

Abstract
Background
Hydrogen sulfide (H2S) has anti-inflammatory and anti-hypertensive effects, and connexins (Cxs) are involved in regulation of immune homeostasis. In this study, we explored whether exogenous H2S prevents hypertensive inflammation by regulating Cxs expression of T lymphocytes in spontaneously hypertensive rats (SHR).
Material/Methods
We treated SHR with sodium hydrosulfide (NaHS) for 9 weeks. Vehicle-treated Wistar-Kyoto rats (WKYs) were used as a control. The arterial pressure was monitored by the tail-cuff method, and vascular function in basilar arteries was examined by pressure myography. Hematoxylin and eosin staining was used to show vascular remodeling and renal injury. The percentage of T cell subtypes in peripheral blood, surface expressions of Cx40/Cx43 on T cell subtypes, and serum cytokines level were determined by flow cytometry or ELISA. Expression of Cx40/Cx43 proteins in peripheral blood lymphocytes was analyzed by Western blot.
Results
Chronic NaHS treatment significantly attenuated blood pressure elevation, and inhibited inflammation of target organs, vascular remodeling, and renal injury in SHR. Exogenous NaHS also improved vascular function by attenuating KCl-stimulated vasoconstrictor response in basilar arteries of SHR. In addition, chronic NaHS administration significantly suppressed inflammation of peripheral blood in SHR, as evidenced by the decreased serum levels of IL-2, IL-6, and CD4/CD8 ratio and the increased IL-10 level and percentage of regulatory T cells. NaHS treatment decreased hypertension-induced Cx40/Cx43 expressions in T lymphocytes from SHR.
Conclusions
Our data demonstrate that H2S reduces hypertensive inflammation, at least partly due to regulation of T cell subsets balance by Cx40/Cx43 expressions inhibition.
MeSH Keywords: Connexins; Hydrogen Sulfide; Hypertension; Inflammation; Rats, Inbred SHR; T-Lymphocytes
Background
Experimental and clinical evidence collected in recent years has demonstrated participation of T lymphocytes in the development of hypertension [1,2]. These studies have also shown that moderate blood pressure (BP) elevation causes activation and proliferation of effector T cells [3]. Once activated, effector T cells (CD4+ and CD8+ T cells) infiltrate into the perivascular regions of blood vessels and kidney [3,4], and release various pro-inflammatory cytokines [4], which promote structural vascular remodeling and renal injury [5–8]. Furthermore, an abnormal alteration or dysfunction of regulatory CD4+CD25+ T lymphocytes (Tregs) is critical for the development of hypertensive inflammation [9]. Increasing evidence also suggests that the immunosuppressive agents targeted toward adaptive immunity successfully ameliorate hypertensive inflammation in some experimental models [10,11], but significant adverse effects of these immunosuppressant drugs may complicate their use in hypertension therapy [11].
Hydrogen sulfide (H2S) has been reported to have important regulatory roles in anti-inflammation and cardiovascular protection [12,13]. Although the role of H2S in the adaptive immune system has long been debated, H2S has been shown to serve as an important endogenous anti-inflammatory mediator in vascular inflammation [14,15]. Recent studies also showed that the H2S inhibited activation and proliferation of lymphocytes, and promoted Tregs differentiation [16–18]. On the other hand, H2S suppresses up-regulation of pro-inflammatory cytokines and up-regulates the expression of anti-inflammatory cytokines such as IL-10 [12,16]. This gaseous signaling molecule also has been proven to be a potential therapeutic agent in pro-inflammatory diseases, but the specific mechanisms by which H2S regulates immune homeostasis remain unclear.
More recently, an increasing number of studies have demonstrated that immune cells activated by inflammatory stimuli can use connexins (Cxs)-based channels to control the activation and proliferation of T lymphocytes and cytokines production by establishing gap junction channels (GJCs) and transferring immuno-relevant signals between T lymphocytes and other immune cells [19]. Of the 4 main Cx proteins in the adaptive immune system, Cx40 and Cx43 are the most important regulators of adaptive immune responses [20]. Data from our laboratory and others have shown that pro-inflammatory stimuli such as pro-inflammatory cytokines, LPS, and hypertension promote T cell proliferation and cytokines production by up-regulating pro-inflammatory Cx43 functional expression and gap junctional intercellular communication in T lymphocytes [19,21–24]. Thus, Cxs-based channels provide novel potential targets for the treatment of hypertensive inflammation.
The above findings demonstrate that H2S signaling and Cxs-mediated GJCs between lymphocytes have diametrically opposite effects during inflammatory response. However, it is not well known whether H2S protects against hypertensive inflammation by regulating Cxs expression on T lymphocytes. Thus, this study was designed to determine if exogenous H2S treatment can prevent hypertensive inflammation, and whether Cx40 and Cx43 on peripheral blood T lymphocytes are involved in this process. These goals were met by detecting vasoconstrictor function, histopathological alteration in target tissues, percentage of peripheral blood T cell subsets, serum levels of cytokines, and expression levels of Cxs in peripheral blood lymphocytes in SHR and WKY rats with and without NaHS treatment.
Material and Methods
Experimental animals and drug treatment
Male SHR and Wistar-Kyoto (WKY) rats (130–200 g body weight) were provided by Vital Beijing River Laboratory Animal Technology Co., Ltd (China). All rats were housed at constant temperature (20–22°C) and humidity (45–55%), with a 12-h light-dark cycle and had free access to standard rat chow and water. Following a 7-day acclimatization period, all rats were subjected to arterial pressure measurement using the tail-cuff method for a further 3 consecutive days to demonstrate the hypertension before starting the protocol. Only SHR exhibiting a blood pressure (BP) of 150 mmHg or above were used. SHR at 9 weeks of age were randomly divided into 2 groups (n=15 in each group): SHR and SHR+NaHS. SHR in the SHR+NaHS group were intraperitoneally injected with 56 μmol/kg−1·day−1 of NaHS (NaHS solution was freshly prepared in normal saline) (Cat. No. 161527; Sigma Aldrich, St. Louis, MO, USA) at the same time daily for 9 weeks. Equal numbers of male WKY rats with the same age and body weight served as normotensive controls (WKY group, n=15), and WKY rats were intraperitoneally injected with the same volume of normal saline once daily over the same time period. After treatment with drug, BP was measured by the tail-cuff method under minimal restraint. All live animal experiments performed in this study complied with Institutional Animal Care and Use Committees (IACUC) (No. A2046-047-02) of the Medical College of Shihezi University.
Measurement of systolic blood pressure
We used a non-invasive tail-cuff apparatus (Chengdu Taimeng Software CO. Ltd., Chengdu, China) without heating to measure the arterial pressure prior to the experiment or at the end of the drug treatment period, as described in a previous report with minor modification [24,25]. Rats had been handled on a regular basis over 3 weeks to reduce stress and were held in a plastic restrainer immediately prior to the BP measurement. The tail cuff with a pneumatic pulse transducer attached was placed around the tail of the rats. Rats were allowed to habituate to this procedure for 7 days before BP measurement. All of the animals were awake and quiet during the BP measurement. The averaged BP of each rat was determined from at least 3 consecutive readings.
Pressure myographic
Rat brains were rapidly removed and placed on a dissecting dish containing ice-cold Krebs’ solution (mmol/L: NaCl, 119; KCl, 4.7; MgSO4 1.2; CaCl2, 2.5; KH2PO4, 1.2; NaHCO3, 25; Glucose 11.1; ethylenediaminetetraacetic acid (EDTA) 0.26, pH 7.4, and gassed with 95% O2, 5% CO2). The basilar arteries (BA) from both sides were dissected, and surrounding connective tissues were removed. BA (400 μm in diameter) were dissected and cut into segments 2–3 mm long, and the 2 ends of arterial segments were cannulated between 2 glass micropipettes (1.2 mm in diameter; World Precision Instruments, LLC, Sarasota, FL, USA) in a microvascular chamber (Pressure Myograph System, DMT, Denmark), and secured using a 12-0 nylon monofilament suture. First, the air in both ends of the glass micropipettes was removed by injecting Krebs’ solution. After one end of the segment was secured on a cannula with 2 nylon sutures, the remaining blood in the vascular lumen was flushed through with Krebs’ solution using a perfusion device at a pressure difference <20 mmHg. The other end was then cannulated. Once the arterial segments were mounted, the arterial segments and the perfusion chamber were transferred to an inverted trinocular microscope equipped with an analog video camera and computer-assisted image capture system (Zeiss Axiovert 40 Microscope, Model 110P) to continuously record the outer diameter of basal cerebral arteries. The bath temperature was held constant at 37C and was continuously monitored with a thermal microprobe placed immediately adjacent to the mounted vessel. The chamber was superfused at 1.5 ml/min with warmed and aerated Krebs’ solution (pH 7.4, aerated with 95% O2 and 5% CO2) and maintained at 37°C. The arterial segments were pressurized with a stepwise increase in transmural pressure that was applied in 10-mmHg increments up to the appropriate working pressure (60 mmHg) with 5 min of equilibration at each pressure, as needed. All experiments were performed under conditions of zero intraluminal flow. The activities of arterial segments were measured by the high-potassium solution, then we washed it with Krebs’ solution for 3 times, and prepared for the experiment after 20 min. The negative-pressure aspirator was started and the Krebs’ solution in the bath was controlled to be 5 ml. First, the blood vessel reached the plateau stage under the Krebs’ solution environment, and then we added 40 mmol/L KCl solution to the bath to completely replace the bath solution. At this point, the vessel contracted and the diameter of the vessel entered the plateau phase (about 3–5 min). The diameter was continuously determined by a video dimension analyzer and recorded using Myoview software (DMT, Denmark). For KCl-induced vascular reactivity experiments, vasoconstriction effects were calculated using the following equation: [vasoconstriction effect=(DPSS–DKCl)/DPSS×100%]. DPSS, the constant vessel diameter in PSS; DPE, the constant vessel diameter following treatment with 40 mmol/L KCl.
Histological analysis
At the end of drug administration, the rats were euthanized by 30 mg/L pentobarbital sodium anesthesia (50 mg/kg, i.p.). The kidneys and BA were weighed and fixed in phosphate buffer (pH 7.4) containing 10% formalin. Formalin-fixed tissues were dehydrated and embedded in paraffin wax and cut into 5-μm-thick sections. Kidney and BA sections were deparaffinized in xylene and rehydrated in graded ethanol solutions (100, 95, 70, and 50% ethanol). The sections were stained with hematoxylin-eosin after washing. Following dehydration and differentiation in alcohol, the slides were photographed with light microscopy (Olympus BX50 microscope; Olympus, Tokyo, Japan). Histological evaluation of renal and vascular injury was performed at 10 different fields (100 or 200× magnification) per section.
Flow cytometry analysis
Whole blood (5 ml) from WKY and SHR rats was mixed using PBS with equal volume and subjected to isolation of the peripheral blood mononuclear cells (PBMCs) by using an isolation kit of mononuclear cells in rat (Cat. No. P8630; Solarbio Science & Technology, Beijing, China). We added 2 ml of FACS™ Lysing solution (Cat. No. 349202; BD Bioscience, NJ, USA) and the mixture was incubated for 10 min at room temperature, thereby removing any remaining RBCs in PBMCs. After PBS rinsing and centrifugation at 1000× g for 10 min, the cells were counted in a hemocytometer chamber and viability of PBMCs was assessed by trypan blue staining (the cell survival rate was determined to be >95%). Surface staining of T lymphocyte subtypes (at least 5×105 PBMCs; all anti-rat CD3, CD4, CD8, and CD25 monoclonal antibodies from Biolegend, Inc., San Diego, CA, USA) was performed according to our previous report [24]. Flow cytometry was used to count percentage of T cell subgroups. Flow cytometry was carried out on a FACSort™ flow cytometer (Becton Dickinson). Populations are expressed as percentage of the total lymphocyte population.
Detection of surface expression of Cxs on T lymphocyte subtypes was performed by flow cytometry as described previously [24]. Briefly, PBMCs were permeabilized using a Cytofix/Cytoperm Kit (BD Biosciences, San Jose, CA) and labeled with anti-Cx40 monoclonal antibody (Santa Cruz Biotechnology, USA) or anti-Cx43 antibody (Abcam, Cambridge, MA, USA), followed by FITC-labeled secondary antibody (Cat. No. 405305, Biolegend, Inc., San Diego, CA, USA). Finally, cells were incubated with anti-CD4 and anti-CD8 antibodies. Two-color immunofluorescence flow cytometry method was used to analyze Cx40/Cx43 expression on CD4+ and CD8+ T lymphocytes.
Serum cytokine measurements by ELISA
Peripheral blood (5 ml) from SHR and WKY rats was collected into plain heparin-coated tubes. The serum was obtained by centrifugation of blood at 800 g for 15 min at 4°C. Double-antibody sandwich enzyme-linked immunosorbent assay (ELISA) was used to measure the concentrations of cytokines (IL-2, IL-6, and IL-10) according to the manufacturer’s instructions of ELISA kits (Cat. No. E-EL-R0013 for IL-2; Elabscience Biotechnology Co., Ltd, Shanghai, China; Cat. No. ER003-96 for IL-6; Cat. No. ER004-96 for IL-10; ExCell Bio Co., Ltd, Shanghai, China). The level of each cytokine in serum was calculated according to the standard curve of each murine recombinant cytokine and expressed in pg/ml.
Western blot
Peripheral blood lymphocytes were lysed with ice-cold total protein lysis buffer (Cat. No. 78510; Pierce Biotechnology Inc., Rockford, IL, USA) containing phenylmethylsulphonyl fluoride (PMSF) for 30 min. Lysed lymphocytes were sonicated and centrifuged at 10 000× g for 20 min at 4°C. The supernatant was collected and the total protein concentration was determined with a BCA protein assay kit (Cat. No. GK5021; Generay Biotechnology, Shanghai, China). Equal amounts of protein (15 μg/lane) for each sample were loaded on a 10% SDS-PAGE gel and transferred to a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA) using a wet transfer apparatus (Bio-Rad Trans Blot SD; Bio-Rad Laboratories, Oxnard, CA, USA) at 80 V for 100 min. Membranes were blocked with 5% non-fat dry milk (Cat. No. 232100; BD, Franklin Lakes, NJ, USA) or 1% BSA (Cat. No. SW3015; Solarbio Science & Technology, Beijing, China) in Tris-buffered saline containing 0.05% Tween-20 (TBST) for 1 h at room temperature and then incubated overnight at 4°C with various anti-Cxs antibodies: anti-Cx43 polyclonal antibody (1: 500) (Cat. No.3512, Cell Signaling Technology, Massachusetts, U.S.A.), anti-Cx40 (1: 500) polyclonal antibody (Cat. No. SC-20466, Santa Cruz Biotechnology, USA), and anti-β-actin monoclonal antibody (Cat. No. TA-09) (1: 1000) (ZSGB. Inc., Beijing, China). The blots were then washed with TBST and incubated with secondary antibodies (Beijing Fir Jinqiao Biotechnology Company, Beijing, China) diluted 1: 10 000 for 1 h. Next, membranes were washed extensively with TBST. The protein bands of interest were visualized using an ECL kit (Cat. No. RPN2109; GE Healthcare Life Sciences, Chalfont, UK) in combination with X-ray film. Band intensity of target protein bands was analyzed using Quantity One software (Bio-Rad, Hercules, CA, USA).
Statistical analysis
All experimental data are shown as the mean ±SEM, and assessed by non-paired t test for the comparison of 2 groups or by one-way analysis of variance (ANOVA). Statistical analysis was performed using GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA, USA), and P<0.05 or P<0.01 (details described in the legend of the each figures) was considered to indicate statistically significant differences.
Results
Exogenous H2S treatment decreases blood pressure in SHR
It has been previously shown that sodium hydrosulfide (NaHS) treatment reduced blood pressure in SHR rats [8]. To verify this previous result, BP was measured by tail-cuff plethysmography in all rats at 18 weeks. BP was 52% higher in the SHR than in the vehicle-treated WKY rats (WKY vs. SHR: (106.70±7.32) vs. (169.70±5.99); P<0.01, Figure 1). However, after treatment with NaHS, BP significantly decreased in SHRs compared with vehicle-treated SHR (P<0.01, Figure 1), and there was no difference in BP between WKY rats and the SHR + NaHS group (WKY vs. SHR + NaHS: (106.70±7.32) vs. (114.00±3.64); P>0.05, Figure 1). The results suggest the role of NaHS in regulating blood pressure.
Figure 1.
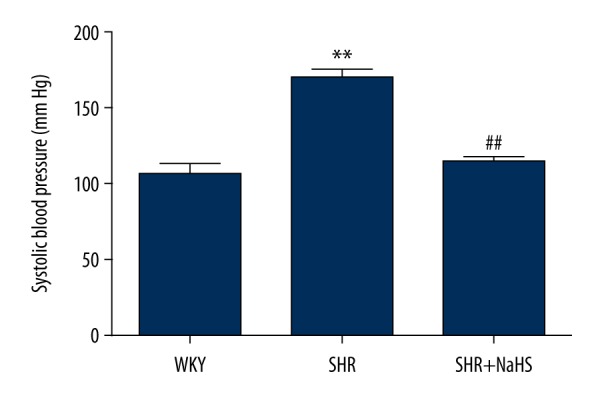
Effect of NaHS on spontaneous hypertension induced increase in blood pressure (BP) in SHR. We treated 9-week-old male SHR and WKY rats with NaHS (56 μmol/kg−1·day−1, i.p.) or the same volume of normal saline and continued every day. At 9 weeks after NaHS injection, systolic blood pressure in WKY, SHR, and NaHS+SHR was measured. SHR had significantly increased blood pressure compared to WKY (** P<0.01). Long-term administration of NaHS significantly reduced BP in the SHR+NaHS group (## P<0.01 vs. SHR). Data analyzed by comparing area under the curve values using one-way ANOVA and the t test, n=15 animals in each group.
Exogenous H2S prevents vascular remodeling and renal injury, and improves the vasomotor function of basilar arteries in SHR
To investigate pathological changes of target organs resulting from hypertension, and to assess the effect of exogenous H2S on renal injury and vascular remodeling (arterial wall thickening) induced by hypertension, the pathological features of BA and kidneys were observed by hematoxylin and eosin (H&E) staining. Figure 2 shows representative H&E staining results of cerebral arteries and the kidney from the WKY, SHR, and SHR+NaHS groups. Under light microscopy, the slices of cerebral arteries in SHR showed increased thickness of the medial wall and severe endothelium injury in BA with more inflammatory cell infiltration compared with that in the WKY rats (Figure 3). The renal tissue of WKY rats revealed normal morphology, and had no significant glomerulus atrophy or inflammatory cell infiltration (Figure 2). The kidney tissues of SHR rats showed severe renal injury marked by severe interstitial inflammatory cell infiltration, tubular dilation, and glomerulus deformation, as well as fibrosis (Figure 2). The renal tubules were also enlarged and infiltrated with inflammatory cells (Figure 2). These results indicate that the SHR exhibited the typical pathological features associated with hypertension, which were consistent with previous studies. However, long-term treatment with exogenous H2S significantly ameliorated arterial thickening, endothelium injury, and the infiltration of inflammatory cells in cerebral arteries and renal tissues compared to SHR (Figure 2).
Figure 2.

Long-term NaHS treatment alleviates vascular remodeling and infiltration of inflammatory cells in BA and kidney tissues of SHR. We treated 9-week-old male SHR and WKY rats with NaHS (56 μmol/kg−1·day−1, i.p.) or the same volume of normal saline and continued every day. At 9 weeks after NaHS injection, kidneys and basal cerebral arteries were harvested and were stained by hematoxylin-eosin. A, cross-sections of BA stained with hematoxylin-eosin staining (magnification×200. scalar bar=4.5 μm). B, longitudinal sections of kidney tissues stained with hematoxylin-eosin staining (magnification×100, scalar bar=4.5 μm) (n=15).
Figure 3.

The inhibitory effect of NaHS on the contractile response in basilar arteries (BA) induced by 40 mM KCl. We treated 9-week-old male SHR and WKY rats with NaHS (56 μmol/kg−1·day−1, i.p.) or the same volume of normal saline and continued every day. At 9 weeks after NaHS injection, BA were isolated and their contractile response were determined as described in “Materials and methods”. Contraction of BA in response to KCl was greater in SHR than in WKY rats (* P<0.05). Long-term NaHS treatment suppressed vascular contraction of cerebral arteries from SHR in response to KCl (# P<0.05 vs. SHR). All data-points are from 7 BA. Results are means ±SEM of 4–6 experiments.
Vascular structural alterations lead to the impairment of cerebral vasodilation [26], and cerebrovascular remodeling during chronic hypertension is an important determinant of cardiovascular and ischemic cerebrovascular diseases [26,27]. Hence, to further test whether long-term H2S treatment improves vascular function or vasoconstrictor responses of peripheral vessels to KCl, basilar arteries (BA) of SHR and WKY rats were stimulated with KCl (40 mmol/L). The representative images in Figure 3 show changes in the diameter of segments (~400 μm in outer diameter) isolated from rat basal cerebral arteries during the establishment of myogenic tone. Myographic measurement showed that the vasoconstrictor responses of BA to KCl were greater in SHR than that of WKY rats (P<0.05, Figure 3). NaHS treatment suppressed vascular contraction of BA from SHR in response to KCl (P<0.05, Figure 3). These findings are consistent with previous studies on the vasorelaxant effect of H2S.
Exogenous H2S alleviates the disorder of peripheral blood T lymphocyte subsets and hypertensive inflammation in SHR
Exogenous H2S is known to exert anti-inflammatory effects both in immune cells and in animal models with inflammatory diseases [16,17,28,29]. In order to characterize T cell profiles in peripheral blood of SHR and to evaluate the effects of NaHS on hypertensive inflammatory response of SHR, we separated monocytes from peripheral blood and analyzed the level of total T lymphocytes (CD3+ T cells) and T cell subsets (CD4+, CD8+ and CD4+CD25+ T cells) by flow cytometry in vehicle control WKY and SHR rats, as well as SHR with long-term NaHS administration. Representative flow cytometry images and bar graph indicating percentage of T cell subsets are shown in Figure 4A. Lymphocytes were first gated based on their FSC/SSC profile, followed by analysis of CD4, CD8, and CD25 expression in a gated population of CD3+ or CD4+ T cells (Figure 4A). A significantly higher percentage of CD3+CD4+ T cells [WKY vs. SHR: (65.85±1.02)% vs. (70.36±0.91)%; P<0.01, Figure 4B] was noted in SHR than those in WKY rats, whereas the frequencies of CD3+CD8+ T cells [WKY vs. SHR: (37.13±0.95)% vs. (32.59±0.85)%; P<0.01, Figure 4B] were significantly lower in the peripheral blood of SHR as compared with WKY rats, which led to an increased CD4/CD8 ratio in SHR [WKY vs. SHR: 1.81±0.07 vs. 2.21±0.07; P<0.01, Figure 4B]. In the meantime, the frequencies of CD4+CD25+ T cells [WKY vs. SHR: (9.88±0.68)% vs. (6.13±0.30)%; P<0.01, Figure 4B] were significantly lower in the peripheral blood of SHR as compared with WKY rats. However, in SHR treated with NaHS, we observed significant reduction of CD4+/CD8+ ratio compared to SHR with vehicle administration (SHR vs. SHR + NaHS: (2.21±0.07) vs. (1.92±0.05); P<0.05, Figure 4B). In particular, long-term NaHS administration in the SHR produced a marked reversal of CD4+CD25+ T cells when compared with that of SHR with vehicle administration (SHR vs. SHR + NaHS: (6.13±0.30)% vs. (8.02±0.11)%; P<0.05, Figure 4B).
Figure 4.

Long-term NaHS treatment reversed the changes of different T lymphocyte subtypes in SHR. We treated 9-week-old male SHR and WKY rats with NaHS (56 μmol/kg−1·day−1, i.p.) or the same volume of normal saline and continued every day. At 9 weeks after NaHS injection, PBMCs were harvested and subjected to flow cytometry analysis. Reported dot plots are generated gating on living PBMCs in the scatter (FSC vs. SSC) dot plot (not shown). A, Representative flow cytometry analysis showing percentages of circulating T lymphocytes subtypes in the peripheral blood of 15 SHR and 15 age-matched WKY rats. B, Bar graph shown are proportion of CD3+, CD4+, CD8+ and CD25+ T cells expressing CD4+ as well as the ratio of CD4+/CD8+ in the peripheral blood of SHR and WKY rats. The vertical axis represents the frequency of various T lymphocyte subtypes. Quantitative analysis of the mean percentage of cells ±SEM. ** P<0.01, compared with the WKY rats; # P<0.05, compared with SHR (n=15 animals in each group).
To further study the effect of H2S on pro-inflammatory and anti-inflammatory cytokines production during hypertension, we used ELISA to examine serum levels of IL-2, IL-6, and IL-10 in the peripheral blood from SHR in the absence or presence of NaHS. As shown in Figure 5, compared with WKY rats, the IL-2 (WKY vs. SHR: (3.58±0.37) pg/ml vs. (5.94±0.92) pg/ml; P<0.05, Figure 5A) and IL-6 (WKY vs. SHR: (8.44±0.65) pg/ml vs. (12.88±1.01) pg/ml; P<0.01, Figure 5B) serum levels were significantly elevated in SHR, and serum levels of anti-inflammatory cytokine (IL-10) in SHR were significantly reduced (WKY vs. SHR: (7.44±0.44) pg/ml vs. (3.75±0.36) pg/ml; P<0.01, Figure 5C). However, long-term administration of NaHS resulted in significantly lower levels of pro-inflammatory cytokines (IL-2 and IL-6) in the SHR + NaHS group than in SHR (P<0.01, Figure 5A, 5B). Importantly, in the SHR + NaHS group, the serum levels of IL-10 were markedly increased compared with those in SHR (SHR vs. SHR + NaHS: (3.75±0.36) pg/ml vs. (5.79±0.36) pg/ml; P<0.01, Figure 5C). Thus, the increased anti-inflammatory cytokine levels and decreased pro-inflammatory cytokines levels imply that H2S attenuates hypertensive pro-inflammatory response in peripheral blood.
Figure 5.

Effect of long-term NaHS treatment on the production of pro-inflammatory and anti-inflammatory cytokines in serum of SHR. We treated 9-week-old male SHR and WKY rats with NaHS (56 μmol/kg−1·day−1, i.p.) or the same volume of normal saline and continued every day. At 9 weeks after NaHS injection, serum level of IL-2 (A), IL-6 (B) and IL-10 (C) were determined as described in “Materials and methods”. Data represented as total amount of cytokine produced in pg/ml in peripheral blood; the results shown are the mean ±SEM; * P<0.05 and ** P<0.01, compared with the WKY rats; ## P<0.01, compared with SHR (n=15 animals in each group).
Exogenous H2S reduced the surface expression of Cxs in CD4+ and CD8+ T lymphocytes from peripheral blood of SHR
Cx40 and Cx43 are widely expressed on different T lymphocytes subsets, and are involved in an intercellular signaling pathway that regulates T cells proliferation and activation, and of pro-inflammatory cytokines production during hypertension [24]. Our previous study found that Cx40 and Cx43 exhibited higher expression on CD4+ and CD8+ T lymphocytes subsets from hypertensive patients compared with healthy controls [24]. Therefore, the expression of Cx40 and Cx43 on the surface of different T cells was investigated in the peripheral blood of SHR and WKY (Figure 6A, 6B). In addition, we also investigated the effect of exogenous H2S on expression of these 2 Cxs on surface of CD4+ and CD8+ T cells of SHR (Figure 6A, 6B). There were significantly increased expression levels of Cx40 in various T lymphocyte subtypes from the peripheral blood of SHR – (7.48±3.45)% for CD4+ T cells and (7.60±3.16)% for CD8+ T cells (P<0.05, Figure 6B) – compared with that from the peripheral blood of WKY rats – (2.58±0.52)% for CD4+ T cells and (1.94±0.49)% for CD8+ T cells. Surface expression of Cx43 was remarkably increased in various T lymphocyte subtypes of SHR – (0.51±0.11)% for CD4+ T cells and (0.41±0.20)% for CD8+ T cells – compared with that of WKY rats – (0.29±0.03)% for CD4+ T cells and (0.10±0.01)% for CD8+ T cells) (P<0.05, Figure 6B). However, long-term treatment with exogenous H2S reversed the elevated expressions of Cx40 and Cx43 in CD4+ – (2.88±0.27)% for Cx40 and (0.08±0.03)% for Cx43) and CD8+ T cells ((1.72±0.40) for Cx40 and (0.08±0.01)% for Cx43) from the peripheral blood of SHR, and their expressions in SHR returned to the levels seen in WKY rats (Figure 6).
Figure 6.

Effect of long-term NaHS treatment on surface expressions of Cx40 and Cx43 in different T lymphocyte subtypes of SHR. A, Representative flow cytometry plots are presented for Cx40 and Cx43 expression levels on gated single-positive CD4+ T lymphocytes or CD8+ T lymphocyte populations in the peripheral blood from 15 SHR and 15 WKY rats. Fresh, resting PBMCs from SHR and WKY rats underwent surface staining with antibodies against CD3, CD4, and CD8 molecules. After surface staining, the cells were fixed, permeabilized, and stained with unlabeled anti-Cx40 or anti-Cx43 plus FITC-labeled secondary antibodies. Based on the CD4+ or CD8+ gate, the cells were further gated based on Cx40 and Cx43 expression levels, and the frequency of CD4+ or CD8+ T cells expressing Cx40 and Cx43 was determined. B, Bar graph shown are the percentage of CD4+ or CD8+ T cell population expressing Cx40 and Cx43. Both Cx40 and Cx43 expression levels are significantly increased in CD4+ or CD8+ T cells of SHR compared with those of WKY rats. Long-term NaHS treatment inhibited the expressions of Cx40 and Cx43 in CD4+ and CD8+ T cells from the peripheral blood of SHR, and their expressions in SHR returned to the levels seen in WKY rats. Values are mean ± SEM. * P<0.05 and ** P<0.01, compared with WKY rats; # P<0.01, compared with SHR rats (n=15 animals in each group).
Effects of H2S on Cxs protein expression in peripheral blood lymphocytes of SHR
Western blotting was used to further observe the effects of NaHS on the protein levels of Cx40 and Cx43 in peripheral blood lymphocytes during hypertension. The results showed that lymphocytes from SHR expressed higher levels of Cx40 and Cx43 than those from WKY rats (P<0.01, Figure 7A, 7B). NaHS markedly decreased the overexpression of Cx40 and Cx43 proteins induced by hypertension in peripheral blood lymphocytes of SHR (P<0.01, Figure 7A, 7B), while their expressions in the SHR+NaHS group were higher than those from WKY rats. Therefore, these results are consistent with observations in different T cells subsets from SHR that express lower Cxs in the presence of NaHS.
Figure 7.

Effect of treatment with NaHS on protein levels of Cx40 and Cx43 in peripheral blood lymphocytes of SHR. We treated 9-week-old male SHR and WKY rats with NaHS (56 μmol/kg−1·day−1, i.p.) or the same volume of normal saline and continued every day. At 9 weeks after NaHS injection, peripheral blood lymphocytes were harvested and examined by Western blot for the expression levels of Cx40 (A) and Cx43 (B). After densitometric analysis, the data were expressed as ratios of Cxs to β-actin. The data represent the mean ±SEM of 3 experiments (n=15 animals in each group). ** P<0.01 vs. WKY rats; ## P<0.01 vs. SHR.
Discussion
The present study investigated the possible therapeutic effect of hydrogen sulfide on hypertenion mediated inflammation. This was done by studying the effects of supplementation of exogenous H2S on blood pressure, target organs, and adaptive immune system in spontaneously hypertensive rats. The major findings of the present study were that the applied exogenous H2S in the form of NaHS reduced the blood pressure, significantly attenuated vascular remodeling and inflammatory injury in kidneys and BA, and improved vasoconstriction and immune homeostasis in SHR. This study supports previous reports of anti-hypertensive and anti-inflammatory effects of H2S and extends the repertoire of H2S to specifically include immune homeostasis regulation in hypertension.
Morphological alterations of cerebral arteries during hypertension is a major target of the hypertensive effects on the brain [27,30]. These structural changes lead to the damage of cerebral vasodilation [26]. Although increased resistance of cerebral arteries may reduce cerebral blood flow during excessive BP elevation, morphological plus functional changes of cerebral arteries during chronic BP elevation play an essential role in the development of ischemic cerebrovascular diseases [27]. Therefore, basilar arteries were used as the primary model of blood vessels in our study. Our results showed significantly higher tail blood pressure, as well as vascular wall thickening accompanied by enhanced vasoconstrictor response in BA of SHR compared to WKY rats, and this is consistent with previous reports [31–33]. Infiltration of innate and adaptive immune cells in the kidneys, arterial wall, and perivascular regions, together with elevated cytokine release, play a key role in the initiation and progression of hypertension, and are a consistent feature in experimental and clinical studies of hypertension [34]. The results of the present study show increased infiltration of immune cells and damage in BA and renal tissues of SHR. These changes of vascular morphology induced by inflammation increase vascular tone and impair arterial relaxation, and thus lead to BP elevation [35]. A growing body of evidence supports the critical role of T lymphocytes in hypertension, and CD4+/CD8+ T cells are the major lymphocyte subpopulation involved in BP control in experimental models of hypertension and in hypertensive patients [4,36].
Aberrant activation of adaptive immune cells, in combination with imbalance of effector and regulatory T cell subsets, cause low-grade inflammation and lead to BP elevation and target organ injury [34]. Indeed, several studies from male hypertensive models (Ang II, DOCA-salt, and SHR) and hypertensive patients found significantly increased vascular and renal infiltration of greater numbers of CD4+/CD8+ T cells and increased the ratio of CD4+/CD8+ [4], and adoptive transfer of CD4+ T cells from pre-eclampsia rats to pregnant rats resulted in a significant BP elevation [37]. Adoptive transfer of CD8+ T cells into Rag1-deficient mice (CD8-deficient mice) also recovered a normal BP increase during Ang II administration [38]. In our study, we also found that increasing CD4+ T cells accumulation and ratio of CD4+/CD8+ occurred in the peripheral blood of SHR, whereas the percentages of CD8+ T cells in the peripheral blood of SHR were reduced. The decrease in the number of activated CD8+ T cells may result from increased CD8+ T cells infiltration into other target organs, but this represents general immunological dysregulation in hypertensive rats, although the causes are not entirely clear from this study. We also observed that CD4+CD25+ T cells were markedly diminished in peripheral blood of the SHR, suggesting there are fewer Tregs present in hypertensive inflammation. On the other hand, taken together with previous studies, the present findings suggest that irregularities of Tregs and effector T lymphocytes contribute to the development of hypertensive inflammation. In addition, our data reveals the elevation of pro-inflammatory cytokines (IL-2 and IL-4) accompanied by a reduction of IL-10 production in serum of SHR. This is consistent with previous reports that several plasma pro-inflammatory cytokines (IL-2, IL-4, IL-6, TNF-α and IFN-γ) are increased in many hypertensive models and in hypertensive patients [1,39]. Among these pro-inflammatory cytokines, IL-6 is fundamental for the development of stress-induced hypertension, while IL-6 deficiency attenuated hypertension caused by high salt and Ang II infusion [40,41]. Above all, these findings, together with our results, provide reasonably definitive evidence that T lymphocytes and cytokines produced by effector T lymphocytes contribute to blood pressure elevation.
H2S is now recognized as a gas with important roles in the cardiovascular system [13]. The association between H2S deficiency and an increase in BP was first investigated in a study on spontaneously hypertensive rats [31]. In animal models with hypertension (SHR and renovascular hypertension), plasma level of H2S and the expression of CSE (the synthase for H2S synthesis) mRNA were significantly lowered in SHR, while exogenous H2S has been shown to decrease BP [31,42]. In exploring the therapeutic effect in SHR after exogenous H2S donor administration for 9 weeks, we observed that long-term NaHS treatment in SHR lowered blood pressure as evidenced by large decreases in vascular remodeling (arterial wall thickening) and improved vascular function, and this is consistent with previous reports [8,13,43]. In the vasculature, H2S is involved in the regulation of vascular tone and vascular remodeling, and is a known vasorelaxant with effects in both larger arteries and arterioles [44,45]. In our study, we also observed that long-term treatment with NaHS restores vascular function of BA to the same level as in the WKY rats. The molecular mechanism of functional improvements in arteries induced by H2S is complicated, with roles for ion (K+, Ca2+ and Cl−) channels, inhibition of proliferation, migration and calcification of vascular smooth muscle cells (VSMCs), and induction of VSMCs apoptosis all being implicated [46,47]. However, this is possibly not the only reason why NaHS reduces vascular tone and blood pressure in the SHR.
Another key aspect of the biology of H2S is its anti-inflammatory effects. Although there are contradictory reports on the roles of H2S in various inflammation processes, to date, all atherosclerotic studies support the anti-inflammatory properties of H2S [15]. Our results also demonstrate that H2S donors can reduce leukocyte infiltration in vascular wall and kidneys [48]. A previous study showed that endogenous H2S drives Tregs differentiation and maintains immune homeostasis in H2S-deficient mice peripheral blood lymphocytes [18]. Mirandola et al. also demonstrated that NaHS induced functional inhibition and cell death of cytotoxic lymphocytes subsets in peripheral blood lymphocytes [16]. However, it is uncertain whether H2S prevents hypertension mediated imbalance of the immune system, and the mechanisms by which H2S regulates hypertensive inflammation are unclear. In comparison with studies from others, an important finding from the present study is that long-term treatment with exogenous H2S significantly attenuates the imbalance between effector and regulatory T cell subsets in SHR by decreasing serum levels of IL-2, IL-6, and CD4/CD8 ratio, and enhancing the level of anti-inflammatory cytokine IL-10 and percentage of Tregs, whereas NaHS administration had no effect on the proportion of CD4+ and CD8+ T cell of peripheral blood in SHR. Tregs suppress cellular immune responses via the anti-inflammatory effects of IL-10 [5]. Several experimental studies demonstrated that adoptive transfer of Tregs reduced vascular inflammation, improved endothelial vasodilatation, and reduced BP elevation in aldosterone- and Ang II-infused male animals [49–51]. Accordingly, we showed that increased Tregs and IL-10 in SHR after exogenous H2S treatment ameliorated vascular dysfunction and counteracted BP elevation and associated kidney and vascular damage by limiting the activation of circulating effector T cells and the expression or production of pro-inflammatory mediators (TNF-α, IL-1β, IL-2, and IL-6).
Our results were obtained from daily intraperitoneally injection of NaHS, but recent studies have reported NaHS forms H2S too rapidly (within seconds) when dissolved [52], and produces only transient (10–30 min) hypotensive effects [53]. Obviously, the brief effect of NaHS is not enough to fully explain the long-term anti-hypertensive and anti-inflammatory effects of NaHS. However, the current literature has shown that H2S can react with methemoglobin to form sulfhemoglobin, which acts as an H2S sink [54], or can be stored in proteins where it may be released upon reduction or under several physiological stimuli [46], and thus lead to a long-term regulatory effect on BP and inflammation. Thus, it is supposed that the administered NaHS may causes an acute effect and then has various longer-term effects. Another possibility regarding this effect is that H2S can enhance activities of specific proteins (i.e., KATP channels, endothelial nitric oxide synthase, and p65 subunit of NF-κB) through protein sulfhydration [46,55]. In addition, the other major mechanisms associated with longer-term anti-hypertensive and anti-inflammatory effects of H2S include the following: (a) NaHS can reverse hypertension induced vascular remodeling by suppressing VSMCs proliferation and collagen formation in the SHR [8,56]; (b) NaHS can also reduce vascular ROS generation and ameliorate oxidative stress by different mechanisms [57–59]; (c) In the case of anti-inflammatory effect, NaHS can lead to significant reduction of pro-inflammatory cytokines, chemokines, and inhibitory effects of monocyte adherence to the vascular endothelium and infiltration of leukocytes to sites of injury/inflammation by modulating phosphatidylinositol-3,4,5-trisphosphate (PIP3)/AMP-activated protein kinase (AMPK)/peroxisome proliferator-activated receptor-γ (PPAR-γ) signaling, NF-κB signaling, and chromatin remodeling of pro-inflammatory cytokine genes [60–62]. Above all, it is possible that NaHS treatment has a longer-lasting effect.
The re-establishment of immune homeostasis under chronic inflammation (e.g., hypertension and diabetes) depends on precise coordination between anti-inflammatory and pro-inflammatory immune cell by Cxs-based channels (GJCs or HCs) that establish direct intercellular or extracellular signaling mechanisms leading to cell-cell or cell-extracellular matrix interaction [19,20]. Growing evidence indicates that Cxs-based channels play an indispensable role in controlling activation, proliferation, and terminal differentiation of T lymphocytes and cytokines production [63]. Cx43 is a positive regulator during pro-inflammatory responses, such as LPS or PHA-induced inflammatory response [21]. Other data from our lab show that increasing Cx40 and Cx43 expression is positively correlated with T lymphocyte proliferation and pro-inflammatory cytokines synthesis in peripheral blood of hypertensive patients and in splenic lymphocytes of SHR [24,64]. Similarly, we also observed an increased Cx40 and Cx43 expression in T cells of peripheral blood in SHR compared with WKY rats. To further explore whether H2S inhibits hypertensive inflammation by affecting surface expression of Cxs on peripheral blood lymphocytes, we examined the effect of NaHS on Cx40 and Cx43 expression on different T cell subsets. We have demonstrated for the first time that NaHS markedly suppressed the protein levels of Cxs in CD4+ and CD8+ T cells, as well as in total peripheral blood lymphocytes of SHR, and this may result in loss of Cx40/Cx43-based channels and consequent spatial remodeling of gap junctions. Our data straightforwardly demonstrate that H2S exerts its anti-inflammatory effect in hypertensive inflammation via down-regulating pro-inflammatory Cxs expression.
The present study has certain limitations. First, the SHR model is analogous to human primary hypertension, and all experimental models of hypertension have an imbalance of different T cell subsets during hypertension, causing low-grade inflammation and target organ damage [4]. However, hypertension is always accompanied with various complications, and the impacts of H2S on anti-inflammatory response are complex. A key question is whether the therapeutic effects of H2S on inflammation induced by the cardiovascular disease can be repeated in other hypertensive or cardiovascular disease models, which has yet to be further explored. Secondly, in our experiments, we did not detect the effect of H2S on hypertension-induced morphological and functional changes in main peripheral resistance arteries, although several other studies have also reported that NaHS can regulate the normal structure and vasorelaxation in resistance arteries [65,66]. Thirdly, we only observed the anti-inflammatory effects of H2S on hypertensive inflammation and did not investigate the detailed anti-inflammatory mechanism of H2S by regulating the function of Cxs-based channels (gap junction channels or HCs) in the context of hypertensive inflammation, which will be the future research direction of our lab. Lastly, how H2S decreases the expressions of Cxs also needs to be better defined.
Conclusions
Taken together, we provide evidence for the first time that exogenous H2S ameliorates BP elevation, hypertensive inflammation, and inflammation-induced target organ damage via inhibiting the cellular cytotoxic response of peripheral blood lymphocytes, by increasing the number of Tregs and the serum level of IL-10, and by decreasing production of pro-inflammatory cytokines. The mechanisms may be at least partially related to H2S in its modulation of Cxs expression. These results provide the possibility for the use of H2S donor drugs in the treatment of hypertension.
Footnotes
Conflict of interest
None.
Source of support: This work was supported by grants from the National Natural Science Foundation of China (No. 81660271 to Ke-tao Ma, 81460098 to Xin-zhi Li, 81560081 to Jun-qiang Si and 81600325 to Liang Zhang) and the International Cooperation Project of Shihezi University (No. GJHZ201603 to Ke-tao Ma)
References
- 1.Schiffrin EL. Immune mechanisms in hypertension and vascular injury. Clin Sci (Lond) 2014;126(4):267–74. doi: 10.1042/CS20130407. [DOI] [PubMed] [Google Scholar]
- 2.Leibowitz A, Schiffrin EL. Immune mechanisms in hypertension. Curr Hypertens Rep. 2011;13(6):465–72. doi: 10.1007/s11906-011-0224-9. [DOI] [PubMed] [Google Scholar]
- 3.Marvar PJ, Vinh A, Thabet S, et al. T lymphocytes and vascular inflammation contribute to stress-dependent hypertension. Biol Psychiatry. 2012;71(9):774–82. doi: 10.1016/j.biopsych.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tipton AJ, Sullivan JC. Sex differences in T cells in hypertension. Clin Ther. 2014;36(12):1882–900. doi: 10.1016/j.clinthera.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Virdis A, Dell’Agnello U, Taddei S. Impact of inflammation on vascular disease in hypertension. Maturitas. 2014;78(3):179–83. doi: 10.1016/j.maturitas.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 6.Rakesh K, Agrawal DK. Cytokines and growth factors involved in apoptosis and proliferation of vascular smooth muscle cells. Int Immunopharmacol. 2005;5(10):1487–506. doi: 10.1016/j.intimp.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 7.He DH, Lin JX, Zhang LM, et al. Early treatment with losartan effectively ameliorates hypertension and improves vascular remodeling and function in a prehypertensive rat model. Life Sci. 2017;173:20–27. doi: 10.1016/j.lfs.2017.01.013. [DOI] [PubMed] [Google Scholar]
- 8.Zhao X, Zhang LK, Zhang CY, et al. Regulatory effect of hydrogen sulfide on vascular collagen content in spontaneously hypertensive rats. Hypertens Res. 2008;31(8):1619–30. doi: 10.1291/hypres.31.1619. [DOI] [PubMed] [Google Scholar]
- 9.Kassan M, Wecker A, Kadowitz P, et al. CD4+CD25+Foxp3 regulatory T cells and vascular dysfunction in hypertension. J Hypertens. 2013;31(10):1939–43. doi: 10.1097/HJH.0b013e328362feb7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bravo Y, Quiroz Y, Ferrebuz A, et al. Mycophenolate mofetil administration reduces renal inflammation, oxidative stress, and arterial pressure in rats with lead-induced hypertension. Am J Physiol Renal Physiol. 2007;293(2):F616–23. doi: 10.1152/ajprenal.00507.2006. [DOI] [PubMed] [Google Scholar]
- 11.Gooch JL, Sharma AC. Targeting the immune system to treat hypertension: Where are we? Curr Opin Nephrol Hypertens. 2014;23(5):473–79. doi: 10.1097/MNH.0000000000000052. [DOI] [PubMed] [Google Scholar]
- 12.Liang YF, Zhang DD, Yu XJ, et al. Hydrogen sulfide in paraventricular nucleus attenuates blood pressure by regulating oxidative stress and inflammatory cytokines in high salt-induced hypertension. Toxicol Lett. 2017;270:62–71. doi: 10.1016/j.toxlet.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Al-Magableh MR, Kemp-Harper BK, Hart JL. Hydrogen sulfide treatment reduces blood pressure and oxidative stress in angiotensin II-induced hypertensive mice. Hypertens Res. 2015;38(1):13–20. doi: 10.1038/hr.2014.125. [DOI] [PubMed] [Google Scholar]
- 14.Jin HF, Liang C, Liang JM, et al. [Effects of hydrogen sulfide on vascular inflammation in pulmonary hypertension induced by high pulmonary blood flow: Experiment with rats]. Zhonghua Yi Xue Za Zhi. 2008;88(32):2235–39. [in Chinese] [PubMed] [Google Scholar]
- 15.Mani S, Untereiner A, Wu L, Wang R. Hydrogen sulfide and the pathogenesis of atherosclerosis. Antioxid Redox Signal. 2014;20(5):805–17. doi: 10.1089/ars.2013.5324. [DOI] [PubMed] [Google Scholar]
- 16.Mirandola P, Gobbi G, Sponzilli I, et al. Exogenous hydrogen sulfide induces functional inhibition and cell death of cytotoxic lymphocytes subsets. J Cell Physiol. 2007;213(3):826–33. doi: 10.1002/jcp.21151. [DOI] [PubMed] [Google Scholar]
- 17.Cao H, Zhou X, Zhang J, et al. Hydrogen sulfide protects against bleomycin-induced pulmonary fibrosis in rats by inhibiting NF-κB expression and regulating Th1/Th2 balance. Toxicol Lett. 2014;224(3):387–94. doi: 10.1016/j.toxlet.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 18.Yang R, Qu C, Zhou Y, Konkel JE, et al. Hydrogen sulfide promotes Tet1- and Tet2-mediated Foxp3 demethylation to drive regulatory T cell differentiation and maintain immune homeostasis. Immunity. 2015;43(2):251–63. doi: 10.1016/j.immuni.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sáez PJ, Shoji KF, Aguirre A, Sáez JC. Regulation of hemichannels and gap junction channels by cytokines in antigen-presenting cells. Mediators Inflamm. 2014;2014:742734. doi: 10.1155/2014/742734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willebrords J, Yanguas SC, et al. Connexins and their channels in inflammation. Crit Rev Biochem Mol Biol. 2016;51(6):413–39. doi: 10.1080/10409238.2016.1204980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oviedo-Orta E, Hoy T, Evans WH. Intercellular communication in the immune system: differential expression of connexin40 and 43, and perturbation of gap junction channel functions in peripheral blood and tonsil human lymphocyte subpopulations. Immunology. 2000;99(4):578–90. doi: 10.1046/j.1365-2567.2000.00991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsue H, Yao J, Matsue K, et al. Gap junction-mediated intercellular communication between dendritic cells (DCs) is required for effective activation of DCs. J Immunol. 2006;176(1):181–90. doi: 10.4049/jimmunol.176.1.181. [DOI] [PubMed] [Google Scholar]
- 23.Oviedo-Orta E, Perreau M, Evans WH, Potolicchio I. Control of the proliferation of activated CD4+ T cells by connexins. J Leukoc Biol. 2010;88(1):79–86. doi: 10.1189/jlb.0909613. [DOI] [PubMed] [Google Scholar]
- 24.Ni X, Wang A, Zhang L, et al. Up-regulation of gap junction in peripheral blood T lymphocytes contributes to the inflammatory response in essential hypertension. PLoS One. 2017;12(9):e0184773. doi: 10.1371/journal.pone.0184773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kubota Y, Umegaki K, Kagota S, et al. Evaluation of blood pressure measured by tail-cuff methods (without heating) in spontaneously hypertensive rats. Biol Pharm Bull. 2006;29(8):1756–58. doi: 10.1248/bpb.29.1756. [DOI] [PubMed] [Google Scholar]
- 26.Zheng LY, Li L, Ma MM, et al. Deficiency of volume-regulated ClC-3 chloride channel attenuates cerebrovascular remodelling in DOCA-salt hypertension. Cardiovasc Res. 2013;100(1):134–42. doi: 10.1093/cvr/cvt156. [DOI] [PubMed] [Google Scholar]
- 27.Kitayama J, Kitazono T, Ooboshi H, et al. Chronic administration of a tyrosine kinase inhibitor restores functional and morphological changes of the basilar artery during chronic hypertension. J Hypertens. 2002;20(11):2205–11. doi: 10.1097/00004872-200211000-00020. [DOI] [PubMed] [Google Scholar]
- 28.Whiteman M, Winyard PG. Hydrogen sulfide and inflammation: The good, the bad, the ugly and the promising. Expert Rev Clin Pharmacol. 2011;4(1):13–32. doi: 10.1586/ecp.10.134. [DOI] [PubMed] [Google Scholar]
- 29.Han Y, Zeng F, Tan G, et al. Hydrogen sulfide inhibits abnormal proliferation of lymphocytes via AKT/GSK3β signal pathway in systemic lupus erythematosus patients. Cell Physiol Biochem. 2013;31(6):795–804. doi: 10.1159/000350097. [DOI] [PubMed] [Google Scholar]
- 30.Zahorul Islam M, Kawaguchi H, Miura N, et al. Hypertension alters the endothelial-dependent biphasic response of bradykinin in isolated Microminipig basilar artery. Microvasc Res. 2017;114:52–57. doi: 10.1016/j.mvr.2017.06.001. [DOI] [PubMed] [Google Scholar]
- 31.Yan H, Du J, Tang C. The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem Biophys Res Commun. 2004;313(1):22–27. doi: 10.1016/j.bbrc.2003.11.081. [DOI] [PubMed] [Google Scholar]
- 32.Zhao Q, Ishibashi M, Hiasa K, et al. Egashira, Essential role of vascular endothelial growth factor in angiotensin II-induced vascular inflammation and remodeling. Hypertension. 2004;44(3):264–70. doi: 10.1161/01.HYP.0000138688.78906.6b. [DOI] [PubMed] [Google Scholar]
- 33.Gendron G, Gobeil F, Jr, Morin J, et al. Contractile responses of aortae from WKY and SHR to vasoconstrictors. Clin Exp Hypertens. 2004;26(6):511–23. doi: 10.1081/ceh-200031826. [DOI] [PubMed] [Google Scholar]
- 34.Idris-Khodja N, Mian MO, Paradis P, Schiffrin EL. Dual opposing roles of adaptive immunity in hypertension. Eur Heart J. 2014;35(19):1238–44. doi: 10.1093/eurheartj/ehu119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodríguez-Iturbe B, Pons H, Quiroz Y, Johnson RJ. The immunological basis of hypertension. Am J Hypertens. 2014;27(11):1327–37. doi: 10.1093/ajh/hpu142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh MV, Chapleau MW, Harwani SC, Abboud FM. The immune system and hypertension. Immunol Res. 2014;59(1–3):243–53. doi: 10.1007/s12026-014-8548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wallace K, Richards S, Dhillon P, et al. CD4+ T-helper cells stimulated in response to placental ischemia mediate hypertension during pregnancy. Hypertension. 2011;57(5):949–55. doi: 10.1161/HYPERTENSIONAHA.110.168344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trott DW, Thabet SR, Kirabo A, et al. Oligoclonal CD8+ T Cells play a critical role in the development of hypertension. Hypertension. 2014;64(5):1108–15. doi: 10.1161/HYPERTENSIONAHA.114.04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens. 2005;19(2):149–54. doi: 10.1038/sj.jhh.1001785. [DOI] [PubMed] [Google Scholar]
- 40.Lee DL, Leite R, Fleming C, et al. Brands, Hypertensive response to acute stress is attenuated in interleukin-6 knockout mice. Hypertension. 2004;44(3):259–63. doi: 10.1161/01.HYP.0000139913.56461.fb. [DOI] [PubMed] [Google Scholar]
- 41.Brands MW, Banes-Berceli AKL, Inscho EW, et al. Interleukin-6 knockout prevents angiotensin II hypertension: Role of renal vasoconstriction and JAK2/STAT3 activation. Hypertension. 2010;56(5):879–84. doi: 10.1161/HYPERTENSIONAHA.110.158071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu M, Liu YH, Goh HS, et al. Hydrogen sulfide inhibits plasma renin activity. J Am Soc Nephrol. 2010;21(6):993–1002. doi: 10.1681/ASN.2009090949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi YX, Chen Y, Zhu YZ, et al. Chronic sodium hydrosulfide treatment decreases medial thickening of intramyocardial coronary arterioles, interstitial fibrosis, and ROS production in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2007;293(4):H2093–100. doi: 10.1152/ajpheart.00088.2007. [DOI] [PubMed] [Google Scholar]
- 44.Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001;20(21):6008–16. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng Y, Ndisang JF, Tang G, et al. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol. 2004;287(5):H2316–23. doi: 10.1152/ajpheart.00331.2004. [DOI] [PubMed] [Google Scholar]
- 46.Liu YH, Lu M, Hu LF, et al. Hydrogen sulfide in the mammalian cardiovascular system. Antioxid Redox Signal. 2012;17(1):141–85. doi: 10.1089/ars.2011.4005. [DOI] [PubMed] [Google Scholar]
- 47.Zavaczki E, Jeney V, Agarwa A, et al. Hydrogen sulfide inhibits the calcification and osteoblastic differentiation of vascular smooth muscle cells. Kidney Int. 2011;80(7):731–39. doi: 10.1038/ki.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zanardo RC, Brancaleone V, Distrutti E, et al. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006;20(12):2118–20. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- 49.Kasal DA, Barhoumi T, Li MW, et al. T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension. 2012;59(2):324–30. doi: 10.1161/HYPERTENSIONAHA.111.181123. [DOI] [PubMed] [Google Scholar]
- 50.Barhoumi T, Kasal DA, Li MW, et al. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension. 2011;57(3):469–76. doi: 10.1161/HYPERTENSIONAHA.110.162941. [DOI] [PubMed] [Google Scholar]
- 51.Matrougui K, Elmageed ZAbd, Kassan M, et al. Natural regulatory T cells control coronary arteriolar endothelial dysfunction in hypertensive mice. Am J Pathol. 2011;178(1):434–41. doi: 10.1016/j.ajpath.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olas B. Hydrogen sulfide in signaling pathways. Clin Chim Acta. 2015;439:212–18. doi: 10.1016/j.cca.2014.10.037. [DOI] [PubMed] [Google Scholar]
- 53.Tomasova L, Drapala A, Jurkowska H, et al. Na2S, a fast-releasing H2S donor, given as suppository lowers blood pressure in rats. Pharmacol Rep. 2017;69(5):971–77. doi: 10.1016/j.pharep.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 54.Al-Magableh MR, Kemp-Harper BK, Hart JL. Hydrogen sulfide treatment reduces blood pressure and oxidative stress in angiotensin II-induced hypertensive mice. Hypertens Res. 2015;38(1):13–20. doi: 10.1038/hr.2014.125. [DOI] [PubMed] [Google Scholar]
- 55.Altaany Z, Ju Y, Yang G, et al. The coordination of S-sulfhydration, S-nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide. Sci Signal. 2014;7(342):ra87. doi: 10.1126/scisignal.2005478. [DOI] [PubMed] [Google Scholar]
- 56.Yang G, Wu L, Bryan S, et al. Cystathionine gamma-lyase deficiency and overproliferation of smooth muscle cells. Cardiovascular Res. 2010;86:487–95. doi: 10.1093/cvr/cvp420. [DOI] [PubMed] [Google Scholar]
- 57.Liang YF, Zhang DD, Yu XJ, et al. Hydrogen sulfide in paraventricular nucleus attenuates blood pressure by regulating oxidative stress and inflammatory cytokines in high salt-induced hypertension. Toxicol Lett. 2017;270:62–71. doi: 10.1016/j.toxlet.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 58.Yan SK, Chang T, Wang H, et al. Effects of hydrogen sulfide on homocysteine-induced oxidative stress in vascular smooth muscle cell. Biochem Biophys Res Commun. 2006;351(2):485–91. doi: 10.1016/j.bbrc.2006.10.058. [DOI] [PubMed] [Google Scholar]
- 59.Wen YD, Wang H, Kho SH, et al. Hydrogen sulfide protects HUVECs against hydrogen peroxide induced mitochondrial dysfunction and oxidative stress. PLoS One. 2013;8(2):e53147. doi: 10.1371/journal.pone.0053147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu XH, Cui LB, Wu K, et al. Hydrogen sulfide as a potent cardiovascular protective agent. Clin Chim Acta. 2014;437:78–87. doi: 10.1016/j.cca.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 61.Wallace JL, Blackler RW, Chan MV, et al. Anti-inflammatory and cytoprotective actions of hydrogen sulfide: Translation to therapeutics. Antioxid Redox Signal. 2015;22(5):398–410. doi: 10.1089/ars.2014.5901. [DOI] [PubMed] [Google Scholar]
- 62.Rios EC, Szczesny B, Soriano FG, et al. Hydrogen sulfide attenuates cytokine production through the modulation of chromatin remodeling. Int J Mol Med. 2015;35(6):1741–46. doi: 10.3892/ijmm.2015.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elgueta R, Tobar JA, Shoji KF, et al. Gap junctions at the dendritic cell-T cell interface are key elements for antigen-dependent T cell activation. J Immunol. 2009;183(1):277–84. doi: 10.4049/jimmunol.0801854. [DOI] [PubMed] [Google Scholar]
- 64.Zhang HC, Zhang ZS, Zhang L, et al. Connexin 43 in splenic lymphocytes is involved in the regulation of CD4+CD25+ T lymphocyte proliferation and cytokine production in hypertensive inflammation. Int J Mol Med. 2018;41(1):13–24. doi: 10.3892/ijmm.2017.3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang R, Szabo C, Ichinose F, et al. The role of H2S bioavailability in endothelial dysfunction. Trends Pharmacol Sci. 2015;36(9):568–78. doi: 10.1016/j.tips.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheng Y, Ndisang JF, Tang G, et al. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol. 2004;287(5):H2316–23. doi: 10.1152/ajpheart.00331.2004. [DOI] [PubMed] [Google Scholar]