

Abstract
Aims
Arrhythmogenic right-ventricular cardiomyopathy (ARVC) is a genetically determined disorder, mostly caused by mutations in genes encoding desmosomal proteins. We evaluated phenotype/genotype characteristics to predict the risk for the first major arrhythmic event in desmosomal-mutation-associated ARVC families.
Methods and results
A cohort of 105 desmosomal-mutation carriers belonging to 39 consecutive ARVC families was evaluated. Serial clinical work-up consisting of history, physical examination, 12-lead/signal-averaged/24 h ambulatory ECG, and two-dimensional echocardiography was performed every 6–12 months. The predictive value of gender and genotype for the first major arrhythmic event was investigated within the cohort using time-to-event analysis. ECG/echocardiographic features were evaluated at the time of event and associated with the outcome using an age-matched nested case–control study within the cohort. Forty-three (41%) participants experienced the primary arrhythmic outcome at median age of 29 (21–46) years. The first event was sustained ventricular tachycardia in 31 and sudden cardiac death in 12. Definite diagnosis according to the 2010 Task Force criteria, showed 57% positive and 100% negative predictive value for the occurrence of arrhythmic outcome. Male gender (hazard ratio = 3.26, 95%CI, 1.63–6.51), predicted the first major arrhythmic event, independently of genotype, on multivariable analysis. Repolarization abnormalities and left-ventricular dysfunction independently associated with clinical disease profile at the time of event.
Conclusion
Male gender, independently of genotype is an arrhythmic risk predictor in ARVC-associated desmosomal-mutation carriers. Repolarization abnormalities and left-ventricular dysfunction are important components of the first event-associated clinical disease profile.
Keywords: Arrhythmogenic right-ventricular cardiomyopathy, Arrhythmia, Sudden death, Risk factors
What's new?
We have studied arrhythmic risk among desmosomal-mutation carriers.
We have encompassed both clinical and genetic information.
We have included ECG/echocardiographic changes observed during follow-up.
Clinical parameters are superior to genotype at risk assessment.
Non-fulfilment of diagnostic criteria is an important low-risk cutoff.
Introduction
Arrhythmogenic right-ventricular cardiomyopathy (ARVC) is a genetically determined disorder and a major cause of sudden death in the young. Life-threatening ventricular arrhythmias arise from the progressively affected heart muscle, which is characterized by myocardial degeneration with fibrous or fibrofatty replacement involving mostly the right ventricle.1,2 In more than 50% of probands, it is associated with mutations in genes encoding desmosomal proteins: plakoglobin (JUP), desmoplakin (DSP), plakophilin-2 (PKP2), desmoglein-2 (DSG2), and desmocollin-2 (DSC2).3 Recent implementation of genetic screening in index cases with suspected or definite ARVC and their relatives has led to an increasing number of mutation carriers with an inherent lifetime arrhythmic risk. Variable penetrance and age-related disease expression challenge prediction of the first major arrhythmic event occurrence, which might be even sudden cardiac death (SCD). The fact that in some ARVC series, over 50% of probands presented with sudden death indicates that risk stratification and appropriate implantable cardioverter defibrillator (ICD) implantation are major priorities.4
The present study was designed to evaluate the impact of phenotype/genotype characteristics on arrhythmic risk in a series of ARVC-associated desmosomal-mutation carriers belonging to consecutively genotyped families. The objectives of this study were two-fold. First, we sought to identify predictors of the first major arrhythmic event during lifetime among baseline characteristics present at birth, such as gender and genotype. Secondly, we sought to investigate an event-associated ECG/echocardiographic profile of patients, independently of baseline characteristics.
Methods
Study population
In this study, we evaluated 39 families of living ARVC probands, recruited from three expertise ARVC centres (Naxos, Athens, and Cyprus), harbouring one or more pathogenic desmosomal mutations following a comprehensive genetic screening. The families were 13 of PKP2, 14 of JUP, 6 of DSC2, and 6 of DSP; one of the DSP families presented digenic heterozygosity with PKP2 mutation. The proband in each family met the 2010 Task Force criteria (TFC) for diagnosis of ARVC. Sequence variants were defined as pathogenic as detailed in the 2010 TFC.5 Genetic screening of available family members for the mutation/s identified in the probands revealed 66 other mutation carriers. The total study population of this cohort included 105 desmosomal-mutation carriers (39 probands and 66 family members) from 39 ARVC families (members per family: median 2, inter-quartile range 2).
All individuals underwent serial cardiac evaluation every 6–12 months during a follow-up of up to 28 years, median 7 (4–12) years and consisted of a detailed personal and family history, physical examination, resting 12-lead ECG, signal-averaged ECG (SAECG), 24 h Holter monitoring, and two-dimensional echocardiography. Clinical events and outcome were recorded by age. Detailed historical data were available from medical records. The diagnosis of ARVC was based on the 2010 TFC and was defined as definite in the presence of two major or one major plus two minor or four minor criteria from different categories.
The progressive nature of ARVC and the heterogeneity in presentation time between probands and family members limits the predictive value of ECG/echocardiographic characteristics at initial clinical evaluation. Therefore an event-associated ECG/echocardiographic profile was investigated. In order to accomplish that, an age-matched nested case–control study within the cohort was used. Subjects who experienced the arrhythmic outcome (cases) were age-matched with an equal number of mutation carriers that remained event-free (controls). The study complies with the Declaration of Helsinki, the locally appointed Ethics Committee approved the research protocol and informed consent was obtained from all participants.
Primary arrhythmic outcome
Primary arrhythmic outcome was defined as the occurrence of the first major arrhythmic event including spontaneous sustained ventricular tachycardia and SCD. Ventricular tachycardia was defined as sustained when lasting more than 30 s, recorded on 12-lead ECG, 24 h Holter monitoring, or stored on intracardiac ICD data. SCD was defined as unexpected death occurring in <1 h from the onset of symptoms. Aborted SCD as a non-self-terminated event was estimated together with SCD. Cardiovascular collapse occurring in the context of severe heart failure was not regarded as SCD.
Electrocardiography and echocardiography
All ECGs were recorded at rest (10 mm/mV in speed 25 mm/s) at the standard lead position. No individual was receiving antiarrhythmic or other drugs known to affect the QRS complex at the time of acquisition of the ECG tracings. Epsilon waves (reproducible low-amplitude signals between the end of QRS complex and the onset of T wave) and T-wave inversion were studied on precordial leads.5 T waves of negative amplitude ≥1 mm (≥0.1 mV) were considered as inverted. QRS complex duration in leads V1–V6 and terminal activation duration of QRS complex (TAD) in leads V1–V3 (from the nadir of S wave to the end of QRS, including R′) were measured according to a standard protocol.6 SAECG was performed using time-domain analysis with a bandpass filter of 40 Hz in individuals with QRS complex duration of <110 ms on standard ECG. A 24 h Holter monitoring was recorded on an outpatient basis. Ventricular extrasystoles and episodes of ventricular tachycardia (≥3 consecutive ventricular complexes at a rate of ≥100 beats/min) were noted. Depolarization abnormalities were defined as the presence of epsilon waves or terminal activation duration of QRS complex ≥55 ms in leads V1–V3 in the absence of complete right bundle branch block (RBBB) or late potentials by SAECG in ≥1 of the three parameters in the absence of QRS complex duration of ≥110 ms on standard ECG.5 Repolarization abnormalities were defined as T-wave inversion in leads V1 and V2 or beyond in individuals >14 years of age in the absence of complete RBBB or T-wave inversion in leads V1, V2, V3, and V4 in individuals >14 years of age in the presence of complete RBBB or T-wave inversion in leads V4, V5, or V6.5
Echocardiography was performed with a 2.5 MHz transducer. Right-ventricular (RV) outflow-tract end-diastolic diameter was measured on parasternal long-axis view (RVOT-PLAX). Wall motion abnormalities (hypokinesia, akinesia, dyskinesia, and aneurysm) of the right and left ventricles were documented. RV dysfunction was determined as the presence of regional akinesia, dyskinesia, or aneurysm plus RVOT-PLAX ≥29 mm (minor criteria).5 Left-ventricular (LV) fraction shortening was measured on parasternal long-axis view. Normal limits of LV fraction shortening were defined by assessment of 164 healthy Greek volunteers (50% males) aged 37 (21–47) years. LV dysfunction was determined as the presence of regional hypokinesia, akinesia, dyskinesia, or fraction shortening <30% (2 SD from normal value).
Statistical analysis
Summary descriptive statistics are reported as median (25th–75th percentile) or frequency counts (%) for continuous and categorical variables, respectively. Categorical variables were compared between groups using the χ2 or Fisher's exact test as appropriate.
The cumulative probability of experiencing the first arrhythmic event during lifetime was determined by using the date of birth as start point for the time-to-event analysis. Subjects were censored at the time of first event or at the time of last clinical evaluation. Kaplan–Meier event-free survival curves stratified by gender and genotype were constructed and compared using the log-rank test. A multivariate Cox proportional hazards regression model was employed to assess the significance and independence of gender and genotype in the prediction of lifetime arrhythmic outcome. Hazard ratios (HR) and 95% confidence intervals are reported.
Relative odds (OR) and their corresponding 95% confidence intervals of experiencing the first major arrhythmic event were estimated according to gender, genotype, electrocardiographic, and echocardiographic parameters, within the nested case–control sample (n = 86) using multiple logistic regression models. ECG/echocardiographic data were analysed at the time of first event or at the time of last clinical evaluation in the event-free subjects. Clinical variables were added to the model in a nested way in order to test for their potential mediating effect. The Hosmer–Lemeshow statistic was calculated to evaluate the model's goodness of fit.
All P-values reported are two-sided and considered significant at the 5% level. SPSS 21 (IBM, Chicago, IL, USA) was used for all analyses.
Results
Genotype/phenotype characteristics of mutation carriers
The study population included 105 desmosomal-mutation carriers (53% male) with the following gene distribution: PKP2, n = 38 (36%); JUP, n = 30 (28%); DSC2, n = 28 (27%); DSP, n = 9 (9%) (Supplementary material online, Table S1). All 30 JUP and one of the 28 DSC2 carriers were homozygous for the mutation; three were digenic carriers of DSP and PKP2.
The last clinical evaluation of 105 mutation carriers revealed repolarization abnormalities in 57 (54%), depolarization abnormalities in 72 (69%), RV dysfunction in 54 (51%), and LV dysfunction in 28 (27%). ECG and/or echocardiographic abnormalities developed during follow-up in 11 (27%) among 40 subjects with a long-term follow-up (>7 years). Seventy-five subjects were classified as definite ARVC by the 2010 TFC. Fourteen fulfilled one major plus at least two minor criteria, whereas the other 61 fulfilled at least two major criteria. Thirty mutation carriers did not fulfill the TFC, the youngest was aged 16 years; nine had minor depolarization abnormalities and three had frequent ventricular extrasystoles (borderline ARVC).
Prophylactic antiarrhythmic drug/beta-blocker therapy was prescribed in 29 (28%) patients and 16 (15%) were implanted with an ICD for primary prevention of life-threatening ventricular arrhythmias.
Primary arrhythmic outcome
Forty-three (41%) mutation carriers (70% male) from 31 different families experienced the primary arrhythmic outcome (Table 1, Supplementary material online, Table S2). The first arrhythmic event was sustained ventricular tachycardia in 31 (appropriate ICD intervention in four) and SCD in 12 (aborted in one). The median age at the time of first arrhythmic event was 29 (21–46) years, with no events occurring before the age of 15. There were 29 primary events among the 39 probands (74%) and 14 primary events among the 66 family members (21%). In the majority of patients, the first event was experienced between the second and fourth decade of life (Figure 1). Seventeen (40%) were mutation carriers of PKP2, 16 (37%) of JUP, 6 (14%) of DSC2, and 4 (9%) of DSP. Forty-two (98%) fulfilled 12-lead ECG criteria; 28 had repolarization and depolarization abnormalities, six had repolarization abnormalities alone, and eight had depolarization abnormalities alone. One patient did not meet 12-lead ECG criteria but fulfilled all three parameters for late potentials on SAECG. Thirty-three individuals (77%) showed RV dysfunction, meeting the revised echocardiographic criteria. Major or minor RV abnormalities were absent in the remaining 10. LV dysfunction was detected in 21 (49%) patients, but only five (12%) of them exhibited moderate-to-severe dysfunction (fraction shortening <25%, ejection fraction <45%). In four of the 21 patients, LV dysfunction first appeared at serial cardiac evaluation.
Table 1.
Clinical characteristics of mutation carriers
| Characteristics | Arrhythmic outcome (n = 43) | No arrhythmic outcome (n = 62) | P-value |
|---|---|---|---|
| Male gender | 30 (70) | 26 (42) | 0.005 |
| Proband status | 29 (67) | 10 (16) | <0.0001 |
| Definite ARVC | 43 (100) | 32 (52) | <0.0001 |
| Repolarization abnormalities | 34 (79) | 23 (37) | <0.0001 |
| Depolarization abnormalities | 37 (86) | 35 (56) | 0.001 |
| RV dysfunction | 33 (77) | 21 (34) | <0.0001 |
| LV dysfunction | 21 (49) | 7 (11) | <0.0001 |
Values are n (%).
LV, left ventricular; RV, right ventricular.
Figure 1.
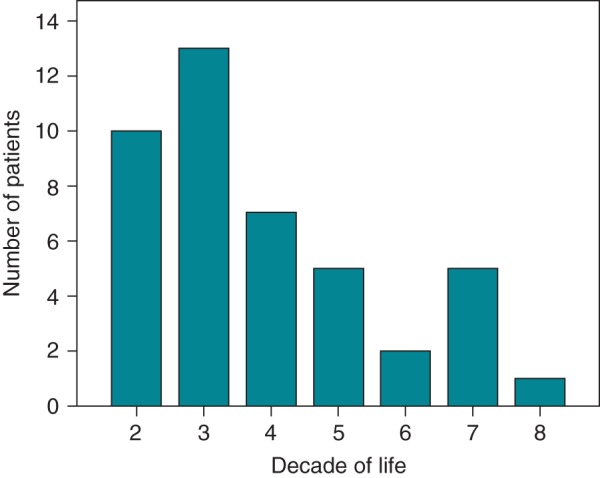
Age distribution, in decades, of primary arrhythmic outcome in ARVC-associated desmosomal mutation carriers.
Sixty-two mutation carriers, aged 50 (37–62) years, the youngest aged 15 years at last follow-up, have not experienced an arrhythmic outcome (Table 1). Subjects with and without major arrhythmic events did not differ with regard to prophylactic antiarrhythmic drug/beta-blocker therapy prescribed for primary prevention of life-threatening arrhythmias (9/43, 21% vs. 20/62, 32%; P = 0.27).
Gender and genotype
Cumulative event-free survival, stratified by gender, was significantly lower in males (Figure 2). By the age of 40 years, cumulative event-free survival was 59 ± 7% for male and 82 ± 6% for female mutation carriers. Increased arrhythmic event occurrence was associated with male gender in the ages under 50 years (25/38 vs. 9/27, P = 0.013), but not in later years (5/18 vs. 4/22, P = 0.7) (Figure 3).
Figure 2.
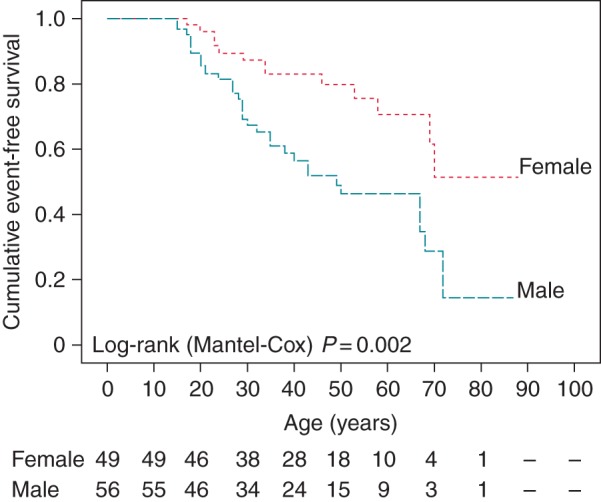
Kaplan–Meier analysis of cumulative arrhythmia event-free survival stratified by gender.
Figure 3.
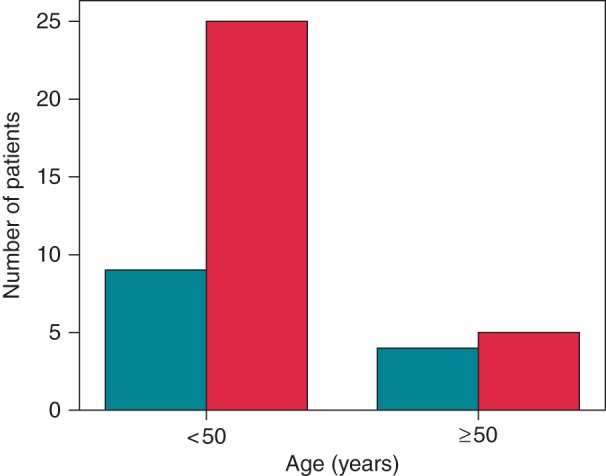
Age distribution of primary arrhythmic outcome in females (turquoise bars) and males (red bars) according to the 50th year of life.
Cumulative event-free survival, stratified by genotype, did not differ among groups of PKP2, JUP, and DSP (PKP2 vs. JUP, P = 0.55; PKP2 vs. DSP, P = 0.71; JUP vs. DSP, P = 0.94), while it was higher in DSC2 group (P = 0.006). By the age of 40 years, cumulative event-free survival was 61 ± 8% for PKP2, 68 ± 9% for JUP, 88 ± 7% for DSC2, and 63 ± 17% for DSP mutation carriers (Figure 4).
Figure 4.
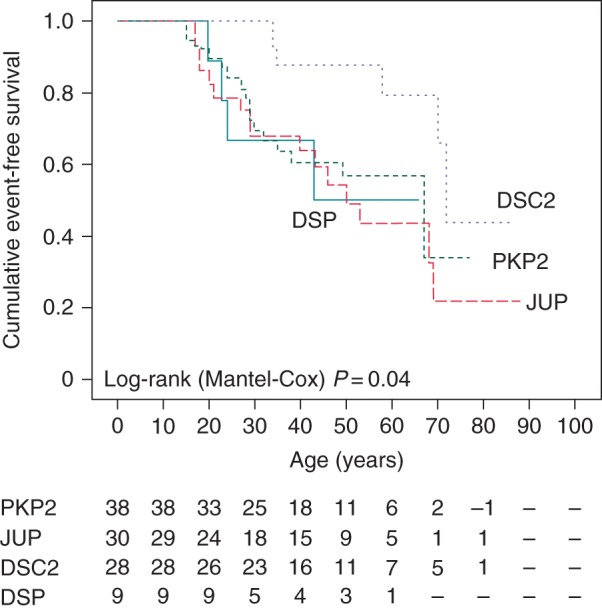
Kaplan–Meier analysis of cumulative arrhythmia event-free survival stratified by genotype. DSC2, desmocollin-2; DSP, desmoplakin; JUP, plakoglobin; PKP2, plakophilin-2.
To assess the significance and independence of the primary arrhythmic outcome predictors, we entered male gender and genotype in a multivariate proportional hazards Cox regression model. Male gender (HR = 3.26, P = 0.001) was the only adverse outcome predictor, whereas DSC2 mutation (HR = 0.33, P = 0.023) had a better lifetime arrhythmic outcome (Table 2).
Table 2.
Baseline predictors of primary arrhythmic outcome
| Variables | Univariate analysis |
Multivariate analysis |
||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P-value | HR | 95% CI | P-value | |
| Male gender | 2.66 | 1.38–5.12 | 0.003 | 3.26 | 1.63–6.51 | 0.001 |
| Genotype | ||||||
| PKP2 | 1.24 | 0.67–2.29 | 0.49 | 1 | ||
| JUP | 1.74 | 0.94–3.25 | 0.08 | 1.42 | 0.71–2.82 | 0.32 |
| DSC2 | 0.32 | 0.13–0.75 | 0.009 | 0.33 | 0.12–0.86 | 0.023 |
| DSP | 1.46 | 0.52–4.13 | 0.48 | 2.01 | 0.65–6.21 | 0.22 |
CI, confidence intervals; DSC2, desmocollin-2; DSP, desmoplakin; HR, hazard ratio; JUP, plakoglobin; PKP2, plakophilin-2.
Event-associated ECG and echocardiographic findings
Repolarization/depolarization abnormalities and RV/LV dysfunction were significantly more common in subjects who experienced arrhythmic outcome (Table 1). All patients who experienced an arrhythmic outcome and 32 of the 62 (52%) without arrhythmic events were diagnosed with definite ARVC by the 2010 TFC (Table 3). Therefore, positive and negative predictive value of a definite diagnosis according to TFC for the occurrence of an arrhythmic outcome was 57 and 100%, respectively.
Table 3.
Results of logistic regression analysis models
| Odds ratio (95% confidence interval) |
|||||
|---|---|---|---|---|---|
| Step 0 | Step 1 | Step 2 | Step 3 | Step 4 | |
| Model 1 | |||||
| Male gender | 3.18 (1.28–7.91)a | 2.20 (0.79–6.07) | 2.77 (0.92–8.35) | 2.70 (0.85–8.60) | 2.91 (0.85–9.91) |
| PKP2 mutation | 1.04 (0.41–2.67) | 1.48 (0.50–4.37) | 0.83 (0.24–2.84) | 1.26 (0.34–4.68) | 1.58 (0.39–6.33) |
| RVD | 7.11 (2.59–19.52)a | 3.50 (1.10–11.13)a | 2.08 (0.58–7.41) | 1.01 (0.24–4.31) | |
| REP | 4.44 (1.31–15.08)a | 6.04 (1.57–23.22)a | 7.81 (1.83–33.26)a | ||
| LVD | 6.33 (1.60–25.02)a | 8.19 (1.97–33.93)a | |||
| DEP | 5.08 (0.96–27.04) | ||||
| Model 2 | |||||
| Male gender | 3.71 (1.44–9.53)a | 2.59 (0.94–7.15) | 2.90 (0.99–8.50) | 3.12 (1.00–9.69) | 3.65 (1.11–12.02)a |
| JUP mutation | 3.67 (1.23–10.92)a | 1.50 (0.42–5.31) | 1.71 (0.46–6.38) | 1.57 (0.40–6.18) | 1.50 (0.38–5.95) |
| RVD | 5.60 (1.86–16.90)a | 2.97 (0.89–9.94) | 1.63 (0.43–6.22) | 0.80 (0.27–3.71) | |
| REP | 4.26 (1.40–12.94)a | 6.67 (1.86–23.98)a | 9.09 (2.23–37.14)a | ||
| LVD | 5.89 (1.54–22.51)a | 7.31 (1.83–29.20)a | |||
| DEP | 4.55 (0.90–23.14) | ||||
| Model 3 | |||||
| Male gender | 3.38 (1.33–8.59)a | 2.50 (0.91–6.85) | 2.63 (0.93–7.46) | 2.87 (0.94–8.71) | 3.30 (1.04–10.49)a |
| DSC2 mutation | 0.23 (0.08–0.71)a | 0.31 (0.10–1.01) | 0.43 (0.12–1.48) | 0.38 (0.09–1.59) | 0.39 (0.09–1.73) |
| RVD | 5.76 (2.10–15.80)a | 3.76 (1.26–11.25)a | 2.02 (0.59–6.94) | 0.98 (0.23–4.18) | |
| REP | 3.30 (1.05–10.33)a | 5.10 (1.38–18.81)a | 6.94 (1.66–28.95)a | ||
| LVD | 5.88 (1.57–22.06)a | 7.07 (1.80–27.70)a | |||
| DEP | 4.58 (0.87–24.13) | ||||
| Model 4 | |||||
| Male gender | 3.30 (1.32–8.27)a | 2.73 (0.98–7.60) | 3.02 (1.03–8.85)a | 3.01 (0.98–9.31) | 3.34 (1.04–10.77)a |
| DSP mutation | 1.23 (0.28–5.38) | 2.47 (0.46–13.26) | 2.94 (0.53–16.26) | 1.43 (0.23–8.92) | 0.93 (0.14–6.10) |
| RVD | 7.51 (2.68–20.99)a | 4.19 (1.36–12.92)a | 2.10 (0.58–7.55) | 0.92 (0.20–4.27) | |
| REP | 4.43 (1.43–13.73)a | 6.56 (1.83–23.59)a | 8.95 (2.19–36.51)a | ||
| LVD | 5.48 (1.35–22.29)a | 7.55 (1.69–33.80)a | |||
| DEP | 4.68 (0.89–24.73) | ||||
DEP, depolarization abnormalities; DSC2, desmocollin-2; DSP, desmoplakin; JUP, plakoglobin homozygocity; LVD, left-ventricular dysfunction; PKP2, plakophilin-2; REP, repolarization abnormalities; RVD, right-ventricular dysfunction.
aResult is significant (P-value < 0.05).
To assess the clinical disease profile at the time of event independently of baseline characteristics, four logistic regression models were created (one to assess each type of genotype) to analyse the age-matched case–control study population. Gender and genotype were initially assessed and ECG/echocardiographic variables were added in a stepwise fashion (Table 3). In the final step, male gender was significantly associated in three models, whereas genotype in none. In all models, repolarization abnormalities and LV dysfunction were significantly and independently associated with the first arrhythmic event with a relative odds range of 6.94–9.09 and 7.07–8.19, respectively.
Discussion
ARVC is a primary heart muscle-disorder characterized clinically by life-threatening ventricular arrhythmias. The genetic background of ARVC is mostly related to mutations in genes encoding desmosomal proteins. Disease expression is often incomplete and mutation-specific genetic testing is recommended for family members following the identification of an ARVC causative mutation in the proband.3 All mutation carriers are potentially at a lifetime risk for developing life-threatening ventricular arrhythmias. Previous risk stratification studies have shown several clinical predictors of arrhythmic outcome such as male gender, depolarization/repolarization abnormalities, moderate-to-severe RV dysfunction, and LV dysfunction.7–13 Most of these studies included severe forms of disease, surrogate ICD outcomes, and unknown genetic background. The goal of this study was to evaluate both genetic and clinical information to assess the arrhythmic risk in ARVC-associated desmosomal-mutation carriers.
Forty-three out of the 105 (41%) mutation carriers belonging to 39 families experienced an arrhythmic outcome, suggesting a marked arrhythmic incidence among these individuals, consistent with results provided by other series.8 The first arrhythmic event is more likely to occur before the fourth decade of life but risk remains until later ages. The high incidence of SCD occurring as initial event reflects the malignant arrhythmic nature of the underlying cardiac pathology. Importantly, positive and negative predictive value of a definite diagnosis for the occurrence of an arrhythmic outcome was 57 and 100%, respectively, suggesting that mutation carriers with no or incomplete phenotype according to the 2010 TFC are at very low risk.
In this study, survival analysis for lifetime major arrhythmic events revealed similar risk between carriers of PKP2, JUP, and DSP mutations, but lower risk for the DSC2 group. Interestingly, although JUP homozygous mutation carriers showed increased disease penetrance and phenotypic expression, the arrhythmic risk did not differ from that of PKP2 heterozygotes. Phenotypic features such as gender, and ECG/echocardiographic findings at the time of event were significantly associated with the occurrence of arrhythmic outcome, while genetic characteristics did not provide additional prognostic information.
Males and females were equally distributed in the study population. Males, however, experienced arrhythmic outcome more frequently and this was identified as an independent predictor for the first arrhythmic event providing a 3.3-fold increase of arrhythmic risk. Physical activity has been shown to be an important modulator of disease phenotype associated with arrhythmic outcome in both desmosomal and gene-elusive ARVC.14 This could be a possible explanation of male preponderance in arrhythmic events considering that men may experience higher intensity exercise although in another study no gender differences were revealed.15 Alternatively, a direct protective role of female sex hormones could be a significant modulator. The latter is further supported by our study, since after the age of 50 years, when menopause usually commences, the arrhythmia event rates were equal in both genders.
ARVC is a progressive disease and ECG/echocardiographic features change during life as observed in this cohort. Therefore using ECG/echocardiographic parameters at initial evaluation as predictors of arrhythmic events later in life might be of limited value. We suggest that ARVC mutation carriers need serial clinical evaluation and lifelong revision of risk stratification. In this study, a serial cardiac assessment was performed every 6–12 months and the ECG/echocardiographic data used in the analysis were those most close to the time of event.
The most common phenotypic feature associated with arrhythmic risk is the characteristic for ARVC 12-lead ECG changes, even in the absence of RV dysfunction on two-dimensional echocardiography. Arrhythmic events in patients exhibiting normal 12-lead ECG are a rare phenomenon.16 In our population, it occurred only in one case in whom the SAECG was the sole abnormal ECG feature. Repolarization abnormalities were independently associated with the arrhythmic outcome. A recent study by Bhonsale et al.,17 predominantly in PKP2 patients, similarly identified repolarization abnormalities as an independent predictor of arrhythmic outcome. Some of our patients who had repolarization abnormalities but not RV/LV dysfunction developed arrhythmic events, a finding permitting speculation that repolarization abnormalities reflect an early subepicardial myocardial injury undetectable by conventional imaging. This hypothesis has been supported by associating repolarization abnormalities with the extent of myocardial injury represented by electroanatomic scar area.18
LV dysfunction was significantly associated with the arrhythmic outcome suggesting that LV involvement signifies a more deleterious myocardial defect. Interestingly in our study, LV dysfunction was mild in most patients who experienced the arrhythmic outcome indicating that even mild LV dysfunction increases arrhythmic risk. This is consistent with data from autopsy and contrast-enhanced CMR studies revealing extensive LV subepicardial/mid-wall involvement to be associated with mild LV dysfunction on conventional imaging.19,20 This highlights a potential role for tissue imaging techniques including contrast-enhanced CMR and electroanatomic voltage-mapping in selected cases.21,22
Study limitations
This study exhibits certain limitations. Thirty out of the 105 individuals were Naxos disease patients. However, data were analysed using multivariable models including the genetic status, specifically JUP homozygosity, which was shown to be non-significant and thus, providing results independent from this variable. Compound/digenic heterozygosity has been associated with increased arrhythmic risk23; there were only three cases of digenic and none of compound heterozygosity and therefore conclusions concerning complex genotype cannot be drawn. The number of ventricular extrasystoles on 24-h Holter study was potentially affected by prophylactic antiarrhythmic/beta-blocker therapy and therefore it was not included in the analysis. Sixteen mutation carriers were implanted an ICD for primary prevention implicating a detection bias in this subgroup. Furthermore, the number of events was not sufficient to statistically assess more parameters as independent predictors of arrhythmic outcome. Proband status was not assessed as an arrhythmic risk predictor since we addressed the first major arrhythmic event during lifetime, which usually precedes the characterization of the patient as a proband. Patients who experienced SCD before the diagnosis were not included in the study. Finally, it is possible that individuals considered event-free by the time this study commences, in the future they might develop clinical events as their follow-up time increases.
Conclusions
In ARVC-associated desmosomal-mutation carriers serial comprehensive clinical screening is of paramount importance for arrhythmic risk stratification, irrespectively of the genetic status. The results indicate that clinical parameters are superior to genotype. Within the context of the 2010 Task Force diagnostic criteria mutation carriers with no or incomplete phenotype are at very low risk. Male gender is an important predictor of primary arrhythmic outcome. Repolarization abnormalities and LV dysfunction were significant components of the first major arrhythmic event-associated disease profile. The results of this study have important implications for management of ARVC families.
Supplementary material
Funding
W.J.M. received a proportion of funding at UCLH/UCL from the Department of Health's NIHR Biomedical Research Centres funding scheme for support of this work.
Conflict of interest: none declared.
Supplementary Material
References
- 1. Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation 1982;65:384–98. [DOI] [PubMed] [Google Scholar]
- 2. Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 1988;318:129–33. [DOI] [PubMed] [Google Scholar]
- 3. Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace 2011;13:1077–109. [DOI] [PubMed] [Google Scholar]
- 4. Quarta G, Muir A, Pantazis A, Syrris P, Gehmlich K, Garcia-Pavia P, et al. Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: impact of genetics and revised task force criteria. Circulation 2011;123:2701–9. [DOI] [PubMed] [Google Scholar]
- 5. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J 2010;31:806–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Protonotarios N, Anastasakis A, Antoniades L, Chlouverakis G, Syrris P, Basso C, et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia on the basis of the revised diagnostic criteria in affected families with desmosomal mutations. Eur Heart J 2011;32:1097–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turrini P, Corrado D, Basso C, Nava A, Bauce B, Thiene G. Dispersion of ventricular depolarization-repolarization: a noninvasive marker for risk stratification in arrhythmogenic right ventricular cardiomyopathy. Circulation 2001;103:3075–80. [DOI] [PubMed] [Google Scholar]
- 8. Corrado D, Leoni L, Link MS, Della Bella P, Gaita F, Curnis A, et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 2003;108:3084–91. [DOI] [PubMed] [Google Scholar]
- 9. Wichter T, Paul M, Wollmann C, Acil T, Gerdes P, Ashraf O, et al. Implantable cardioverter/defibrillator therapy in arrhythmogenic right ventricular cardiomyopathy: single-center experience of long-term follow-up and complications in 60 patients. Circulation 2004;109:1503–8. [DOI] [PubMed] [Google Scholar]
- 10. Hulot JS, Jouven X, Empana JP, Frank R, Fontaine G. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 2004;110:1879–84. [DOI] [PubMed] [Google Scholar]
- 11. Pinamonti B, Dragos AM, Pyxaras SA, Merlo M, Pivetta A, Barbati G, et al. Prognostic predictors in arrhythmogenic right ventricular cardiomyopathy: results from a 10-year registry. Eur Heart J 2011;32:1105–13. [DOI] [PubMed] [Google Scholar]
- 12. Corrado D, Calkins H, Link MS, Leoni L, Favale S, Bevilacqua M, et al. Prophylactic implantable defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and no prior ventricular fibrillation or sustained ventricular tachycardia. Circulation 2010;122:1144–52. [DOI] [PubMed] [Google Scholar]
- 13. Bhonsale A, James CA, Tichnell C, Murray B, Gagarin D, Philips B, et al. Incidence and predictors of implantable cardioverter-defibrillator therapy in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy undergoing implantable cardioverter-defibrillator implantation for primary prevention. J Am Coll Cardiol 2011;58:1485–96. [DOI] [PubMed] [Google Scholar]
- 14. Sawant AC, Bhonsale A, Te Riele AS, Tichnell C, Murray B, Russell SD, et al. Exercise has a disproportionate role in the pathogenesis of arrhythmogenic right ventricular dysplasia/cardiomyopathy in patients without desmosomal mutations. J Am Heart Assoc 2014;3: 10.1161/JAHA.114.001471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol 2013;62:1290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. te Riele AS, James CA, Bhonsale A, Groeneweg JA, Camm CF, Murray B, et al. Malignant arrhythmogenic right ventricular dysplasia/cardiomyopathy with a normal 12-lead electrocardiogram: a rare but underrecognized clinical entity. Heart Rhythm 2013;10:1484–91. [DOI] [PubMed] [Google Scholar]
- 17. Bhonsale A, James CA, Tichnell C, Murray B, Madhavan S, Philips B, et al. Risk stratification in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. Circ Arrhythm Electrophysiol 2013;6:569–78. [DOI] [PubMed] [Google Scholar]
- 18. Zorzi A, Migliore F, Elmaghawry M, Silvano M, Marra MP, Niero A, et al. Electrocardiographic predictors of electroanatomic scar size in arrhythmogenic right ventricular cardiomyopathy: implications for arrhythmic risk stratification. J Cardiovasc Electrophysiol 2013;24:1321–7. [DOI] [PubMed] [Google Scholar]
- 19. Protonotarios A, Patrianakos A, Spanoudaki E, Kochiadakis G, Michalodimitrakis E, Vardas P. Left dominant arrhythmogenic cardiomyopathy: a morbid association of ventricular arrhythmias and unexplained infero-lateral T-wave inversion. J Electrocardiol 2013;46:352–5. [DOI] [PubMed] [Google Scholar]
- 20. Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol 2008;52:2175–87. [DOI] [PubMed] [Google Scholar]
- 21. te Riele AS, Bhonsale A, James CA, Rastegar N, Murray B, Burt JR, et al. Incremental value of cardiac magnetic resonance imaging in arrhythmic risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol 2013;62:1761–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Migliore F, Zorzi A, Silvano M, Bevilacqua M, Leoni L, Marra MP, et al. Prognostic value of endocardial voltage mapping in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ Arrhyth Electrophysiol 2013;6:167–76. [DOI] [PubMed] [Google Scholar]
- 23. Rigato I, Bauce B, Rampazzo A, Zorzi A, Pilichou K, Mazzotti E, et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet 2013;6:533–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.