

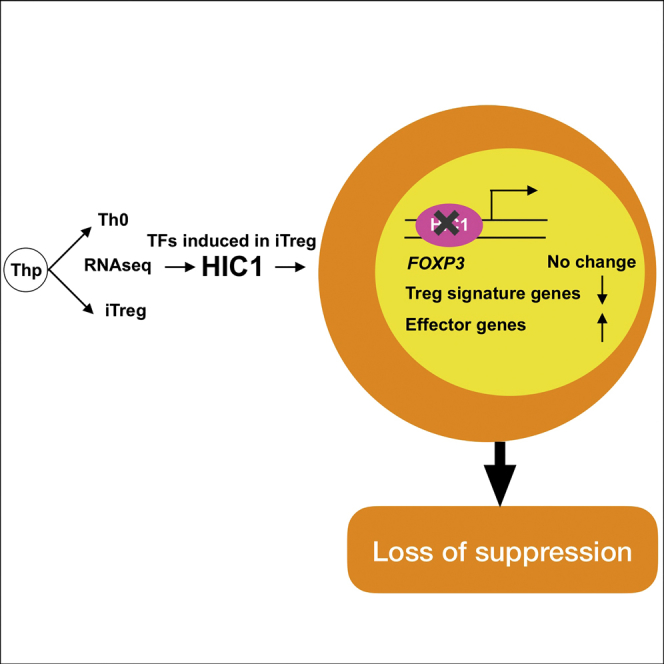
Summary
Regulatory T (Treg) cells are critical in regulating the immune response. In vitro induced Treg (iTreg) cells have significant potential in clinical medicine. However, applying iTreg cells as therapeutics is complicated by the poor stability of human iTreg cells and their variable suppressive activity. Therefore, it is important to understand the molecular mechanisms of human iTreg cell specification. We identified hypermethylated in cancer 1 (HIC1) as a transcription factor upregulated early during the differentiation of human iTreg cells. Although FOXP3 expression was unaffected, HIC1 deficiency led to a considerable loss of suppression by iTreg cells with a concomitant increase in the expression of effector T cell associated genes. SNPs linked to several immune-mediated disorders were enriched around HIC1 binding sites, and in vitro binding assays indicated that these SNPs may alter the binding of HIC1. Our results suggest that HIC1 is an important contributor to iTreg cell development and function.
Keywords: iTreg, suppression, HIC1, FOXP3, regulatory SNP, RNA-seq, ChIP-seq, expression kinetics, T cells, transcriptional regulation
Graphical Abstract
Highlights
-
•
Hypermethylated in cancer 1 (HIC1) is upregulated in iTreg cells
-
•
HIC1-deficient iTreg cells express FOXP3 but have reduced suppressive ability
-
•
Autoimmune-disease-associated SNPs are enriched within HIC1 binding loci
-
•
HIC1 is an important regulator of iTreg development and function
Ullah et al. find that HIC1 is induced during human iTreg cell differentiation. HIC1 binds to and regulates the expression of key genes during iTreg differentiation. Several autoimmune-disease-associated SNPs are enriched near HIC1 ChIP-seq peaks.
Introduction
Immune homeostasis at the site of inflammation is maintained primarily by peripherally induced FOXP3+ regulatory T (Treg) cells that develop from CD4+ T cells in the presence of interleukin (IL)-2 and transforming growth factor (TGF)-β (Yadav et al., 2013). These cells can also be induced in vitro when a naive CD4+ T cell is activated in the presence of IL-2, TGF-β, and retinoic acid (RA) (Coombes et al., 2007, Sun et al., 2007). In vitro induced Treg cells are called iTreg cells (Abbas et al., 2013). Identification and understanding the functions of factors important for the development of Treg cells are crucial for developing T cell-based therapies (Bluestone et al., 2015). During the past decade, we have learned much about the mechanism of Treg cell development, particularly in mice. A network of transcription factors (TFs), including Foxp3, the Ikaros family of TFs, Nr4a nuclear receptors, c-Rel, Nfat, Smad factors, Stat5, and Runx factors, act in concert, leading to Treg differentiation (Iizuka-Koga et al., 2017).
Although other TFs regulate Treg cell differentiation and function, FOXP3 is the key factor associated with iTreg cells. Deletion of FOXP3 results in severe autoimmunity in humans and mice (Bennett et al., 2001, Fontenot et al., 2003). Additionally, in mice, ectopic expression of Foxp3 confers suppressive ability to effector T cells (Fontenot et al., 2003). Recent studies suggest that additional factors are involved in Treg lineage specification. For instance, analysis of co-expression networks of 24 cell types of the mouse immune system suggested that regulation of Foxp3-bound genes in Treg cells is independent of Foxp3 expression (Vandenbon et al., 2016). Also, ectopic expression of FOXP3 in effector T cells failed to induce the expression of most of Treg signature genes (Hill et al., 2007, Sugimoto et al., 2006). Moreover, disrupting Foxp3 in mice does not revert Treg cells to conventional T cells (Kuczma et al., 2009). In humans, T cell receptor (TCR) stimulation leads to transient expression of FOXP3 (Allan et al., 2007) without any suppressive function. Furthermore, in rheumatoid arthritis patients, Treg cells show unaltered FOXP3 expression despite their severely compromised suppressive ability (Nie et al., 2013). Thus, besides FOXP3, additional lineage-specific factors contribute to Treg cell suppressive function.
iTreg cells represent a reasonable model to study the factors contributing to the development of Treg cells, as these cells have properties of immune suppression in vivo and in vitro (DiPaolo et al., 2007, Huter et al., 2008, Lu et al., 2010, Hippen et al., 2011). Besides expressing high Foxp3, both polyclonal and antigen-specific iTreg cells suppress effector cell response in mouse models (DiPaolo et al., 2007, Huter et al., 2008). However, although human iTreg cells are suppressive in vitro, their regulatory effects in vivo have been controversial. iTreg cells induced by TGF-β and IL-2 were not suppressive, whereas those generated with additional factors, namely RA (Lu et al., 2010) and rapamycin (Hippen et al., 2011), were suppressive in xenogenic graft versus host disease (GVHD). Although the in vivo suppressive ability of RA-induced iTreg cells has also been questioned (Schmidt et al., 2016, Shevach and Thornton, 2014), there is continued interest in understanding the mechanisms of iTreg development because of their great potential in clinical applications (Kanamori et al., 2016). Furthermore, the conserved noncoding sequence 1 (CNS1) region on the FOXP3 locus serves as response element for TGF-β-SMAD signaling pathway and is required for the generation of peripheral Treg cells (Tone et al., 2008). The CNS1 region also harbors RA response element (Xu et al., 2010), suggesting that RA signaling may potentiate efficient Treg generation in the periphery, especially in the intestine, in which stromal cells and CD103+ dendritic cells (DCs) within mesenteric lymph node (mLN) and intestine express high levels of RA synthesizing the enzyme retinaldehyde dehydrogenase (RALDH2) (Hammerschmidt et al., 2008). Therefore, studying RA-induced iTreg cells may be functionally very relevant for intestinal Treg cells. In the present study, we comprehensively analyzed the transcriptomes of RA-induced iTreg cells across ten time points of development and identified hypermethylated in cancer 1 (HIC1; also called ZBTB29) as a TF specifically upregulated during iTreg cell differentiation. Here, we report the role of HIC1 in iTreg cell development and function.
Results
RNA Sequencing Analysis Identifies Gene Expression Signature of iTreg Cell Priming
To determine how naive T cells acquire a regulatory phenotype, we performed a kinetic analysis of the transcriptome of developing iTreg cells. RNA samples for sequencing were collected across ten time points during differentiation (Figure S1A). About 90% of the iTreg cells were positive for FOXP3 (Figure S1B) and could suppress the proliferation of responder cells (Figure S1C).
In total, 2,927 genes were differentially expressed (DE) at one or more time points during iTreg cell development (Table S1). Eighty percent of the DE genes were protein coding (Figure 1A). Transcriptional regulators were among the most common functional classes within the DE genes, second only to enzymes (Figure 1B). Although a few genes were already DE before 2 hr post-cell activation, most of the changes in gene expression were observed from 6 hr after induction of differentiation (Figure 1C). Unlike protein-coding genes, noncoding genes were predominantly downregulated early on, up to 24 hr (Figure 1C). As expected, several factors involved in Treg cell lineage specification (e.g., FOXP3, IKZF4, NR4A nuclear receptors, REL, NFAT, SMAD factors, and RUNX1) were DE in iTreg cells (Figure 1D; Table S1) (Lu et al., 2014, Ono et al., 2007, Visekruna et al., 2010, Wu et al., 2006). Pathway analysis of DE genes suggested that although at early time points (2–24 hr), signaling through cytokine-cytokine receptors, including TGF-β signaling and RAR/RXR signaling, was primarily enriched, at later time points, especially at 48 hr, several metabolism-related pathways, including cholesterol biosynthesis and glucose metabolism, were enriched (Figure S1D; Table S1).
Figure 1.

RNA-Seq Analysis Identifies Gene Expression Signature of iTreg Cells
(A) Pie chart showing the distribution of different RNA species among the genes differentially expressed (DE) during iTreg differentiation.
(B) Figure showing the functional annotations of DE genes. The annotations were obtained from Ingenuity Pathway Analysis (IPA).
(C) Bar chart showing the number of DE genes at respective time point (hours) for protein-coding (top) and noncoding (bottom) transcripts. Light green denotes genes upregulated in iTreg cells at the given time point for the first time in the time course, whereas dark green shows number of genes that were upregulated at already earlier time points. Similarly, light blue shows genes downregulated in iTreg cells for the first time, whereas dark blues shows genes downregulated at earlier time points.
(D) Heatmap showing the expression of the top 50 DE genes across the time points of differentiation (0.5–72 hr). The first nine columns show the log2 fold change (FC) (iTreg/Th17), while the last nine columns show the expression (reads per kilobase of transcript per million mapped reads [RPKM]) of those genes in iTreg cells at different time points.
(E) Heatmap of the log2 fold change of TFs that were DE in both iTreg cell (left) and Th17 cell differentiation (right). The time points are indicated at the bottom.
(F) Expression profile (mean ± SE) of HIC1 expression (RNA-seq) in Th0, Th1, Th2, Th17, and iTreg cells differentiated from cord blood are shown as a line plot. TPM, transcript per million.
iTreg cells were metabolically similar to ex vivo Treg cells: several glycolytic enzymes (e.g., PGK1, PGAM1, ENO2, ENO3, ALDOC) were upregulated in iTreg cells (Table S1) (Procaccini et al., 2016). Moreover, unlike Th0 cells, iTreg cells used fatty acid oxidation (FAO) as a parallel source of energy, as suggested by upregulation of several enzymes of FAO (e.g., ACAA2, ACADVL, ACSL1, ACSL4, EC12, GPD2) in iTreg cells. Conversely, enzymes catalyzing the synthesis of fatty acid (e.g., FASN, ACAT2) or ACOT2, which catalyze the hydrolysis of acyl-CoAs to the free fatty acid and coenzyme A, were downregulated in iTreg cells.
We overlapped genes DE during iTreg cell priming in this study with the findings of DE genes from three other studies on Treg cells. First, compared with a recent human study (Ferraro et al., 2014) analyzing CD4+ CD25hi CD127lo Treg and conventional T cells, 126 of 359 Treg-specific upregulated genes (e.g., FOXP3, IL2R, IKZF4, CTLA4, DUSP4) were DE in our data (Table S1). Similarly, compared with mouse Treg signature genes (Hill et al., 2007), 139 of 411 human orthologs of 603 mouse genes were DE in our data (Table S1). Finally, compared with a study in which the authors analyzed Treg cell-specific FOXP3-dependent and FOXP3-independent genes (Sugimoto et al., 2006), 104 of 287 Treg-related genes were DE in our data (Table S1). In conclusion, besides confirming a number of key genes previously associated with Treg cells, using the time-series design, we identified hundreds of genes DE during human iTreg cell priming that were not previously associated with Treg cell differentiation or function.
Genome-wide association studies (GWAS) have identified many risk loci associated with autoimmune diseases and various cancers. Autoimmune-disease-associated SNPs can be found near or over the TF binding site on the noncoding regions of the genome and modulate TF binding or epigenetic state (Farh et al., 2015). We determined if disease-associated SNPs extracted from the National Human Genome Research Institute (NHGRI) GWAS catalog (Welter et al., 2014) are enriched in genomic locations near (±100 kb) iTreg DE genes. Interestingly, the top diseases whose SNPs were enriched (adjusted p value < 0.1) include several autoimmune diseases (e.g., inflammatory bowel disease [IBD], celiac disease [CeD], Crohn’s disease [CrD], and rheumatoid arthritis [RhA]) (Figure S1E). The analysis suggests that the SNPs may cause aberrant expression of iTreg signature genes in population that in turn may lead to autoimmune pathologies.
To identify potential TFs required for the development and function of iTreg cells, we studied the expression of all the TFs during iTreg development. To identify iTreg-specific factors, we compared the iTreg RNA sequencing (RNA-seq) data with our recently published human Th17 data (Tuomela et al., 2016). Seventy-eight TFs were DE in both iTreg cells (compared with Th0) as well as Th17 cells (compared with Th0), and 149 and 84 TFs were DE only in the iTreg or Th17 cell subsets, respectively (Figure 1E; Table S2). SMAD factors, NFAT factors, and EGR1 were among the TFs upregulated only in iTreg cells, whereas IRF factors, STAT3, STAT2, FOXO1, NOTCH1, and EGR2 were upregulated only in Th17 cells (Table S2). Many known iTreg cell-related genes (e.g., FOXP3, IKZF3, IKZF4, RUNX1, FOSL1, FOSL2) were also upregulated in Th17 cells, albeit to lower levels or transiently and, therefore, were not in iTreg-specific TFs. Of 78 common factors, the majority behaved similarly (i.e., were either up or down in both subsets) (Figure 1E). However, there was a set of 9 TFs (i.e., HIC1, GFI1, PDLIM1, STAT4, NFKBID, EPAS1, ZBTB32, NR4A1, and NR4A2) that were upregulated in iTreg cells but downregulated in Th17 cells (Figure 1E; Table S2). HIC1 had the opposite expression profile in the two subsets across all time points. Interestingly, SIRT1, a gene negatively regulated by HIC1 (Chen et al., 2005), was among the 10 TFs (i.e., MAF, RB1, KLF11, TAF4B, BATF3, ARID5B, SIRT1, AHR, SMARCA2, and KLF4) upregulated in Th17 cells but downregulated in iTreg cells (Table S2). Furthermore, HIC1 was uniquely upregulated only in iTreg cells, though it had expression similar to Th0 in all Th cell subsets, including Th1/2 (unpublished data) (Figure 1F).
To understand the development of transcriptome during iTreg cell differentiation, we investigated the transcription factor binding sites (TFBS) on the promoter of DE genes at each time point. On the promoter of upregulated genes, EGR1/2, the Ikaros family of TFs, and RA receptors were enriched at multiple time points across differentiation (Figure S2A; Table S2). FOXP3, beta-catenin, and ARID3A/B sites were enriched specifically on the promoters of downregulated genes. Notably, FOXP3 binding sites were enriched only at 24 and 48 hr (Figure S2A), indicating that FOXP3 is not among the earliest regulators of iTreg differentiation. The binding of FOXP3 on the promoters of downregulated genes is consistent with its role as a transcriptional repressor (Arvey et al., 2014). Surprisingly, HIC1 was among the TFs whose binding sites were enriched already at 0.5 hr, along with TCR-induced TFs, including RelA-p65. Furthermore, HIC1 binding sites were enriched primarily on the promoters of upregulated genes, suggesting that in iTreg cells, HIC1 acts as a transcriptional activator besides its known role as a repressor (Pinte et al., 2004).
HIC1 Is Induced by RA, and It Contributes to the Suppressive Activity of iTreg Cells
HIC1 was clearly upregulated at the protein level in iTreg cells, compared with Th0 cells. Importantly, HIC1 protein was detected as early as 12 hr, which preceded FOXP3 expression in iTreg cells (Figure 2A), suggesting that HIC1 is required early on in iTreg cell development program. To determine which differentiation factors induce HIC1 expression during early iTreg cell differentiation, we activated naive cells in the presence of different combinations of factors used for T helper (Th) and iTreg cell differentiation. Interestingly, all combinations containing RA resulted in upregulation of HIC1 expression (Figure S2B), indicating that HIC1 in iTreg cells is induced by RA. Furthermore, we observed a dose-dependent increase in HIC1 expression with increasing concentration of RA, confirming that RA induces HIC1 expression in iTreg cells (Figure 2B). HIC1 was similarly upregulated in human iTreg cell generated from peripheral blood (Figure S2C) as well as in mouse iTreg cells differentiated from splenic CD4+CD62L+ naive cells (Figure S2D).
Figure 2.

HIC1 Is Induced by RA, and It Contributes to the Suppressive Activity of iTreg Cells
(A) Cells were stimulated in Th0 or iTreg conditions for the indicated time points (hours), followed by HIC1 and FOXP3 measurement by WB. A representative blot from two biological replicates is shown.
(B) Cells were activated in presence of indicated concentrations of RA for 24 hr followed by measurement of HIC1 expression by WB. A representative image as well as mean of three biological replicates are shown. Error bars represent SE.
(C) Cells were nucleofected with three siRNAs against HIC1 (si1–3) and one non-targeting (NT), followed by a rest for 24 hr. Cells were then cultured for 72 hr in iTreg culture conditions, and HIC1 expression was measured by WB. A representative blot is shown at the bottom, and the bar chart shows the mean ± SE of four biological replicates. Significance was measured by two-tailed paired t test. ∗p < 0.05, ∗∗p < 0.01.
(D) FOXP3 expression was measured in HIC1-sufficient (red) and HIC1-deficient (blue) cells after 72 hr of culturing in iTreg conditions. Gray color shows isotype control. The bar charts show percentage FOXP3-positive cells (left) and median fluorescence intensity (MFI) (right) of three biological replicates (mean ± SE).
(E) Histogram plots showing the proliferation of responder cells at a responder/suppressor ratio of 1:0.5 after 72 hr of activation in absence (responder only) or presence of HIC1-sufficient (NT) or HIC1-deficient (siHIC1) iTreg cells. The number on the histogram plot shows the percentage of proliferating cells. The line plot shows the percentage of proliferating responder cells at different responder/suppressor ratios. The shaded area shows the minimum and maximum value of the three replicates. Significance was measured using two-tailed paired t test. ∗p < 0.05.
(F) Same as (E), except that the responder cells were cultured in the presence of activated T cells (Th0) instead of iTreg cells.
To study the function of HIC1 in iTreg cells, we silenced HIC1 with small interfering RNA (siRNA). Three previously published HIC1-specific siRNAs (Kumar, 2014) successfully knocked down HIC1 expression (Figure 2C). Because siRNA1 and siRNA3, overlapping different regions of the HIC1 sequence, were more efficient, we decided to use them as a pooled siRNA cocktail in the subsequent knockdown experiments. HIC1-deficient cells were normal in terms of both the percentage and extent of FOXP3 protein expression (Figure 2D). More important, however, the suppressive ability of these cells was severely reduced at all responder/suppressor ratios (Figure 2E). Conversely, silencing HIC1 had no effect on the suppressive ability of Th0 cells (Figure 2F). These findings suggested that HIC1 is an important contributor to the suppressive function of iTreg cells, and its role is independent of FOXP3.
To study if HIC1 is functionally relevant for Treg cells in vivo, we analyzed transcriptome data from published in vivo Treg cell studies from human and mouse. The expression of HIC1 in the CD4+ CD25+ Treg cells sorted from human peripheral blood was comparable with CD3+ CD4+ CD45RA+ CCR7+ CD27+ naive CD4+ T cells (Mold et al., 2010) both in the adult and in the fetus (Figure S2E: data plotted from GEO: GSE25087), suggesting that HIC1 is not upregulated in Treg cells in the blood. Because HIC1 is induced in response to RA, we postulated that HIC1 is induced in intestinal Treg cells, in which RA is available in the milieu. Analysis of data from a mouse study (GEO: GSE41229) revealed that Hic1 was indeed upregulated in the Treg cells from mLN but not in the Treg cells from spleen (Figure S2F) (Keerthivasan et al., 2014), suggesting a role for HIC1 in intestinal homeostasis.
HIC1 Deficiency Alters iTreg Cell Transcriptome and Prepares the Cells for Alternative Fates
To elucidate the mechanism of HIC1 action in iTreg cells, we performed RNA-seq on HIC1-deficient iTreg cells in three biological replicates at 48 and 72 hr post-cell differentiation. HIC1 expression was reduced in knockdown samples (Figure S3A). In total, 1,205 and 447 genes were DE at 48 and 72 hr, respectively (false discovery rate [FDR] < 0.05), indicating that HIC1 is a major regulator of iTreg cell priming (Table S3). The top DE genes (FDR < 0.05, |log2[fold change (FC)]| > 1) were similarly regulated at the two time points (Figure 3A). HIC1-deficient cells had altered expression of a variety of immune-related genes, including cytokines, cytokine receptors, and TFs (Figures 3B and 3C). Among the upregulated immune response-related genes at one or more times were seven HLA genes, cytokine/chemokine (IL1B, CXCL8, CXCL9, CXCL10), and their receptors (CCR1, CCR2, CCR5, CXCR3, and CXCR6) (Table S3). Among the downregulated cytokine/chemokine and their receptors were several tumor necrosis factor (TNF) factors and their receptors, as well as IL2 and IFNG. Among the TFs, ETV7, CREB3L3, CIITA, and PDLIM1 were upregulated, and TEAD4, IRF8, RORC, EGR2, and ETV4 were downregulated at both time points (Figure 3C; Table S3). CIITA upregulation was consistent with the increased expression of many HLA genes in HIC1-deficient cells. CIITA is required for Th1/2 differentiation (Patel et al., 2004, Patel et al., 2005), suggesting that increased expression of CIITA in HIC1-deficient iTreg cells may drive the cells to effector lineages. To further confirm this idea, we investigated TBET and GATA3 expression in HIC1-deficient iTreg cells. HIC1-deficient cells were cultured under iTreg cell condition for 3 days, and the expression of TBET and GATA3 was measured by western blotting (WB). Interestingly, both TBET and GATA3 were significantly upregulated (Figures 3D and S3C), suggesting that HIC1 deficiency makes the cells poised to differentiate to alternative lineages. Because of increased TBET expression, we hypothesized that HIC1-deficient iTreg cells when reactivated would produce higher interferon (IFN)-γ levels. Indeed, IFN-γ expression was significantly upregulated when HIC1-deficient iTreg cells were reactivated (Figures 3E and S3D). To investigate if there was a global loss of iTreg cell gene signature upon HIC1 silencing, we performed gene set enrichment analysis (GSEA). We defined the top (48 hr, FDR = 0.05, log2[FC] > 2) genes upregulated in iTreg cell condition in our time-series data as “iTreg cell signature genes” (163 genes). We ranked genes DE upon HIC1 silencing at 48 hr using Signal2Noise metric (Experimental Procedures). We then asked if iTreg cell signature genes lie at the top or bottom of the ranked list. The majority of iTreg cell signature genes were more abundant in the control samples than in the HIC1-deficient samples (Figure 3F), indicating that iTreg cell signature genes were lost in HIC1-deficient cells.
Figure 3.

HIC1 Deficiency Alters iTreg Cell Transcriptome and Positions the Cells for Alternative Fates
(A–C) Heatmaps showing the expression of top genes DE (FDR < 0.05, |log2[FC]| > 1) upon HIC1 silencing. siHIC1 and NT columns show expression (RPKM) in HIC1-deficient and HIC1-sufficient conditions, respectively. FC columns show fold change (iTreg/Th0) of the expression: (A) top DE genes (FDR = 0.05, |log2[FC]| > 1) that were common to the two time points; (B) cytokines and chemokines and their receptors at 48 and 72 hr; (C) TFs at 48 and 72 hr.
(D) NT- or siHIC1-treated cells were cultured in iTreg condition for 3 days, followed by WB. Blots were quantitated in ImageJ (NIH). Normalization was done by GAPDH. Log2(FC) was calculated with respect to NT samples. Significance was determined using two-tailed paired t test. ∗p < 0.05. Data from four biological replicates are shown.
(E) Boxplot showing IFN-γ expression in NT- or siHIC1-treated iTreg cells when reactivated after 72 hr with PMA and ionomycin. Data from three biological replicates are shown. Significance was determined using two-tailed paired t test. ∗p < 0.05.
(F) iTreg signature genes were top upregulated genes (log2FC > 2) in iTreg conditions compared with Th0 at 48 hr (Table S1). Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) was then used to test where the genes from this set lie in the ranked HIC1 knockdown (KD) 48 hr data. Each vertical line in the plot shows one gene. Lines toward the red indicate genes that are enriched in NT sample, and those toward the blue indicate enrichment in siHIC1 samples.
(G) Bar chart showing the NHGRI diseases and traits whose SNPs are enriched around (±100 kb) genes DE upon HIC1 silencing. All the diseases are enriched at an FDR of 0.05, and those that cross the dotted line are enriched at an FDR of 0.01.
We studied disease-associated SNPs near (±100 kb) genes DE upon HIC1 silencing. Almost all the autoimmune diseases that were enriched around iTreg DE genes were also enriched in genes DE upon HIC1 silencing (Figure 3G). The analysis suggests that HIC1 indeed regulates the expression of a significant fraction of immune related genes in iTreg cells.
Although FOXP3 protein expression was unaffected by HIC1 silencing (Figure 2D), FOXP3 mRNA was slightly upregulated at 72 hr (Table S3). Furthermore, HIC1 silencing did not alter the expression of different FOXP3 isoforms (Figure S3E). Pathway analysis of genes DE upon HIC1 silencing suggested that many signaling pathways, particularly VDR/RXR activation as well as Nur77 signaling pathways, are perturbed in HIC1-deficient cells (Figure S3F). TFBS enrichment analysis on the promoters of DE genes in HIC1-deficient cells suggested that many of the TFBS that were enriched on the promoters of upregulated iTreg genes (Figure S2A) were also enriched on the promoters of genes downregulated upon HIC1 silencing (Figure S3G; Table S3). This category of TFs includes EGR1, EGR3, HIF1A, Ikaros, AHR, and REST. Taken together, the transcriptome of HIC1-deficient iTreg cells suggests that HIC1 deficiency alters the transcriptome of iTreg cells and prepares the cells for effector-like functions.
Chromatin Immunoprecipitation Sequencing Analysis Identifies Genome-wide Binding Sites of HIC1 during iTreg Cell Differentiation
Fractionation studies showed that HIC1 translocated to the nucleus during iTreg differentiation at both the 72 and 120 hr time points (Figure 4A). This was further validated by fluorescent microscopy, with which we found HIC1 translocation already at 24 hr (Figure S4A). We performed chromatin immunoprecipitation sequencing (ChIP-seq) analysis in iTreg cells (72 hr). Library complexity analysis revealed that the ChIP-seq libraries had 15 million to 20 million unique reads (Figure S4B). ChIP-seq analysis identified 16,424 and 31,604 HIC1 binding peaks in the two replicates, with 6,628 peaks common between the two replicates (irreproducibility discovery rate [IDR] analysis [Li et al., 2011], q < 0.1; Table S4). Approximately two thirds of HIC1 binding sites were located in intronic and distal intergenic regions, suggesting that HIC1 potentially regulates the expression of genes by binding to these distal regulatory elements (Figure 4B). About 18% of the binding sites were located at 5ʹUTR, the transcription start site (TSS), and promoter regions (Figure 4B). Further analysis of distance between peaks and nearest TSS revealed that 49% of the HIC1 peaks were found within 10 kb up- or downstream of the nearest TSS (Figure 4C), indicating that HIC1 is likely recruited to these regions in iTreg cells to directly regulate the transcriptional activity of the neighboring genes. Moreover, de novo TF motif analysis at HIC1 peaks revealed binding sites for several TFs, with motifs for AP1, ETS1, HIC1, RUNX1, and NFKB2 being the top five (Figure 4D; Table S4).
Figure 4.

ChIP-Seq Analysis Identifies Genome-wide Binding Sites of HIC1 during iTreg Cell Differentiation
(A) Blots showing HIC1 localization from fractionation experiments (representative of three replicates) with Th0 and iTreg cells at the indicated times. ⍺-Tubulin and LSD1 were used as loading controls for cytoplasmic and nuclear fractions, respectively.
(B) Pie chart showing the genomic distribution of HIC1 peaks at 72 hr of iTreg polarization detected using MACS on ChIP-seq data.
(C) Histogram plot showing the distribution of HIC1 peaks around transcriptional start sites.
(D) Top five motifs detected in de novo motif discovery using the Homer tool. Motif discovery was done on peaks with IDRs < 0.1.
(E) Venn diagram showing the overlap of HIC1 binding sites from ChIP-seq experiments and genes that are DE upon HIC1 silencing. The associated bar chart shows the direction of differential expression of overlapped genes (direct targets). TFs among the direct targets are also plotted separately.
(F) Heatmap showing the expression of HIC1 direct targets upon HIC1 silencing: the top (logFC < –1 or > 1) DE genes (left) as well as TFs (right).
(G) University of California, Santa Cruz (UCSC), Genome Browser shots for the key genes that are upregulated (top) or downregulated (bottom) upon HIC1 silencing.
(H) Pathway analysis of direct targets genes. Analysis was done using Ingenuity Pathway Analysis (IPA).
To identify direct and indirect targets of HIC1, ChIP-seq results were overlapped with the RNA-seq of HIC1-deficient iTreg cells at 48 and 72 hr (Table S3). Altogether, 449 genes were directly regulated (had a neighboring HIC1 binding peaks and were DE upon HIC1 silencing) by HIC1, with 57 and 299 genes regulated specifically at 48 and 72 hr, respectively, and 93 genes at both time points (Figure 4E, left; Table S4). Functional annotation of HIC1 direct targets suggested that they include transcriptional regulators as well as kinases and phosphatases (Figures S4C and S4D). On the basis of enrichment of HIC1 binding sites on the promoters of iTreg DE genes (Figure S2A), we predicted that HIC1 would bind to both up- and downregulated genes. Indeed, HIC1 was bound near the promoters of both up- and downregulated genes in iTreg cells (Figure 4E, right; Table S3). HIC1 directly bound and positively regulated the expression of several important Treg cell-associated genes, including DUSP2, DUSP5, TIGIT, and CTLA4. Interestingly, HIC1 also bound and negatively regulated genes involved in effector T cell functions, such as CCR2, CCR5, CXCR3, and CXCR6 (Figures 4F and 4G).
To study the connectivity of HIC1 to its direct and indirect target TFs (Figures S5A and S5B), we constructed a HIC1-TF interaction network (Supplemental Experimental Procedures). The TFs were highly connected and formed an integrated network (Figure S5C). TFs that positively regulate Treg cell differentiation and function, including ID2, SMAD3, NR4A, and EGR family TFs, CREM, REL and IRF8, were among the highly connected nodes in the network and may form positive regulatory module. Conversely, TFs CIITA, E2F1, and JUN may form a negative regulatory module during iTreg cell differentiation. Several Th cells related to signaling pathways, including TCR signaling, Th1/Th2 pathways, NF-kB signaling, CXCR4 signaling, and STAT3 signaling, were enriched among HIC1 direct target genes (Figure 4H). Altogether, these findings strongly indicate that HIC1 uses both positive and negative gene regulatory modules to program iTreg cell differentiation.
SNPs Associated with Autoimmune Diseases Overlap with HIC1 Binding Sites and Alter HIC1 DNA Binding Ability
A SNP at the HIC1 motif could result in a gain or loss of HIC1 binding that may alter the target gene expression and contribute to disease. Therefore, we examined autoimmune-associated SNPs that specifically fall within the HIC1 peak (Supplemental Experimental Procedures). Interestingly, SNPs associated with 6 of 11 autoimmune diseases tested were enriched within the peak regions with maximum number of SNPs found for CrD followed by RhA and ulcerative colitis (UC) (Figure 5A). The analysis suggested that HIC1 ChIP-seq peaks did harbor SNPs that are relevant in autoimmune disease. To take it to the next level, we asked if autoimmune-disease-associated SNPs fall exactly within the HIC1 motif. Three proxy SNPs in linkage disequilibrium (LD) with four lead SNPs overlapped with HIC1 motif within the peak regions (Figure 5B) potentially disrupting the binding of HIC1 to the target sites. Of the three SNPs, one each fell into promoter (PLAU), intronic (IRF5), and intergenic (TRAF1) regions (Figure 5B). Interestingly, PLAU (plasminogen activator, urokinase) was identified as a key Treg gene that regulated FOXP3 expression and was required for suppressive activity (He et al., 2012).
Figure 5.

SNPs Associated with Autoimmune Diseases Overlap with HIC1 Binding Sites and Alter the HIC1 DNA Binding Ability
(A) Enrichment of autoimmune-associated SNPs at HIC1 binding sites compared with a random set of background SNPs.
(B) SNPs overlapping the HIC1 motif (Experimental Procedures). rsID, overlapping SNP ID; proxyTo, lead SNP to which overlapping SNP is a proxy; Chr, chromosome; location, SNP location; major, major allele; minor, minor allele; disease, disease to which lead SNP is attributed; motif, motif sequence (SNPs in red); motifStart, location of motif start; motifStrand, strand info.
(C) Two DNA sequences with HIC1 binding site (HIC1 wild-type [WT]) and corresponding negative control DNA sequences in which the HIC1 motif has been mutated (HIC1 MT) were used as controls (Table S5). Data shown here are a representative of three biological replicates. Significance was determined with one-tailed paired t test. ∗p < 0.05.
To confirm whether these SNPs could change HIC1 binding, we performed DNA affinity precipitation assays (DAPAs). HIC1 was bound to oligonucleotides with reference genome as bait sequence; however, the binding was reduced in all the oligonucleotides with mutations corresponding to disease-associated SNPs (Figure 5C). Thus, we conclude that disease-associated SNPs alter HIC1 binding to DNA in vitro. These findings suggest a potential mechanism for how a disease-associated genetic variant modulates HIC1-mediated gene regulation and affects the expression of its target gene.
Discussion
HIC1 is a member of the POK/ZBTB protein family that includes several TFs. HIC1 has been extensively studied in the context of cancer. It acts as a tumor suppressor, and it is involved in several major processes of carcinogenesis, including cell growth and survival and cell migration (Rood and Leprince, 2013). HIC1 is probably most relevant for the function of intestinal Treg cells, as the Treg cells from blood had HIC1 level comparable with that of naive cells. A recent in vivo study showed that HIC1 expression indeed was restricted to intestinal immune cells. HIC1 deletion did not affect FOXP3 expression in CD4+ T cells, but it was required for intestinal homeostasis, as T cell specific deletion of HIC1 led to increased Th17 bias in lamina propria and mLN (Burrows et al., 2017). Our findings, along with these studies, suggest that HIC1 may play an important role in intestinal homeostasis by maintaining Treg cell suppressive ability to sustain tolerance to innocuous antigens.
NR4A receptors were among the TFs that, like HIC1, were upregulated in iTreg cells but downregulated in Th17 cells. NR4A1-3 receptors are required for the thymic Treg cell developmental program via directly binding to the promoter of Foxp3 (Sekiya et al., 2013). Other potential TFs that may regulate the development and function of iTreg cells are PDLIM1, NFKBID, EPAS1, and ZBTB32, as they had profiles similar to HIC1. PDLIM1 and NFKBID are particularly interesting, as they regulate NF-κB signaling by sequestering p65 subunit in cytoplasm (PDLIM1) (Ono et al., 2015) or by inhibiting NF-κB in the nucleus by acting as atypical IκB (NFKBID) (Schuster et al., 2013). EPAS1 is a direct STAT6 target and it regulated STAT6-mediated RUNX1 expression by direct binding to RUNX1 promoter (O’Shea et al., 2011). ZBTB32, which is another member of HIC1 family, controls the proliferative burst of natural killer (NK) cells in response to infection (Beaulieu et al., 2014).
A recent study reported a RARE element conserved between mouse and human on the promoter of HIC1 (Hassan et al., 2017). Moreover, we analyzed the HIC1 flanking regions for TFBS using MATCH tool in TRANSFAC and found a few RARE elements within an intron of the SMG6 gene downstream of HIC1. Hassan et al. (2017) found that the RARE sites on the promoter and downstream interact to enhance the transcription of HIC1. These data suggest that RA directly regulates HIC1 expression by directly binding to RARE elements near HIC1.
Several mechanisms may explain the lack of suppressive ability in HIC1-deficient iTreg cells. HIC1 silencing led to upregulation of several TFs, including CIITA, TBET, GATA3, BATF, and MAF, which support effector T cell differentiation. ChIP-seq data show that HIC1 directly binds to the upstream of these TFs and, therefore, may directly suppress their expression in iTreg. Similar to B cells (Zeng et al., 2016), HIC1 repressed CIITA expression in iTreg cells (Figure 3C). Importantly, constitutive expression of CIITA in CD4 T cells leads to regulation and induction of Th2 cytokine production (Patel et al., 2005) and expression of major histocompatibility complex (MHC) class II molecules. Consistent with increased CIITA expression, several HLA genes were also upregulated upon HIC1 deficiency (Figure 3B). Thus, in line with these findings, our results suggest that increased HIC1 expression in iTreg cells may limit the Th1/2 inflammatory response by keeping CIITA expression low.
HIC1 may also regulate the suppressive ability of iTreg cells through RORC. RORC-expressing Treg cells have enhanced suppressive capacity in intestinal inflammation (Yang et al., 2016). RA in the intestinal microenvironment may facilitate the induction of RORC. In germ-free mice in which the intestinal microbiota is compromised, both Treg cell numbers and their efficacy decline as exhibited by reduced expression of CTLA4, ICOS, and IL10 (Tanoue et al., 2016). Interestingly, in our data, CTLA4 expression was reduced upon HIC1 silencing both at 48 and 72 hr (Figure S3B). We show that RORC is a direct target of HIC1, as the latter binds to RORC promoter during iTreg cell development, and RORC was downregulated in HIC1-silenced cells at both times (Figures 3A and 3C), suggesting that HIC1 is a positive regulator of RORC.
Several glycolytic enzymes, including glucokinase (GCK), the enzyme required for the first step of glycolysis, were significantly downregulated in HIC1-deficient cells (Table S3), suggesting that these cells lost the “iTreg type” metabolic profile given that Treg cells preferentially use glycolysis as a source of energy (Michalek et al., 2011).
To date, only a dozen HIC1 direct target genes have been identified (Rood and Leprince, 2013). We found that HIC1 was bound in the vicinity of several genes, including CTLA4, PD1, TIGIT, EBI3, EGR2/3, NR4A receptors, ID2/3, and IRF8, that contribute to Treg cell function (Collison et al., 2007, Curran et al., 2010, Joller et al., 2014, Miyazaki et al., 2014, Morita et al., 2016, Sekiya et al., 2013). The analysis of SNPs within the HIC1 motif identified PLAU as a candidate that can have perturbed expression because of altered HIC1 binding to its promoter. PLAU was not DE in iTreg cells, but its receptor PLAUR was upregulated in iTreg cells at 72 hr compared with Th0. Further studies are required to understand the regulation of PLAU expression by HIC1 and the role of the SNP. Together, our results identify HIC1 as a crucial factor regulating iTreg development and function. Mechanistically, HIC1 binds to the promoters of TFs required for Th1/2/17 cell development and represses their transcription in iTreg cells.
Experimental Procedures
CD4+ Cell Isolation and Differentiation to iTreg Cells
CD4+ T cells were isolated from human umbilical cord blood as described previously (Hawkins et al., 2013). CD25 depletion was performed using LD columns and a CD25 depletion kit (Miltenyi Biotec). CD4+CD25− cells from multiple donors (three or more) were activated directly or pooled before activation with plate-bound anti-CD3 (500 ng/24-well culture plate well; Immunotech) and soluble anti-CD28 (500 ng/mL; Immunotech) at a density of 2 × 106 cells/mL of X-vivo 15 serum-free medium (Lonza). For iTreg differentiation, the medium was supplemented with IL-2 (12 ng/mL), TGF-β (10 ng/mL) (both from R&D Systems), all-trans retinoic acid (ATRA) (10 nM; Sigma-Aldrich), and human serum (10%) and cultured at 37°C in 5% CO2. Control Th0 cells were stimulated with plate-bound anti-CD3 soluble anti-CD28 X-vivo 15 serum-free medium without cytokines.
Suppression Assays
To evaluate the ability of a candidate population to suppress the proliferation of an effector population (responder cells), we used a mixed lymphocyte reaction (MLR). Responder cells (Tres) were CD4+CD25− cells isolated from a peripheral blood buffy coat with the Dynal CD4+ Isolation Kit (Invitrogen) and CD25 depletion kit (Miltenyi Biotec). To reduce the variability between different responder cell populations, a set of responder cells were isolated and stored at –80°C in freezing medium (90% fetal calf serum [FCS] and 10% DMSO). On day 1 of the assay, responder cells were labeled with cell trace violet (EF670/CTV; Thermo Fisher Scientific). Fifty thousand responder cells were cultured in different wells in the presence of Th0 or iTreg cells in ratios of 1:1, 1:0.5, 1:0.25, 1:0.125, and 1:0. The division of responder cells was analyzed by dye dilution at the end of the assay (day 4). Suppression was calculated at 1:0.25 or 1:5 ratios. The percentage suppression was calculated using the following formula: % suppression = [% of dividing cells (Tres-iTreg)/% of dividing cells in Tres] × 100.
siRNA-Mediated Gene Knockdown
Four million cells were transfected with 300 pmol of siRNA in 100 μl of OptiMEM media using Amaxa nucleofector device (Lonza). For details, see Supplemental Experimental Procedures.
Intracellular Staining and Flow Cytometry
Intracellular staining was performed using buffer sets of Human Regulatory T Cell Staining Kit (eBioscience/Thermo Fisher Scientific), following the manufacturer’s protocol. For intracellular staining of IFN-γ, HIC1-deficient and HIC1-sufficient iTreg cells were first activated with phorbol myristate acetate (PMA) and ionomycin for 4 hr followed by treatment of brefeldin A for 90 min. The antibodies are listed in Supplemental Experimental Procedures.
RNA Isolation, RNA-Seq Sample Preparation, and Data Analysis
RNA was isolated using the RNeasy Mini Kit (QIAGEN). Libraries for RNA-seq were prepared using the Illumina TruSeq Stranded mRNA Sample Preparation Kit. The reads were aligned using tophat2, and counts were determined using HTSeq-count. DE calling was performed using edgeR. For details, please see Supplemental Experimental Procedures.
Chromatin Immunoprecipitation Assay and ChIP-Seq
ChIP assay was performed as described previously (Hawkins et al., 2013). For details, see Supplemental Experimental Procedures.
DAPA
DAPA experiments were performed as described previously with minor modifications (Hawkins et al., 2013). Details can be found in Supplemental Experimental Procedures.
Analysis of TFBS
Overrepresentation of TFBS on the promoters of DE genes was performed using the commercial version of an FMatch tool at the TRANSFAC database (Release 2017.2). For details, see Supplemental Experimental Procedures.
Pathway Analysis
Pathway analysis was performed using Ingenuity Pathway Analysis (IPA) (https://www.ingenuity.com; May 2016). Both overrepresentation (positive Z score) and underrepresentation (negative Z score) were calculated. Benjamini-Hochberg corrected p values < 0.05 was considered to indicate significance.
GSEA
GSEA was performed using the tool from the Broad Institute (Subramanian et al., 2005). For details, see Supplemental Experimental Procedures.
Statistical Methods
For comparison of means, two-tailed paired t tests were performed, and p values < 0.05 were considered to indicate significance unless otherwise stated. The Benjamini-Hochberg method was used for multiple hypothesis correction whenever applicable. The statistical methods used for SNP analysis enrichment analysis, pathways analysis, and other procedures are described the respective sections.
Ethical Approval
The use of the blood of unknown donors was approved by the Ethics Committee of the Hospital District of Southwest Finland (24.11.1998 article 323). Mouse studies were conducted according to the guidelines of Provincial Government of Southern Finland and handled in accordance with the institutional animal care policies of University of Turku (license KEK/2010-1512). Female wild-type mice aged 8–12 weeks of C57BL/6 background were used for the experiments in this study.
Acknowledgments
This study was supported by Academy of Finland (AoF) grants 256355, 258313, 259942, 292482, and 294337; by the AoF Centre of Excellence in Molecular Systems Immunology and Physiology Research 2012–2017 grant 250114; AoF Terva grant 314444; and by grants from the Sigrid Jusélius Foundation (SJF), the Paulo Foundation, and the Finnish Cancer Foundation. This work was supported by the AOF and the German Research Foundation (DFG; WI1722/12-1) Immunology Initiative “Systems Biology Approach to Molecular Mechanisms of Human TGF-β Induced iTreg Cell Differentiation and the Role of iTreg in Multiple Sclerosis” to R.L., H.L., and H.W., as well as DFG Collaborative Research Centre CRC/SFB128 “Initiating/Effector versus Regulatory Mechanisms in Multiple Sclerosis – Progress towards Tackling the Disease,” project A09, to H.W. and C.C.G. U.B. is funded by the Cluster of Excellence Cells in Motion (CiM). L.L.E. has received grants from the European Research Council (ERC; grant 677943), the European Union’s Horizon 2020 research and innovation program (grant 675395), the AoF (grants 296801 and 304995), the Juvenile Diabetes Research Foundation (JDRF; grant 2-2013-32), the Finnish Funding Agency for Innovation (TEKES; grant 1877/31/2016), and SJF. We thank all the volunteer blood donors and personnel of Turku University Hospital, Department of Obstetrics and Gynecology, Maternity Ward Hospital District of Southwest Finland for the cord blood collection. Lysates from mouse Th0/iTreg cells were kindly provided by Tanja Buchacher and Mohd Moin Khan. We acknowledge Marjo Hakkarinen for technical help with the experiments and Bridget Palmer for critical reading of the manuscript. This study was supported by the Finnish Functional Genomics Centre, the University of Turku, Åbo Akademi University, and Biocenter Finland.
Author Contributions
U.U. designed the study, analyzed the data, prepared the figures, and wrote the manuscript. S.B.A.A. designed and performed the experiments, analyzed and interpreted the data, and contributed to writing the manuscript. S.K.T. designed and performed the experiments, analyzed data, prepared figures, and wrote part of the manuscript. O.D. designed and performed the experiments and analyzed the data. S.R. analyzed RNA-seq data and prepared figures. K.K. analyzed ChIP-seq data and prepared figures. C.C.G. provided expertise in setting up iTreg cultures. S.L. designed and performed the experiments and analyzed the data. K.B. designed and performed the experiments and analyzed the data. J.T. performed experiments and analyzed data. U.B. provided expertise in setting up iTreg cultures. D.C. analyzed networks. L.L.E. supervised D.C. H.L. supervised S.R. and K.K. H.W. supervised C.C.G. and U.B. O.R. designed the experiments, analyzed the data, and wrote the manuscript. R.L. designed and supervised the study and wrote the manuscript.
Declaration of Interests
C.C.G. has received speaking honoraria and travel expenses for attending meeting for Biogen, Genzyme, Novartis Pharma, and Bayer Health Care. Her work is funded by the German Ministry for Education and Research (BMBF; 01GI1603A) and the German Research Foundation (DFG; GR3946/3-1 and SFB128 A09). H.W. receives honoraria for acting as a member of scientific advisory boards and as consultant for Biogen, Evgen, MedDay Pharmaceuticals, Merck Serono, Novartis, Roche Pharma, and Sanofi-Genzyme and speaking honoraria and travel support from Alexion, Biogen, Cognomed, F. Hoffmann-La Roche, Gemeinnützige Hertie-Stiftung, Merck Serono, Novartis, Roche Pharma, Sanofi-Genzyme, Teva, and WebMD Global. H.W. acts as a paid consultant for Abbvie, Actelion, Biogen, IGES, Novartis, Roche, Sanofi-Genzyme, and the Swiss Multiple Sclerosis Society. Research by H.W. is funded by the BMBF (01FI1601E, 01GI1603A, and O1GI1603D), DFG (SFB128 A09, A10, Z02, V and SFB1009 A03), Else Kröner Fresenius Foundation, Fresenius Foundation, Hertie Foundation, NRW Ministry of Education and Research, Interdisciplinary Center for Clinical Studies (IZKF) Muenster and RE Children’s Foundation, Biogen, GlaxoSmithKline, Roche Pharma, and Sanofi-Genzyme. All other authors have no financial interest related to this work.
Published: February 20, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and five tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.01.070.
Data and Software Availability
The accession numbers for the RNA-seq and ChIP-seq data reported in this paper are GEO: GSE90570 and GSE99889, respectively.
Supplemental Information
References
- Abbas A.K., Benoist C., Bluestone J.A., Campbell D.J., Ghosh S., Hori S., Jiang S., Kuchroo V.K., Mathis D., Roncarolo M.G. Regulatory T cells: recommendations to simplify the nomenclature. Nat. Immunol. 2013;14:307–308. doi: 10.1038/ni.2554. [DOI] [PubMed] [Google Scholar]
- Allan S.E., Crome S.Q., Crellin N.K., Passerini L., Steiner T.S., Bacchetta R., Roncarolo M.G., Levings M.K. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int. Immunol. 2007;19:345–354. doi: 10.1093/intimm/dxm014. [DOI] [PubMed] [Google Scholar]
- Arvey A., van der Veeken J., Samstein R.M., Feng Y., Stamatoyannopoulos J.A., Rudensky A.Y. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat. Immunol. 2014;15:580–587. doi: 10.1038/ni.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu A.M., Zawislak C.L., Nakayama T., Sun J.C. The transcription factor Zbtb32 controls the proliferative burst of virus-specific natural killer cells responding to infection. Nat. Immunol. 2014;15:546–553. doi: 10.1038/ni.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett C.L., Christie J., Ramsdell F., Brunkow M.E., Ferguson P.J., Whitesell L., Kelly T.E., Saulsbury F.T., Chance P.F., Ochs H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- Bluestone J.A., Buckner J.H., Fitch M., Gitelman S.E., Gupta S., Hellerstein M.K., Herold K.C., Lares A., Lee M.R., Li K. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015;7:315ra189. doi: 10.1126/scitranslmed.aad4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows K., Antignano F., Bramhall M., Chenery A., Scheer S., Korinek V., Underhill T.M., Zaph C. The transcriptional repressor HIC1 regulates intestinal immune homeostasis. Mucosal Immunol. 2017;10:1518–1528. doi: 10.1038/mi.2017.17. [DOI] [PubMed] [Google Scholar]
- Chen W.Y., Wang D.H., Yen R.C., Luo J., Gu W., Baylin S.B. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437–448. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Collison L.W., Workman C.J., Kuo T.T., Boyd K., Wang Y., Vignali K.M., Cross R., Sehy D., Blumberg R.S., Vignali D.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- Coombes J.L., Siddiqui K.R.R., Arancibia-Cárcamo C.V., Hall J., Sun C.-M., Belkaid Y., Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran M.A., Montalvo W., Yagita H., Allison J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. U S A. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPaolo R.J., Brinster C., Davidson T.S., Andersson J., Glass D., Shevach E.M. Autoantigen-specific TGFbeta-induced Foxp3+ regulatory T cells prevent autoimmunity by inhibiting dendritic cells from activating autoreactive T cells. J. Immunol. 2007;179:4685–4693. doi: 10.4049/jimmunol.179.7.4685. [DOI] [PubMed] [Google Scholar]
- Farh K.K., Marson A., Zhu J., Kleinewietfeld M., Housley W.J., Beik S., Shoresh N., Whitton H., Ryan R.J.H., Shishkin A.A. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–343. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro A., D’Alise A.M., Raj T., Asinovski N., Phillips R., Ergun A., Replogle J.M., Bernier A., Laffel L., Stranger B.E. Interindividual variation in human T regulatory cells. Proc. Natl. Acad. Sci. U S A. 2014;111:E1111–E1120. doi: 10.1073/pnas.1401343111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot J.D., Gavin M.A., Rudensky A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt S.I., Ahrendt M., Bode U., Wahl B., Kremmer E., Förster R., Pabst O. Stromal mesenteric lymph node cells are essential for the generation of gut-homing T cells in vivo. J. Exp. Med. 2008;205:2483–2490. doi: 10.1084/jem.20080039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan H.M., Kolendowski B., Isovic M., Bose K., Dranse H.J., Sampaio A.V., Underhill T.M., Torchia J. Regulation of active DNA demethylation through RAR-mediated recruitment of a TET/TDG complex. Cell Rep. 2017;19:1685–1697. doi: 10.1016/j.celrep.2017.05.007. [DOI] [PubMed] [Google Scholar]
- Hawkins R.D., Larjo A., Tripathi S.K., Wagner U., Luu Y., Lönnberg T., Raghav S.K., Lee L.K., Lund R., Ren B. Global chromatin state analysis reveals lineage-specific enhancers during the initiation of human T helper 1 and T helper 2 cell polarization. Immunity. 2013;38:1271–1284. doi: 10.1016/j.immuni.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F., Chen H., Probst-Kepper M., Geffers R., Eifes S., Del Sol A., Schughart K., Zeng A.-P., Balling R. PLAU inferred from a correlation network is critical for suppressor function of regulatory T cells. Mol. Syst. Biol. 2012;8:624. doi: 10.1038/msb.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J.A., Feuerer M., Tash K., Haxhinasto S., Perez J., Melamed R., Mathis D., Benoist C. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. 2007;27:786–800. doi: 10.1016/j.immuni.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Hippen K.L., Merkel S.C., Schirm D.K., Nelson C., Tennis N.C., Riley J.L., June C.H., Miller J.S., Wagner J.E., Blazar B.R. Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. Am. J. Transplant. 2011;11:1148–1157. doi: 10.1111/j.1600-6143.2011.03558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huter E.N., Punkosdy G.A., Glass D.D., Cheng L.I., Ward J.M., Shevach E.M. TGF-beta-induced Foxp3+ regulatory T cells rescue scurfy mice. Eur. J. Immunol. 2008;38:1814–1821. doi: 10.1002/eji.200838346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka-Koga M., Nakatsukasa H., Ito M., Akanuma T., Lu Q., Yoshimura A. Induction and maintenance of regulatory T cells by transcription factors and epigenetic modifications. J. Autoimmun. 2017;83:113–121. doi: 10.1016/j.jaut.2017.07.002. [DOI] [PubMed] [Google Scholar]
- Joller N., Lozano E., Burkett P.R., Patel B., Xiao S., Zhu C., Xia J., Tan T.G., Sefik E., Yajnik V. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. 2014;40:569–581. doi: 10.1016/j.immuni.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamori M., Nakatsukasa H., Okada M., Lu Q., Yoshimura A. Induced regulatory T cells: their development, stability, and applications. Trends Immunol. 2016;37:803–811. doi: 10.1016/j.it.2016.08.012. [DOI] [PubMed] [Google Scholar]
- Keerthivasan S., Aghajani K., Dose M., Molinero L., Khan M.W., Venkateswaran V., Weber C., Emmanuel A.O., Sun T., Bentrem D.J. β-Catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in T cells. Sci. Transl. Med. 2014;6:225ra28. doi: 10.1126/scitranslmed.3007607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczma M., Podolsky R., Garge N., Daniely D., Pacholczyk R., Ignatowicz L., Kraj P. Foxp3-deficient regulatory T cells do not revert into conventional effector CD4+ T cells but constitute a unique cell subset. J. Immunol. 2009;183:3731–3741. doi: 10.4049/jimmunol.0800601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S. P53 induction accompanying G2/M arrest upon knockdown of tumor suppressor HIC1 in U87MG glioma cells. Mol Cell Biochem. 2014;395:281–290. doi: 10.1007/s11010-014-2137-9. [DOI] [PubMed] [Google Scholar]
- Li Q., Brown J.B., Huang H., Bickel P.J. Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat. 2011;5:1752–1779. [Google Scholar]
- Lu L., Zhou X., Wang J., Zheng S.G., Horwitz D.A. Characterization of protective human CD4 + CD25 + FOXP3 + regulatory T cells generated with IL-2, TGF- b and retinoic acid. PLoS ONE. 2010;5:e15150. doi: 10.1371/journal.pone.0015150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Wang J., Gu J., Lu H., Li X., Qian X., Liu X., Wang X., Zhang F., Lu L. Rapamycin regulates iTreg function through CD39 and Runx1 pathways. J. Immunol. Res. 2014;2014:989434. doi: 10.1155/2014/989434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek R.D., Gerriets V.A., Jacobs S.R., Macintyre A.N., MacIver N.J., Mason E.F., Sullivan S.A., Nichols A.G., Rathmell J.C. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M., Miyazaki K., Chen S., Itoi M., Miller M., Lu L.F., Varki N., Chang A.N., Broide D.H., Murre C. Id2 and Id3 maintain the regulatory T cell pool to suppress inflammatory disease. Nat. Immunol. 2014;15:767–776. doi: 10.1038/ni.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mold J.E., Venkatasubrahmanyam S., Burt T.D., Michaëlsson J., Rivera J.M., Galkina S.A., Weinberg K., Stoddart C.A., McCune J.M. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science. 2010;330:1695–1699. doi: 10.1126/science.1196509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K., Okamura T., Inoue M., Komai T., Teruya S., Iwasaki Y., Sumitomo S., Shoda H., Yamamoto K., Fujio K. Egr2 and Egr3 in regulatory T cells cooperatively control systemic autoimmunity through Ltbp3-mediated TGF-β3 production. Proc. Natl. Acad. Sci. U S A. 2016;113:E8131–E8140. doi: 10.1073/pnas.1611286114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie H., Zheng Y., Li R., Guo T.B., He D., Fang L., Liu X., Xiao L., Chen X., Wan B. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-α in rheumatoid arthritis. Nat. Med. 2013;19:322–328. doi: 10.1038/nm.3085. [DOI] [PubMed] [Google Scholar]
- O’Shea J.J., Lahesmaa R., Vahedi G., Laurence A., Kanno Y. Genomic views of STAT function in CD4+ T helper cell differentiation. Nat. Rev. Immunol. 2011;11:239–250. doi: 10.1038/nri2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M., Yaguchi H., Ohkura N., Kitabayashi I., Nagamura Y., Nomura T., Miyachi Y., Tsukada T., Sakaguchi S. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446:685–689. doi: 10.1038/nature05673. [DOI] [PubMed] [Google Scholar]
- Ono R., Kaisho T., Tanaka T. PDLIM1 inhibits NF-κB-mediated inflammatory signaling by sequestering the p65 subunit of NF-κB in the cytoplasm. Sci. Rep. 2015;5:18327. doi: 10.1038/srep18327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D.R., Kaplan M.H., Chang C.H. Altered Th1 cell differentiation programming by CIITA deficiency. J. Immunol. 2004;173:5501–5508. doi: 10.4049/jimmunol.173.9.5501. [DOI] [PubMed] [Google Scholar]
- Patel D.R., Li W., Park J.S., Sofi M.H., Gourley T.S., Hangoc G., Kaplan M.H., Chang C.H. Constitutive expression of CIITA directs CD4 T cells to produce Th2 cytokines in the thymus. Cell. Immunol. 2005;233:30–40. doi: 10.1016/j.cellimm.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Pinte S., Stankovic-Valentin N., Deltour S., Rood B.R., Guérardel C., Leprince D. The tumor suppressor gene HIC1 (hypermethylated in cancer 1) is a sequence-specific transcriptional repressor: definition of its consensus binding sequence and analysis of its DNA binding and repressive properties. J. Biol. Chem. 2004;279:38313–38324. doi: 10.1074/jbc.M401610200. [DOI] [PubMed] [Google Scholar]
- Procaccini C., Carbone F., Di Silvestre D., Brambilla F., De Rosa V., Galgani M., Faicchia D., Marone G., Tramontano D., Corona M. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity. 2016;44:406–421. doi: 10.1016/j.immuni.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rood B.R., Leprince D. Deciphering HIC1 control pathways to reveal new avenues in cancer therapeutics. Expert Opin. Ther. Targets. 2013;17:811–827. doi: 10.1517/14728222.2013.788152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A., Eriksson M., Shang M.M., Weyd H., Tegnér J. Comparative analysis of protocols to induce human CD4+Foxp3+ regulatory T cells by combinations of IL-2, TGF-beta, retinoic acid, rapamycin and butyrate. PLoS ONE. 2016;11:e0148474. doi: 10.1371/journal.pone.0148474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster M., Annemann M., Plaza-sirvent C., Schmitz I. Atypical IkB proteins—nuclear modulators of NF-kB signaling. Cell Commun. Signal. 2013;11:1–11. doi: 10.1186/1478-811X-11-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya T., Kashiwagi I., Yoshida R., Fukaya T., Morita R., Kimura A., Ichinose H., Metzger D., Chambon P., Yoshimura A. Nr4a receptors are essential for thymic regulatory T cell development and immune homeostasis. Nat. Immunol. 2013;14:230–237. doi: 10.1038/ni.2520. [DOI] [PubMed] [Google Scholar]
- Shevach E.M., Thornton A.M. tTregs, pTregs, and iTregs: similarities and differences. Immunol. Rev. 2014;259:88–102. doi: 10.1111/imr.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto N., Oida T., Hirota K., Nakamura K., Nomura T., Uchiyama T., Sakaguchi S. Foxp3-dependent and -independent molecules specific for CD25+CD4+ natural regulatory T cells revealed by DNA microarray analysis. Int. Immunol. 2006;18:1197–1209. doi: 10.1093/intimm/dxl060. [DOI] [PubMed] [Google Scholar]
- Sun C.-M., Hall J.A., Blank R.B., Bouladoux N., Oukka M., Mora J.R., Belkaid Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoue T., Atarashi K., Honda K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 2016;16:295–309. doi: 10.1038/nri.2016.36. [DOI] [PubMed] [Google Scholar]
- Tone Y., Furuuchi K., Kojima Y., Tykocinski M.L., Greene M.I., Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat. Immunol. 2008;9:194–202. doi: 10.1038/ni1549. [DOI] [PubMed] [Google Scholar]
- Tuomela S., Rautio S., Ahlfors H., Öling V., Salo V., Ullah U., Chen Z., Hämälistö S., Tripathi S.K., Äijö T. Comparative analysis of human and mouse transcriptomes of Th17 cell priming. Oncotarget. 2016;7:13416–13428. doi: 10.18632/oncotarget.7963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbon A., Dinh V.H., Mikami N., Kitagawa Y., Teraguchi S., Ohkura N., Sakaguchi S. Immuno-Navigator, a batch-corrected coexpression database, reveals cell type-specific gene networks in the immune system. Proc. Natl. Acad. Sci. U S A. 2016;113:E2393–E2402. doi: 10.1073/pnas.1604351113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visekruna A., Huber M., Hellhund A., Bothur E., Reinhard K., Bollig N., Schmidt N., Joeris T., Lohoff M., Steinhoff U. c-Rel is crucial for the induction of Foxp3(+) regulatory CD4(+) T cells but not T(H)17 cells. Eur. J. Immunol. 2010;40:671–676. doi: 10.1002/eji.200940260. [DOI] [PubMed] [Google Scholar]
- Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L., Parkinson H. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Borde M., Heissmeyer V., Feuerer M., Lapan A.D., Stroud J.C., Bates D.L., Guo L., Han A., Ziegler S.F. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126:375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- Xu L., Kitani A., Stuelten C., McGrady G., Fuss I., Strober W. Positive and negative transcriptional regulation of the Foxp3 gene is mediated by access and binding of the Smad3 protein to enhancer I. Immunity. 2010;33:313–325. doi: 10.1016/j.immuni.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M., Stephan S., Bluestone J.A. Peripherally induced tregs—role in immune homeostasis and autoimmunity. Front. Immunol. 2013;4:232. doi: 10.3389/fimmu.2013.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B.-H., Hagemann S., Mamareli P., Lauer U., Hoffmann U., Beckstette M., Föhse L., Prinz I., Pezoldt J., Suerbaum S. Foxp3(+) T cells expressing RORγt represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol. 2016;9:444–457. doi: 10.1038/mi.2015.74. [DOI] [PubMed] [Google Scholar]
- Zeng S., Yang Y., Cheng X., Zhou B., Li P., Zhao Y., Kong X., Xu Y. HIC1 epigenetically represses CIITA transcription in B lymphocytes. Biochim. Biophys. Acta. 2016;1859:1481–1489. doi: 10.1016/j.bbagrm.2016.10.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.