

Abstract
Background: Risk of the outcome is a mathematical determinant of the absolute treatment benefit of an intervention, yet this can vary substantially within a trial population, complicating the interpretation of trial results.
Methods: We developed risk models using Cox or logistic regression on a set of large publicly available randomized controlled trials (RCTs). We evaluated risk heterogeneity using the extreme quartile risk ratio (EQRR, the ratio of outcome rates in the lowest risk quartile to that in the highest) and skewness using the median to mean risk ratio (MMRR, the ratio of risk in the median risk patient to the average). We also examined heterogeneity of treatment effects (HTE) across risk strata.
Results: We describe 39 analyses using data from 32 large trials, with event rates across studies ranging from 3% to 63% (median = 15%, 25th–75th percentile = 9–29%). C-statistics of risk models ranged from 0.59 to 0.89 (median = 0.70, 25th–75th percentile = 0.65–0.71). The EQRR ranged from 1.8 to 50.7 (median = 4.3, 25th–75th percentile = 3.0–6.1). The MMRR ranged from 0.4 to 1.0 (median = 0.86, 25th–75th percentile = 0.80–0.92). EQRRs were predictably higher and MMRRs predictably lower as the c-statistic increased or the overall outcome incidence decreased. Among 18 comparisons with a significant overall treatment effect, there was a significant interaction between treatment and baseline risk on the proportional scale in only one. The difference in the absolute risk reduction between extreme risk quartiles ranged from −3.2 to 28.3% (median = 5.1%; 25th–75th percentile = 0.3–10.9).
Conclusions: There is typically substantial variation in outcome risk in clinical trials, commonly leading to clinically significant differences in absolute treatment effects. Most patients have outcome risks lower than the trial average reflected in the summary result. Risk-stratified trial analyses are feasible and may be clinically informative, particularly when the outcome is predictable and uncommon.
Keywords: Risk prediction, heterogeneity of treatment effect, subgroup analysis, personalized medicine, patient-centered outcomes research
Introduction
A fundamental incongruity in evidence-based medicine (EBM) is that evidence is derived from groups of people yet medical decisions are made for individuals. Popular approaches to EBM have encouraged the direct application of average effects estimated in clinical trials to guide decision making for individuals, as though all patients meeting trial inclusion criteria are likely to experience similar effects from treatments. This simplistic attitude has proven remarkably durable and compelling, despite the variation in patient characteristics and outcomes seen in clinical practice.1
The most commonly used method of examining whether treatment effects vary in a trial population is to serially divide patients into subgroups based on potentially relevant pre-treatment characteristics. The main problem with this conventional approach is that there are too many potentially influential characteristics. This leads to myriad ‘one-variable-at-a-time’ subgroup analyses, which are typically both underpowered and vulnerable to false-positive results due to multiple comparisons.2,3 It can also be difficult to understand how to apply such analyses to individuals in clinical practice, because patients have multiple characteristics that vary from one another simultaneously.
In part for these reasons, subgroup analyses are usually ‘exploratory’ and rarely actionable, leaving the clinician to assume that all patients meeting trial inclusion criteria should be similarly treated. EBM is thus methodologically canalized to ‘one-size-fits-all’ recommendations, a problem increasingly recognized even as EBM has become the dominant paradigm.4–6 This remains a central challenge to be addressed if EBM is to become more personalized and patient-centred.4–6
We recently proposed a framework for assessing heterogeneity of treatment effect (HTE) that seeks to address these issues.7 The framework prioritizes the analysis and reporting of multi-variable risk-based HTE and suggests that other subgroup analyses should be explicitly labelled either as primary subgroup analyses (well-motivated by prior evidence and intended to produce clinically actionable results) or secondary (exploratory) subgroup analyses (performed to inform future research). Whereas other recommendations or guidance documents have (appropriately) emphasized the risks of overinterpreting the results of subgroup analyses,8,9 and the different goals of such analyses,10 our framework is novel in that it also suggests that presenting summary results without examining and reporting how treatment effects change across subgroups with heterogeneous outcome risk is under-utilizing trial data and tantamount to incompletely reporting trial results.
Despite compelling theoretical arguments, a risk-modelling approach is rarely applied. Empirical evidence for its importance remains anecdotal and there are concerns about the feasibility of routine and broad application of this analytical approach in datasets collected in typical randomized trials. To address these concerns, we examined the distribution of outcome risk across a broad range of trials and examine how the effects of therapy were related to this risk.
Methods
We searched for publicly available individual participant datasets of randomized clinical trials from the National Heart, Lung, and Blood Institute (NHLBI),11 the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK),12 the journal Trials and GlaxoSmithKline.13 We required that eligible studies had enrolled at least 1000 participants (some subcohorts entered in our analyses had fewer than 1000 participants) randomized to at least two treatment groups, and had a binary (or time-to-event) clinical (i.e. not surrogate) outcome.
Predicting outcome risk using baseline covariates
Risk modelling for each trial was informed by examining previously developed published predictive models ‘matched’ to each trial on the basis of the index condition of the population and the primary outcome.14 We identified risk predictors that had been used in the published models and the corresponding variables in the trial datasets. Because trial datasets were often not fully compatible with externally developed predictive models, we developed ‘internal models’ on the trial data using risk predictors that were as close as possible to those in published models. To verify that the use of internal models would not bias estimates of HTE across risk groups, we performed a series of simulations described in a separate publication.15 Briefly, the simulations revealed that, across a range of scenarios, analyses based on internal models developed on trial participants yield results similar to analyses based on external models developed on non-trial participants sampled from the same population.
All available established risk predictors were entered into a regression model to predict the primary outcome for all patients in the trial. Both trial arms were used in model development, without using the treatment assignment indicator, to avoid differential model fit between the trial arms, potentially inducing a spurious risk-by-treatment interaction.15 To minimize model complexity for trials for which there were many established predictors, non-significant risk predictors were ranked in order of significance and removed until no more than 20 variables were entered into the model (this was needed in only 3 of the 32 trials). No other formal variable selection process or attempt at model re-specification was performed.
In trials with non-statistically significant overall treatment effects for the primary outcome and a statistically significant treatment effect for a binary (or time-to-event) clinical secondary outcome, an additional regression model was fit to predict the secondary outcome. When treatment effects for multiple secondary outcomes were statistically significant, we selected the outcome identified as most clinically relevant in the published trial report.
To minimize bias due to missing data, multivariate normal multiple imputation was used when a complete case analysis would exclude more than 5% of trial participants. Risk factors with missing information from more than 20% of trial participants were not used in analyses.
The statistical analysis model (Cox proportional hazards regression for time-to-event outcomes or logistic regression for binary outcomes) was selected on the basis of the primary analysis of the clinical trial and determined by the nature of the trial data. In general, we included variables as main effects in their original scale, unless published predictive models specified the use of interactions or variable transformations.
Model performance was assessed with respect to discrimination, calibration and overfitting. Discriminatory ability was quantified using the c-statistic.16 Calibration was assessed visually using calibration plots. Overfitting was assessed with bootstrap validation.17 We report the number of events per variable in each trial as an indicator for the risk of overfitting.
We evaluated the distribution of predicted risk in the overall study population and separately in each treatment arm. Visual examination of the risk distribution was facilitated by the use of box plots of the predicted risk of the outcome. In addition, we plotted histograms of the empirical distribution of predicted risk in each study to assess how closely the distribution conformed to the truncated log-normal (for risk predicted by proportional hazard models) or the logistic-normal distribution (for risk predicted by logistic regression models).
To describe and quantify risk heterogeneity using clinically interpretable metrics, we used two indexes, the extreme quartile risk ratio (EQRR) and the median-to-mean risk ratio (MMRR). To calculate the EQRR, we stratified the trial population into equal-sized quartiles according to the baseline predicted risk from the model.18 We then calculated the ratio of the predicted outcome risk in the extreme quartiles (high-risk quartile outcome probability divided by the low-risk quartile outcome probability, EQRRpredicted). We also calculated the same index based on the observed outcome rate (EQRRobserved) within strata defined by predicted risk. Greater EQRR values indicate greater risk heterogeneity in the risk-stratified patient population. The MMRR is a clinically interpretable measure of skewness calculated as the ratio of the median predicted outcome probability to the mean predicted outcome probability. As the MMRR deviates from one, it reflects the degree to which the summary (average) result may not reflect the effects in the ‘typical’ patient in the trial. We also calculated Pearson’s median skewness coefficient [3*(mean-median)/standard deviation], a more common measure of skeweness.
We also examined the relationship between the outcome prevalence and the c-statistic, and the EQRR and MMRR, visually and using linear regression.
Evaluating HTE over predicted outcome risk
Additionally, we analysed the relationship between treatment effect and predicted outcome risk. We estimated treatment effects within each risk quartile on relative and absolute scales. Specifically, we estimated relative treatment effects using logistic regression (using odds ratios as the measure of effect) or Cox regression (using hazard ratios as the measure of effect); we estimated absolute treatment effects using linear probability models for binary outcomes (using absolute risk reduction as the measure of effect). For time-to-event analyses, we calculated absolute risk reduction as the difference in Kaplan-Meier survival probabilities between the intervention and comparator treatment arms.19 We tested the null hypothesis of no HTE over predicted outcome risk using a product term (‘interaction’) between the fitted value of the linear predictor (from the risk model) and the treatment assignment indicator. We also compared relative and absolute risk reduction between the extreme risk quartiles in each trial. We summarized these metrics for the subset of trials with statistically significant overall treatment effects, i.e. those trials showing statistically significant benefit or harm on either a primary or a secondary outcome.
Statistical analyses were performed using SAS version 9.3,20 R open-source software version 3.1.2 (The R Foundation for Statistical Computing) and Stata version 13.1 (Stata Corp., College Station, TX).
Results
A total of 32 trials met our inclusion criteria (Table 1). Most trials were in the field of cardiovascular disease, including trials evaluating interventions in atrial fibrillation, coronary heart disease, acute myocardial infarction, heart failure, hypertension and acute stroke. We also included trials of other conditions, such as prediabetes, acute kidney failure, chronic hepatitis C and prostatic hyperplasia. The number of patients in the analysed trial cohorts ranged from 715 to 33 357, and totalled 180 291. Trials had been conducted over a span of several decades; the earliest trial had been published in 1979 and the latest in 2008. Of note, our trials generally did not include interventions with harms anticipated to affect the primary outcome (e.g. as in carotid endarterectomy, which both prevents and causes stroke).
Table 1.
Description of trials
| Trial acronym | Year of publication | Patients randomized (n) | Patient population/ index condition | Intervention | Comparator | Primary outcomea | Secondary outcomea |
|---|---|---|---|---|---|---|---|
| ACCORDb23 | 2008 | 10251 | Type 2 diabetes mellitus | Intensive strategy | Standard treatment | First occurrence of a major CVD event | All-cause mortality (‐) |
| AFFIRM51 | 2002 | 4060 | Atrial fibrillation/risk of stroke or death | Rate control therapy | Rhythm control therapy | All-cause mortality | Not assessed |
| ALLHAT HTNb,c24 | 2002 | 33357 | Hypertension | Amlodipine or lisinopril | Chlorthalidone | Fatal CHD or nonfatal MI combined | Combined CVD events (‐) |
| ALLHAT LLT52 | 2002 | 10355 | Hypercholesterolaemia/hypertension | Pravastatin | Usual care | All-cause mortality | Not assessed |
| AMIS30 | 1980 | 4524 | Myocardial infarction | Aspirin | Placebo | All-cause mortality | Not assessed |
| ATN29 | 2008 | 1124 | Acute kidney failure/sepsis | Intensive renal-replacement therapy | Less intensive renal- replacement therapy | All-cause mortality (60-day) | Not assessed |
| BARI53 | 1996 | 1829 | Coronary artery disease/severe angina | Percutaneous transluminal coronary balloon angioplasty | Coronary artery bypass grafting | Cardiac mortality | Not assessed |
| BESTb25 | 2001 | 2708 | Advanced heart failure/congestive heart failure | Bucindolol hydrocholoride | Placebo | All-cause mortality | Death due to cardiovascular causes (+) |
| BHAT54 | 1982 | 3837 | Acute myocardial infarction | Propranolol | Placebo | All-cause mortality (+) | Not assessed |
| CAST55 | 1991 | 1498 | Myocardial infarction | Class I and Ib ant-iarrhythmic agents | Placebo | All-cause mortality or cardiac arrest (‐) | Not assessed |
| CPPT56 | 1984 | 3806 | Hypercholesterolaemia | Cholestyramine | Placebo | CHD death and/or definite nonfatal myocardial infarction (+) | Not assessed |
| DCCTb21 | 1993 | Prevention: 726 intervention: 715 | Type 1 diabetes mellitus | Intensive diabetes therapy | Conventional diabetes therapy | Appearance and/or progression of retinopathy and other complications (+) | Not assessed |
| DIGb26 | 1997 | 6800 | Heart failure | Digoxin | Placebo | All-cause mortality | Hospitalization for worsening heart failure (+) |
| DPPc33 | 2002 | 3234 | At risk for diabetes mellitus | 1) metformin 2) intensive lifestyle intervention | Placebo | Development of diabetes (+) | Not assessed |
| ENRICHD57 | 2003 | 2481 | Acute myocardial infarction | Cognitive behaviour therapy-based intervention | Usual medical care | All-cause mortality or recurrent myocardial infarction | Not assessed |
| FAVORIT58 | 2011 | 4110 | Stable kidney transplant | Multivitamin plus folic acid, vitamin B12 and vitamin B6 | Treatment with an identical multivitamin alone | Arteriosclerotic cardiovascular disease outcome | Not assessed |
| FUTURA59 | 2010 | 2026 | Unstable angina/ | Low-dose unfractionated heparin | Standard-dose unfractionated heparin | Peri-PCI major bleeding, minor bleeding, major vascular access site complications | Not assessed |
| Non–STEMI | |||||||
| HALTC60 | 2008 | 1050 | Chronic hepatitis c | Indefinite pegylated interferon alpha-2a (beyond 3.5 years of use) | Pegylated interferon alpha-2a, discontinue use after 3.5 years | Progression to cirrhosis | Not assessed |
| HDFP61 | 1979 | 10940 | Hypertension | Stepped care antihypertensive therapy | Referred care | All-cause mortality (+) | Not assessed |
| HEMOc62 | 2002 | 1846 | Haemodialysis | High-dose/high-flux dialysis | Standard-dose/low-flux dialysis | All-cause mortality | Not assessed |
| ISTb22 | 1997 | 19435 | Acute stroke | Unfractionated heparin, aspirin | Placebo | Death within 14 days, death/dependency at 6 months | Not assessed |
| MAGIC63 | 2002 | 6213 | Acute myocardial infarction | Intravenous magnesium sulphate | Placebo | All-cause mortality | Not assessed |
| MRFIT64 | 1982 | 12866 | At risk for coronary heart disease | Stepped-care treatment, counselling, dietary advice | Usual care | Death from coronary heart disease | Not assessed |
| MTOPSd32 | 2003 | 3047 | Benign prostatic hyperplasia | 1)doxazosin, 2) finasteride or 3) combination therapy | Placebo | Clinical progression of benign prostatic hyperplasia (+) | Not assessed |
| OAT65 | 2006 | 2166 | Congestive heart failure | Routine PCI and stenting with optimal medical therapy | Optimal medical therapy alone | Mortality, recurrent MI, and hospitalization for CHF | Not assessed |
| PEACE66 | 2004 | 8290 | Coronary artery disease | Trandolapril | Placebo | Death from cardiovascular causes or non-fatal MI | Not assessed |
| ROCb,c67,68 | HS: 2011 | HS:895 | Hypovolaemic shock | Hypertonic saline solution | Normal saline solution | 28-day survival, 6-month neurological outcome based on the extended Glasgow Outcome Scale | Not assessed |
| TBI: 2010 | TBI: 1331 | Traumatic brain injury | |||||
| SHEP69 | 1991 | 4736 | Hypertension | Chlorthalidone/atenolol antihypertensive drug regimen | Placebo | Non-fatal and fatal stroke (+) | Not assessed |
| SOLVDe27,28 | Prevention: 1992 | Prevention: 4228 intervention: 2569 | Congestive heart failure | Enalapril | Placebo | All-cause mortality (T+) | Death or hospitalization for heart failure (P+) |
| intervention: 1991 | |||||||
| TIMI-II70 | 1989 | 3262 | Acute myocardial infarction | Invasive strategy | Conservative strategy | All-cause mortality or non-fatal MI | Not assessed |
| 32 trials | 180291 | 18 positive treatment effects | |||||
| (33 cohorts) | From 14 trials |
ALLHAT HTN, antihypertensive trial; ALLHAT LLT, lipid-lowering trial; HEMO used 2-by-2 factorial design; IST investigated two primary outcomes; primary outcome treatment effect for SOLVD was significant for the left ventricular dysfunction cohort (T) and the secondary outcome treatment effect was positive for the asymptomatic cohort (P); summary results are from 39 risk distributions; DCCT has two cohorts whereas all other trials have one cohort; PCI, percutaneous coronary intervention.
aSummary treatment effect on outcome is statistically insignificant unless indicated by sign: (+) indicates positive treatment effect, (‐) indicates treatment harm.
bIndicates two risk distributions.
cIndicates two treatment arms.
dIndicates three treatment arms.
eIndicates three risk distributions (if not specified, assume one risk distribution).
One trial had more than one patient cohort (DCCT21), one trial had more than one primary outcome (IST22) and five trials had non-statistically significant results for their primary outcome but significant results for a secondary outcome (ACCORD,23 ALLHAT HTN,24 BEST,25 DIG,26 SOLVD27,28). Thus, we developed a total of 39 separate risk models. The median number of risk factors used in these models was 10 (average = 10.9; range = 4–20) (Table 2). The median number of events per variable was 51.3 (average = 107.0; range = 12.5–907.1), suggesting that models were unlikely to overfit the data. The median c-statistic was 0.69 (average = 0.70; range = 0.59–0.89). Bootstrap validation produced optimism-corrected c-statistics in the range of 0.58 to 0.88 (median = 0.68, 25th–75th percentile = 0.64–0.70). The difference between original and optimism-corrected c-statistics ranged from 0.001 to 0.02 (median = 0.007, 25th–75th percentile = 0.004–0.009), again suggesting the absence of substantial overfitting.
Table 2.
Summary of results for 39 risk distributions
| Median | 25th–75th percentile | Mean | Range | |
|---|---|---|---|---|
| Overall event rate | 0.15 | 0.09–0.29 | 0.20 | 0.03–0.63 |
| Model risk predictors | 10 | 7–16 | 10.9 | 4–20 |
| Events per variable | 51.3 | 32.3–84.7 | 107.0 | 12.5–907.1 |
| c-statistic | 0.69 | 0.65–0.71 | 0.70 | 0.59–0.89 |
| EQRR observed | 4.3 | 3.0–6.1 | 6.1 | 1.8–50.7 |
| EQRR predicted | 4.0 | 3.1–5.4 | 5.3 | 1.9–35.2 |
| MMRR | 0.86 | 0.80–0.92 | 0.84 | 0.42–1.04 |
| PMSC | 0.74 | 0.60–0.86 | 0.70 | −0.24–1.56 |
EQRR, extreme quartile risk ratio; MMRR, median-to-mean risk ratio; PMSC, Pearson’s median skewness coefficient.
Distribution of predicted outcome risk in large randomized trials
The median overall event rate across the trials was 15% (average = 20%; range = 3–63%). Summary statistics describing the risk heterogeneity of the population are shown in Table 2. The median EQRRobserved was approximately 4, but more than a quarter of all analyses had an EQRRobserved over 6 and the range extended to 50. Values of EQRRpredicted corresponded closely to the observed values. Whereas the median MMRR was 0.86 (indicating that the typical patient was at 86% the outcome risk compared with the average), this index ranged as low as 0.4—and only twice exceeded 1 (ATN,29 IST22 6-month outcome), both times for trials with high outcome rates (52.6% in ATN and 62.6% in IST).
We found the overall outcome rate in the trial and the c-statistic were strong predictors of the risk distribution. In linear regression, the outcome rate and c-statistic were shown to strongly predict the EQRR (R2 = 0.86) and the MMRR (R2 = 0.78) (Table 3). As discrimination improved, and as the overall outcome rate was lower, EQRR increased and MMRR decreased in a predictable fashion. Indeed, we found that knowing these two parameters (overall outcome incidence and c-statistic) essentially determine the full distribution of predicted risk, because the risk distributions were close to the log-normal (for risk predicted using Cox models) or logistic-normal shape (for risk predicted using logistic regression models) (Figure 1). This can be seen by comparing the histograms and kernel densities of the predicted values (in black) against the log-normal (red) or logistic-normal densities (blue) fit to the same values via maximum likelihood, which were fairly similar in most studies.
Table 3.
Regression model results
| log EQRR predicted |
MMRR |
|||||
|---|---|---|---|---|---|---|
| Estimate (SE) | t-Value | P-value | Estimate (SE) | t-Value | P-value | |
| Intercept | −3.88 (0.39) | −10.05 | <0.0001 | 1.80 (0.12) | 15.47 | <0.0001 |
| Overall event rate | −1.94 (0.24) | −7.98 | <0.0001 | 0.66 (0.07) | 9.06 | <0.0001 |
| c-statistic | 8.27 (0.57) | 14.41 | <0.0001 | −1.57 (0.17) | −9.05 | <0.0001 |
| R-square | 0.86 | 0.78 | ||||
EQRR, extreme quartile risk ratio; MMRR, median-to-mean risk ratio; SE, standard error.
Figure 1.

Risk distributions. The histograms show the distribution of the predicted risk for the outcome of interest. Curves shown in red are fitted to the distribution of predictions generated by Cox models; curves shown in blue are fitted to the distribution of predictions generated by logistic models. Fitted log-normal curves and fitted logistic-normal curves are also shown for the Cox- and logistic regression-generated curves, respectively. As can be seen, these log-normal and logistic-normal curves approximate very well the red and blue fitted curves. Note: The FUTURA Trial is not included in this figure since we could not export individual-level patient predictions from the site in which the data were housed.
HTE over-predicted outcome risk
Among the 18 trials with statistically non-significant results, two trials showed statistically significant HTE over the estimated linear predictor from the risk model. In the AMIS trial,30 high-risk patients with acute myocardial infarction appeared to get more benefit from aspirin than low-risk patients (P = 0.02) on the proportional scale; in IST,22 for the combined outcome of death or dependency at 6 months, low-risk patients appeared to obtain more benefit than high-risk patients (P = 0.04) on the proportional scale.
In the 14 trials with statistically significant results, 18 unique treatment comparisons were analysed. Although the relative treatment effects appeared to decrease over risk quantiles in some trials (e.g. BEST, CPPT and MTOPS [Figure 2a]) and increase over risk quantiles in others (ACCORD, CAST and DPP [Figure 2a]), overall there was no apparent relationship between baseline risk and the hazard (or odds) ratio of treatment across trials. The median ratio of the hazard or odds ratio in the fourth quartile over that in the first quartile was 1.02 (25th–75th percentile = 0.70–1.21) (Table 4). We found a statistically significant interaction between treatment and the estimated linear predictor on the proportional scale only in one of 18 analyses -(DPP, metformin vs placebo; high-risk patients experienced greater benefit than low-risk patients; P = 0.0008). Despite the absence of ‘statistically significant’ HTE on the proportional scale, absolute risk reduction estimates varied substantially over predicted outcome risk and were generally higher in high-risk strata, ranging from −1.4% to 18.3% (median = 4.7%; 25th–75th percentile = 0.8–6.1%) in the first quartile of predicted risk and from 0.8% to 35.0% (median = 9.0%; 25th–75th percentile = 3.3–19.8%) in the fourth quartile. The difference in the absolute risk reduction between the extreme-risk quartiles ranged from −3.2% to 28.3% (median = 5.1%; 25th–75th percentile = 0.3–10.9) across studies. Figure 2b displays these absolute effects graphically.
Figure 2.
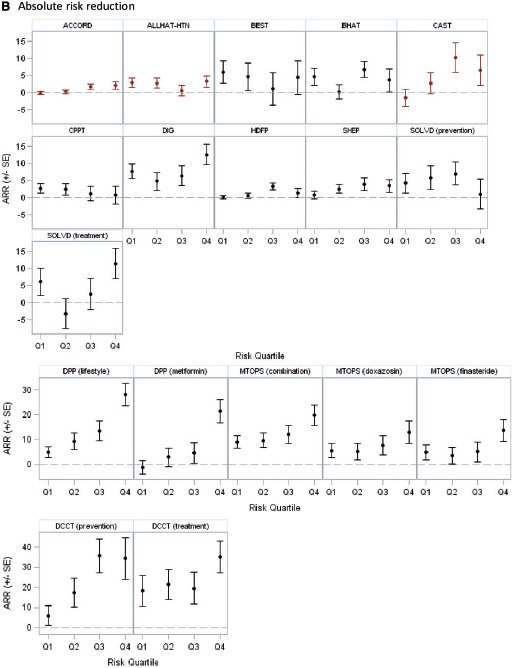
B: Absolute risk reduction across risk quartiles Red markers indicate that the treatment arms were switched (intervention was harmful). In Figure 2B, the scale for absolute risk reduction is different for DPP, MTOPS, and DCCT.
Figure 2.
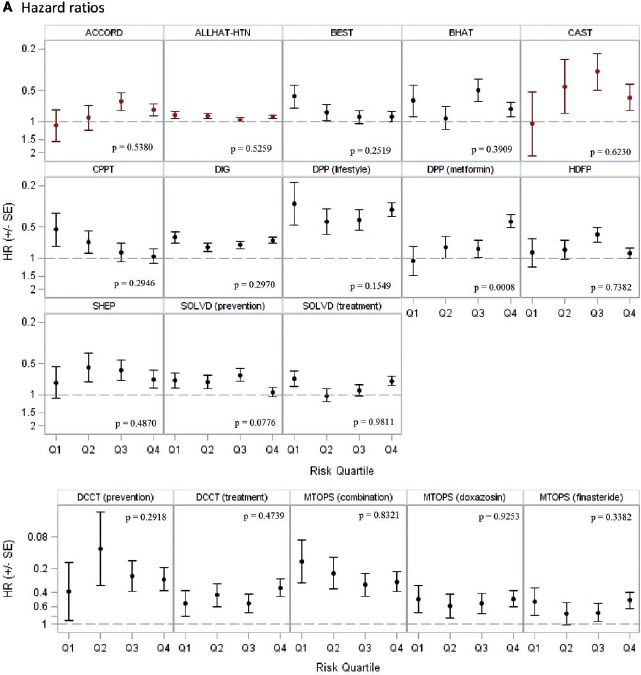
A: Hazard or odds ratios across risk quartiles Hazard ratios are shown for all trials except HDFP, which displays odds ratios. Red markers indicate that the treatment arms were switched (intervention was harmful). The scale for hazard ratio axis is different for DCCT and MTOPS.
Table 4.
Summary of results for 18 positive treatment comparisons (14 trials)
| Median | IQR | Mean | Range | |
|---|---|---|---|---|
| Hazard (or odds) ratio Q1 | 0.63 | 0.52–0.87 | 0.66 | 0.16–1.10 |
| Hazard (or odds) ratio Q4 | 0.69 | 0.44–0.90 | 0.64 | 0.27–0.96 |
| Extreme quartile relative hazard ratio (Q4/Q1) | 1.02 | 0.70–1.21 | 1.05 | 0.41–1.82 |
| Absolute risk reduction Q1 (%) | 4.73 | 0.83–6.06 | 4.50 | −1.43–18.27 |
| Absolute risk reduction Q4 (%) | 9.04 | 3.25–19.84 | 12.01 | 0.77–34.99 |
| Extreme quartile absolute risk reduction difference (Q4-Q1) | 5.10 | 0.33–10.91 | 7.51 | −3.23–28.33 |
Q, quartile; IQR, inter-quartile range.
Discussion
Our results show that clinically significant risk heterogeneity is common even in phase III ‘efficacy’ trials, which are often characterized as enrolling relatively homogeneous populations. Whereas statistically significant HTE on the proportional scale was unusual in this set of trials, in which interventions generally did not have anticipated harms on the primary outcome, variability in risk often gave rise to substantial HTE on the absolute risk scale. Though it is most common to test for heterogeneity on the proportional scale, absolute risk reduction (and its inverse, the number needed to treat) are generally considered the most relevant scales for clinical decision making.31 We did not use formal criteria to assess clinically important HTE, but it is noteworthy that, among treatment comparisons with statistically significant overall results, 25% showed differences in absolute risk differences greater than 10% between the extreme quartiles of predicted risk. We considered our analysis of two trials (MTOPS32 and DPP33), encompassing 5 of our 18 treatment comparisons, to be of sufficient clinical interest to report in separate clinical manuscripts.34,35 These papers join a growing list of papers showing clinically important variation in benefits when trial results are risk stratified, typically showing that an identifiable subgroup of higher-risk patients often account for most of the treatment benefit.36–46
Another consistent finding was that the median predicted outcome risk in these trials was lower than the mean predicted risk (i.e. MMRR < 1). Because the summary results of trials reflect the arithmetical mean risk, rather than the median risk, this implies that the typical patient is often at somewhat lower risk—and sometimes at much lower risk—than one might infer from the overall result. When proportional effects are similar across risk groups, summary results may have a tendency to overestimate the degree of benefit on the absolute scale.5,47 These concerns are especially germane when outcomes rates are predictable and outcome rates relatively low.
Whereas several trials in our database of trials exhibited large heterogeneity in predicted outcome risk, overall the results of our analyses were somewhat less extreme than previous published examples might have suggested.36–45 There are several explanations for this observation. First, risk heterogeneity may be somewhat restricted in large phase III randomized studies if they tend to enroll homogeneous patient populations. Second, because we wanted to limit the risk of overfitting models to data, we favoured simpler models, which generally had modest discriminatory ability. Finally, previously published examples might be ‘cherry-picked’ for extreme results and clinical significance. It is also important to recognize that expressing heterogeneity of risk using a finer grouping of predicted risk (e.g. quintiles or deciles) would yield ratios that are more extreme than the EQRRs reported here.
The observation that indices that describe the distribution of predicted risk are predictable based on the c-statistic, and the overall event rate of each trial, are as telling as the specific examples in our study. The predictability of the risk distribution derives from the fact that the linear predictor from the risk model conforms fairly closely to a normal distribution,48 yielding distributions of risk that (to a good approximation) conform to log-normal (for risk estimates derived from Cox models) or logistic-normal distributions (for risk estimates derived from logistic regression models). This relationship permits us to anticipate the degree of risk heterogeneity (i.e. EQRR) and the skewness (i.e. MMRR) based on knowledge of the outcome rate and the discrimination (c-statistic) of the model—provided that the risk model is well calibrated. For example, using our simple linear regression results, we would anticipate that, when the outcome rate is 10% and the c-statistic is 0.8, the EQRR will be approximately 13 and the MMRR will be approximately 0.6. When risk differs 13-fold between large population subsets, the overall treatment effect estimated for the trial population is not clinically interpretable. When the median risk is 40% lower than the mean risk, it also seems likely that the average effects may not be easily translated even to typical patients in the same trial. Higher c-statistics and lower outcome prevalence would lead to even more skewed distributions, implying greater risk heterogeneity.
Thus, it does not take extreme assumptions to yield risk distributions that would make overall clinical trial results misleading for many patients. The relationship also implies that a risk-stratified approach might be especially important and clinically informative when the outcome is predictable, based on easily available clinical information, and the overall outcome rate is low. This conclusion is consistent with clinical intuition, because when the outcome is rare and predictable by baseline covariates, it is possible to identify very-low-risk patients who are unlikely to benefit from therapy. Analyses of HTE over-predicted risk are also more likely to be useful for risky or costly therapies, when identifying patients who are unlikely to benefit may be of especially high interest.
Despite the fact that only one trial (DPP) showed a ‘statistically significant’ interaction between the linear predictor of risk and the treatment assignment indicator, we would urge caution in interpreting the ostensible consistency of effects on the multiplicative scale. We note that the true relationship between risk and effect is underdetermined by the data. Indeed, trial results may often be statistically consistent with homogeneous effects on both the additive and the multiplicative scales across risk groups—despite the mathematical incompatibility of these models and the potential clinical importance of the different inferences the models may yield. We believe that consistency of effects across any of these scales is unlikely to represent the ‘true’ relationship between the risk of the outcome and the effect of a therapy.
Our study has several limitations. We acknowledge that the use of quartiles is arbitrary, and tends to underestimate heterogeneity, compared with using finer strata of predicted risk or assuming a smooth function of predicted risk. We present our data in quartiles to facilitate comparisons across analyses, based on a previously suggested framework.18 Heterogeneity may be slightly overestimated based on model overfitting or underestimated based on underfitting; more careful model building (e.g. exploring non-linearity and interactions in the risk models) could have given the impression of more extreme risk heterogeneity. We did not explore non-linear relationships between risk and treatment effects, which may have revealed additional HTE. Additionally, we tried to standardize our modelling approach but we used only a single model for each trial. Different models may fit the data equally well, yet results regarding HTE may be sensitive to the specific variables included in the models and whether any of these variables are treatment effect modifiers. Whereas different models may yield different results, the degree to which any particular covariate modifies treatment is typically unknown—and when there is a strong a priori reason to believe that a particular covariate is likely to modify a treatment effect (apart from its influence on risk) then the relationship of the covariate with the treatment effect should also be examined separately. Finally, we used a convenience sample of large trials, which does not represent the full spectrum of clinical conditions or, specifically, those conditions for which risk modelling may be most informative. A risk-modelling approach may be especially informative when treatment can both prevent and cause the primary outcome of interest (presumably via different mechanisms).5,6,39,49 In such conditions, the risks of therapy may outweigh the benefits in very low-risk patients, and more treatment effect heterogeneity would be anticipated.
Despite these limitations, our results suggest that clinically important differences in effect across predicted risk are likely to be common in trials with statistically significant average treatment effects. A common assumption (of unclear validity) is consistency of treatment effects across risk groups on the proportional scale, but the only way of testing this assumption is to actually perform such risk-stratified analyses. Even when analyses fail to reject the null of proportional effects across different risk strata, the results of risk-stratified analyses can demonstrate clinically important risk differences which would otherwise be obscured. Nevertheless, risk-stratified analyses of clinical trials are still rarely planned as part of the initial study design; if reviewers, editors and regulators expected (or required) such analyses to be routinely conducted, the approach would be more widely adopted.50
In summary, predicted risk distributions from Cox regression and logistic regression are largely determined based on c-statistic and outcome rates. Clinically significant risk heterogeneity is common even in large ‘efficacy’ trials—particularly when outcome rates are low and c-statistics are high. The median risk in these trials is generally lower than the average risk. Statistically significant HTE on the relative risk scale is unusual, but clinically significant heterogeneity in absolute effects appears to be common. A risk stratified approach to trial analysis is feasible and may be most clinically informative when an uncommon outcome is predictable by baseline covariates.
Key Messages
Outcome risk is a mathematical determinant of the treatment effect yet can vary substantially across a trial population, making it unclear how treatment effects might vary in the trial population.
Using simple risk models based on baseline patient characteristics, among a sample of trials from publicly available sources, we found that outcome rates in the highest risk quartile were as high as 50-fold those in the lowest risk quartile; in fully a quarter of the trials, this ratio exceeded 6.
Because outcome risk in the trials was generally skewed (log-normal or logistic-normal), with a small group of high-risk patients accounting for a large number of outcomes, the outcome risk in most patients was almost always less than that reflected by the trial summary results.
Whereas we did not often detect treatment effect heterogeneity on the proportional scale across patients at different baseline risk in this set of trials, substantial differences in absolute treatment effects were common; differences in absolute treatment effects between the extreme quartiles of risk exceeded 10% in a quarter of trials that showed benefit.
Displaying results across subgroups defined by risk is feasible and can lead to clinically important findings.
Funding
This work was supported by a Patient-Centered Outcomes Research Institute (PCORI) Pilot Project Program Award (grant number IP2PI000722), a PCORI Methods Research Award (grant number ME-1306‐03758) and the National Institutes of Health (grant numbers U01NS086294, UL1 TR001064). All statements in this paper are solely those of the authors and do not necessarily represent the views of the PCORI, its Board of Governors, the PCORI Methodology Committee or the National Institutes of Health.
Conflict of interest: No authors have any disclosures to report
Supplementary Material
Acknowledgements
This article was prepared using research materials from Action to Control Cardiovascular Risk in Diabetes (ACCORD), Atrial Fibrillation Follow-Up Investigation of Rhythm Management (AFFIRM), Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT), Aspirin-Myocardial Infarction Study (AMIS), Bypass Angioplasty Revascularization Investigation (BARI), Beta-Blocker Evaluation in Survival Trial (BEST), Beta-Blocker Heart Attack Trial (BHAT), Cardiac Arrhythmia Suppression Trial (CAST), Digitalis Investigation Group (DIG), Enhancing Recovery in Coronary Heart Disease Patients (ENRICHD), Hypertension Detection and Follow-Up Program (HDFP), Lipid Research Clinics (LRC), Coronary Primary Prevention Trial (CPPT), Magnesium in Coronaries (MAGIC), Multiple Risk Factor Intervention Trial for the Prevention of Coronary Heart Disease (MRFIT), Occluded Artery Trial (OAT), Prevention of Events With Angiotensin-Converting Enzyme Inhibitor Therapy (PEACE), Resuscitation Outcomes Consortium (ROC), Hypertonic Saline Trial Shock Study (HS) and Traumatic Brain Injury Study (TBI), Systolic Hypertension in the Elderly Program (SHEP), Studies of Left Ventricular Dysfunction (SOLVD) and Thrombolysis in Myocardial Ischemia Trial II (TIMI II) obtained from the National Heart, Lung, and Blood Institute Biologic Specimen and Data Repository Information Coordinating Center, and does not necessarily reflect the opinions or views of the study investigators or the National Heart, Lung, and Blood Institute. The Acute Renal Failure Trial Network (ATN), Diabetes Control and Complications Trial (DCCT), Diabetes Prevention Program (DPP), Folic Acid for Vascular Outcome Reduction in Transplantation Trial (FAVORIT), Hepatitis C Antiviral Long-Term Treatment Against Cirrhosis (HALT-C), Hemodialysis Study (HEMO), Medical Therapy of Prostatic Symptoms (MTOPS), and Stress Incontinence Surgical Treatment Efficacy Trial (SISTEr) were conducted by study Investigators and supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The data from the trials reported here were supplied by the NIDDK Central Repositories. This manuscript was not prepared in collaboration with Investigators of these studies and does not necessarily reflect the opinions or views of Investigators, the NIDDK Central Repositories or the NIDDK. Additional research material from the Fondaparinux Trial With Unfractionated Heparin During Revascularization in Acute Coronary Syndromes (FUTURA) was provided by GlaxoSmithKline. This paper does not necessarily reflect the opinions or views of the study Investigators or GlaxoSmithKline. We would like to acknowledge members of the Stakeholder Panel for their input during all stages of this project and for their assistance in disseminating our research findings. The Stakeholder Panel includes: Bray Patrick-Lake, MFS, Co-chair at NIH Advisory Committee to the Director Working Group on the Precision Medicine Initiative, Director of Stakeholder Engagement at Clinical Trials Transformation and President of the PFO Medical Research Foundation; Joseph Cappelleri, PhD, MPH, Senior Director of Statistics, World Wide Pharmaceutical Operations, Pfizer Inc.; and Robert Dubois, MD, PhD; Chief Science Officer, National Pharmaceutical Council. We would like to thank the International Stroke Trial Investigators for providing access to the International Stroke Trial (IST) data. We would also like to thank Jennifer S. Lutz, MA, (PACE, Tufts Medical Center, Boston, MA) for technical support for this project and assistance with manuscript preparation.
Supplementary Data
Supplementary data are available at IJE online.
References
- 1. Rothwell PM. Can overall results of clinical trials be applied to all patients? Lancet 1995;345:1616–19. [DOI] [PubMed] [Google Scholar]
- 2. Brookes ST, Whitley E, Peters TJ, Mulheran PA, Egger M, Davey Smith G. Subgroup analyses in randomised controlled trials:quantifying the risks of false-positives and false-negatives. Health Technol Assess 2001;5:1–56. [DOI] [PubMed] [Google Scholar]
- 3. Brookes ST, Whitely E, Egger M, Davey Smith G, Mulheran PA, Peters TJ. Subgroup analyses in randomized trials:risks of subgroup-specific analyses; power and sample size for the interaction test. J Clin Epidemiol 2004;57:229–36. [DOI] [PubMed] [Google Scholar]
- 4. Kravitz RL, Duan N, Braslow J. Evidence-based medicine, heterogeneity of treatment effects, and the trouble with averages. Milbank Q 2004;82:661–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kent DM, Hayward RA. Limitations of applying summary results of clinical trials to individual patients: the need for risk stratification. JAMA 2007;298:1209–12. [DOI] [PubMed] [Google Scholar]
- 6. Rothwell PM, Mehta Z, Howard SC, Gutnikov SA, Warlow CP. Treating individuals 3: from subgroups to individuals: general principles and the example of carotid endarterectomy. Lancet 2005;365:256–65. [DOI] [PubMed] [Google Scholar]
- 7. Kent DM, Rothwell PM, Ioannidis JP, Altman DG, Hayward RA. Assessing and reporting heterogeneity in treatment effects in clinical trials: a proposal. Trials 2010;11:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun X, Briel M, Walter SD, Guyatt GH. Is a subgroup effect believable? Updating criteria to evaluate the credibility of subgroup analyses. BMJ 2010;340:c117. [DOI] [PubMed] [Google Scholar]
- 9. Sun X, Ioannidis JP, Agoritsas T, Alba AC, Guyatt G. How to use a subgroup analysis: users’ guide to the medical literature. JAMA 2014;311:405–11. [DOI] [PubMed] [Google Scholar]
- 10. Varadhan R, Segal JB, Boyd CM, Wu AW, Weiss CO. A framework for the analysis of heterogeneity of treatment effect in patient-centered outcomes research. J Clin Epidemiol 2013;66:818–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. National Heart Lung and Blood Institute. NHBLI Biologic Specimen and Data Repository Information Coordinating Center. https://biolincc.nhlbi nih gov/home (2 March 2015, date last accessed).
- 12. National Institute of Diabetes and Dignestive and Kidney Diseases. NIDDK Central Data Repository. https://www.niddkrepository.org (2 March 2015, date last accessed).
- 13. GlaxoSmithKline. GSK Clinical Study Data. https://www.clinicalstudydatarequest.com (2 March 2015, date last accessed).
- 14. Wessler BS, Lai YL, Kramer W et al. Clinical prediction models for cardiovascular disease: Tufts predictive analytics and comparative effectiveness clinical prediction model database. Circ Cardiovasc Qual Outcomes 2015;8:368–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burke JF, Hayward RA, Nelson JP, Kent DM. Using internally developed risk models to assess heterogeneity in treatment effects in clinical trials. Circ Cardiovasc Qual Outcomes 2014;7:163–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harrell FE Jr, Lee KL, Califf RM, Pryor DB, Rosati RA. Regression modelling strategies for improved prognostic prediction. Stat Med 1984;3:143–52. [DOI] [PubMed] [Google Scholar]
- 17. Harrell FE. Regression Modeling Strategies: with Applications to Linear Models, Logistic Regression, and Survival Analysis. New York, NY: Springer, 2001. [Google Scholar]
- 18. Ioannidis JP, Lau J. Heterogeneity of the baseline risk within patient populations of clinical trials: a proposed evaluation algorithm. Am J Epidemiol 1998;148:1117–26. [DOI] [PubMed] [Google Scholar]
- 19. Altman DG, Andersen PK. Calculating the number needed to treat for trials where the outcome is time to an event. BMJ 1999;319:1492–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. SAS Institute Inc. Base SAS® 9.3 Procedures Guide. Cary, NC: SAS Institute Inc., 2011. [Google Scholar]
- 21. Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med 1993;329:977–86. [DOI] [PubMed] [Google Scholar]
- 22. International Stroke Trial (IST) : a randomised trial of aspirin, subcutaneous heparin, both, or neither among 19435 patients with acute ischaemic stroke. International Stroke Trial Collaborative Group. Lancet 1997;349:1569–81. [PubMed] [Google Scholar]
- 23. Gerstein HC, Miller ME, Byington RP et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008;358:2545–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002;288:2981–97. [DOI] [PubMed] [Google Scholar]
- 25. Beta-Blocker Evaluation of Survival Trial Investigators. A trial of the beta-blocker bucindolol in patients with advanced chronic heart failure. N Engl J Med 2001;344:1659–67. [DOI] [PubMed] [Google Scholar]
- 26. Digitalis Investigation Group. The effect of digoxin on mortality and morbidity in patients with heart failure. N Engl J Med 1997;336:525–33. [DOI] [PubMed] [Google Scholar]
- 27. SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. The SOLVD Investigators. N Engl J Med 1992;327:685–91. [DOI] [PubMed] [Google Scholar]
- 28. SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. The SOLVD Investigators. N Engl J Med 1991;325:293–302. [DOI] [PubMed] [Google Scholar]
- 29. Palevsky PM, Zhang JH, O’Connor TZ et al. Intensity of renal support in critically ill patients with acute kidney injury. N Engl J Med 2008;359:7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aspirin Myocardial Infarction Study (AMIS). A randomized, controlled trial of aspirin in persons recovered from myocardial infarction. JAMA 1980;243:661–69. [PubMed] [Google Scholar]
- 31. Rothman KJ, Greenland S, Walker AM. Concepts of interaction. Am J Epidemiol 1980;112:467–70. [DOI] [PubMed] [Google Scholar]
- 32. McConnell JD, Roehrborn CG, Bautista OM et al. The long-term effect of doxazosin, finasteride, and combination therapy on the clinical progression of benign prostatic hyperplasia. N Engl J Med 2003;349:2387–98. [DOI] [PubMed] [Google Scholar]
- 33. Knowler WC, Barrett-Connor E, Fowler SE et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002;346:393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sussman JB, Kent DM, Nelson JP, Hayward RA. Improving diabetes prevention with benefit based tailored treatment: risk based reanalysis of Diabetes Prevention Program. BMJ 2015;350:h454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kozminski MA, Wei JT, Nelson J, Kent DM. Baseline characteristics predict risk of progression and response to combined medical therapy for benign prostatic hyperplasia (BPH). BJU Int 2015;115:308–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kent DM, Ruthazer R, Griffith JL et al. A percutaneous coronary intervention-thrombolytic predictive instrument to assist choosing between immediate thrombolytic therapy versus delayed primary percutaneous coronary intervention for acute myocardial infarction. Am J Cardiol 2008;101:790–95. [DOI] [PubMed] [Google Scholar]
- 37. Thune JJ, Hoefsten DE, Lindholm MG et al. Simple risk stratification at admission to identify patients with reduced mortality from primary angioplasty. Circulation 2005;112:2017–21. [DOI] [PubMed] [Google Scholar]
- 38. Eli Lilly & Co. Xigris: drotrecogin alfa (activated): PV 3420. AMP Indianapolis: Eli Lilly & Co., 2001. [Google Scholar]
- 39. Kent DM, Hayward RA, Griffith JL et al. An independently derived and validated predictive model for selecting patients with myocardial infarction who are likely to benefit from tissue plasminogen activator compared with streptokinase. Am J Med 2002;113:104–11. [DOI] [PubMed] [Google Scholar]
- 40. Antman EM, Cohen M, Bernink PJ et al. The TIMI risk score for unstable angina/non-ST elevation MI: A method for prognostication and therapeutic decision making. JAMA 2000;284:835–42. [DOI] [PubMed] [Google Scholar]
- 41. Morrow DA, Antman EM, Snapinn SM, McCabe CH, Theroux P, Braunwald E. An integrated clinical approach to predicting the benefit of tirofiban in non-ST elevation acute coronary syndromes. Application of the TIMI Risk Score for UA/NSTEMI in PRISM-PLUS. Eur Heart J 2002;23:223–29. [DOI] [PubMed] [Google Scholar]
- 42. Cannon CP, Weintraub WS, Demopoulos LA et al. Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med 2001;344:1879–87. [DOI] [PubMed] [Google Scholar]
- 43. Kovalchik SA, Tammemagi M, Berg CD et al. Targeting of low-dose CT screening according to the risk of lung-cancer death. N Engl J Med 2013;369:245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van der Leeuw J, Oemrawsingh RM, van der Graaf Y et al. Prediction of absolute risk reduction of cardiovascular events with perindopril for individual patients with stable coronary artery disease - Results from EUROPA. Int J Cardiol 2014;182C:19499. [DOI] [PubMed] [Google Scholar]
- 45. Dorresteijn JA, Visseren FL, Ridker PM et al. Estimating treatment effects for individual patients based on the results of randomised clinical trials. BMJ 2011;343:d5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Califf RM, Woodlief LH, Harrell FE Jr. et al. Selection of thrombolytic therapy for individual patients: development of a clinical model. GUSTO-I Investigators. Am Heart J 1997;133:630–39. [DOI] [PubMed] [Google Scholar]
- 47. Vickers AJ, Kent DM. The Lake Wobegon effect: Why most patients are at below-average risk. Ann Intern Med 2015;162:886–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Royston P, Altman DG. Visualizing and assessing discrimination in the logistic regression model. Stat Med 2010;29:2508–20. [DOI] [PubMed] [Google Scholar]
- 49. Hayward RA, Kent DM, Vijan S, Hofer TP. Multivariable risk prediction can greatly enhance the statistical power of clinical trial subgroup analysis. BMC Med Res Methodol 2006;6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hayward RA, Kent DM, Vijan S, Hofer TP. Reporting clinical trial results to inform providers, payers, and consumers. Health Aff (Millwood) 2005;24:1571–81. [DOI] [PubMed] [Google Scholar]
- 51. Wyse DG, Waldo AL, DiMarco JP et al. A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med 2002;347:1825–33. [DOI] [PubMed] [Google Scholar]
- 52. Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT-LLT). Major outcomes in moderately hypercholesterolemic, hypertensive patients randomized to pravastatin vs usual care: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT-LLT). JAMA 2002;288:2998–3007. [DOI] [PubMed] [Google Scholar]
- 53. Bypass Angioplasty Revascularization Investigation (BARI) Investigators. Comparison of coronary bypass surgery with angioplasty in patients with multivessel disease. The Bypass Angioplasty Revascularization Investigation (BARI) Investigators. N Engl J Med 1996;335:217–25. [DOI] [PubMed] [Google Scholar]
- 54. The beta-Blocker Heart Attack Trial (BHAT). A randomized trial of propranolol in patients with acute myocardial infarction. I. Mortality results. JAMA 1982;247:1707–14. [DOI] [PubMed] [Google Scholar]
- 55. Echt DS, Liebson PR, Mitchell LB et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med 1991;324:781–88. [DOI] [PubMed] [Google Scholar]
- 56. The Lipid Research Clinics Coronary Primary Prevention Trial. Results. I. Reduction in incidence of coronary heart disease. JAMA 1984;251:351–64. [DOI] [PubMed] [Google Scholar]
- 57. Berkman LF, Blumenthal J, Burg M et al. Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Heart Disease Patients (ENRICHD) Randomized Trial. JAMA 2003;289:3106–16. [DOI] [PubMed] [Google Scholar]
- 58. Bostom AG, Carpenter MA, Kusek JW et al. Homocysteine-lowering and cardiovascular disease outcomes in kidney transplant recipients: primary results from the Folic Acid for Vascular Outcome Reduction in Transplantation trial. Circulation 2011;123:1763–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Steg PG, Jolly SS, Mehta SR et al. Low-dose vs standard-dose unfractionated heparin for percutaneous coronary intervention in acute coronary syndromes treated with fondaparinux: the FUTURA/OASIS-8 randomized trial. JAMA 2010;304:1339–49. [DOI] [PubMed] [Google Scholar]
- 60. Di Bisceglie AM, Shiffman ML, Everson GT et al. Prolonged therapy of advanced chronic hepatitis C with low-dose peginterferon. N Engl J Med 2008;359:2429–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hypertension Detection and Follow-up Program Cooperative Group. Five-year findings of the hypertension detection and follow-up program. I. Reduction in mortality of persons with high blood pressure, including mild hypertension. Hypertension Detection and Follow-up Program Cooperative Group. JAMA 1979;242:2562–71. [PubMed] [Google Scholar]
- 62. Eknoyan G, Beck GJ, Cheung AK et al. Effect of dialysis dose and membrane flux in maintenance hemodialysis. N Engl J Med 2002;347:2010–19. [DOI] [PubMed] [Google Scholar]
- 63. Magnesium in Coronaries (MAGIC) Trial. Early administration of intravenous magnesium to high-risk patients with acute myocardial infarction in the Magnesium in Coronaries (MAGIC) Trial: a randomised controlled trial. Lancet 2002;360:1189–96. [DOI] [PubMed] [Google Scholar]
- 64. Multiple Risk Factor Intervention Trial Research Group. Multiple risk factor intervention trial. Risk factor changes and mortality results. JAMA 1982;248:1465–77. [PubMed] [Google Scholar]
- 65. Hochman JS, Lamas GA, Buller CE et al. Coronary intervention for persistent occlusion after myocardial infarction. N Engl J Med 2006;355:2395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Braunwald E, Domanski MJ, Fowler SE et al. Angiotensin-converting-enzyme inhibition in stable coronary artery disease. N Engl J Med 2004;351:2058–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bulger EM, May S, Kerby JD et al. Out-of-hospital hypertonic resuscitation after traumatic hypovolemic shock: a randomized, placebo controlled trial. Ann Surg 2011;253:431–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bulger EM, May S, Brasel KJ et al. Out-of-hospital hypertonic resuscitation following severe traumatic brain injury: a randomized controlled trial. JAMA 2010;304:1455–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. SHEP Cooperative Research Group. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). JAMA 1991;265:3255–64. [PubMed] [Google Scholar]
- 70. TIMI Study Group. Comparison of invasive and conservative strategies after treatment with intravenous tissue plasminogen activator in acute myocardial infarction. Results of the thrombolysis in myocardial infarction (TIMI) phase II trial.N Engl J Med 1989;320:618–27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.