylsilane (TMS) was used as internal standard. The purity of target compounds was determined by HPLC using Varian 100-5 C18 250 × 4.6 mm column with UV detection (288 nm) (40% acetonitrile/60% methanol or 38% acetonitrile/62% H2O/2% acetic acid, pH 3.0 two solvent systems) to be ≥ 95%.
Method A
To a solution of cholesterol (1, 10.00 mmol) in tetrahydrofuran (THF) (50.00 mL) was added NaH (40.00 mmol) and 1,4-dibromobutane (40.00 mmol). The mixture was refluxed for 48 h, water was then added drop by drop. The reaction mixture was extracted with ethyl acetate (50.00 mL × 2) and the combined organic phase was dried over anhydrous sodium sulfate. The solvent was removed and the residue was purified by column chromatography to give the intermediate (1.32 g, 25%). 1H NMR (400 MHz, CDCl3) δ 5.40 – 5.33 (m, 1H), 3.52 (t, J = 6.3 Hz, 2H), 3.47 (t, J = 6.8 Hz, 2H), 3.22 – 3.09 (m, 1H), 2.44 – 2.32 (m, 1H), 2.24 – 2.14 (m, 1H), 2.11 – 1.79 (m, 6H), 1.79 – 1.68 (m, 2H), 1.68 – 0.81 (m, 34H), 0.70 (s, 3H).
The prepared intermediate from above (1.50 mmol) and phthalimide (3.00 mmol) were dissolved in dimethyl formaldehyde (DMF) (15.0 mL). Potassium carbonate (3.00 mmol) was added to the solution. The mixture was stirred at 70 °C for 5 h, water (15.00 mL) was then added. The reaction mixture was extracted with ethyl acetate (30.00 mL × 2). After dried with anhydrous sodium sulfate, the solvent was removed and the residue was purified by column chromatography to intermediate 2-(4-(((3S,9S,10R,13R,14S,17R)-10,13-dimethyl-17-((R)-6-methylheptan-2-yl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl)oxy)butyl)isoindoline-1,3-dione(3) as a white solid (0.79 g, 90%). 1H NMR (400 MHz, CDCl3) δ 7.95 – 7.79 (m, 2H), 7.79 – 7.65 (m, 2H), 5.35 (m, 1H), 3.74 (t, J = 7.2 Hz, 2H), 3.51 (t, J = 6.7 Hz, 2H), 3.20 – 3.15 (m, 1H), 2.45 – 2.31 (m, 1H), 2.19 (m, 1H), 2.10 – 1.71 (m, 7H), 1.70 – 0.79 (m, 35H), 0.68 (s, 3H).
2-(4-(((4S,6aR,6bS,8aS,12aS)-5′,6a,8a,9-tetramethyl-1,3,3′,4,4′,5,5′,6,6a,6b,6′,7,8,8a,8b,9,11a,12,12a,12b-icosahydrospiro[naphtho[2′,1′:4,5]indeno[2,1-b]furan-10,2′-pyran]-4-yl)oxy)butyl)isoindoline-1,3-dione (4) was prepared starting with diosgenin (2, 5.00 mmol) following Method A. 1H NMR (400 MHz, CDCl3) δ 7.90 – 7.82 (m, 2H), 7.77 – 7.69 (m, 2H), 5.41 – 5.27 (m, 1H), 4.43 (dd, J = 15.0, 7.5 Hz, 1H), 3.83 – 3.66 (m, 2H), 3.60 – 3.30 (m, 4H), 3.20 – 3.05 (m, 1H), 2.45 – 2.30 (m, 1H), 2.25– 2.11 (m, 1H), 2.10 – 0.88 (m, 32H), 0.87 – 0.74 (m, 6H).
Method B
To a suspension of 3 (1.34 mmol) in ethanol (20.00 mL) was added hydrazine (5.00 eq). The mixture was refluxed for 5 h, and then the solvent was removed. Dichloromethane (DCM) (30.00 mL) was added and filtered to remove the precipitate. The filtrate was collected and the solvent was removed to give the amine intermediate (0.65 g). Without further purification, to the amine was added DCM (5.00 mL), ethyl dimethylaminopropyl carbodiimide (EDC) (0.80 mmol), and 2-(2-(2-azidoethylamino)-2-oxoethoxy)-acetic acid (5, 0.80 mmol). The mixture was stirred at room temperature (rt) overnight. Water (10.00 mL) was added and the mixture was then extracted with DCM (15.00 mL × 2), and the combined organic phase was dried over anhydrous sodium sulfate. After removing solvents, the residue was purified by column chromatography to give N-(2-azidoethyl)-2-(2-((4-(((3S,9S,10R,13R,14S,17R)-10,13-dimethyl-17-((R)-6-methylheptan-2-yl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl)oxy)butyl)amino)-2-oxoethoxy)acetamide (7) as a white solid (0.22 g, 26%). 1H NMR (400 MHz, CDCl3) δ 6.90 (s, 1H), 6.61 (s, 1H), 5.41 – 5.32 (m, 1H), 4.11– 4.05 (m, 4H), 3.58 – 3.43 (m, 6H), 3.42 – 3.31 (m, 2H), 3.22 – 3.10 (m, 1H), 2.42 – 2.32 (m, 1H), 2.25 – 2.15 (m, 1H), 2.08 – 1.76 (m, 5H), 1.73 – 0.77 (m, 37H), 0.70 (s, 3H).
Reaction of 3 (0.40 mmol) with 2-(2-(4-azidobutylamino)-2-oxoethoxy)-acetic acid (6, 0.40 mmol) following Method B yielded N-(6-azidohexyl)-2-(2-((4-(((3S,9S,10R,13R,14S,17R)-10,13-dimethyl -17-((R)-6-methylheptan-2-yl)-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-3-yl)oxy)butyl)amino)-2-oxoethoxy)acetamide (8) (0.18 g, 65%). 1H NMR (400 MHz, CDCl3) δ 6.62 (s, 1H), 6.55 (s, 1H), 5.40 – 5.30 (m, 1H), 4.11– 4.02 (m, 4H), 3.60 – 3.45 (m, 2H), 3.42 – 3.23 (m, 5H), 3.22 – 3.10 (m, 1H), 2.42 – 2.30 (m, 1H), 2.26 – 2.14 (m, 1H), 2.10– 1.78 (m, 5H), 1.75 – 0.78 (m, 46H), 0.68 (s, 3H).
N-(2-azidoethyl)-2-(2-oxo-2-((4-(((4S,6aR,6bS,8aS,12aS)-5′,6a,8a,9-tetramethyl-1,3,3′,4,4′,5,5′,6,6a,6b,6′,7,8,8a,8b,9,11a,12,12a,12b-icosahydrospiro[naphtho[2′,1′:4,5]indeno[2,1-b]furan-10,2′-pyran]-4-yl)oxy)butyl)amino)ethoxy)acetamide (9) was prepared following Method B starting from 4. 1H NMR (400 MHz, CDCl3) δ 6.95 (s, 1H), 6.64 (s, 1H), 5.35 – 5.20 (m, 1H), 4.41 – 4.24 (m, 1H), 4.00 (d, J = 5.4 Hz, 4H), 3.56 – 2.99 (m, 11H), 2.34– 2.24 (m, 1H), 2.23 – 2.02 (m, 2H), 2.00 – 1.87 (m, 2H), 1.86 – 1.74 (m, 3H), 1.73 – 1.30 (m, 16H), 1.28 – 0.89 (m, 10H), 0.72 (t, J = 3.2 Hz, 6H).
N-(6-azidohexyl)-2-(2-oxo-2-((4-(((4S,6aR,6bS,8aS,12aS)-5′,6a,8a,9-tetramethyl-1,3,3′,4,4′,5,5′,6,6a,6b,6′,7,8,8a,8b,9,11a,12,12a,12b-icosahydrospiro[naphtho[2′,1′:4,5]indeno[2,1-b]furan-10,2′-pyran]-4-yl)oxy)butyl)amino)ethoxy)acetamide (10) was prepared following Method B starting from 4. 1H NMR (400 MHz, CDCl3) δ 6.62 (s, 1H), 6.55 (s, 1H), 5.39 – 5.34 (m, 1H), 4.43 (dd, J = 14.9, 7.5 Hz, 1H), 4.10 – 4.00 (m, 4H), 3.61 – 3.23 (m, 11H), 3.23 – 3.05 (m, 1H), 2.45 – 2.30 (m, 1H), 2.28– 2.11 (m, 1H), 2.10 – 0.86 (m, 39H), 0.81 (t, J = 3.2 Hz, 6H).
2-(2-((2-azidoethyl)amino)-2-oxoethoxy)acetic acid (12, 0.41 g, 2.00 mmol) and HOBt (0.33 g, 2.40 mmol) were dissolved in dry DCM (20.00 mL) under ice bath followed by the addition of EDCI (0.46 g, 2.40 mmol) and stirring for 30 min. 1-Butylmine (0.18 g, 2.40 mmol) and TEA (0.34 mL) dissolved in dry DCM (10.00 mL) were added into the solution directly. The mixture was stirred for 3 h at room temperature. The DCM solution was washed with 1 N aqueous citric acid, saturated NaHCO3 and brine for 3 times, dried over anhydrous sodium sulfate. After removing solvents, the residue was purified by column chromatography to give N-(2-azidoethyl)-2-(2- (butylamino)-2-oxoethoxy) acetamide (13) as a white solid (0.24 g, 46%). 1H NMR (400 MHz, CDCl3) δ 6.77 (s, 1H), 6.33 (s, 1H), 4.03 – 4.00 (m, 4H), 3.28 – 3.23 (m, 2H), 1.50 – 1.43 (m, 2H), 1.35 – 1.25 (m, 2H), 1.21 – 1.15 (m, 2H),0.89 (t, J = 7.3 Hz, 3H).
Method C
Compound 7 (1.00 eq) and (1E,4Z,6E)-5-hydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl) -4-(prop-2-ynyl)hepta-1,4,6-trien-3-one (11, 2.00 eq) were dissolved in THF:water (1:1, 10.00 mL). Sodium ascorbate (0.040 eq) and copper sulfate (0.020 eq) were added. The reaction mixture was stirred at room temperature for 24 h, DCM (10.00 mL) was then added. The organic layer was separated and washed with water followed by brine, dried over anhydrous sodium sulfate, and concentrated in vacuum. The residue was purified by flash column chromatography (DCM/methanol: 20/1) to afford 17MO as orange solid. 1H NMR (400 MHz, CDCl3) δ 7.79 – 7.50 (m, 3H), 7.45 (s, 1H), 7.13 – 6.74 (m, 8H), 6.67 (d, J = 15.6 Hz, 1H), 5.28 – 5.18 (m, 1H), 4.92 – 4.70 (m, 1H), 4.52 – 4.35 (m, 2H), 4.12 – 3.55 (m, 16H), 3.47 – 3.13 (m, 6H), 3.11 – 2.97 (m, 1H), 2.35 – 2.18 (m, 1H), 2.15 – 2.00 (m, 1H), 1.99 – 1.66 (m, 5H), 1.55 – 0.87 (m, 26H), 0.84 (d, J = 6.6 Hz, 3H), 0.79 (dd, J = 1.7, 6.6 Hz, 6H), 0.59 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 194.1, 183.2, 169.6, 168.6, 149.1, 148.4, 147.1, 145.7, 143.0, 140.8, 127.7, 126.5, 124.3, 123.2, 121.8, 121.7, 121.6, 117.5, 115.0, 110.5, 110.2, 79.2, 79.1, 71.0, 70.8, 67.5, 56.8, 56.2, 56.1, 50.2, 42.3, 39.8, 39.5, 39.1, 39.0, 38.7, 38.7, 37.2, 36.9, 36.2, 35.8, 31.9, 31.9, 28.5, 28.2, 28.0, 27.5, 26.4, 24.3, 23.8, 22.8, 22.5, 21.1, 19.3, 18.7, 11.8.
Compound 17MD was prepared starting from 9 following Method C
1H NMR (400 MHz, CDCl3) δ 7.70 –7.42 (m, 4H), 7.10 – 6.75 (m, 8H), 6.70 (d, J = 15.8 Hz, 1H), 5.24 (d, J = 4.7 Hz, 1H), 4.95 – 4.85 (m, 1H), 4.55 – 4.40 (m, 2H), 4.33 (q, J = 7.4 Hz, 1H), 4.11 (s, 1H), 3.99 – 3.64 (m, 13H), 3.44 – 3.19 (m, 7H), 3.10 – 2.97 (m, 1H), 2.30 – 2.20 (m, 1H), 2.13 – 2.00 (m, 1H), 1.96 – 1.85 (m, 2H), 1.83 – 0.82 (m, 31H), 0.72 (d, J = 6.9 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 194.3, 183.1, 169.4, 168.5, 149.1, 148.4, 147.1, 145.5, 142.7, 140.9, 140.8, 127.7, 126.5, 124.2, 123.0, 121.8, 121.4, 121.3, 117.7, 115.1, 115.1, 110.5, 110.2, 109.3, 107.6, 80.8, 79.0, 71.1, 71.0, 70.8, 67.5, 66.8, 62.1, 56.5, 56.1, 56.1, 50.1, 41.6, 40.3, 39.8, 39.1, 39.0, 38.9, 38.9, 37.2, 37.0, 32.1, 31.8, 31.4, 31.4, 30.3, 28.8, 28.4, 27.4, 26.4, 20.8, 19.4, 17.1, 16.2, 14.5.
Compound 17SC was prepared starting from 11 following Method C
1H NMR (400 MHz, CDCl3) δ 7.68 – 7.52 (m, 2H), 7.35 (s, 1H), 7.10 – 7.01 (m, 3H), 6.99 – 6.93 (m, 2H), 6.89 – 6.79 (m, 3H), 6.64 (d, J = 15.8 Hz, 2H), 6.45 – 6.35 (m, 1H), 4.64 (t, J = 7.2 Hz, 1H), 4.45 – 4.30 (m, 2H), 4.00 – 3.81 (m, 11H), 3.72 – 3.64 (m, 2H), 3.38 – 3.18 (m, 4H), 1.49 – 1.43 (m, 2H), 1.30 – 1.24 (m, 2H), 0.88 – 0.82 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 193.3, 182.1, 168.1, 167.3, 148.0, 147.2, 145.9, 144.4, 144.0, 141.7, 126.8, 125.6, 123.2, 122.1, 122.0, 120.7, 116.7, 114.0, 113.9, 109.3, 108.9, 106.7, 70.1, 69.8, 62.3, 55.1, 55.1, 37.9, 37.8, 37.7, 30.6, 28.7, 19.1, 12.7.
Compound 17SD was prepared starting from 9 following Method C
1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.29 (s, 1H), 6.65 (s, 1H), 5.30 – 5.22 (m, 1H), 4.56 – 4.50 (m, 1H), 4.39 – 4.30 (m, 1H), 4.02 – 3.92 (m, 4H), 3.82 – 3.74 (m, 1H), 3.52 – 2.97 (m, 10H), 2.37 – 0.57 (m, 48H). 13C NMR (100 MHz, CDCl3) δ 198.8, 169.3, 168.3, 140.9, 126.8, 121.4, 109.3, 80.8, 79.1, 71.1, 70.9, 67.5, 66.8, 62.2, 56.5, 50.1, 49.7, 41.6, 40.3, 39.8, 39.2, 39.0, 38.8, 37.2, 37.1, 37.0, 32.1, 31.9, 31.5, 31.4, 30.3, 29.7, 28.8, 28.5, 27.4, 26.5, 22.7, 20.9, 19.4, 18.4, 17.1, 16.3, 14.5.
Compound 21MO was prepared starting with 8 following Method C
1H NMR (400 MHz, CDCl3) δ 7.70 – 7.55 (m, 2H), 7.53 (s, 1H), 7.10 – 6.57 (m, 10H), 5.29 – 5.22 (m, 1H), 4.91(s, 1H), 4.32 – 4.13 (m, 2H), 4.05 (s, 1H), 4.00 – 3.91 (m, 4H), 3.83 (d, J = 10.1 Hz, 6H), 3.49 – 2.91 (m, 10H), 2.30 – 2.20 (m, 1H), 2.08 (t, J = 12.3 Hz, 1H), 2.01 – 1.64 (m, 7H), 1.59 – 0.87 (m, 33H), 0.84 (d, J = 6.5 Hz, 3H), 0.79 (dd, J = 1.8, 6.6 Hz, 6H), 0.60 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 194.3, 183.1, 168.7, 168.6, 149.0, 148.4, 147.1, 147.1, 145.5, 142.6, 140.8, 140.8, 127.7, 126.6, 124.2, 123.3, 122.2, 121.7, 117.6, 115.0, 115.0, 110.1, 79.2, 79.2, 71.2, 71.1, 67.4, 56.8, 56.2, 56.1, 50.2, 42.3, 39.8, 39.5, 39.2, 38.9, 38.9, 38.7, 38.6, 37.2, 36.9, 36.2, 35.8, 31.9, 31.9, 29.8, 29.2, 28.5, 28.2, 28.0, 27.4, 26.5, 25.8, 25.7, 24.3, 23.8, 22.8, 22.5, 21.1, 19.4, 18.7, 11.8.
Compound 21MD was prepared starting from 10 following Method C
1H NMR (400 MHz, CDCl3) δ 7.68 – 7.51 (m, 2H), 7.44 (s, 1H), 7.14 – 6.76 (m, 8H), 6.76 – 6.40 (m, 2H), 5.30 – 5.20 (m, 1H), 4.95 – 4.75 (m, 1H), 4.33 (q, J = 7.4 Hz, 1H), 4.21 (q, J = 7.0 Hz, 2H), 4.03 (s, 1H), 3.98 – 3.88 (m, 4H), 3.83 (d, J = 10.1 Hz, 6H), 3.52 – 2.90 (m, 12H), 2.32 – 2.15 (m, 1H), 2.15 – 1.99 (m, 1H), 1.98 – 1.85 (m, 2H), 1.85 – 0.96 (m, 31H), 0.96 – 0.81 (m, 7H), 0.71 (d, J = 5.6 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 194.4, 183.1, 168.7, 168.6, 149.1, 148.4, 147.2, 145.4, 142.5, 140.8, 127.7, 126.5, 124.2, 123.2, 122.1, 121.4, 117.7, 115.1, 115.1, 110.2, 109.3, 107.6, 80.8, 79.1, 71.2, 71.1, 67.4, 66.8, 65.8, 62.1, 56.5, 56.1, 56.1, 50.1, 41.6, 40.3, 39.8, 39.1, 38.9, 38.7, 37.2, 37.0, 32.1, 31.8, 31.4, 31.4, 30.3, 29.7, 29.2, 28.8, 28.4, 27.4, 26.5, 25.9, 25.8, 20.8, 19.4, 17.1, 16.2, 15.2, 14.5.
Biological assays
MC65 cells were kindly provided by Dr. George M. Martin at the University of Washington, Seattle and were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Life Technologies, Inc., Grand Island, NY) supplemented with 10% of heat-inactivated fetal bovine serum (FBS) (Hyclone, Logan, UT), 1 μg/mL TC and 0.2 mg/mL G418 (Invitrogen) and maintained at 37 °C in a fully humidified atmosphere containing 5% CO2. Neuronal N2a cells were purchased from American Type Cell Culture (ATCC, Manassas, VA) and were cultured in Minimum Essential Medium (MEM) supplemented with 10% FBS and maintained at 37 °C in a fully humidified atmosphere containing 5% CO2. Primary cortical neurons isolated from E17 rats were seeded on poly-D-lysine coated coverslips in 24-well plates (2 × 104/well) and cultured in neurobasal medium supplemented with B27 and Glutamax.
Rescue assay in MC65 cells
MC65 cells were washed twice with PBS, resuspended in Opti-MEM, and seeded in 96-well plates (4×104 cells/well). Indicated compounds were then added, and cells were incubated at 37 °C under -TC conditions for 72 h. Then, 10 μL of MTT solution (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, 5 mg/mL in PBS) were added and the cells were incubated for another 4 h. Cell medium was then removed, and the remaining formazan crystals produced by the cellular reduction of MTT were dissolved in 100 μL of DMSO. Absorbance at 570 nm was immediately recorded using a FlexStation 3 plate reader (Molecular Devices, CA). Values were expressed as a percentage relative to those obtained in the +TC controls. Each data point was averaged from six replicates and the experiments were independently repeated at least three times.
Neuritic outgrowth of N2a cells and mouse primary cortical neurons
Neuronal N2a cells were seeded in 24-well plates (1 × 104 cells/well) and cultured in MEM supplemented with 0.5% FBS in the presence of testing compounds for 48 h. The medium was removed and cells were stained with Neurite Outgrowth Staining kit (Life Technologies, Carlsbad, CA) per the manufacturer’s instruction. Images were recorded for each well using 10× objective (20 images around the center of each well) by Zeiss Axiovert 200M fluorescence microscopy. Images were then analyzed by Image J to quantify the neurite length.
Primary cortical neurons from E17 rat embryos were treated with DMSO or compounds at the first day in vitro (DIV 1) and fixed at DIV5 with 4% PFA in PBS. Neurites were stained with beta III tubulin and nuclei were visualized after staining with DAPI. Images were taken for each well using 20× objective. At least 150 neurons in each group were randomly selected. The neurite length was traced and quantified using Image J.
Brain mitochondrial isolation and oxygen consumption determination
The Institutional Animal Care and Use Committees (IACUC) of the McGuire VA Medical Center and Virginia Commonwealth University approved this protocol. Brain cortex tissue was collected as discarded tissue from C57BL/6 mice after the mouse was deeply anesthetized with sodium pentobarbital (100 mg/kg, i.p.) and euthanized by removal of the heart for other experiments. Harvested brain tissue was placed into 5 mL MSM buffer (210 mM Mannitol, 70 mM Sucrose, 5.0 mM MOPS, 1.0 mm EDTA, pH 7.4) at 4 °C, finely minced, and incubated with Subtilisin A (1 mg/g tissue) for 1 min. Another 5 mL MSM buffer including 0.2% BSA was added to the incubated tissue that was then homogenized by one stroke using a Teflon pestle. The homogenate was centrifuged at 600xg for 10 min at 4 °C. The supernatant was then centrifuged at 5000xg for 10 min at 4 °C to spin down the mitochondria. The mitochondrial pellet was washed once with MSM buffer, then resuspended in 100–200 μL of MSM buffer. Total protein concentration was measured by the Lowry method using bovine serum albumin (BSA) as a standard. Oxygen consumption in mitochondria was measured by a Clark-type oxygen electrode at 30 °C using glutamate + malate (complex I substrates) and succinate + rotenone (complex II substrates) in the presence or absence of testing compounds.
ETC Assays
ETC assays were conducted following reported procedure.59 Frozen and thawed mouse brain mitochondria were solubilized in 5% cholate (pH 7.2), then diluted with KME (100 mM KCl, 50 mM MOPS, and 1 mM EGTA) buffer to 0.1% protein concentration. Complex I activity was measured by monitoring the rate of NADH consumption using spectrophotometer (Hewlett-Packard model 8453) at 340 nm in the presence of 100 μM antimycin A, 1 mM decylubiquinone, and 150 μM NADH to 40 μg/mL mitochondria protein with or without 7.5 μM rotenone. Complex I activity was expressed as rotenone-sensitive rate (Complex I activity without rotenone – complex I activity with rotenone). Complex II activity was measured as the rate of oxidation of dichlorophenol-indophenol (DCPIP) at 600 nm in the presence of mitochondria (2 ug/ml) and 1 mM oxidized decylubiquinone and 1 M succinate. Complex II activity was expressed as thenoyltrifluoroacetone (TTFA) sensitive rate (Complex II activity without TTFA – complex II activity with TTFA). Complex III activity was measured as the initial rate of the reduction of cytochrome c at 550 nm on addition of 40 μM reduced decylubiquinone and 1 mM cytochrome c to 3 ug/ml mitochondrial protein with or without 100 μM antimycin A. Complex III activity was expressed as antimycin A sensitive rate (Complex III activity without antimycin A – complex III activity with antimycin A). Complex IV activity was measured as the first-order rate of consumption of 1 mM reduced cytochrome C at 550 nm over 7.5 minutes in the presence of mitochondria (2 ug/ml). Complex IV activity was expressed as the difference between the measured activity and the background activity.
Assays to eliminate PAINS concerns
Curcumin has been reported as one of the PAINs structures. Although our compounds are totally different than curcumin and our research is focused on the biological roles of the membrane anchor of the bivalent compounds, they do contain the curcumin moiety. To rule out any concerns of PAINs in the observed biological activity, we conducted experiments to include controls (17SC and 17SD, and in some experiments we also included curcumin itself) and compared the results with the designed bivalent compounds. These experiments include Rescue assay of MC65 cells, Neurite outgrowth assay in N2a cells, and mouse brain mitochondria assays. From these assays, we did not observe any significant effects from the control compounds that contain the curcumin moiety. This clearly indicate that the observed biological activity is solely due to the presence of the bivalent ligands, not the interference of the curcumin moiety, thus eliminating the concerns for these compounds as PAINs.
Model membrane assays
1, 2-dipalmitoyl-sn-phosphatidylcholine (DPPC) was obtained from Avanti Polar Lipids (Birmingham, Alabama, USA). Other chemicals were obtained from Sigma Chemical Co. (Madrid, Spain) and used as received unless otherwise indicated. The effects of bivalent compounds on the different phases adopted by DPPC, namely Lβ′ (gel phase with the chains tilted with respect to the normal to the bilayer plane) to Pβ′ (rippled gel phase with tilted chains with respect to the normal to the bilayer plane) and from Pβ′ to Lα (fluid phase) transitions of the DPPC membranes was measured by DSC and the calorimetric profiles of the thermotropic gel-to-liquid-crystalline transition of pure DPPC and those with bivalent compounds at different molar ratios.
Preparation of samples for DSC
Samples were prepared by dissolving the desired amount of DPPC in chloroform/methanol (2:1). 17MN, 17MO and 17MD were also prepared in the same solvents and mixed with the phospholipid in the desired ratio. The mixture was then dried under a nitrogen stream and finally under vacuum. Samples (2 mg of phospholipid) were hydrated by dispersing them in 0.5 mL of 100 mM NaCl, 25 mM Hepes pH 7.4 buffer and multilamellar vesicles were prepared by vortexing at 55 °C. The samples and the same volume of buffer (used as reference) were degassed for 10 min before loading to the calorimeter. Thermograms were recorded by using a Microcal VP scanning calorimeter (Microcal, Northampton, MA, USA). All of the samples were scanned out over a 10–60 °C temperature range at a heating rate of 60 °C/h. Thermogram data were recorded and analysed using Microcal Origin 5.0 software. The traces were normalized, depending on the DPPC concentration of each sample. The thermal behaviour of liposomes was evaluated by determining the linear onset and completion temperatures of the pre-transition and the main transition (Tc) and by calculating the ΔH corresponding to the main transition of DPPC in the samples.
X-ray diffraction
Samples for X-ray diffraction were prepared as described above for DSC studies. After centrifugation at 13,000 xg, the pellets were collected. Simultaneous SAXD and WAXD measurements were carried out using a modified Kratky compact camera (MBraun-Graz-Optical Systems, Graz Austria), incorporating two coupled linear position sensitive detectors (PSD, MBraun, Garching, Germany) to monitor the s-ranges [s=2 sin θ/λ, 2θ=scattering angle, λ=1.54 Å] between 0.0075–0.07 and 0.20–0.29 Å−1, respectively. Nickel-filtered Cu KR X-rays were generated by a Philips PW3830 X-ray generator operating at 50 kV and 30 mA. The detector position was calibrated using Ag-stearate (small-angle region, d-spacing at 48.8 Å) and lupolen (wide angle region, d-spacing at 4.12 Å) as reference materials. Sample pellets were placed in a steel holder with cellophane windows, which provided good thermal contact with the Peltier heating unit. X-ray diffraction profiles were obtained for 10 min exposure times after 10 min of temperature equilibration.
Acknowledgments
The work was supported in part by the NIA of the NIH under award number R01AG041161 (SZ), Alzheimer’s Drug Discovery Foundation 20150601 (SZ), Award No. 18-2 from the Commonwealth of Virginia’s Alzheimer’s and Related Disease Research Award Fund administered by the Center on Aging, School of Allied Health Professions, Virginia Commonwealth University (SZ), VCU Presidential Research Quest Fund (SZ), R01 NS083385 from NINDS (XZ), AARG-16-443584 from Alzheimer’s Association (XZ), Office of Research and Development, Medical Research Service Merit Review Award (2IO1BX001355-01A2) Department of Veterans Affairs (EJL), a Grant-in-aid (15GRNT24480123) from the American Heart Association (QC), 1R21AG054975-01 (QC), and the Pauley Heart Center, Virginia Commonwealth University (QC, EJL), Grant 386 from University of Murcia (JCG). We thank Dr. Glen E. Kellogg in the Department of Medicinal Chemistry, Virginia Commonwealth University for helpful discussions.
Abbreviations
- AD
Alzheimer’s disease
- Aβ
beta-amyloid
- AβOs
Aβ oligomers
- APP
Amyloid precursor protein
- ADP
adenosine diphosphate
- ATP
adenosine triphosphate
- DCM
Dichloromethane
- DMEM
Dulbecco’s modified eagle’s medium
- DMF
Dimethyl formaldehyde
- DPPC
1,2-Dipalmitoyl-sn-glycero-phosphocholine
- DSC
Differential scanning calorimetry
- EDC
Ethyl dimethylaminopropyl carbodiimide
- ETC
electron transport chain
- FBS
Fetal bovine serum
- IACUC
Institutional animal care and use committees
- MEM
Minimum essential medium
- MTT
(3-(4,5-dimethylthiazol-2-yl)-3,5-diphenyltetrazolium bromide
- OXPHOS
oxidative phosphorylation
- PMA
Phosphpmolybdic acid
- SAXD
small angle X-ray diffraction
- TC
tetracycline
- TFA
Trifluoroacetic acid
- THF
Tetrahydrofuran
- TLC
Thin-layer chromatography
- TMS
Tetramethylsilane
- WAXD
wide angle X-ray diffraction
Footnotes
Author Contributions:
Experiments were completed by LH, KL, VGM, YJ, XM, AT, QC. Experiment design, data analysis, writing, and editing were completed by QC, XZ, EL, JCG, BX, SZ. All authors have given approval to the final version of the manuscript.
Supporting Information
Molecular formula strings (CSV)
References
- 1.Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2016;12:459–509. doi: 10.1016/j.jalz.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J, Higgins G. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 4.Avila J. Role of Tau Protein in Both Physiological and Pathological Conditions. Physiol Rev. 2004;84:361–384. doi: 10.1152/physrev.00024.2003. [DOI] [PubMed] [Google Scholar]
- 5.Zhu X, Su B, Wang X, Smith MA, Perry G. Causes of Oxidative Stress in Alzheimer Disease. Cell Mol Life Sci. 2007;64:2202–2210. doi: 10.1007/s00018-007-7218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calsolaro V, Edison P. Neuroinflammation in Alzheimer’s Disease: Current Evidence and Future directions. Alzheimers Dement. 2016;12:719–732. doi: 10.1016/j.jalz.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 7.Krstic D, Madhusudan A, Doehner J, Vogel P, Notter T, Imhof C, Manalastas A, Hilfiker M, Pfister S, Schwerdel C, Riether C, Meyer U, Knuesel I. Systemic Immune Challenges Trigger and Drive Alzheimer-Like Neuropathology in Mice. J Neuroinflammation. 2012;9:151–173. doi: 10.1186/1742-2094-9-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Wang W, Li L, Perry G, Lee H-g, Zhu X. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochim Biophys Acta, Mol Basis Dis. 2014;1842:1240–1247. doi: 10.1016/j.bbadis.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carreiras M, Mendes E, Perry M, Francisco A, Marco-Contelles J. The Multifactorial Nature of Alzheimer’s Disease for Developing Potential Therapeutics. Curr Top Med Chem. 2013;13:1745–1770. doi: 10.2174/15680266113139990135. [DOI] [PubMed] [Google Scholar]
- 10.Decker M. Design of Hybrid Molecules for Drug Development. Elsevier; United Kingdom: 2017. [Google Scholar]
- 11.Galdeano C, Viayna E, Arroyo P, Bidon-Chanal A, Ramon Blas J, Munoz-Torrero D, Javier Luque F. Structural Determinants of the Multifunctional Profile of Dual Binding Site Acetylcholinesterase Inhibitors as Anti-Alzheimer Agents. Curr Pharm Des. 2010;16:2818–2836. doi: 10.2174/138161210793176536. [DOI] [PubMed] [Google Scholar]
- 12.Morphy R, Rankovic Z. Designing Multiple Ligands – Medicinal Chemistry Strategies and Challenges. Curr Pharm Des. 2009;15:587–600. doi: 10.2174/138161209787315594. [DOI] [PubMed] [Google Scholar]
- 13.Ariga T, McDonald MP, Yu RK. Role of Ganglioside Metabolism in the Pathogenesis of Alzheimer’s Disease--a Review. J Lipid Res. 2008;49:1157–1175. doi: 10.1194/jlr.R800007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cordy JM, Hooper NM, Turner AJ. The Involvement of Lipid Rafts in Alzheimer’s Disease. Mol Membr Biol. 2006;23:111–122. doi: 10.1080/09687860500496417. [DOI] [PubMed] [Google Scholar]
- 15.Kim SI, Yi JS, Ko YG. Amyloid Beta Oligomerization Is Induced by Brain Lipid Rafts. J Cell Biochem. 2006;99:878–889. doi: 10.1002/jcb.20978. [DOI] [PubMed] [Google Scholar]
- 16.Oda A, Tamaoka A, Araki W. Oxidative Stress up-Regulates Presenilin 1 in Lipid Rafts in Neuronal Cells. J Neurosci Res. 2010;88:1137–1145. doi: 10.1002/jnr.22271. [DOI] [PubMed] [Google Scholar]
- 17.Kholodenko BN, Hoek JB, Westerhoff HV. Why Cytoplasmic Signalling Proteins Should Be Recruited to Cell Membranes. Trends Cell Biol. 2000;10:173–178. doi: 10.1016/s0962-8924(00)01741-4. [DOI] [PubMed] [Google Scholar]
- 18.Rajendran L, Knolker HJ, Simons K. Subcellular Targeting Strategies for Drug Design and Delivery. Nat Rev Drug Discovery. 2010;9:29–42. doi: 10.1038/nrd2897. [DOI] [PubMed] [Google Scholar]
- 19.Schwyzer R. Membrane-Assisted Molecular Mechanism of Neurokinin Receptor Subtype Selection. EMBO J. 1987;6:2255–2259. doi: 10.1002/j.1460-2075.1987.tb02498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwyzer R. How Do Peptides Interact with Lipid Membranes and How Does This Affect Their Biological Activity? Braz J Med Biol Res. 1992;25:1077–1089. [PubMed] [Google Scholar]
- 21.Boonyarattanakalin S, Martin SE, Dykstra SA, Peterson BR. Synthetic Mimics of Small Mammalian Cell Surface Receptors. J Am Chem Soc. 2004;126:16379–16386. doi: 10.1021/ja046663o. [DOI] [PubMed] [Google Scholar]
- 22.Hussey SL, He E, Peterson BR. A Synthetic Membrane-Anchored Antigen Efficiently Promotes Uptake of Antifluorescein Antibodies and Associated Protein a by Mammalian Cells. J Am Chem Soc. 2001;123:12712–12713. doi: 10.1021/ja017087o. [DOI] [PubMed] [Google Scholar]
- 23.Hussey SL, Peterson BR. Efficient Delivery of Streptavidin to Mammalian Cells: Clathrin-Mediated Endocytosis Regulated by a Synthetic Ligand. J Am Chem Soc. 2002;124:6265–6273. doi: 10.1021/ja0258733. [DOI] [PubMed] [Google Scholar]
- 24.Linning P, Haussmann U, Beyer I, Weidlich S, Schieb H, Wiltfang J, Klafki HW, Knolker HJ. Optimisation of Bace1 Inhibition of Tripartite Structures by Modification of Membrane Anchors, Spacers and Pharmacophores - Development of Potential Agents for the Treatment of Alzheimer’s Disease. Org Biomol Chem. 2012;10:8216–8235. doi: 10.1039/c2ob26103k. [DOI] [PubMed] [Google Scholar]
- 25.Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, Braxmeier T, Schwille P, Schulz JB, Schroeder C, Simons M, Jennings G, Knolker HJ, Simons K. Efficient Inhibition of the Alzheimer’s Disease Beta-Secretase by Membrane Targeting. Science. 2008;320:520–523. doi: 10.1126/science.1156609. [DOI] [PubMed] [Google Scholar]
- 26.Schieb H, Weidlich S, Schlechtingen G, Linning P, Jennings G, Gruner M, Wiltfang J, Klafki HW, Knolker HJ. Structural Design, Solid-Phase Synthesis and Activity of Membrane-Anchored Beta-Secretase Inhibitors on Abeta Generation from Wild-Type and Swedish-Mutant App. Chemistry. 2010;16:14412–14423. doi: 10.1002/chem.201002878. [DOI] [PubMed] [Google Scholar]
- 27.Braxmeier T, Friedrichson T, FRÖHNER W, Jennings G, Munick M, Schlechtingen G, Schroeder C, KNÖLKER HJ, Simons K, Zerial M. Tripartite Conjugates Containing a Structure Interacting with Cell Membrane Rafts and Their Use. 2,005,097,199. WO Patent. 2005 Oct 20;
- 28.Saathoff JM, Liu K, Chojnacki JE, He L, Chen Q, Lesnefsky EJ, Zhang S. Mechanistic Insight of Bivalent Compound 21mo as Potential Neuroprotectant for Alzheimer’s Disease. Molecules. 2016;21:412–420. doi: 10.3390/molecules21040412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chojnacki JE, Liu K, Saathoff JM, Zhang S. Bivalent Ligands Incorporating Curcumin and Diosgenin as Multifunctional Compounds against Alzheimer’s Disease. Bioorg Med Chem. 2015;23:7324–7331. doi: 10.1016/j.bmc.2015.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu K, Chojnacki JE, Wade EE, Saathoff JM, Lesnefsky EJ, Chen Q, Zhang S. Bivalent Compound 17mn Exerts Neuroprotection through Interaction at Multiple Sites in a Cellular Model of Alzheimer’s Disease. J Alzheimers Dis. 2015;47:1021–1033. doi: 10.3233/JAD-150242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu K, Gandhi R, Chen J, Zhang S. Bivalent Ligands Targeting Multiple Pathological Factors Involved in Alzheimer’s Disease. ACS Med Chem Lett. 2012;3:942–946. doi: 10.1021/ml300229y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu K, Guo TL, Chojnacki J, Lee HG, Wang X, Siedlak SL, Rao W, Zhu X, Zhang S. Bivalent Ligand Containing Curcumin and Cholesterol as Fluorescence Probe for Abeta Plaques in Alzheimer’s Disease. ACS Chem Neurosci. 2012;3:141–146. doi: 10.1021/cn200122j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lenhart JA, Ling X, Gandhi R, Guo TL, Gerk PM, Brunzell DH, Zhang S. “Clicked” Bivalent Ligands Containing Curcumin and Cholesterol as Multifunctional Abeta Oligomerization Inhibitors: Design, Synthesis, and Biological Characterization. J Med Chem. 2010;53:6198–6209. doi: 10.1021/jm100601q. [DOI] [PubMed] [Google Scholar]
- 34.Portoghese PS. From Models to Molecules: Opioid Receptor Dimers, Bivalent Ligands, and Selective Opioid Receptor Probes. J Med Chem. 2001;44:2259–2269. doi: 10.1021/jm010158+. [DOI] [PubMed] [Google Scholar]
- 35.Cavalli A, Bolognesi ML, Minarini A, Rosini M, Tumiatti V, Recanatini M, Melchiorre C. Multi-Target-Directed Ligands to Combat Neurodegenerative Diseases. J Med Chem. 2008;51:347–372. doi: 10.1021/jm7009364. [DOI] [PubMed] [Google Scholar]
- 36.Frautschy SA, Cole GM. Why Pleiotropic Interventions Are Needed for Alzheimer’s Disease. Mol Neurobiol. 2010;41:392–409. doi: 10.1007/s12035-010-8137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin Inhibits Formation of Amyloid Beta Oligomers and Fibrils, Binds Plaques, and Reduces Amyloid in Vivo. J Biol Chem. 2005;280:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 38.Papadopoulos V, Lecanu L. Caprospinol: Discovery of a Steroid Drug Candidate to Treat Alzheimer’s Disease Based on 22r-Hydroxycholesterol Structure and Properties. J Neuroendocrinol. 2012;24:93–101. doi: 10.1111/j.1365-2826.2011.02167.x. [DOI] [PubMed] [Google Scholar]
- 39.Tohda C, Lee YA, Goto Y, Nemere I. Diosgenin-Induced Cognitive Enhancement in Normal Mice Is Mediated by 1,25d(3)-Marrs. Sci Rep. 2013;3:3395–3402. doi: 10.1038/srep03395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sopher BL, Fukuchi K, Kavanagh TJ, Furlong CE, Martin GM. Neurodegenerative Mechanisms in Alzheimer Disease. A Role for Oxidative Damage in Amyloid Beta Protein Precursor-Mediated Cell Death. Mol Chem Neuropathol. 1996;29:153–168. doi: 10.1007/BF02814999. [DOI] [PubMed] [Google Scholar]
- 41.Hong HS, Maezawa I, Yao N, Xu B, Diaz-Avalos R, Rana S, Hua DH, Cheng RH, Lam KS, Jin LW. Combining the Rapid Mtt Formazan Exocytosis Assay and the Mc65 Protection Assay Led to the Discovery of Carbazole Analogs as Small Molecule Inhibitors of Abeta Oligomer-Induced Cytotoxicity. Brain Res. 2007;1130:223–234. doi: 10.1016/j.brainres.2006.10.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takalo M, Salminen A, Soininen H, Hiltunen M, Haapasalo A. Protein Aggregation and Degradation Mechanisms in Neurodegenerative Diseases. Am J Neurodegener Dis. 2013;2:1–14. [PMC free article] [PubMed] [Google Scholar]
- 43.Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting Basal Levels of Autophagy in the Nervous System Enhances Longevity and Oxidant Resistance in Adult Drosophila. Autophagy. 2008;4:176–184. doi: 10.4161/auto.5269. [DOI] [PubMed] [Google Scholar]
- 44.Maher P, Akaishi T, Schubert D, Abe K. A Pyrazole Derivative of Curcumin Enhances Memory. Neurobiol Aging. 2010;31:706–709. doi: 10.1016/j.neurobiolaging.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 45.Liu Y, Dargusch R, Maher P, Schubert D. A Broadly Neuroprotective Derivative of Curcumin. J Neurochem. 2008;105:1336–1345. doi: 10.1111/j.1471-4159.2008.05236.x. [DOI] [PubMed] [Google Scholar]
- 46.Nelson KM, Dahlin JL, Bisson J, Graham J, Pauli GF, Walters MA. The Essential Medicinal Chemistry of Curcumin. J Med Chem. 2017;60:1620–1637. doi: 10.1021/acs.jmedchem.6b00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeKosky ST, Scheff SW. Synapse Loss in Frontal Cortex Biopsies in Alzheimer’s Disease: Correlation with Cognitive Severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 48.Dickson TC, Vickers JC. The Morphological Phenotype of Beta-Amyloid Plaques and Associated Neuritic Changes in Alzheimer’s Disease. Neuroscience. 2001;105:99–107. doi: 10.1016/s0306-4522(01)00169-5. [DOI] [PubMed] [Google Scholar]
- 49.Jackson M, Gentleman S, Lennox G, Ward L, Gray T, Randall K, Morrell K, Lowe J. The Cortical Neuritic Pathology of Huntington’s Disease. Neuropathol Appl Neurobiol. 1995;21:18–26. doi: 10.1111/j.1365-2990.1995.tb01024.x. [DOI] [PubMed] [Google Scholar]
- 50.Liberski PP, Budka H. Neuroaxonal Pathology in Creutzfeldt-Jakob Disease. Acta Neuropathol. 1999;97:329–334. doi: 10.1007/s004010050995. [DOI] [PubMed] [Google Scholar]
- 51.Mattila PM, Rinne JO, Helenius H, Roytta M. Neuritic Degeneration in the Hippocampus and Amygdala in Parkinson’s Disease in Relation to Alzheimer Pathology. Acta Neuropathol. 1999;98:157–164. doi: 10.1007/s004010051064. [DOI] [PubMed] [Google Scholar]
- 52.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical Basis of Cognitive Alterations in Alzheimer’s Disease: Synapse Loss Is the Major Correlate of Cognitive Impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 53.Xu J, Lacoske MH, Theodorakis EA. Neurotrophic Natural Products: Chemistry and Biology. Angew Chem, Int Ed Engl. 2014;53:956–987. doi: 10.1002/anie.201302268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuboyama T, Tohda C, Komatsu K. Neuritic Regeneration and Synaptic Reconstruction Induced by Withanolide A. Br J Pharmacol. 2005;144:961–971. doi: 10.1038/sj.bjp.0706122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tohda C, Kuboyama T, Komatsu K. Search for Natural Products Related to Regeneration of the Neuronal Network. Neurosignals. 2005;14:34–45. doi: 10.1159/000085384. [DOI] [PubMed] [Google Scholar]
- 56.Svenda J, Sheremet M, Kremer L, Maier L, Bauer JO, Strohmann C, Ziegler S, Kumar K, Waldmann H. Biology-Oriented Synthesis of a Withanolide-Inspired Compound Collection Reveals Novel Modulators of Hedgehog Signaling. Angew Chem, Int Ed Engl. 2015;54:5596–5602. doi: 10.1002/anie.201500112. [DOI] [PubMed] [Google Scholar]
- 57.Moosavi F, Hosseini R, Saso L, Firuzi O. Modulation of Neurotrophic Signaling Pathways by Polyphenols. Drug Des, Dev Ther. 2016;10:23–42. doi: 10.2147/DDDT.S96936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tohda C, Urano T, Umezaki M, Nemere I, Kuboyama T. Diosgenin Is an Exogenous Activator of 1,25d(3)-Marrs/Pdia3/Erp57 and Improves Alzheimer’s Disease Pathologies in 5xfad Mice. Sci Rep. 2012;2:535–545. doi: 10.1038/srep00535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, Sabbah HN, Hoppel CL. Cardiac Mitochondria in Heart Failure: Decrease in Respirasomes and Oxidative Phosphorylation. Cardiovasc Res. 2008;80:30–39. doi: 10.1093/cvr/cvn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barry J, Fritz M, Brender JR, Smith PE, Lee DK, Ramamoorthy A. Determining the Effects of Lipophilic Drugs on Membrane Structure by Solid-State Nmr Spectroscopy: The Case of the Antioxidant Curcumin. J Am Chem Soc. 2009;131:4490–4498. doi: 10.1021/ja809217u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perez-Lara A, Ausili A, Aranda FJ, de Godos A, Torrecillas A, Corbalan-Garcia S, Gomez-Fernandez JC. Curcumin Disorders 1,2-Dipalmitoyl-Sn-Glycero-3-Phosphocholine Membranes and Favors the Formation of Nonlamellar Structures by 1,2-Dielaidoyl-Sn-Glycero-3-Phosphoethanolamine. J Phys Chem B. 2010;114:9778–9786. doi: 10.1021/jp101045p. [DOI] [PubMed] [Google Scholar]
- 62.Chapman D, Urbina J. Biomembrane Phase Transitions. Studies of Lipid-Water Systems Using Differential Scanning Calorimetry. J Biol Chem. 1974;249:2512–2521. [PubMed] [Google Scholar]
- 63.Gomez-Fernandez JC, Goni FM, Bach D, Restall CJ, Chapman D. Protein-Lipid Interaction. Biophysical Studies of (Ca2+ + Mg2+)-Atpase Reconstituted Systems. Biochim Biophys Acta. 1980;598:502–516. doi: 10.1016/0005-2736(80)90031-0. [DOI] [PubMed] [Google Scholar]
- 64.Rappolt M, Hickel A, Bringezu F, Lohner K. Mechanism of the Lamellar/Inverse Hexagonal Phase Transition Examined by High Resolution X-Ray Diffraction. Biophys J. 2003;84:3111–3122. doi: 10.1016/S0006-3495(03)70036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rand RP, Luzzati V. X-Ray Diffraction Study in Water of Lipids Extracted from Human Erythrocytes: The Position of Cholesterol in the Lipid Lamellae. Biophys J. 1968;8:125–137. doi: 10.1016/S0006-3495(68)86479-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tardieu A, Luzzati V, Reman FC. Structure and Polymorphism of the Hydrocarbon Chains of Lipids: A Study of Lecithin-Water Phases. J Mol Biol. 1973;75:711–733. doi: 10.1016/0022-2836(73)90303-3. [DOI] [PubMed] [Google Scholar]
- 67.Castellani R, Hirai K, Aliev G, Drew KL, Nunomura A, Takeda A, Cash AD, Obrenovich ME, Perry G, Smith MA. Role of Mitochondrial Dysfunction in Alzheimer’s Disease. J Neurosci Res. 2002;70:357–360. doi: 10.1002/jnr.10389. [DOI] [PubMed] [Google Scholar]
- 68.Greenough MA, Camakaris J, Bush AI. Metal Dyshomeostasis and Oxidative Stress in Alzheimer’s Disease. Neurochem Int. 2013;62:540–555. doi: 10.1016/j.neuint.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 69.Perrin RJ, Fagan AM, Holtzman DM. Multimodal Techniques for Diagnosis and Prognosis of Alzheimer’s Disease. Nature. 2009;461:916–922. doi: 10.1038/nature08538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lecanu L, Rammouz G, McCourty A, Sidahmed EK, Greeson J, Papadopoulos V. Caprospinol Reduces Amyloid Deposits and Improves Cognitive Function in a Rat Model of Alzheimer’s Disease. Neuroscience. 2010;165:427–435. doi: 10.1016/j.neuroscience.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 71.Lecanu L, Tillement L, Rammouz G, Tillement JP, Greeson J, Papadopoulos V. Caprospinol: Moving from a Neuroactive Steroid to a Neurotropic Drug. Expert Opin Invest Drugs. 2009;18:265–276. doi: 10.1517/13543780902762827. [DOI] [PubMed] [Google Scholar]