

Abstract
Subsets of mature B cell neoplasms are linked to infection with intracellular pathogens such as Epstein-Barr virus (EBV), hepatitis C virus (HCV), or Helicobacter pylori. However, the association between infection and the immunoglobulin-secreting (Ig-secreting) B proliferative disorders remains largely unresolved. We investigated whether the monoclonal IgG (mc IgG) produced by patients diagnosed with monoclonal gammopathy of undetermined significance (MGUS) or multiple myeloma (MM) targets infectious pathogens. Antigen specificity of purified mc IgG from a large patient cohort (n = 244) was determined using a multiplex infectious-antigen array (MIAA), which screens for reactivity to purified antigens or lysates from 9 pathogens. Purified mc IgG from 23.4% of patients (57 of 244) specifically recognized 1 pathogen in the MIAA. EBV was the most frequent target (15.6%), with 36 of 38 mc IgGs recognizing EBV nuclear antigen-1 (EBNA-1). MM patients with EBNA-1–specific mc IgG (14.0%) showed substantially greater bone marrow plasma cell infiltration and higher β2-microglobulin and inflammation/infection–linked cytokine levels compared with other smoldering myeloma/MM patients. Five other pathogens were the targets of mc IgG: herpes virus simplex-1 (2.9%), varicella zoster virus (1.6%), cytomegalovirus (0.8%), hepatitis C virus (1.2%), and H. pylori (1.2%). We conclude that a dysregulated immune response to infection may underlie disease onset and/or progression of MGUS and MM for subsets of patients.
Keywords: Autoimmunity, Immunology
Keywords: B cell receptor, Immunoglobulins
A dysregulated immune response to EBV may underlie the onset or progression of monoclonal gammopathy of undetermined significance and multiple myeloma for subsets of patients.
Introduction
Multiple myeloma (MM) is preceded by a clonal but asymptomatic stage termed monoclonal gammopathy of undetermined significance (MGUS) (1). MGUS may progress over time towards smoldering myeloma (SM) and overt MM stages. The causes of MGUS remain unknown, but disease progression to SM and MM is associated with numerous and increasingly damaging genetic alterations in clonal plasma cells (2–4).
In MGUS, SM, and MM, clonal plasma cells produce large quantities of monoclonal immunoglobulin (mc Ig). Since these antibodies are assumed not to have functional activity, antigen-driven (Ag-driven) stimulation as an early pathogenic mechanism has not been thoroughly investigated in the context of MGUS and MM. However, it is established that viral and bacterial infection in patients can lead to the production of oligoclonal or mc Ig (5–7). In addition, infection with intracellular oncogenic viruses such as Epstein-Barr virus (EBV) and hepatitis C virus (HCV) or bacteria such as Helicobacter pylori can lead to solid cancers and B cell malignancies, including Hodgkin lymphoma, non-Hodgkin lymphoma, and B cell chronic lymphocytic leukemia (8–11). Malignant transformation may occur directly via B cell infection and transformation, or indirectly via chronic Ag-driven stimulation of the B cell receptor (BCR) that has identical heavy and light chain specificity for secreted mc Ig. Our previous studies of HCV-positive patients who presented with MGUS or MM showed that the mc Ig produced in these cases almost always target the virus, especially the oncogenic core protein (12, 13). These results imply that an abnormal plasma cell response to infection could be the initiating event of some MGUS, SM, or MM cases. Notably, a report demonstrated that efficient antiviral treatment of a patient with chronic HCV infection led to MM regression (14). Thus, rapid and reliable detection of infection-initiated MGUS, SM, and MM cases might lead to major advances in patient management that would target the associated infection as well as clonal plasma cells.
Here, we provide potentially new evidence supporting a role for infectious pathogens in MGUS and MM pathogenesis. Purified mc IgGs from a large cohort of patients (n = 244) diagnosed with MGUS, SM, or MM were analyzed by using a multiplexed infectious-Ag array (MIAA) assay (15). The MIAA determines the serological status and specificity of purified mc IgG for 9 pathogens selected for their capacity for latent and chronic infection (15). We report that 23.4% of patients had a purified mc IgG specific for 1 infectious pathogen on the array. Of these, EBV nuclear antigen-1 (EBNA-1) was the most frequent target.
Results
Patient characteristics and serological status.
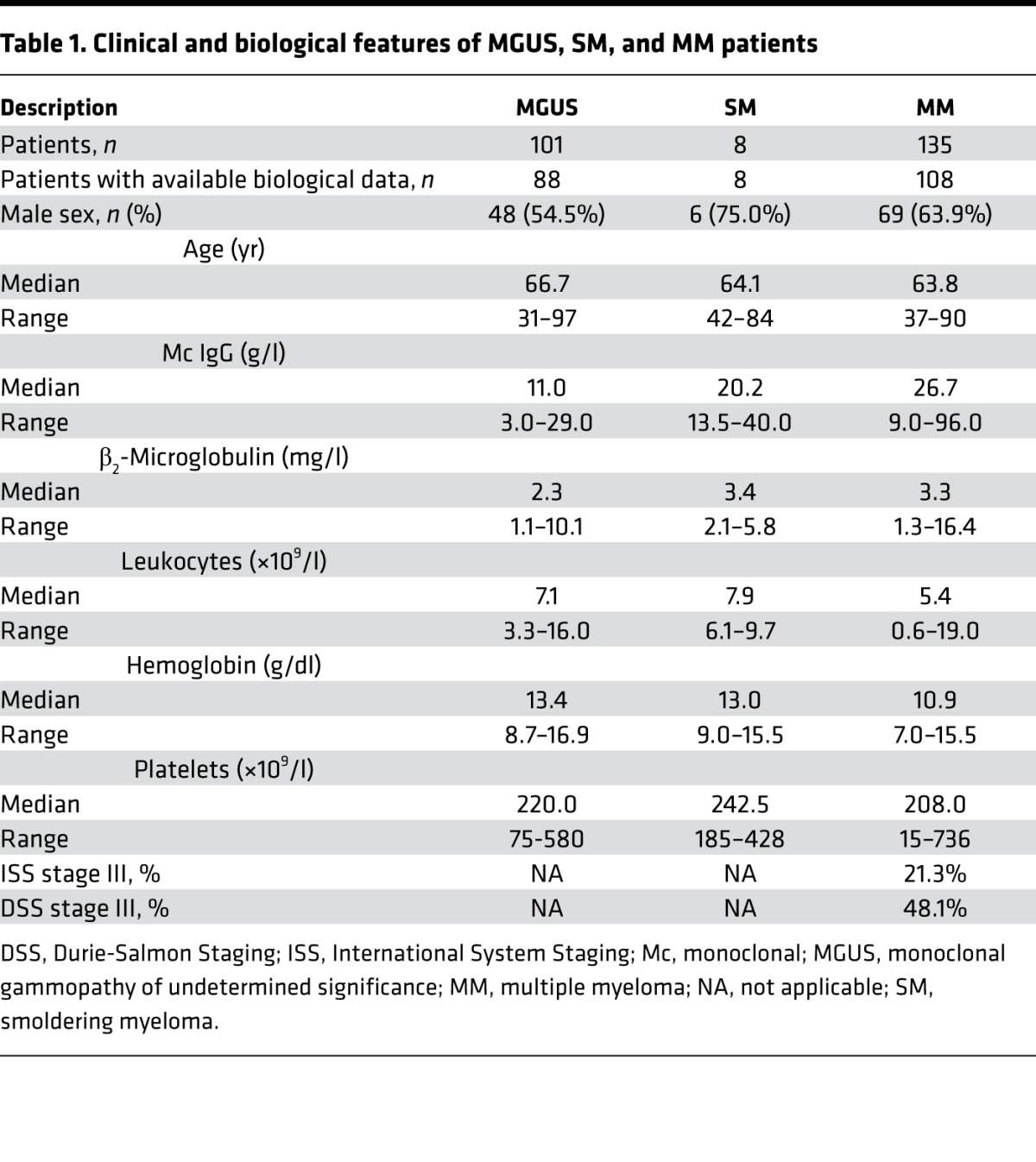
In this retrospective study, we analyzed in parallel the serum and purified mc IgGs from 244 patients diagnosed with MGUS (n = 101), SM (n = 8), and MM (n = 135). Annotated clinical data were available for 204 of 244 patients (88 MGUS, 8 SM, and 108 MM). The biological and clinical characteristics of the 204 patients are shown in Table 1. The median age of the MGUS, SM, and MM patients at the time of diagnosis was 66.7, 64.1, and 63.8 years, respectively. The male/female ratio was 54.5% for MGUS, and higher for SM and MM patients: 75.0% and 63.9%, respectively. The International Staging System (ISS) and Durie-Salmon Staging (DSS) scores indicated that 21.3% of the MM patients presented with ISS stage III at the time of diagnosis (DSS stage III: 48.1%).
Table 1. Clinical and biological features of MGUS, SM, and MM patients.
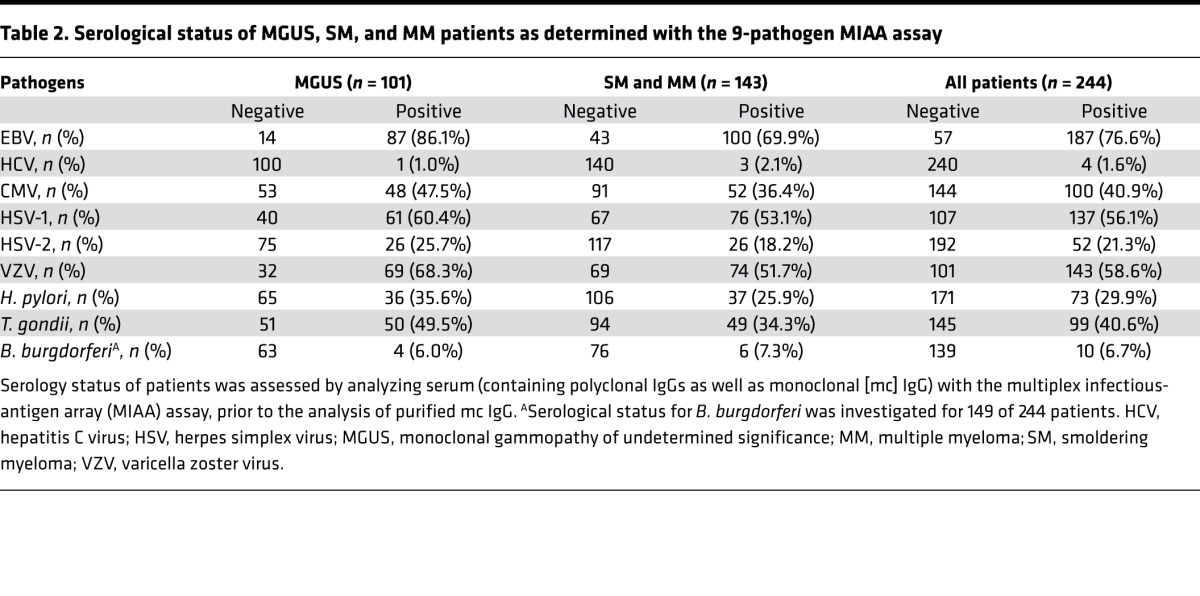
We hypothesized that mc IgG from a significant number of patients may be specific for common human pathogens. Therefore, the sera from all 244 patients were first analyzed for polyclonal serological status by using the MIAA assay. The MIAA assay tests for reactivity to lysates and/or purified Ag representing 9 infectious agents (Figure 1) (15). The MIAA assay used in this study allows testing for panels of commercially available Ag from EBV, HCV, cytomegalovirus (CMV), herpes simplex virus-1 (HSV-1), HSV-2, varicella zoster virus (VZV), H. pylori, Toxoplasma gondii, and Borrelia burgdorferi. The serological status of MGUS, SM, and MM patients as assessed by the MIAA assay is summarized in Table 2; the serological status for B. burgdorferi was investigated for 149 of 244 patients. Overall, there were lower rates of positive polyclonal serology for SM/MM compared with MGUS patients, likely due to reduced normal B cell functionality and decreased levels of circulating polyclonal IgG, IgA, and IgM antibodies in later-stage disease.
Figure 1. Description of the MIAA assay.

(A) The multiplex infectious-antigen array (MIAA) assay consists of microarray slides that contain 16 identical pads; 1 pad (represented enlarged) is shown in detail, with Ag or lysate spotted in triplicate. For each patient, serum and purified monoclonal (mc) IgGs are examined in parallel using the MIAA assay; each type of sample is tested in duplicate or more (some samples were tested 6 times within 1 experiment). Fluorescence signal is used to determine the serological status of each sample (15). (B) Human serum samples containing polyclonal IgGs specific for each of the 9 infectious pathogens (= positive controls, CTRL+) were used to set up the assay and assess reproducibility. Human serum control samples that did not contain IgG specific for 1 or several pathogens (= negative controls, CTRL–) served to evaluate nonspecific binding and to determine the fluorescence threshold of specific positivity for each pathogen, Ag, or lysate (L), or mix of Ag (M). Shown is the net fluorescence intensity of the samples after subtracting background fluorescence (F–B). (C) Detail of negative controls and thresholds of positivity. Three fluorescence thresholds of specific positivity were used in all MIAA experiments: 500 for hepatitis C virus (HCV), H. pylori, and T. gondii; 1,000 for CMV, herpes simplex virus-1 (HSV-1), and HSV-2; and 1,400 for EBV, varicella zoster virus (VZV), and B. burgdorferi. Signals below these thresholds were considered to be negative. Dots may be superimposed; black horizontal bars represent the means of results obtained for a pathogen, Ag, or lysate. Positive and negative controls are run in every MIAA experiment, as internal controls.
Table 2. Serological status of MGUS, SM, and MM patients as determined with the 9-pathogen MIAA assay.
Specificity of recognition of purified mc IgG with the MIAA assay.
Our next goal was to evaluate the possibility that the mc IgG, produced by clonal MGUS, SM, or MM B cells, is specific for infectious pathogens represented on the MIAA. To do so, mc IgGs from 244 serum samples were purified individually as previously described (refs. 12, 13, 15, and Figure 2A). The degree of purity of each mc IgG was verified by isoelectric focusing (IEF) and immunoblotting (Figure 2B). In some cases, the purity of the mc IgG preparations was also confirmed by mass spectrometry, as described (Figure 2, C and D, and ref. 16). Each purified mc IgG was then applied to the patterned MIAA arrays to determine Ag specificity.
Figure 2. Purification of mc IgGs, and verification of purity of the mc IgGs.

(A) For all 244 patients, monoclonal (mc) IgGs were purified as previously reported (12, 13, 15). (B) The purity of mc IgGs was verified using isoelectric focusing and immunoblotting; 3 examples (patients P16, P63, and P205) are shown. For certain patients, purified mc IgG (C) and polyclonal IgGs (D) were also studied by mass spectrometry. Only one IgG3 isotype (red circle) was observed in the mc IgG, while the polyclonal IgGs contained 3 isotypes: IgG1 (green circle), IgG2 (blue circle), and IgG3 (red circle), demonstrating the high purity of the mc IgG. The different peaks observed for each isotype correspond to the cumulative mass of the different hexoses carried by the IgG Fc fragment. Experiments were repeated twice.
Results obtained with the MIAA assay, described in Figures 3–9 and Supplemental Figure 1 (supplemental material available online with this article; https://doi.org/10.1172/jci.insight.95367DS1), showed that purified mc IgG isolated from almost one quarter of patients (57 of 244, 23.4%) specifically recognized a single pathogen lysate or recombinant protein on the array. Among these 57 patients, the incidence across disease stage was not significantly different. In total, 6 pathogens were recognized by purified mc IgG. The most frequently recognized pathogen was EBV: 38 patients (18 MGUS, 4 SM, and 16 MM) had an mc IgG that targeted EBV, as assessed by the MIAA assay (Figures 3 and 4, and Supplemental Figure 1). In this group of patients (38 of 244 patients, 15.6%), we evaluated the most common EBV Ags. The majority (36 of 38, or 94.7%) of EBV-reactive mc IgGs recognized EBNA-1 (Figure 3). Only 2 mc IgGs were specific for EBV viral capsid Ag (VCA) (Figure 4). The other infectious pathogens targeted by mc IgGs were HSV-1, in 7 cases (5 MGUS, 2 MM) (Figure 5); VZV, in 4 cases (4 MM) (Figure 6); CMV, in 2 cases (1 MGUS, 1 SM) (Figure 7); H. pylori, in 3 cases (1 MGUS, 2 MM) (Figure 8); and HCV, in 3 cases (3 MM) (Figure 9 and Supplemental Figure 1). HCV-specific mc IgGs targeted the core protein (2 cases) or NS-4 (1 case), as previously reported (12, 13). HSV-2 and T. gondii were not recognized by the purified mc IgGs in this study. Of note, although CMV infects 50% or more of the population, only 2 of 244 (0.8%) purified mc IgGs recognized CMV.
Figure 3. EBV-specific mc IgG as determined by the MIAA assay.

For each patient, serum and purified monoclonal (mc) IgGs were incubated in parallel in the multiplex infectious-antigen array (MIAA) assay; results of hybridization are shown for patients P16 (A and B) and P63 (C and D). (A and B) For P16, serum contained IgG that recognized EBV nuclear antigen-1 (EBNA-1), EBV viral capsid antigen (VCA), herpes simplex virus-1 (HSV-1), HSV-2, and varicella zoster virus (VZV) glycoprotein E (gE), whereas the purified mc IgG recognized EBNA-1 only. (C and D) For P63, serum contained IgG that recognized EBNA-1, EBV VCA, HSV-1, HSV-2, and T. gondii, whereas the purified mc IgG recognized EBNA-1 only. (B and D) Quantification of the fluorescence signals generated by the serum and purified mc IgG of patients P16 (B) and P63 (D) as assessed with the MIAA assay. (E) Results obtained for the 36 patients found to have an EBNA-1–specific mc IgG. For each patient, the results obtained for EBV with the patient’s serum (S) and purified mc IgG are shown in dark blue (EBNA-1) or light blue (VCA). The fluorescence values shown for EBNA-1 or EBV VCA were obtained after subtraction of the threshold of specific positivity for each pathogen, protein, or lysate (1,400 for EBV). Dots may be superimposed; horizontal bars represent the means of results obtained for a pathogen, Ag, or lysate. Experiments were performed at least twice.
Figure 4. Results of 2 patients with EBV VCA–specific mc IgG.

For each patient, results obtained with the serum (S) and the purified monoclonal (mc) IgG are represented. (A) For patients P10 and P50, the serum contained IgG that recognized EBV nuclear antigen-1 (EBNA-1) and EBV viral capsid antigen (VCA), but the purified mc IgG recognized only EBV VCA. (B) Detail of results obtained for patient P10: serum P10 contained IgG that recognized B. burgdorferi, CMV, EBNA-1, EBV VCA, H. pylori, herpes simplex virus-2 (HSV-2), T. gondii, and varicella zoster virus (VZV), whereas the purified mc IgG recognized EBV VCA only. (C) Detail of results obtained for patient P50: serum P50 contained IgG that recognized CMV, EBNA-1, EBV VCA, HSV-2, and VZV, whereas the purified mc IgG recognized EBV VCA only. The fluorescence values shown for each pathogen, Ag, or lysate were obtained after subtraction of the threshold of specific positivity of the pathogen, Ag, or lysate (1,400 for EBV). Dots may be superimposed; horizontal bars represent the means of results obtained for a pathogen, Ag, or lysate. Experiments were performed at least twice.
Figure 5. HSV-1–specific mc IgG as determined by the MIAA assay.

(A) Results of the multiplex infectious-antigen array (MIAA) assay for the 7 patients found to have a herpes simplex virus-1–specific (HSV-1–specific) monoclonal (mc) IgG. For each patient, the results obtained for HSV-1 lysate and glycoprotein G (gG) with the patient’s serum (S) and with purified mc IgG (mc) are shown. (B) Detail of results obtained for patient P9: P9 serum contained IgG that recognized EBV nuclear antigen-1 (EBNA-1), EBV viral capsid antigen (VCA), CMV, HSV-1, varicella zoster virus (VZV), and T. gondii, whereas the purified mc IgG recognized only HSV-1 gG. The fluorescence values shown for each pathogen, Ag, or lysate were obtained after subtraction of the threshold of specific positivity of the pathogen, Ag, or lysate (1,000 for HSV-1). Dots may be superimposed; horizontal bars represent the means of results obtained for a pathogen, Ag, or lysate. Experiments were performed at least twice.
Figure 6. VZV-specific mc IgGs as determined by the MIAA assay.

(A) Results of the multiplex infectious-antigen array (MIAA) assay for the 4 patients found to have a varicella zoster virus–specific (VZV-specific) monoclonal (mc) IgG. For each patient, the results obtained for VZV glycoprotein E (gE) and ORF-26 with the patient’s serum (S) and purified mc IgG (mc) are shown. (B) Detail of results obtained for patient P109: P109 serum contained IgG that recognized CMV, EBV nuclear antigen-1 (EBNA-1), herpes simplex virus-1 (HSV-1), VZV gE, and VZV ORF-26, whereas purified mc IgG recognized only VZV gE. The fluorescence values shown for each pathogen, Ag, or lysate were obtained after subtraction of the threshold of specific positivity of the pathogen, Ag, or lysate (1,400 for VZV). Dots may be superimposed; black horizontal bars represent the means of results obtained for a pathogen, Ag, or lysate. Experiments were performed at least twice.
Figure 7. CMV-specific mc IgGs as determined by the MIAA assay.

(A) Results of the multiplex infectious-antigen array (MIAA) assay for the 2 patients found to have a CMV-specific monoclonal (mc) IgG. For each patient, the results obtained for the CMV lysate and a mix of 5 CMV Ags (see Methods) with the patient’s serum (S) and purified mc IgG are shown. (B) Detail of results obtained for patient P84: serum P84 contained IgG that recognized CMV, EBV nuclear antigen-1 (EBNA-1), EBV viral capsid antigen (VCA), H. pylori, herpes simplex virus-1 (HSV-1), HSV-2, and varicella zoster virus (VZV) ORF-26, whereas the purified mc IgG recognized only the CMV lysate. (C) Detail of results obtained for patient P112: serum P112 contained IgG that recognized B. burgdorferi, CMV, EBNA-1, EBV VCA, H. pylori, HSV-1, HSV-2, T. gondii, and VZV, whereas the purified mc IgG recognized only the CMV Ag mix. The fluorescence values shown for each pathogen, Ag, or lysate were obtained after subtraction of the threshold of specific positivity of the pathogen, Ag, or lysate (1,000 for CMV). Dots may be superimposed; horizontal bars represent the means of results obtained for a pathogen, Ag, or lysate. Experiments were performed at least twice.
Figure 8. H.pylori–specific mc IgGs as determined by the MIAA assay.

(A) Results of the multiplex infectious-antigen array (MIAA) assay for the 3 patients with a H. pylori–specific monoclonal (mc) IgG. For each patient, the results obtained for H. pylori lysate 1 and lysate 2 with the patient’s serum (S) and purified mc IgG are shown. (B) The results obtained for patient P186 are detailed: serum P186 contained IgG that recognized CMV, EBV nuclear antigen-1 (EBNA-1), EBV viral capsid protein (VCA), H. pylori, herpes simplex virus-1 (HSV-1), HSV-2, T. gondii, and varicella zoster virus (VZV) ORF-26, whereas the purified mc IgG recognized only the H. pylori lysates (dark green: mix of lysates 1 and 2; medium green: lysate 1; light green: lysate 2). Note that certain H. pylori proteins and Ags are likely present both in lysate 1 and lysate 2. The fluorescence values shown for each pathogen, Ag, or lysate were obtained after subtraction of the threshold of specific positivity of the pathogen, Ag, or lysate (500 for H. pylori). Dots may be superimposed; horizontal bars represent the mean of results obtained for a pathogen, Ag, or lysate. Experiments were performed at least twice. (C) The immunoblot assay was performed using the commercial kit Helico Blot 2.1 (MP Biomedicals), which consisted of a Western blot made from bacterial lysate of H. pylori strain ATCC 49503. The test strip contained H. pylori Ags with molecular weights of 116 kDa (CagA), 89 kDa (VacA), 65 kDa (urease B), 60 kDa (heat shock protein [HSP]), 37 kDa (H. pylori undetermined protein [UP]), 35 kDa (H. pylori UP), and 30 kDa (urease A) as separate bands. The assay was performed and interpreted according to the instructions of the manufacturers. Experiments were performed at least twice.
Figure 9. HCV-specific mc IgGs as determined by the MIAA assay.

Results of the multiplex infectious-antigen array (MIAA) assay for the 3 patients with an hepatitis C virus–specific (HCV-specific) monoclonal (mc) IgG. For each patient, the results obtained for HCV core, NS-3, or NS-4 proteins with the patient’s serum and purified mc IgG are shown. Serum contained IgG that recognized the HCV core (P205, P206, P207) and NS-3 (P206) or NS-4 (P205, P207), whereas the purified mc IgG recognized only the HCV core (P205, P206) or NS4 (P207). The fluorescence values shown for each HCV protein were obtained after subtraction of the threshold of specific positivity (500 for HCV). Dots may be superimposed; horizontal bars represent means of results obtained for HCV core, NS3, or NS4. Experiments were performed at least twice.
Confirmation of the specificity of Ag recognition of purified mc IgG.
The high prevalence of mc IgG specific for EBNA-1 prompted us to perform further analysis, with the goal of identifying specific epitopes. Dot blots with recombinant full-length EBNA-1 confirmed a positive reaction for 32 of 38 patients (Figure 10A). We focused on an immunodominant B cell public epitope within the EBNA-1 sequence, PGRRPFF (residues 400–406); this epitope was reported as being recognized by polyclonal antibodies from 86.3% of individuals (17). To evaluate whether it is also a target for mc IgG, we employed a dot blot assay based upon synthetic peptides derived from the PGRRPFF sequence (relevant peptide, RRPPPGRRPFFHPVA, Figure 10B). A site-substituted synthetic (irrelevant) peptide (RRPPFGRRFFFHPVA) served as a negative control. However, only 2 mc IgGs gave a strongly positive signal for the PGRRPFF peptide (Figure 10B). The positivity of EBNA-1–specific mc IgG was verified and confirmed by peptide competition experiments with the MIAA assay, where the relevant EBNA-1 PGRRPFF peptide significantly inhibited the recognition of EBNA-1 recombinant protein by the EBNA-1–specific mc IgG, in a dose-dependent manner (Figure 10C).
Figure 10. Confirmation of the specificity of EBNA-1 recognition of purified mc IgGs.

(A) Dot blotting assays with purified recombinant EBV nuclear antigen-1 (EBNA-1) were first performed in parallel with serum and purified monoclonal (mc) IgG from 3 patients: P169 and P190, patients with EBNA-1–specific mc IgG as assessed by the multiplex infectious-antigen array (MIAA) assay, as positive controls; and patient P160, whose mc IgG did not recognize EBNA-1 as assessed by the MIAA assay, as a negative control. EBNA-1 dot blotting was then performed with the purified mc IgGs of 30 additional patients with EBNA-1–specific mc IgG as assessed by the MIAA assay. (B) Dot blotting assays performed by coating relevant (RRPPPGRRPFFHPVA) or irrelevant (RRPPFGRRFFFHPVA) EBNA-1–derived peptides and studying reactivity with serum and purified mc IgGs. Amino acids of the relevant epitope are in bold and underlined. The modified amino acids in the irrelevant peptide are shown in blue. Signal was obtained only with relevant peptide, but only 2 of 24 of the purified mc IgGs that could be tested bound to the relevant peptide. The serum of patient P98 does not contain IgG specific for EBNA-1. Patient P1 is shown as an example of serum containing polyclonal IgGs that bound to the relevant peptide, while the purified mc IgG did not bind. (C) Dose-dependant inhibition of recognition of EBNA-1 protein by purified mc IgG specific for EBNA-1 in the presence of the relevant peptide in the MIAA assay. Recombinant EBNA-1 protein was spotted on the array. A purified mc IgG found to be EBNA-1 specific with the MIAA assay and to recognize the relevant EBNA-1–derived peptide RRPPPGRRPFFHPVA with the dot blot assay, was preincubated with different concentrations of this peptide for 1 hour at room temperature before adding the mc IgG to the MIAA pad. The irrelevant peptide RRPPFGRRFFFHPVA was used as a negative control. Results are means ± SD of triplicates. *P < 0.05 by ANOVA. Experiments were performed at least twice.
Our next goal was to identify peptide epitopes in the HSV-1 proteome that are recognized by the HSV-1–specific mc IgG. To do so, we used high-throughput combinatorial phage random peptide display methodology. We employed the fUSE5-based phage peptide library that displays 109 cyclic random peptides (CX7C), where C represents cysteine and X represents any random residue (18). We selected index patient P56, characterized by a strong positive reaction of purified mc IgG against the HSV-1 lysate in the MIAA assay. The purified mc IgG was immobilized on Protein A/G beads and incubated with the phage display peptide library, which was precleared with Protein A/G beads. Bound phages were eluted, amplified, and enriched for high-specificity binding by using a selection procedure (18), which was repeated 3 times prior to sequencing of DNA inserts encoding the displayed peptide (Figure 11A). The most prevalent cluster included 14 related peptides (Figure 11A). An online BLAST search of the HSV-1 proteome resulted in a match for the sequence HL[SEDLGG]VPALR in HSV-1 large tegument protein (UL36) (residues 1498–1510). Further confirmation of the specificity of the purified mc IgG from the index patient for HSV-1 sequence HL[SEDLGG]VPALR was obtained by dot blot assay using the relevant peptide or a site-substituted, irrelevant synthetic peptide (EAVLHLSEDLGGRPAERQYVP) as a negative control. Furthermore, the HL[SEDLGG]VPALR sequence was recognized by all 7 cases where the mc IgG was positive for HSV-1 reactivity on the MIAA (Figure 11B). Thus, the VPALR sequence is a common BCR epitope for patients with HSV-1–specific mc IgG.
Figure 11. Confirmation of the specificity of HSV-1 recognition of purified mc IgGs.

(A, left): The purified monoclonal (mc) IgG from herpes simplex virus-1–positive (HSV-1+) index patient P56 was immobilized on Protein A/G beads and used for 4 rounds (R1–R4) of phage display peptide purification, with a CX7C library; the Fd-tet lacking peptide inserts served as a negative control. After the fourth round of selection, bacterial colonies were picked at random for sequencing. Results are expressed as mean ± SEM of duplicate wells. (A, right) Alignment of 14 related sequences, used for a BLAST alignment identifying the sequence VPALR from HSV-1 tegument protein, UL36 (in blue, aa 1498–1510). (B) Dot blotting with HSV-1 lysate and relevant (EAVLHLSEDLGGVPALRQYVP) and irrelevant (EAVLHLSEDLGGRPAERQYVP) HSV-1–derived peptides was performed for serum and purified mc IgG from the 7 patients found to have HSV-1–specific mc IgG with the multiplex infectious-antigen array (MIAA) assay. Patient P1, whose mc IgG did not recognize HSV-1 using the MIAA assay, was used as a negative control. Amino acids of the relevant epitope are in bold and underlined. The modified amino acids of the irrelevant peptide are in blue. Dot blots were performed at least twice.
Characteristics of patients with EBNA-1–specific mc IgG.
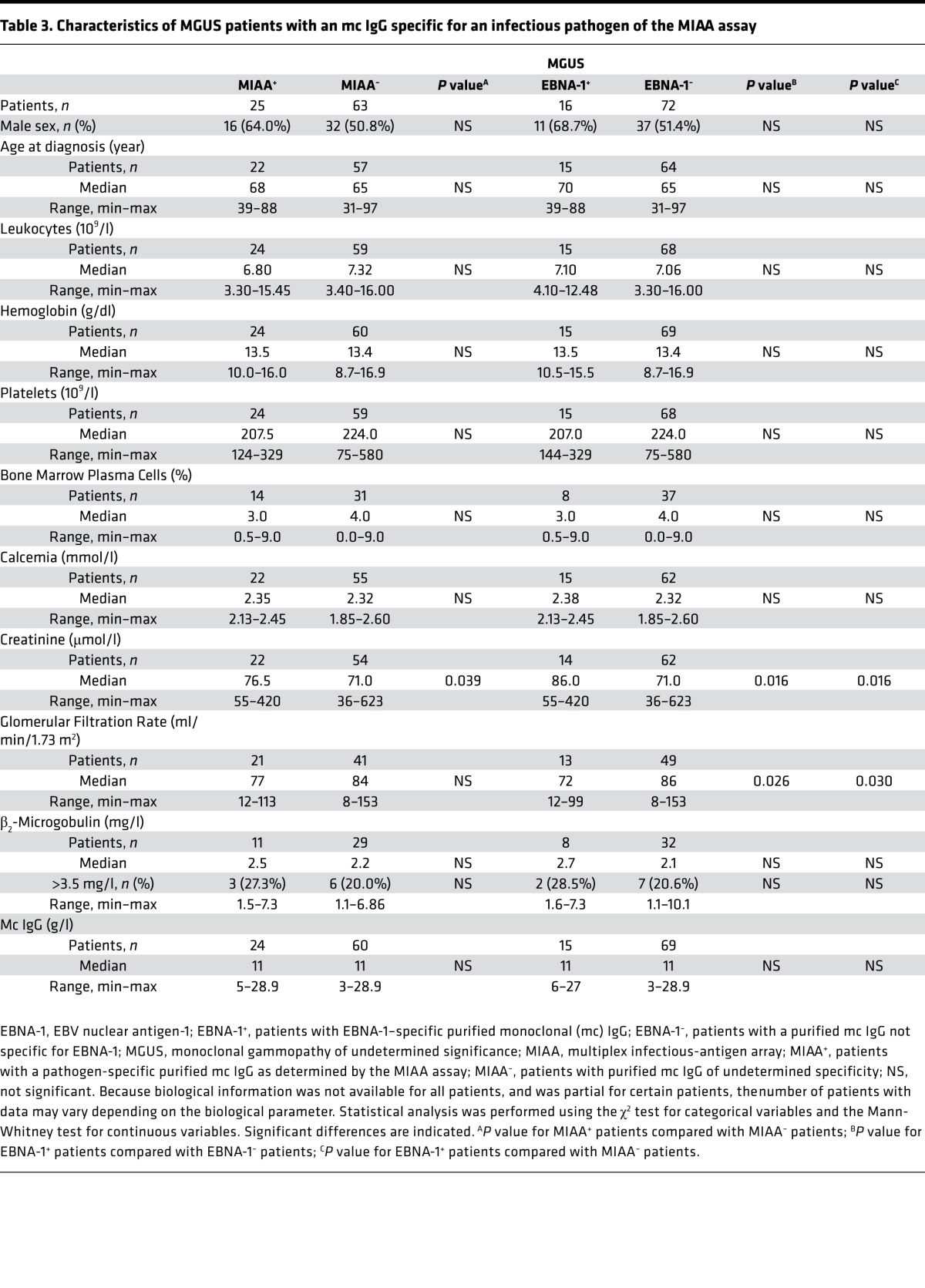
Having identified 14.7% (36 of 244) cases with EBNA-1–specific mc IgG, we next compared the clinical characteristics of these patients with the 168 other cases with clinical and biological data. First, we compared the 25 MGUS patients with mc IgG specific for one pathogen of the MIAA assay (MIAA+ patients) with MGUS patients with mc IgGs that were nonreactive for pathogens in the MIAA (MIAA– patients) (Table 3). There was a trend for mildly reduced renal function (serum creatinine [SCr]: 76.5 vs. 71.0 μmol/l, P = 0.039) for MIAA+ MGUS patients. We then compared the 16 MGUS patients with EBNA-1–specific mc IgG with the 72 MGUS patients whose mc IgG did not recognize EBNA-1 (Table 3). MGUS cases with EBNA-1–specific mc IgG differed from other MGUS patients by presenting with a mildly reduced renal function (SCr: 86.0 vs. 71.0 μmol/l, P = 0.016; glomerular filtration rate [GFR]: 72.0 vs. 86.0 ml/min, P = 0.026).
Table 3. Characteristics of MGUS patients with an mc IgG specific for an infectious pathogen of the MIAA assay.
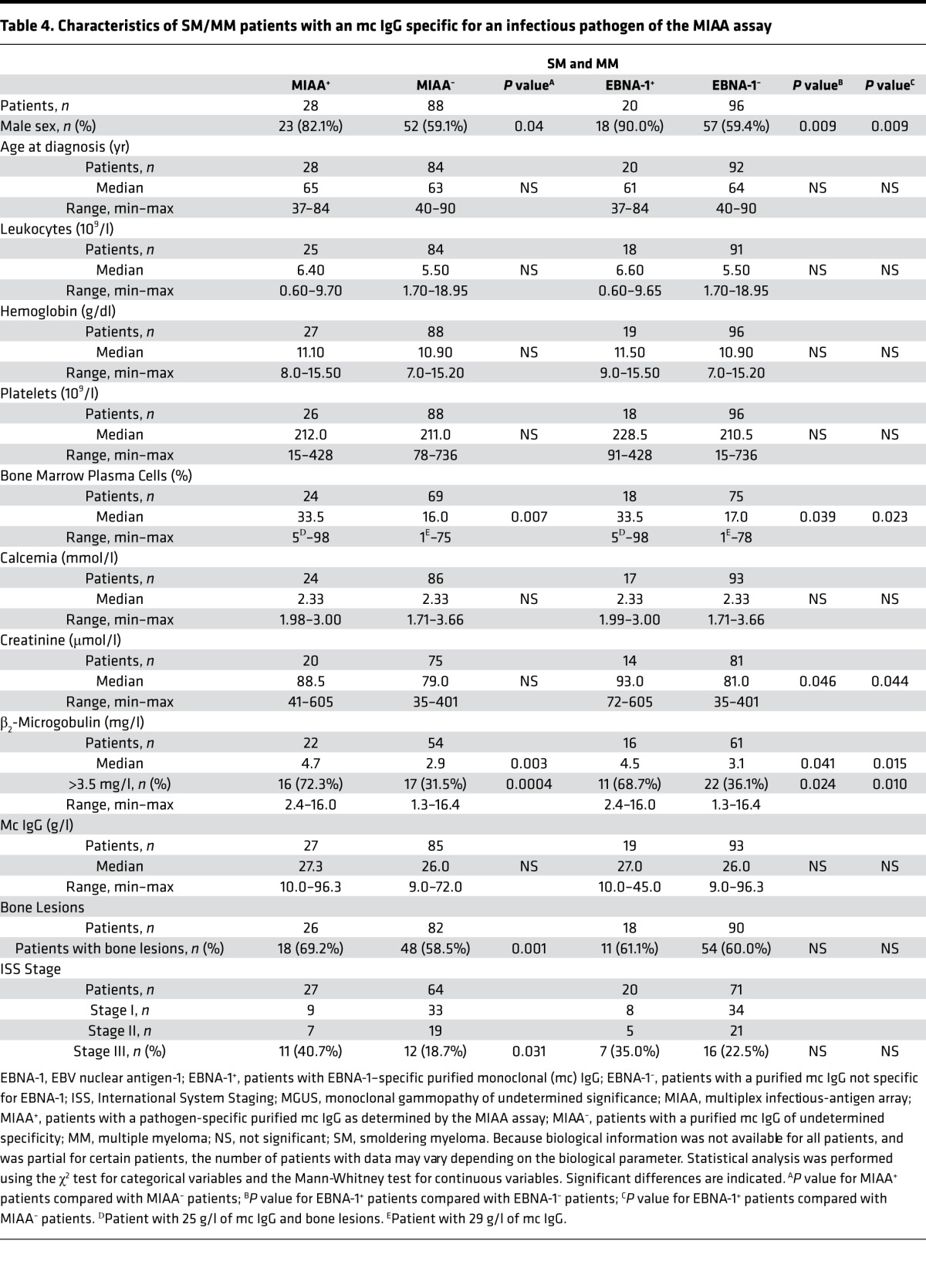
Moreover, we compared SM/MM patients with a positive reaction to a pathogen on the MIAA (MIAA+ SM/MM patients) with MIAA– SM/MM patients (Table 4). MIAA+ SM/MM patients presented more frequently with stage III MM (ISS score) (44.4% vs. 18.7%, P = 0.039). We then compared the 20 SM/MM patients with EBNA-1–specific mc IgG (EBNA-1+ patients) to the 96 SM/MM patients whose mc IgG did not react with any MIAA Ags (Table 4). At the time of diagnosis, 4 parameters distinguished SM/MM patients with EBNA-1–specific mc IgG: male sex (18 of 20 patients), significantly more plasma cells in bone marrow (33.5% vs. 17.0%, P = 0.039), higher β2-microglobulin (4.5 vs. 3.1 mg/l, P = 0.041), and reduced renal function (SCr: 93.0 vs. 81.0 μmol/l, P = 0.046).
Table 4. Characteristics of SM/MM patients with an mc IgG specific for an infectious pathogen of the MIAA assay.
Inflammation status of MGUS, SM, and MM patients.
Cytokines linked to inflammation, including interleukin 6 (IL-6), IL-10, and hepatocyte growth factor (HGF), have been reported as risk factors in MM (19–22). Other cytokines, notably IL-22, IL-26, and IL-33, are known to be important to mount adequate antimicrobial immune responses (23–26). We evaluated the levels of these molecules in the serum of 64 patients, to test whether pathogen-reactive neoplastic B cells are specifically associated with inflammation and infection. These patients represented different disease stages (34 MGUS, 4 SM, and 26 MM) and included MIAA+, EBNA-1+, and MIAA– cases. Results reported in Figure 12, A–C, show that HGF, IL-6, and IL-10 were significantly elevated in both MGUS and SM/MM, compared with controls (9 healthy donors). Infection-linked IL-22, IL-26, and IL-33 levels varied (Figure 12, D–G). Regarding myeloma, one should note that although increases were modest, MIAA+ patients had significantly higher levels of IL-22 than MIAA– patients, and EBNA-1+ patients had higher levels of IL-22 and IL-33 than EBNA-1– patients (Figure 12, D–F). In MGUS, IL-33 was significantly elevated for MIAA+ patients and EBNA-1+ patients, compared with healthy donors (Figure 12G).
Figure 12. Cytokine levels in serum of MGUS and SM/MM patients with infectious pathogen–specific or EBNA-1–specific mc IgG.

Six cytokines were quantified in serum using Luminex technology (Bio-Plex 200) with Bio-Plex Pro Human Cytokine Panel kits (Bio-Rad) in 3 different groups: healthy donors (HD, n = 9), monoclonal gammopathy of undetermined significance (MGUS) patients (n = 34), and a group of 4 smoldering myeloma (SM) patients and 26 multiple myeloma (MM) patients (designated as MM; n = 30). The MGUS and SM/MM groups were subdivided according to the pathogen specificity of monoclonal (mc) IgG: patients with mc IgG specific for one of the infectious pathogens from the multiplex infectious-antigen array (MIAA) assay are designated as MIAA+ patients; all others are designated as MIAA– patients. Patients with EBV nuclear antigen-1–specific (EBNA-1–specific) mc IgG are designated as EBNA-1+ patients; all others are designated as EBNA-1–. The cytokines measured were (A) HGF, (B) IL-6, (C) IL-10, (D) IL-22, (E) IL-26, and (F) IL-33. Cytokine levels were measured in serum from MIAA+/– and EBNA1+/– MM patients. (G) Serum IL-33 levels of MIAA+/– and EBNA1+/– MGUS patients. Note that the scales for IL-33 concentrations are different in F and G. For a given patient, results are means of technical duplicate measurements, expressed in pg/ml. Median values of groups of patients are shown as bars; means are indicated with the + symbol. Normal cytokine values in blood of healthy donors are as follows: HGF, 63–1,283 pg/ml; IL-6, < 9 pg/ml (in our group of HD, one individual had 165.5 pg/ml IL-6); IL-10, < 2 pg/ml; IL-22, 0 pg/ml; IL-26 and IL-33, normal values are not defined. *P < 0.05, **P < 0.01, and ***P < 0.001 by ANOVA.
Discussion
It is well established that the incidence of MGUS and MM increases with age (27). Thus, these conditions are a growing concern in aging populations worldwide. Although extensive genetic studies have been performed in MM, genetic evaluation does not predict disease evolution nor the effectiveness of therapy (28). Moreover, MGUS patients are often maintained on a watchful-waiting phase and there are presently no interventions in place to slow or halt disease progression to MM. Our study provides evidence that chronic Ag stimulation, here of infectious origin, underlies the initiation of MGUS and MM. We report that 6 infectious pathogens, including carcinogenic viruses (EBV, HCV, and HSV) and bacteria (H. pylori), are the candidate targets for approximately 23% of mc IgGs in MGUS, SM, and MM. This result suggests novel pathogenic mechanisms in these diseases, and offers new opportunities for improving patient diagnosis and treatment. Because only 9 infectious pathogens were tested with the present MIAA assay, the frequency of MGUS and MM patients with pathogen-specific mc IgG is likely greater than 23%. One objective of our future on-going studies is to identify additional infectious pathogens and Ags associated with MGUS and MM.
In our patient cohorts, the most frequent target of purified mc IgG was EBNA-1, which was found for 15.6% of patients. In this study, we were not able to study clonal plasma cells to search for evidence of EBV infection and eventual direct transformation. However, our previous study of herpes virus DNA in samples from 11 patients diagnosed with myeloma or plasma cell leukemia had found only 1 patient with a significantly elevated copy number of the EBV DNA polymerase in clonal plasma cells (29). The specificity of the mc IgG was not investigated for these patients. Further studies of the role of EBV in myeloma are warranted, but indirect cell transformation via chronic stimulation appears likely to be more frequent in myeloma than direct transformation following cell infection by EBV.
The PGRRPFF sequence, previously identified as a frequent EBNA-1 IgG epitope in the general population (17), was identified as the target of mc IgG from 2 EBNA-1–reactive cases. Further characterization of linear or conformational epitopes, recognized by mc IgG from the remaining group of patients with EBNA-1–specific mc IgG, will be required to determine if there are EBNA-1 epitope hotspots that are overrepresented in the BCR specificity of neoplastic B cells in MGUS and MM. One might speculate that epitopes identified outside of the EBNA-1 dimer interface would maximize its ability to serve as a bivalent ligand for the BCR and promote BCR aggregation, signaling, and internalization (30).
Remarkably, peptides incorporating the VPALR sequence in UL36, the tegument protein critical for HSV-1 capsid assembly, were recognized by purified mc IgG from all 7 patients with HSV-1–specific mc IgG. Thus, this sequence may represent an HSV-1 epitope hotspot that triggers abnormal BCR signaling during early-stage disease at the onset of MGUS. Although the limited structural information available regarding UL36 does not include the VPALR epitope, recent evidence indicates that dimerization may also be a feature of recombinant UL36 proteins (31).
Based upon the biological and clinical presentation, and increased levels of infection-linked cytokines (IL-22, IL-33) in blood, EBNA-1–associated MM may identify a distinct and possibly more severe form of the disease spectrum. Studies of larger cohorts are required to fully characterize EBNA-1–associated MM, and to determine whether these patients may carry specific genetic alterations. In particular, the high incidence of males in the EBNA-1–associated MM group might indicate hereditary, X-linked susceptibility to an abnormal immune response to EBV infection and EBV-associated MGUS and myeloma, as recently reported for certain cases of B cell lymphomas (32–34).
Overall, our findings imply that chronic stimulation by infectious Ag may promote MGUS and MM in certain patient subsets. Importantly, some of the identified infectious pathogens (HSV, HCV, H. pylori) can be effectively treated. This observation has obvious clinical consequences, since the detection of MGUS or SM patients with an mc IgG specific for a treatable pathogen would allow the proposal of antiviral or antibiotic treatment for patients. If an underlying chronic infection were cleared early enough in disease progression, it could perhaps offer the tantalizing possibility to prevent MGUS transition towards SM and overt MM for the first time. In such cases, addition of antiviral or antibiotic treatment to current protocols might indeed induce disease regression and/or improve response to standard treatments, as reported for interferon-γ treatment in HCV-associated MM (14). Similarly, eradication of H. pylori is known to result in tumor regression of H. pylori–associated malignancies (35).
A few aspects of this work merit further comment. First, based upon frequency, EBV infection may contribute to MM pathogenesis in subsets of patients (36). The main antigenic target, EBNA-1, is expressed in 3 types of latency observed in EBV-associated B cell malignancies; it is essential for EBV persistence and transcription (37–39). Existing drugs target the main signaling molecules or pathways altered by EBNA-1, notably p53/TP53, ubiquitin- and proteasome-dependent proteolysis, and the NF-κB pathway (40–42). Proteasome inhibitors that restore the presentation of EBNA-1–derived epitopes should be particularly useful in EBNA-1–associated MM, via the destruction of EBNA-1–expressing cells and subsequent reduction in EBNA-1 stimulation of clonal plasma cells (43–45). Proteasome inhibitors may also act directly on clonal plasma cells infected by EBV.
Second, other important antigenic drivers of MGUS and MM are currently being discovered. A recent report suggested that lyso-glucosylceramide 1 (LGL1) and lyso-phosphatidylcholine (LPC) may be the target of up to 30% of mc IgGs in plasma-cell proliferative disorders (46). Although mc IgGs were not purified in that intriguing study, the results are consistent with the notion that chronic Ag stimulation and an abnormal immune response may contribute to the initiation and/or progression of MGUS and myeloma.
In conclusion, infectious pathogens and Ag, and particularly EBNA-1, are frequent targets of purified mc IgGs in MGUS, SM, and MM. An abnormal immune response to infection, especially to EBV, may therefore be part of MM pathogenesis in subsets of patients. Future, larger studies shall determine whether EBNA-1–associated MM is a more severe form of the disease. Future studies aiming to identify the additional EBNA-1 epitopes that are targeted by purified mc IgGs, and the mechanisms that underlie the abnormal immune response to EBV in patients with clonal plasma cell disorders, will also provide valuable insights into the pathogenesis of these malignant diseases.
Methods
Patients.
We examined 244 patients presenting with mc IgG: 101 MGUS, 8 SM, and 135 MM diagnosed at the French University Hospitals (Centres Hospitaliers Universitaires, or CHUs) of Dijon, Nantes, Paris Saint-Antoine, and Tours, over the 2010–2016 period.
Purification of mc IgG.
After clotting, patient blood samples were centrifuged at 2,200 g (15 minutes, 4°C) and serum aliquots were stored at –80°C or –20°C, depending on collecting site. IgG concentration was measured with an immuno-nephelometric assay performed on a Beckman Immage Analyzer (Beckman Coulter). Purification of mc IgG and verification of purity was performed as previously described (12, 13, 15). Protein concentration was determined by using a Nanodrop Spectro-photometer ND-1000. Mass spectrometry was performed for 4 samples to validate purity, as previously described (16).
Analysis of mc IgG purity by mass spectrometry.
Purified mc IgGs were digested with trypsin and glycopeptides were isolated from peptides by using 2 methods, i.e., reversed-phase high-performance liquid chromatography and a protocol involving the commercial ProteoExtract Glycopeptide Enrichment Kit (EMD-Millipore) as indicated. All fractions were concentrated for analysis by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-ToF-MS) to confirm that all (glyco)peptides originated from human IgG. Analysis was performed by using an UltraFleXtreme operated in positive ion, reflective mode (16).
The MIAA assay.
The MIAA assay used in this study has been described previously (15). The assay allows testing for panels of commercially available Ag and/or lysates from EBV, HCV, CMV, HSV-1, HSV-2, VZV, H. pylori, T. gondii, and B. burgdorferi.
MIAA Ags and lysates.
Ags were purchased from Abcam, Advanced Biotechnologies Inc., and ImmunoDiag. Lysates were supplied by Advanced Biotechnologies Inc. and EastCoast Bio. For EBV, 2 Ags were used: VCA p23, full-length protein, EBV recombinant protein EBNA-1, and purified viral lysate. For CMV, a mix of 5 Ags was used: region 297–510 of CMV glycoprotein pp65; CMV pp28 (UL99) immunodominant region; CMV pp52 (UL44) immunodominant region; glycoprotein B immunodominant region; and CMV pp38 (UL80a) immunodominant region as well as a purified viral lysate. For T. gondii, we used 1 Ag, the p24 (GRA1) protein, and a purified tachyzoite lysate. For HCV, 3 Ags were used: core protein, composed of 119 residues (aa 1–119); NS-3 protein recombinant fragment subtype 1c (residues 1192–1459); and NS-4 recombinant protein, composed of 118 residues (aa 2–119). For H. pylori, we used 2 purified bacterial lysates (strain 49503) and recombinant proteins (urease and CagA) for immunoblotting. For HSV-1, we used glycoprotein G and a purified viral lysate (MacIntyre strain). For HSV-2, we used glycoprotein G and a purified viral lysate (G strain). For VZV, 2 Ags were used: envelop glycoprotein E, and ORF26 protein. For B. burgdorferi, we used a mix of 2 Ags: region 158–296 of B. burgdorferi p41 recombinant protein, and the VIsE full-length protein. Ags were diluted in phosphate-buffered saline (PBS) from 1 to 16 μM, and lysates were diluted from 10 to 400 μg/ml. To avoid aggregates, lysates were ultrasonicated prior to dilution in PBS.
Processing of the microarray slides and analysis of mc IgG Ag specificity by MIAA.
Preparation of the MIAA assay was performed as previously described (15). The arrays consisted of 8 × 8 matrices that included (a) 10 Ags: 2 for EBV, 3 for HCV, 1 for T. gondii, 1 for HSV-1, 1 for HSV-2, and 2 for VZV; (b) 7 lysates: EBV, CMV, HSV-1, HSV-2, T. gondii, and 2 for H. pylori; (c) 3 mixes: 1 of 5 CMV Ags, 1 of 2 B. burgdorferi Ags, and 1 of 2 H. pylori lysates; and (d) 2 negative controls: PBS and PBS with 0.1% bovine serum albumin (BSA). For hybridization, IgG concentrations were adjusted to 400 μg/ml for serum and from 50 to 200 μg/ml for purified mc IgGs. Samples (80 μl) were incubated for 2 hours at room temperature. Competition was performed by preincubation of the purified mc IgG with relevant or irrelevant peptides at different concentrations for 1 hour at room temperature. After washing, slides were incubated with a labeled secondary antibody (0.2 μg/ml DyLight 680–labeled goat anti–human IgG (H+L), from SeraCare; catalog 5230-0342). Fluorescence signal, detected with the Odyssey infrared imaging system scanner at 21-μm resolution (LI-COR Biosciences), was used to determine the serological status of each sample. Specific fluorescence was quantified by using the GenePix Pro 4 Microarray Acquisition & Analysis Software (Molecular Devices) (15).
MIAA controls and thresholds of specific positivity.
A first series of negative controls, not containing any antibody, were spotted on the MIAA assay: these controls were PBS, and PBS with BSA (15). A second series of negative controls were human serum samples that did not contain antibodies specific for 1 or several of the 9 pathogens, Ag, or lysates in the MIAA. The 2 categories of negative controls served to evaluate nonspecific binding and allowed us to determine the fluorescence threshold of specific positivity for each pathogen, Ag, or lysate of the MIAA. Note that a negative-control serum may be negative, for example, for antibodies specific for HSV-1, HSV-2, VZV, HCV, H. pylori, T. gondii, or B. burgdorferi but contain polyclonal Ig specific for EBV and polyclonal Ig specific for CMV. Then, human serum samples known to contain polyclonal Ig specific for 1 or more of the 9 infectious pathogens of the MIAA were used as positive controls. Altogether, 3 fluorescence thresholds of specific positivity were validated and used thereafter for all MIAA experiments: 500 for HCV, H. pylori, and T. gondii; 1,000 for CMV, HSV-1, and HSV-2; and 1,400 for EBV, VZV, and B. burgdorferi. Fluorescent signals below these thresholds were considered to be negative.
EBV and HSV-1 dot blotting assays.
For dot blotting assays, nitrocellulose membranes (Amersham) were spotted with recombinant EBNA-1 protein, with viral lysate for HSV-1 or with relevant and irrelevant EBV- or HSV-1–derived peptides, then dried. Saturation of the membranes was done by incubation in Tween-PBS-5% dry milk (TPM) for 2 hours. Membranes were incubated with the serum from the patient or purified mc IgG diluted in TPM (0.1 g/l) for 1 hour at room temperature. After washing, membranes were incubated for 1 hour with Peroxidase AffiniPure donkey anti–human IgG (H+L) (catalog 709-035-149, Jackson ImmunoResearch) at a 1:1,000 dilution. Revelation of Ag-antibody complexes was performed by chemiluminescence using a Thermo Fisher Scientific kit.
H. pylori immunoblotting assay.
The immunoblotting assay was performed by using the commercial kit Helico Blot 2.1 (MP Biomedicals). Helico Blot 2.1 consisted of a Western blot made from bacterial lysate of H. pylori strain ATCC 49503, as well as current infection marker (CIM) recombinant Ag. The test strip contained H. pylori Ags with molecular weights of 116 kDa (CagA), 89 kDa (VacA), 37 kDa, 35 kDa, 30 kDa (urease A), and 19.5 kDa as separate lines. CIM was located at the bottom of the strip as an independent band. The assay was done and interpreted according to the instructions of the manufacturers.
Antibody epitope-mapping with a phage display random peptide library.
The fUSE 5 phage display library used for this study is based upon 7 residues flanked by cysteines (CX7C) into the bacteriophage capsid protein pIII, with a combinatorial complexity of 109 unique sequences (18, 47–50). In brief, 109 transducing units of the library was precleared by incubation for 1 hour at 4°C with Protein A/G beads. The precleared library (supernatant) was added to fresh tubes containing 1 μg of purified mc IgG from HSV-1+ patient P56 immobilized on Protein A/G beads, followed by a 2 hours of incubation at 4°C. Samples were washed 3 times with PBS plus 0.05% Tween (PBS-Tween). Phage particles were eluted from beads with 0.1 M glycine, pH 3.0 for 2 minutes, neutralized with 1 M Tris–HCl (pH 9.0), and infected with E. coli strain K91. Serial dilutions of phage-infected K91 were plated onto Luria-Bertani (LB) agar plates containing kanamycin (100 μg/ml) and tetracycline (40 μg/ml), and grown overnight at 37°C. Phages were recovered from the bacterial media by PEG/NaCl precipitation for subsequent rounds of panning. After 4 rounds of selection, bacterial colonies were picked at random for PCR amplification and peptide-insert sequencing. DNA sequence analysis software was used to translate the sequences obtained.
Peptides.
EBNA-1–derived peptides (relevant, RRPPPGRRPFFHPVA; irrelevant, RRPPFGRRFFFHPVA) and HSV-1–derived peptides (relevant, EAVLHLSEDLGGVPALRQYVP; irrelevant, EAVLHLSEDLGGRPAERQYVP) were synthesized and purified at 98% by Covalab. Each soluble peptide was dissolved in 50 μl of DMSO, diluted in water, aliquoted, and stored at –20°C until use.
Cytokine quantification.
For 64 patients (34 MGUS, 4 SM, and 26 MM) and 9 healthy donors, frozen aliquots of serum were used to quantify 6 cytokines linked to inflammation or infection using Luminex technology (Bio-Plex 200) with Bio-Plex Pro Human Cytokine Panel kits (Bio-Rad), following the manufacturer’s instructions.
Statistics.
Patient parameters were expressed as medians and ranges, and/or means ± SD. The χ2 test was used for categorical variables, the Mann-Whitney test was performed for continuous variables, and ANOVA was used to compare peptide competition results and cytokine levels. A P value below 0.05 was considered statistically significant.
Study approval.
The study was promoted by the University Hospital of Nantes (RC12 0085) with the approval of the local ethics committee (Comité Consultatif de Protection des Personnes dans la Recherche Biomédicale (CCPRB), Nantes, France) and the Commission Nationale de l’Informatique et des Libertés (CNIL, Paris, France; CNIL 912335). Written informed consent was obtained from patients in the relevant clinical departments of the different CHUs, in the Biological Resource Center Ferdinand Cabanne BB-0033-00044 (Dijon, France), and in the blood bank for healthy volunteers (HV) enrolled by the Etablissement Français du Sang (EFS, Nantes, France). A convention has been signed between our laboratory (CRCINA, INSERM U1232) and the blood bank (EFS Pays de La Loire).
Author contributions
SH, EBC, AB, DF, MCV, RP, WA, and JH designed the research, analyzed data, and wrote the initial manuscript draft. CC, MCV, ADN, BSW, HP, PW, and JH performed experiments and edited the manuscript. AT, CR, DC, LG, PM, EP, and FG contributed patient samples and data. All authors gave final approval of the version to be submitted for publication and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the article are appropriately investigated and resolved.
Supplementary Material
Acknowledgments
We thank Tomasz Kula and Stephen J. Elledge (Department of Genetics, Harvard Medical School, Boston, Massachusetts, USA) for their help in selecting EBNA-1 epitope sequences and for useful discussions. We are grateful to Sophie Allain and Nicolas Mennesson (Nantes, France) for expert technical help, and to Marianne Coste-Burel (Nantes, France) for providing useful reagents. We thank all the colleagues from the Departments of Hematology or Internal Medicine of the University Hospitals of Dijon, Nantes, Paris Saint-Antoine, and Tours, who contributed to the diagnosis and care of patients in this study. We also thank Emy Komatsu and Luka Markovic (Winnipeg, Manitoba, Canada) for technical help with mass spectrometric measurements. We thank Helen Pickersgill (Life Science Editors) for professional English editing of the draft manuscript. We also thank the Biological Resource Center Ferdinand Cabanne BB-0033-00044 (Dijon, France).
The study was supported by grant number 2010-088 from Institut National du Cancer (INCa), to SH and EBC (2010–2012); by a grant from the Comités Départementaux of Loire-Atlantique, Maine et Loire, Vendée and Finistère from the Ligue Nationale contre le Cancer, to EBC (2013–2014); by a grant from the Cancéropôle Grand Ouest and Région Pays de la Loire, to SH (2015–2016); by a grant from the Cancéropôle Grand Ouest and Région Centre, to EP (2015–2016); by NIH P50GM085273 to BW; and awards from the Gillson-Longenbaugh Foundation to RP and WA. This work has been funded, in part, by a UNM Cancer Center Support Grant (P30 CA118100) to RP and WA. The INCa financed the salary of DF (2011-2012), the Cancéropôle Grand Ouest and Région Pays de la Loire financed the salary of AB (HII-GO project, 2015-2016) and the Academic Science Education and Research Training-Institutional Research and Academic Career Development Award (ASERT IRACDA, grant number NIGMS K12 GM088021) supported the salary of Alfreda Nelson.
Version 1. 10/05/2017
Electronic publication
Funding Statement
Number not provided by the funding Institution
Number not provided by the funding Institution
Number not provided by the funding Institution
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2017;2(19):e95367. https://doi.org/10.1172/jci.insight.95367.
Contributor Information
Adrien Bosseboeuf, Email: adrien.bosseboeuf@univ-nantes.fr.
Delphine Feron, Email: delphine.feron@gmail.com.
Anne Tallet, Email: anne.tallet@univ-tours.fr.
Cédric Rossi, Email: cedric.rossi66@gmail.com.
Cathy Charlier, Email: cathy.charlier@univ-nantes.fr.
Laurent Garderet, Email: laurent.garderet@sat.aphp.fr.
Denis Caillot, Email: denis.caillot@chu-dijon.fr.
Philippe Moreau, Email: philippe.moreau@chu-nantes.fr.
Marina Cardó-Vila, Email: mcardovila@salud.unm.edu.
Renata Pasqualini, Email: rpasqual@salud.unm.edu.
Wadih Arap, Email: warap@salud.unm.edu.
Hélène Perreault, Email: Helene.Perreault@umanitoba.ca.
Eric Piver, Email: piver_e@univ-tours.fr.
François Girodon, Email: francois.girodon@chu-dijon.fr.
Jean Harb, Email: jean.harb@univ-nantes.fr.
Edith Bigot-Corbel, Email: edith.bigot@univ-nantes.fr.
Sylvie Hermouet, Email: sylvie.hermouet@univ-nantes.fr.
References
- 1.Kyle RA. Monoclonal gammopathy of undetermined significance. Natural history in 241 cases. Am J Med. 1978;64(5):814–826. doi: 10.1016/0002-9343(78)90522-3. [DOI] [PubMed] [Google Scholar]
- 2.Dhodapkar MV. MGUS to myeloma: a mysterious gammopathy of underexplored significance. Blood. 2016;128(23):2599–2606. doi: 10.1182/blood-2016-09-692954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kyle RA, Greipp PR. Smoldering multiple myeloma. N Engl J Med. 1980;302(24):1347–1349. doi: 10.1056/NEJM198006123022405. [DOI] [PubMed] [Google Scholar]
- 4.Rajkumar SV, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–e548. doi: 10.1016/S1470-2045(14)70442-5. [DOI] [PubMed] [Google Scholar]
- 5.Drouet E, et al. Oligo-monoclonal immunoglobulins frequently develop during concurrent cytomegalovirus (CMV) and Epstein-Barr virus (EBV) infections in patients after renal transplantation. Clin Exp Immunol. 1999;118(3):465–472. doi: 10.1046/j.1365-2249.1999.01084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Babel N, Schwarzmann F, Pruss A, Volk HD, Reinke P. Monoclonal gammopathy of undetermined significance (MGUS) is associated with an increased frequency of Epstein-Barr Virus (EBV) latently infected B lymphocytes in long-term renal transplant patients. Transplant Proc. 2004;36(9):2679–2682. doi: 10.1016/j.transproceed.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 7.Rajkumar SV, Kyle RA, Plevak MF, Murray JA, Therneau TM. Helicobacter pylori infection and monoclonal gammopathy of undetermined significance. Br J Haematol. 2002;119(3):706–708. doi: 10.1046/j.1365-2141.2002.03912.x. [DOI] [PubMed] [Google Scholar]
- 8.Plummer M, de Martel C, Vignat J, Ferlay J, Bray F, Franceschi S. Global burden of cancers attributable to infections in 2012: a synthetic analysis. Lancet Glob Health. 2016;4(9):e609–e616. doi: 10.1016/S2214-109X(16)30143-7. [DOI] [PubMed] [Google Scholar]
- 9.Seifert M, Scholtysik R, Küppers R. Origin and pathogenesis of B cell lymphomas. Methods Mol Biol. 2013;971:1–25. doi: 10.1007/978-1-62703-269-8_1. [DOI] [PubMed] [Google Scholar]
- 10.Franceschi S, et al. Infection with hepatitis B and C viruses and risk of lymphoid malignancies in the European Prospective Investigation into Cancer and Nutrition (EPIC) Cancer Epidemiol Biomarkers Prev. 2011;20(1):208–214. doi: 10.1158/1055-9965.EPI-10-0889. [DOI] [PubMed] [Google Scholar]
- 11.Saha A, Robertson ES. Epstein-Barr virus-associated B-cell lymphomas: pathogenesis and clinical outcomes. Clin Cancer Res. 2011;17(10):3056–3063. doi: 10.1158/1078-0432.CCR-10-2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hermouet S, Corre I, Gassin M, Bigot-Corbel E, Sutton CA, Casey JW. Hepatitis C virus, human herpesvirus 8, and the development of plasma-cell leukemia. N Engl J Med. 2003;348(2):178–179. doi: 10.1056/NEJM200301093480219. [DOI] [PubMed] [Google Scholar]
- 13.Bigot-Corbel E, Gassin M, Corre I, Le Carrer D, Delaroche O, Hermouet S. Hepatitis C virus (HCV) infection, monoclonal immunoglobulin specific for HCV core protein, and plasma-cell malignancy. Blood. 2008;112(10):4357–4358. doi: 10.1182/blood-2008-07-167569. [DOI] [PubMed] [Google Scholar]
- 14.Panfilio S, et al. Regression of a case of multiple myeloma with antiviral treatment in a patient with chronic HCV infection. Leuk Res Rep. 2013;2(1):39–40. doi: 10.1016/j.lrr.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feron D, et al. Multiplexed infectious protein microarray immunoassay suitable for the study of the specificity of monoclonal immunoglobulins. Anal Biochem. 2013;433(2):202–209. doi: 10.1016/j.ab.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 16.Komatsu E, et al. Characterization of immunoglobulins through analysis of N-glycopeptides by MALDI-TOF MS. Methods. 2016;104:170–181. doi: 10.1016/j.ymeth.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Xu GJ, et al. Viral immunology. Comprehensive serological profiling of human populations using a synthetic human virome. Science. 2015;348(6239):aaa0698. doi: 10.1126/science.aaa0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mintz PJ, et al. Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat Biotechnol. 2003;21(1):57–63. doi: 10.1038/nbt774. [DOI] [PubMed] [Google Scholar]
- 19.Seidel C, Børset M, Turesson I, Abildgaard N, Sundan A, Waage A. Elevated serum concentrations of hepatocyte growth factor in patients with multiple myeloma. The Nordic Myeloma Study Group. Blood. 1998;91(3):806–812. [PubMed] [Google Scholar]
- 20.Iwasaki T, Hamano T, Ogata A, Hashimoto N, Kitano M, Kakishita E. Clinical significance of vascular endothelial growth factor and hepatocyte growth factor in multiple myeloma. Br J Haematol. 2002;116(4):796–802. doi: 10.1046/j.0007-1048.2002.03364.x. [DOI] [PubMed] [Google Scholar]
- 21.Jurczyszyn A, et al. HGF, sIL-6R and TGF-β1 play a significant role in the progression of multiple myeloma. J Cancer. 2014;5(7):518–524. doi: 10.7150/jca.9266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang XS, et al. Inflammatory markers and development of symptom burden in patients with multiple myeloma during autologous stem cell transplantation. Clin Cancer Res. 2014;20(5):1366–1374. doi: 10.1158/1078-0432.CCR-13-2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gimeno Brias S, Stack G, Stacey MA, Redwood AJ, Humphreys IR. The role of IL-22 in viral infections: paradigms and paradoxes. Front Immunol. 2016;7:211. doi: 10.3389/fimmu.2016.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12(5):383–390. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- 25.Stephen-Victor E, Fickenscher H, Bayry J. IL-26: an emerging proinflammatory member of the IL-10 cytokine family with multifaceted actions in antiviral, antimicrobial, and autoimmune responses. PLoS Pathog. 2016;12(6):e1005624. doi: 10.1371/journal.ppat.1005624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. 2016;16(11):676–689. doi: 10.1038/nri.2016.95. [DOI] [PubMed] [Google Scholar]
- 27.Therneau TM, et al. Incidence of monoclonal gammopathy of undetermined significance and estimation of duration before first clinical recognition. Mayo Clin Proc. 2012;87(11):1071–1079. doi: 10.1016/j.mayocp.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boise LH, Kaufman JL, Bahlis NJ, Lonial S, Lee KP. The Tao of myeloma. Blood. 2014;124(12):1873–1879. doi: 10.1182/blood-2014-05-578732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hermouet S, et al. Qualitative and quantitative analysis of human herpesviruses in chronic and acute B cell lymphocytic leukemia and in multiple myeloma. Leukemia. 2003;17(1):185–195. doi: 10.1038/sj.leu.2402748. [DOI] [PubMed] [Google Scholar]
- 30.Gauld SB, Dal Porto JM, Cambier JC. B cell antigen receptor signaling: roles in cell development and disease. Science. 2002;296(5573):1641–1642. doi: 10.1126/science.1071546. [DOI] [PubMed] [Google Scholar]
- 31.Scrima N, Lepault J, Boulard Y, Pasdeloup D, Bressanelli S, Roche S. Insights into herpesvirus tegument organization from structural analyses of the 970 central residues of HSV-1 UL36 protein. J Biol Chem. 2015;290(14):8820–8833. doi: 10.1074/jbc.M114.612838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rigaud S, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444(7115):110–114. doi: 10.1038/nature05257. [DOI] [PubMed] [Google Scholar]
- 33.Li FY, Chaigne-Delalande B, Su H, Uzel G, Matthews H, Lenardo MJ. XMEN disease: a new primary immunodeficiency affecting Mg2+ regulation of immunity against Epstein-Barr virus. Blood. 2014;123(14):2148–2152. doi: 10.1182/blood-2013-11-538686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Izawa K, et al. Inherited CD70 deficiency in humans reveals a critical role for the CD70-CD27 pathway in immunity to Epstein-Barr virus infection. J Exp Med. 2017;214(1):73–89. doi: 10.1084/jem.20160784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papagiannakis P, Michalopoulos C, Papalexi F, Dalampoura D, Diamantidis MD. The role of Helicobacter pylori infection in hematological disorders. Eur J Intern Med. 2013;24(8):685–690. doi: 10.1016/j.ejim.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 36.Mameli G, et al. Epstein-Barr virus infection is associated to patients with multiple myeloma and monoclonal gammopathy of undetermined significance. Leuk Lymphoma. 2017;58(2):466–469. doi: 10.1080/10428194.2016.1190976. [DOI] [PubMed] [Google Scholar]
- 37.Zucca E, Bertoni F. The spectrum of MALT lymphoma at different sites: biological and therapeutic relevance. Blood. 2016;127(17):2082–2092. doi: 10.1182/blood-2015-12-624304. [DOI] [PubMed] [Google Scholar]
- 38.Küppers R. B cells under influence: transformation of B cells by Epstein-Barr virus. Nat Rev Immunol. 2003;3(10):801–812. doi: 10.1038/nri1201. [DOI] [PubMed] [Google Scholar]
- 39.Kang MS, Kieff E. Epstein-Barr virus latent genes. Exp Mol Med. 2015;47:e131. doi: 10.1038/emm.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sivachandran N, Sarkari F, Frappier L. Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through disruption of PML nuclear bodies. PLoS Pathog. 2008;4(10):e1000170. doi: 10.1371/journal.ppat.1000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frappier L. Contributions of Epstein-Barr nuclear antigen 1 (EBNA1) to cell immortalization and survival. Viruses. 2012;4(9):1537–1547. doi: 10.3390/v4091537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tellam J, et al. Targeting of EBNA1 for rapid intracellular degradation overrides the inhibitory effects of the Gly-Ala repeat domain and restores CD8+ T cell recognition. J Biol Chem. 2001;276(36):33353–33360. doi: 10.1074/jbc.M104535200. [DOI] [PubMed] [Google Scholar]
- 43.Zou P, Kawada J, Pesnicak L, Cohen JI. Bortezomib induces apoptosis of Epstein-Barr virus (EBV)-transformed B cells and prolongs survival of mice inoculated with EBV-transformed B cells. J Virol. 2007;81(18):10029–10036. doi: 10.1128/JVI.02241-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Destro F, et al. Proteasome inhibitors induce the presentation of an Epstein-Barr virus nuclear antigen 1-derived cytotoxic T lymphocyte epitope in Burkitt’s lymphoma cells. Immunology. 2011;133(1):105–114. doi: 10.1111/j.1365-2567.2011.03416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hui KF, Lam BH, Ho DN, Tsao SW, Chiang AK. Bortezomib and SAHA synergistically induce ROS-driven caspase-dependent apoptosis of nasopharyngeal carcinoma and block replication of Epstein-Barr virus. Mol Cancer Ther. 2013;12(5):747–758. doi: 10.1158/1535-7163.MCT-12-0811. [DOI] [PubMed] [Google Scholar]
- 46.Nair S, Branagan AR, Liu J, Boddupalli CS, Mistry PK, Dhodapkar MV. Clonal immunoglobulin against lysolipids in the origin of myeloma. N Engl J Med. 2016;374(6):555–561. doi: 10.1056/NEJMoa1508808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vidal CI, et al. An HSP90-mimic peptide revealed by fingerprinting the pool of antibodies from ovarian cancer patients. Oncogene. 2004;23(55):8859–8867. doi: 10.1038/sj.onc.1208082. [DOI] [PubMed] [Google Scholar]
- 48.Jaalouk DE, et al. The original Pathologische Anatomie Leiden-Endothelium monoclonal antibody recognizes a vascular endothelial growth factor binding site within neuropilin-1. Cancer Res. 2007;67(20):9623–9629. doi: 10.1158/0008-5472.CAN-07-2737. [DOI] [PubMed] [Google Scholar]
- 49.Cardó-Vila M, et al. From combinatorial peptide selection to drug prototype (II): targeting the epidermal growth factor receptor pathway. Proc Natl Acad Sci U S A. 2010;107(11):5118–5123. doi: 10.1073/pnas.0915146107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mintz PJ, et al. Discovery and horizontal follow-up of an autoantibody signature in human prostate cancer. Proc Natl Acad Sci U S A. 2015;112(8):2515–2520. doi: 10.1073/pnas.1500097112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.