

Abstract
Rationale:
Pleomorphic liposarcoma (PLS), is a rare subtype of liposarcoma, and is considered to be of the highest malignancy grade.
Patient concerns:
We aimed to analyze the clinical features, diagnosis, treatment, and recurrence of the 6 cases of PLS.
Diagnoses:
Six cases with confirmed pathological PLS presented at out hospital from January 2003 to January 2017. The postoperative pathology of 5 cases confirmed PLS, and the other was confirmed as PLS with well-differentiated liposarcoma.
Interventions:
All 6 patients underwent complete tumor resection at the time of the first definite diagnosis, and one of them had underwent 3 cycles of chemotherapy treatment.
Outcomes:
There were 4 cases with local recurrence and surgery was repeated after the first radical excision. One case was not recurrent after 27 months post-operation, and the other was lost. The shortest recurrence time of all of these cases was 4 months, and the longest was 29 months after the first radical surgery.
Lessons:
PLS is a rare and high-grade malignancy with high recurrence, poor prognosis, and its treatment is still highly controversial. More studies are required to determine the appropriate treatment and therapeutic strategies to improve the survival rate of patients with PLS, as the disease is associated with frequent relapse.
Keywords: liposarcoma, operative, prognosis, recurrence, sarcoma, surgical procedures
1. Introduction
Liposarcoma, which develops in adipose tissue, is one of the most common soft tissue sarcomas. From a histopathological point of view, liposarcoma can be divided into 5 categories according to the 1994 World Health Organization guidelines. These include well-differentiated (WD; including the lipoma-like, sclerosing, and inflammatory subtypes), myxoid, round cell (poorly differentiated myxoid), pleomorphic, and dedifferentiated. However, according to the recent morphological features and cytogenetic data, it is divided into 3 principal groups: atypical lipomatous tumor/WD and dedifferentiated liposarcoma; myxoid-round cell liposarcoma; and pleomorphic liposarcoma (PLS).[1] PLS is very rare, and accounts for only 5% to 10% of lipomatous tumors.[1] However, it is considered to be of the highest malignancy grade, with high invasion, metastasis, and recurrence.[2,3] Meanwhile, appropriate therapeutic strategies for PLS are controversial. Herein, we attempted to generate more significant information of PLS, through an analysis of the clinical features, methods of diagnosis, and treatment, and an examination of the clinical recurrence in 6 patients with PLS.
2. Methods
2.1. Clinical information
The clinical data from 6 PLS cases were collected and analyzed. The age of the patients ranged from 46 to 82 years, with a mean age 65.40 ± 13.48 years. The group included 4 men and 2 women with PLS, that was confirmed by pathological examination and who attended our hospital from January 2003 to January 2017. One patient had an excision of lipoma on the posterior occipital region in another hospital 6 months ago before attending our hospital, but the biopsy was not checked for pathology. One other patient had a history of asthma for almost 60 years, 1 had a high risk of grade 2 hypertension, and the other cases had no definite histories.
All of the patients and/or their families gave their informed, written consents after receiving a detailed explanation of the study. The study design, the manner of data collection, the analysis, and reporting of all data were approved by the Ethics Committee of Fuling Central Hospital of Chongqing City.
2.2. Clinical features
PLS was characterized by the growth of a progressively painless mass in all patients. The symptoms of the patients were different and dependent in the anatomic site of the tumor. One of the 2 retroperitoneal cases, presented with anorexia and abdominal distension, whereas the other patient had a persistent pain in the left lower abdomen due to the compression from the pelvic tumor. However, these symptoms were ignored until the mass was big enough or there were some other serious manifestations. Tumors of the other 3 cases were located more superficially on the surface, with 1 experiencing skin rupture. In addition, the time period from diagnosis to surgical intervention was very short, because of the rapid growth of tumor. However, the duration from the patients in the text first suspecting the tumor to the definite diagnosis at the hospital, ranged from 1 month to a longest period of 3 years. A computed tomography (CT) scan had been performed for all the patients at the first visit to our hospital. Scans in 5 cases showed round, soft-tissue solid shadows, with still sharp borders; and the other case was similar but with an irregular shape and a sharp border. The enhanced CT showed an uneven, but slightly enhanced strengthening in the tumor shadows (Fig. 1A). Ultrasonography investigations showed that there are 4 hypoechoes and 2 mixed echoes among the 6 cases, all with irregular morphology and unclear boundaries. Five cases demonstrated a high impedance of arterial spectrums.
Figure 1.
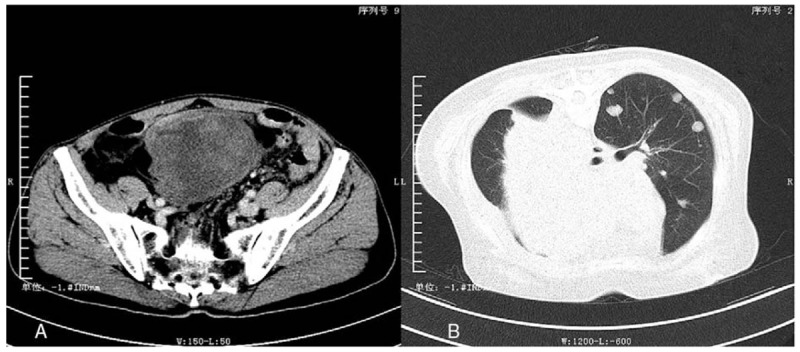
The enhanced computed tomography (CT) of primary pleomorphic liposarcoma (PLS) located in lower abdomen (A) and the CT scan of metastatic lesions (B). A, The picture is the enhanced CT of case no. 4. There was a large soft-tissue shadow in lower abdomen, with heterogeneous microenhancement and clear boundary. B, The picture is the CT scan of the case no. 5 before her last surgery. There were couples of unequal-sized metastatic tumors in the left side of lung and a giant metastatic tumor in the mediastinum.
2.3. Treatment
All 6 patients underwent complete tumor resection at the time of the first definite diagnosis (Table 1). One patient had a colon resection, which had a poor blood supply due to the compression of splenic flexure of colon. There were approximately 80 mL of slightly bloody ascites found during the operation, and in which no dysplastic cells were detected. Another patient had the right kidney and right adrenal gland removed due to the invasion of the right kidney, which was found during operation to be obviously squeezed, deformed, and displaced with hydronephrosis. Three cases had complete tumor resection on the surface, with 1 experiencing skin rupture. The above 5 cases were not treated with chemotherapy and/or radiotherapy after operation. A wide excision of the tumor mass, which also included a total abdominal hysterectomy and a bilateral salpingo-oophorectomy, was performed for the final case with a left fallopian tube PLS. There were approximately 150 mL of slightly bloody ascites found during operation, in which dysplastic cells were detected. Following the surgery, a 21-day period of chemotherapy treatment was initiated, with ifosfamide [2 g, days 1–3, intravenous injection (ivgtt)], and epirubicin (60 mg day 1; 50 mg day 2, ivgtt). The progress of the chemotherapy treatment went as expected. She only underwent 3 cycles of chemotherapy treatment and then refused the subsequent treatment.
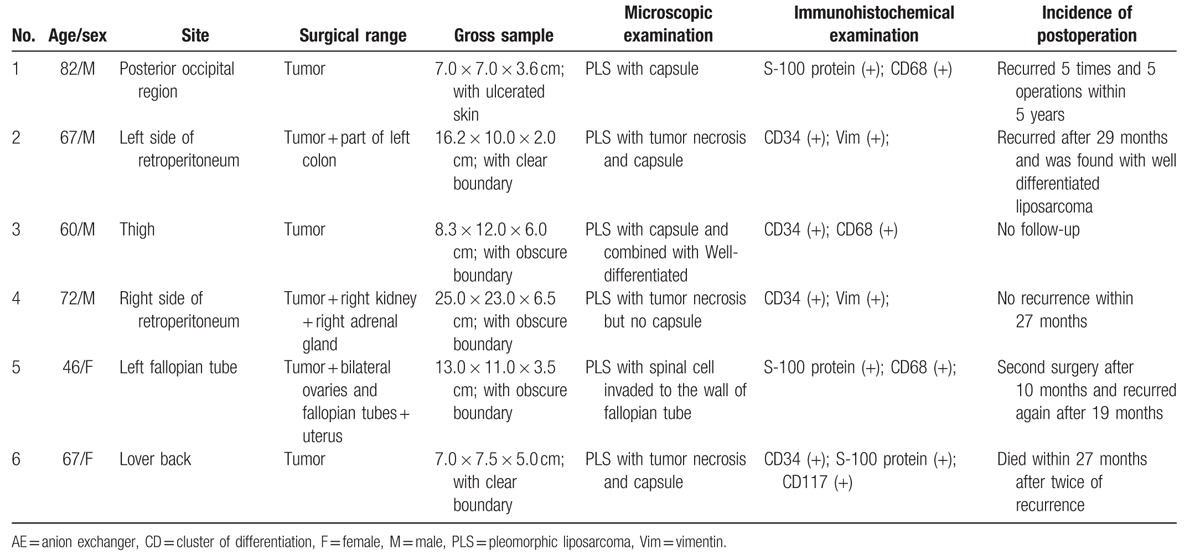
Table 1.
Summary of clinical and follow-up data from 6 patients with pleomorphic liposarcoma.
3. Results
3.1. Pathological results
In this study, tumors in the retroperitoneal, abdominal, and pelvic cavities were larger than those near the body surface such as the subcutaneous and intermuscular tissues. Gross examinations of the specimens following the first surgery showed that the smallest size was approximately 7.0 × 7.0 × 3.6 cm, and the largest was approximately 25.0 × 23.0 × 6.5 cm, accompanied with white, yellow, or a yellowish-white section, that was firm with lobulated borders. However, the tumors from 2 cases did not have clear borders, and they were closely adhered to the right renal adipose capsule (No. 4) and dermis (No. 1), respectively.
The histopathology revealed that the tumors were high-grade PLS, with multiple and high metatypic cells. There were multiple mono-/multinucleate giant cells, bizarre cells, lipoblasts (Fig. 2). The lipoblasts were characterized by heterotypic, hyperchromatic nuclei with uni-/multivacuolated cytoplasm. Furthermore, the surrounding lymph nodes and peripheral vessels were not infiltrated. Four cases had complete envelopes, whereas the others did not. Five cases had no invasion to the surrounding tissues, whereas 1 case (No. 5) had tumor invasion to the fallopian tube wall, and its histopathology revealed that the smooth muscle area had been invaded by pleomorphic cells, especially spindle cells. In addition, 5 cases were confirmed as PLS, whereas the remaining case (No. 2) was PLS accompanied with a WD liposarcoma. Tumor cells were detected at the margins only in case No. 6.
Figure 2.
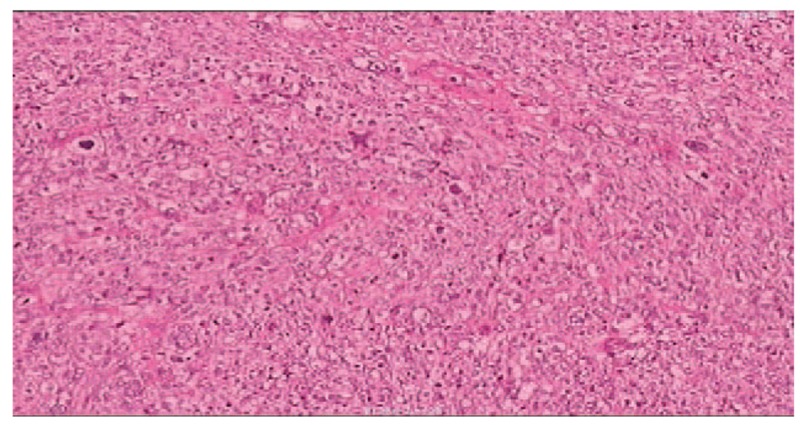
The pathology of pleomorphic liposarcoma (PLS). The tumor cells were bizarre and diffuse distribution. There were multinuclear giant cells with deep-dyed big nucleolus, pathological karyokinesis, and granular and alveolate cytoplasm (HE ×200).
Immunohistochemistry showed that 4 cases were CD34 (+), 3 cases were S-100 protein (+) and CD68 (+), 2 cases were vimentin (+), and 1 was CD117 (+). However, all 6 cases were negative for smooth muscle antibody (SMA), anion exchanger (AE1/AE3), and desmin, whereas the index of ki-67 was 10% to 30% (Table 1).
3.2. Prognosis and outcome
There were 4 cases with local recurrence and surgery was repeated after the first radical excision. The details were as follows: 1 case (No. 1), relapsed 4 months after surgery, and the patient underwent 5 radical surgeries in the original location over 5-year period due to tumor recurrence. There was no recurrence 6 months after the last surgery. Another case (No. 6), relapsed and the patient underwent a radical surgery 12 months after the first operation. Eleven months later, a palliative resection had been performed because of recurrence with pulmonary and mediastinal metastases. The third operative histopathology revealed that striated muscle tissue was infiltrated by tumor cells. Following this surgery, another 2 operations were performed due to bleeding and infection. Eventually, the patient died due to multiple organ failure which occurred 27 months after the first diagnosis, and he was hospitalized for 132 days. For case No. 5, chemotherapy was stopped after the third period due to detection of metatypic cells in ascites. However, the patient accepted surgery again due to the recurrence after 10 months. And a further CT of the chest and abdomen from our outpatient department showed there was no obvious evidence of metastasis 3 months later. The histopathology results for the above 3 cases were PLS, without changes to degree of malignancy or a mixture with other liposarcoma cells. However, 1 case in the study (No. 2) was confirmed as WD liposarcoma in the second surgery specimen. The patient in case No. 3 was lost to follow-up after the first treatment in our hospital. The patient in case No. 4 went to the hospital due to hypertension and the re-examinations of records showed no recurrence after 27 months postoperation, and the patient was lost later to follow-up. In conclusion, the shortest recurrence time of all of these cases was 4 months (No. 1), and the longest was 29 months (No. 2) after the first radical surgery. As indicated, PLS relapse occurred frequently and demonstrated repeated relapses.
4. Discussion
PLS is a pleomorphic, highly malignant liposarcoma, with various unique lipoblasts in the histopathology. It is very rare, and only counts for 5% to 10% of liposarcoma cases.[4] From January 2003 to January 2017, there were a total of 89 liposarcoma cases in our hospital, including 6 PLS cases, which accounted for 6.74% of liposarcoma patients. PLS mostly appeared in middle age and older patients, with equal sex distributions. However, some cases have been reported in younger patients, and the youngest recorded was 8 years old.[5–7] In our study, there were 4 men and 2 women, and the mean age was 65.40 ± 13.48 years (range, 46–82 years).
PLS is characterized as the growth of a progressively painless mass, and it is easily ignored until the mass is big enough or there are some other compressive manifestations. PLS could occur in various organs, but the most common sites are proximal extremities, especially in the lower extremities, and in other uncommon sites including the retroperitoneum, the abdominal wall, the chest wall, the mesentery, the pelvic cavity, the spermatic cord, the mediastinum, the parietal pleura, the pericardium, the foot, the spine, the head, and neck region.[1,8–12] In the current study, there were 3 PLS cases that occurred in deep soft tissues, and 3 that were found in superficial tissues, and mostly in subcutaneous adipose tissues. There were 2 cases in retroperitoneum, and 1 in the fallopian tube, which is probably the first case of PLS observed in the fallopian tube. One case each was located in the occiput, lower back, and thigh. PLS is a type of tumor with rapid growth characteristics, such that the time course duration for preoperative care was very short. Taken together, PLS in superficial tissue was found more easily, and the duration from initial detection to definite diagnosis at the hospital varied from 1 month to a longest period of 3 years.
Accessory examinations such as CT, ultrasound, and magnetic resonance imaging are critical for surgical methods by assessing the size of tumor and the degree of tumor infiltration into the surrounding tissues. However, it is difficult to distinguish PLS from other liposarcoma and sarcoma using these accessory examinations. The definite diagnosis of PLS still depends on the pathological examination, which reveals highly metatypic cells with granular and/or foamy small vacuoles in cytoplasm, mono-/poly-nuclear giant cells with a deep-dyed large nucleolus, and pathological karyokinesis, and also scattered highly specific spindle cells.[13]
Surgery, especially radical resection, is the main treatment for PLS. It is an important palliative intervention for patients with PLS with distant metastasis and local invasion to relieve the compression symptoms. However, the local recurrence rate is very high.[14] It is reported that radical resection usually requires removal of the surrounding organs, the most common of which is kidney, followed by the colon.[15] In our study, 1 case involved a resected right kidney and right adrenal gland, another case resected colon, another resected bilateral ovaries, fallopian tubes, and uterus, and 1 recurrent case involved resected sigmoid colon with some of the small intestine. In addition, chemotherapy is still controversial regarding any curative effect on PLS. A study of Miura et al[16] found that conventional chemotherapy was not beneficial to sarcoma patients, and it could not improve patient survival rates. Currently, there is still no standardized treatment approaches reported for PLS.[17] At present, radical surgical resection is the best and main treatment for PLS, with chemotherapy and radiotherapy in multimodality treatment strategies still remaining controversial.[18] Cassier et al[19] reported a retrospective study examining radiotherapy in WD liposarcoma in a total of 283 patients, and suggested that radiotherapy could help to reduce the postoperative recurrence rate, but it could not improve the survival rate. A study by Le Grange et al[20] suggested that preoperative radiotherapy could reduce the size of tumor in 80% of patients with myxoid liposarcoma and fibrosarcoma, whereas the size of the sarcoma tumor increased in a few patients. In addition, it was reported that in a young woman who received radiotherapy for an epithelioid sarcoma on her left calf, the tumor relapsed later and its pathology was diagnosed as PLS. It has been suggested that the change of histopathology of this tumor was caused by radiotherapy.[21] In our study, the first histopathology examination finding of No. 3 was WD liposarcoma, whereas the second histopathology examination finding of No. 2 was also well-differentiated liposarcoma. This may suggest that PLS could be combined with other types of differentiated liposarcoma, and even transform with them.
In addition, even though the anatomical relationship between PLS and the surrounding tissues was not entirely clear, the surrounding tissue was seldom invaded by PLS. However, it is reported that PLS is aggressive and highly metastatic. The total metastases rate was approximately 20%, whereas the metastatic rates to the lung, the liver, and to the bone and pancreas were 82%, 18%, and 18%, respectively.[2,22] In addition, the 1-, 3-, and 5-year survival rates for PLS were 93%, 75%, and 29%, respectively.[2,13] The study by Hornick et al[13] on 57 PLS cases, reported that the closer the tumor mass was to the center position of the trunk, the worse the prognosis was. In addition, the larger the tumor size (>10 cm), the deeper the location, or if there was necrosis present, the worse the prognosis was, as evident by more frequent relapses, and dramatically decreased survival time.
5. Conclusion
PLS is a type of rare and high-grade malignancy, with a high recurrence rate and poor prognosis. The diagnosis is dependent on histopathological examination of tumor biopsies. In addition, it is common to observe relapses, and the standard treatment for PLS remains controversial. Consequently, more studies are required to develop treatment and therapeutic strategies for PLS.
Acknowledgments
Thanks for the understanding of these patients and their families.
Footnotes
Abbreviations: AE = anion exchanger, CT = computed tomography, ivgtt = intravenous injection, PLS = pleomorphic liposarcoma, SMA = smooth muscle antibody, Vim = vimentin, WD = well-differentiated.
There are no funds. The authors declare that they have no conflict of interests.
References
- [1].Gebhard S, Coindre JM, Michels JJ, et al. Pleomorphic liposarcoma: clinicopathologic, immunohistochemical, and follow-up analysis of 63 cases: a study from the French Federation of Cancer Centers Sarcoma Group. Am J Surg Pathol 2002;26:601–16. [DOI] [PubMed] [Google Scholar]
- [2].Ghadimi MP, Liu P, Peng T, et al. Pleomorphic liposarcoma: clinical observations and molecular variables. Cancer 2011;117:5359–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tseng WW, Somaiah N, Lazar AJ, et al. Novel systemic therapies in advanced liposarcoma: a review of recent clinical trial results. Cancers (Basel) 2013;5:529–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Peterson JJ, Kransdorf MJ, Bancroft LW, et al. Malignant fatty tumors: classification, clinical course, imaging appearance and treatment. Skeletal Radiol 2003;32:493–503. [DOI] [PubMed] [Google Scholar]
- [5].Rudzinski E, Mawn L, Kuttesch J, et al. Orbital pleomorphic liposarcoma in an eight-year-old boy. Pediatr Dev Pathol 2011;14:339–44. [DOI] [PubMed] [Google Scholar]
- [6].Ahmed Z, Shah HU, Yaqoob N, et al. Pleomorphic liposarcoma in a ten year old child. J Pak Med Assoc 2004;54:533–4. [PubMed] [Google Scholar]
- [7].Saeed M, Plett S, Kim GE, et al. Radiological-pathological correlation of pleomorphic liposarcoma of the anterior mediastinum in a 17-year-old girl. Pediatr Radiol 2010;40suppl 1:S68–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mumert ML, Walsh MT, Jensen EM, et al. Pleomorphic liposarcoma originating from intracranial dura mater. J Neurooncol 2010;97:149–53. [DOI] [PubMed] [Google Scholar]
- [9].Wang L, Ren W, Zhou X, et al. Pleomorphic liposarcoma: a clinicopathological, immunohistochemical and molecular cytogenetic study of 32 additional cases. Pathol Int 2013;63:523–31. [DOI] [PubMed] [Google Scholar]
- [10].Wang JG, Wei ZM, Liu H, et al. Primary pleomorphic liposarcoma of pericardium. Interact Cardiovasc Thorac Surg 2010;11:325–7. [DOI] [PubMed] [Google Scholar]
- [11].Brcic L, Jakovcevic A, Vuletic LB, et al. Pleomorphic liposarcoma of the foot: a case report. Diagn Pathol 2008;3:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Morales-Codina AM, Martin-Benlloch JA, Corbellas Aparicio M. Primary pleomorphic liposarcoma of the spine. Case report and review of the literature. Int J Surg Case Rep 2016;25:114–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hornick JL, Bosenberg MW, Mentzel T, et al. Pleomorphic liposarcoma: clinicopathologic analysis of 57 cases. Am J Surg Pathol 2004;28:1257–67. [DOI] [PubMed] [Google Scholar]
- [14].Choi C, Park JH, Lee CG, et al. Successful salvage treatment of myxoid liposarcoma with multiple peritoneal seeding using helical tomotherapy-based intraperitoneal radiotherapy: a case report. BMC Res Notes 2015;8:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lee SY, Goh BK, Teo MC, et al. Retroperitoneal liposarcomas: the experience of a tertiary Asian center. World J Surg Oncol 2011;9:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Miura JT, Charlson J, Gamblin TC, et al. Impact of chemotherapy on survival in surgically resected retroperitoneal sarcoma. Eur J Surg Oncol 2015;41:1386–92. [DOI] [PubMed] [Google Scholar]
- [17].Sherman KL, Wayne JD, Chung J, et al. Assessment of multimodality therapy use for extremity sarcoma in the United States. J Surg Oncol 2014;109:395–404. [DOI] [PubMed] [Google Scholar]
- [18].Singer S, Antonescu CR, Riedel E, et al. Histologic subtype and margin of resection predict pattern of recurrence and survival for retroperitoneal liposarcoma. Ann Surg 2003;238:358–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cassier PA, Kantor G, Bonvalot S, et al. Adjuvant radiotherapy for extremity and trunk wall atypical lipomatous tumor/well-differentiated LPS (ALT/WD-LPS): a French Sarcoma Group (GSF-GETO) study. Ann Oncol 2014;25:1854–60. [DOI] [PubMed] [Google Scholar]
- [20].Le Grange F, Cassoni AM, Seddon BM. Tumour volume changes following pre-operative radiotherapy in borderline resectable limb and trunk soft tissue sarcoma. Eur J Surg Oncol 2014;40:394–401. [DOI] [PubMed] [Google Scholar]
- [21].Orosz Z, Rohonyi B, Luksander A, et al. Pleomorphic liposarcoma of a young woman following radiotherapy for epithelioid sarcoma. Pathol Oncol Res 2000;6:287–91. [DOI] [PubMed] [Google Scholar]
- [22].Malleo G, Crippa S, Partelli S, et al. Pleomorphic liposarcoma of the axilla metastatic to the pancreas. Dig Surg 2009;26:262–3. [DOI] [PubMed] [Google Scholar]