

Glioblastoma can suppress immunity by using surface PD-L1 on extracellular vesicles to block T cell receptor–mediated T cell activation.
Abstract
Binding of programmed death ligand-1 (PD-L1) to programmed cell death protein-1 (PD1) leads to cancer immune evasion via inhibition of T cell function. One of the defining characteristics of glioblastoma, a universally fatal brain cancer, is its profound local and systemic immunosuppression. Glioblastoma has also been shown to generate extracellular vesicles (EVs), which may play an important role in tumor progression. We thus hypothesized that glioblastoma EVs may be important mediators of immunosuppression and that PD-L1 could play a role. We show that glioblastoma EVs block T cell activation and proliferation in response to T cell receptor stimulation. PD-L1 was expressed on the surface of some, but not of all, glioblastoma-derived EVs, with the potential to directly bind to PD1. An anti-PD1 receptor blocking antibody significantly reversed the EV-mediated blockade of T cell activation but only when PD-L1 was present on EVs. When glioblastoma PD-L1 was up-regulated by IFN-γ, EVs also showed some PD-L1–dependent inhibition of T cell activation. PD-L1 expression correlated with the mesenchymal transcriptome profile and was anatomically localized in the perinecrotic and pseudopalisading niche of human glioblastoma specimens. PD-L1 DNA was present in circulating EVs from glioblastoma patients where it correlated with tumor volumes of up to 60 cm3. These results suggest that PD-L1 on EVs may be another mechanism for glioblastoma to suppress antitumor immunity and support the potential of EVs as biomarkers in tumor patients.
INTRODUCTION
Glioblastoma is a devastating and universally fatal cancer that evades therapy because of its complex and adaptive cellular composition and its ability to rapidly develop resistance to conventional and targeted therapeutics (1). Novel strategies that aim to prime the patient’s immune system against cancerous tissue have recently gained momentum in the clinic (2–4). In this context, glioblastoma has long been recognized as a local and systemic immunosuppressive neoplasm by releasing anti-inflammatory cytokines, reducing levels of antigen-presenting molecules, and engaging immune checkpoint–activating regulatory receptors on T cells (2, 5). There are increasing efforts to target glioblastoma clinically using immune therapies including immune checkpoint blockade, which has shown unprecedented activity in some tumors (6–8). Immune checkpoints refer to a broad spectrum of immune inhibitory pathways hardwired in the immune system that are pivotal in the balanced regulation of physiological T cell responses (3). One of the most prominent molecules used by tumor cells to engage T cell immune checkpoints is programmed death ligand 1 (PD-L1) (9, 10). PD-L1 expressed on the surface of tumor and antigen-presenting cells binds to programmed cell death protein 1 (PD1), which is expressed by activated T cells. In normal biology, this machinery is used to control T cell activation. However, in tumors, this leads to blockade of T cell activation and protects tumor cells from T cell–mediated killing. It is now clear that tumors use various mechanisms to hijack immune checkpoints to evade immune recognition, primarily against tumor neoantigen-specific T cells (3, 4).
Glioblastoma is a heterogeneous brain tumor, with three major transcriptionally defined subtypes [proneural (P), classical, and mesenchymal (M)] (11). Gene expression analysis suggests that these subtypes may use distinct immunosuppressive mechanisms, with M glioblastoma showing elevated expression of genes with immune functions, including significant elevation of PD-L1 when compared with P glioblastoma, suggesting that specific transcriptional subtypes may respond differentially to immune-based therapies (12). We now recognize that glioblastoma heterogeneity can be maintained by extracellular vesicles (EVs) shed by tumor cells (13). EVs, including exosomes and microvesicles, resemble small cellular surrogates that act as major conduits in cell-cell communication. When released from tumors, EVs locally and systemically transfer host cell biological materials (including proteins, RNA, and DNA) that facilitate tumor progression, angiogenesis, and immune tolerance (14–16). However, tumor EVs carry multiple layers of immune modulatory capacities, and the knowledge of how these influence infiltrating lymphocytes within the tumor microenvironment is still rudimental. Here, we show that glioblastoma-derived EVs block T cell receptor (TCR)–mediated T cell activation. Mechanistically, we show that, for some glioblastomas, this is partially rescued by treatment with an anti-PD1 antibody that blocks ligand binding and that PD-L1 on EVs can directly ligate PD1. We also show that glioblastoma EVs bind to the surface of T cells. PD-L1 expression correlates with the mesenchymal gene expression subtype and is enriched in cells located in the perinecrotic and pseudopalisading niches of glioblastomas. Furthermore, PD-L1 DNA is present in circulating EVs from glioblastoma patients, where it correlates with glioblastoma volumes of up to 60 cm3. These findings highlight novel roles of glioblastoma-derived EVs with the potential for both local and systemic suppression of antitumor immunity.
RESULTS
We sought to determine whether EVs derived from glioblastoma stem-like cells (GSCs) can mediate T cell immunosuppression. We asked whether EVs isolated from human GSCs could inhibit CD4+ or CD8+ T cells when activated through the TCR. Figure 1A shows that EVs from human GSCs suppressed the activation of both CD4+ and CD8+ T cells by anti-CD3 to mimic antigen recognition through the TCR. This was initially performed in peripheral blood mononuclear cells (PBMCs) from healthy human donors; thus, anti-CD3 alone can be used for T cell stimulation because costimulation is provided by other cells within the assay. Data are shown as percent change in activation/proliferation for clarity to correct for patient-to-patient variability. In addition, to ensure that the observed EV-mediated suppression of T cell activation occurred in the context of TCR recognition of an antigen bound to the major histocompatibility complex, we used mouse CD8+ T cells that react to the gp33 epitope, isolated from transgenic P14 mice. To present the gp33 antigen, mouse dendritic cells (DCs) were pulsed with gp33 peptide and then co-incubated with gp33+ CD8+ T cells in the presence or absence of mouse glioma EVs. Figure 1B shows that CD8+ T cell activation mediated by DC presentation of an antigen was also inhibited by EVs. Therefore, these findings demonstrate that glioma EVs suppressed T cell activation mediated by TCR reaction with anti-CD3 or mediated by DC presentation of antigen.
Fig. 1. Glioblastoma EVs inhibit T cell activation in an antigen-specific manner.
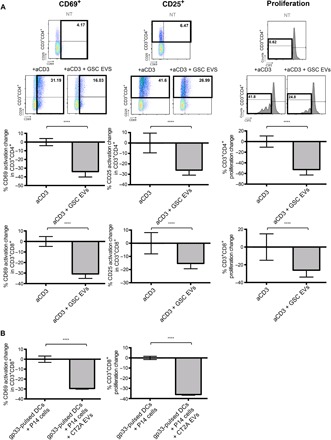
(A) EVs from human GSC cultures inhibit both CD4+ and CD8+ T cell activation and proliferation. PBMCs (isolated from eight human volunteers, n = 8) were treated with anti-CD3 (500 ng/ml) to activate TCR signaling in the presence or absence of GSC EVs (5 μg/ml; isolated from four different GSCs, that is, n = 4) for 2 days. Top: Dot plots of CD69 and CD25 and proliferation flow cytometry data. Bottom: Percent changes of CD69 (left) and CD25 (middle) compared to anti-CD3 alone and percent change of proliferating cells (right) compared to anti-CD3 treatment alone after 3 days for CD4+ and CD8+ T cells, measured by carboxyfluorescein diacetate succinimidyl ester (CFSE) content. (B) EVs from CT2A glioma cells inhibited CD8+ T cells in an antigen-specific manner. Percent CD69 expression change (left) of CD8+ T cells isolated from transgenic P14 mice reacting against gp33 peptide presented by DC ± CT2A EVs; proliferation change (right) of gp33 antigen–specific P14 CD8+ T cells ± CT2A EVs measured by CFSE (n = 4). Statistical analysis was performed by two-tailed Student’s t test (****P < 0.0001). Bars represent means ± SD of each of the eight T cell preparations after incubation with one EV preparation from each of the four gender-matched GSC EVs.
Glioblastoma is known to express PD-L1 and is infiltrated by PD1–expressing tumor-infiltrating lymphocytes (17). PD-L1 functions by binding to PD1 and inhibiting TCR-related pathways (3). We thus wanted to know whether the observed mechanism of EV-mediated suppression of T cell activation involved PD-L1. First, we examined PD-L1 expression on GSC EVs by Western blotting. This showed that there was variability in the expression of PD-L1, which was readily detectable in some GSCs and their EVs (PD-L1high) (Fig. 2A), in cells freshly explanted from glioblastoma patients [primary cell cultures (PCCs)] (Fig. 2B), and in murine glioblastoma CT2A cells and their EVs (fig. S1A). However, PD-L1 was also expressed at low or nondetectable levels in some GSCs, PCCs, and their EVs (PD-L1low). This was confirmed by electron microscopy, which also showed that PD-L1 was present on the surface of PD-L1high EVs but was barely detectable on the surface of PD-L1low EVs (fig. S1, B and C). The overall EV size distribution did not reveal significant differences between PD-L1high and PD-L1low vesicles with an average diameter of 120 to 130 nm and a range of 50 to 170 nm (fig. S1D).
Fig. 2. Glioblastoma EVs contain PD-L1 and block TCR-mediated T cell activation.
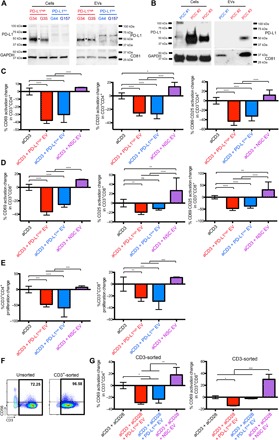
(A) Detection of PD-L1 by Western blot in glioblastoma EVs. Four GSCs were analyzed for their cellular and EV PD-L1 expression (red, PD-L1high; blue, PD-L1low). GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (B) PD-L1 is found in primary cell cultures (PCCs) from glioblastoma patients and EVs isolated from these cell cultures. (C and D) PD-L1high and PD-L1low GSC EVs inhibit CD4+ (C) and CD8+ (D) T cell activation. Percent CD69 and CD25 and CD69+CD25+ expression change compared to anti-CD3 for 2 days ± glioblastoma EVs (5 μg/ml) from four different GSCs. Anti-CD3–stimulated PBMCs were from eight human volunteers ± PD-L1high/low EVs, and NSC EVs were added to anti-CD3–stimulated PBMCs from three human volunteers. (E) GSC EVs can inhibit T cell proliferation. Percent change of proliferating cells compared to anti-CD3 treatment after 3 days for CD4+ and CD8+ T cells, measured by CFSE content (aCD3 ± PD-L1high/low EVs, n = 7; NSC EVs, n = 3). (F) T cell inhibition is partially mediated by a direct effect on T cells. Left: unsorted PBMCs. Right: CD3+ cells are enriched after sorting. (G) CD3+CD4+ (left) and CD3+CD8+ (right) cells (n = 3) after treatment. Statistical analysis was performed by one-way analysis of variance (ANOVA), with post hoc Bonferroni’s correction (****P < 0.0001, ***P < 0.001, **P < 0.01, and *P < 0.05; ns, not significant.). Examples of the flow cytometry data are available at http://harveycushing.bwh.harvard.edu/chiocca-lab/.
Next, we tried to determine whether PD-L1 on EVs played a role in the observed T cell immunosuppression. T cell activation in PBMCs was then performed in the presence or absence of EVs derived from PD-L1high GSCs, PD-L1low GSCs, and neural stem cells (NSCs). There was significant down-regulation of CD69, CD25, and double-positive CD69+CD25+ activation markers on anti-CD3–activated CD4+ (Fig. 2C) and CD8+ (Fig. 2D) T cells exposed to either PD-L1high or PD-L1low GSC EVs when compared to NSC EVs. In addition, there was significant down-regulation of PD1, a chronic activation/exhaustion marker (fig. S1E). These observed changes in activation markers also correlated with functional changes because there was a significant down-regulation of anti-CD3–stimulated CD4+ and CD8+ T cell proliferation when exposed to either PD-L1high or PD-L1low GSC EVs versus NSC EVs (Fig. 2E). NSCs were PD-L1low, as shown by Western blot (fig. S1F). To ensure that the observations were specific to T cells and not due to effects on other cells within the PBMC population in the assay, we then performed similar experiments in isolated CD3+ cells. T cell activation was inhibited by GSC-derived EVs in this assay, confirming a direct effect of EVs on T cells (Fig. 2, F and G).
As an additional control, we showed that glioblastoma-derived EVs did not alter activation marker expression in unstimulated T cells (data seen in downloadable figure, http://harveycushing.bwh.harvard.edu/chiocca-lab/) or change the overall CD3+ population. In a time-course assay, CD69 expression did not change significantly at the 24-, 48-, and 72-hour time points, whereas CD25 expression increased at 48 and 72 hours compared to 24 hours (fig. S1G). We conclude that the observed effects at the selected time point for the assay (48 hours) were not an artifact of the natural evolution of the temporal expression of marker activation, in agreement with published studies (18, 19). Furthermore, the effects of EVs were dose-dependent with an IC50 (median inhibitory concentration) of approximately 25 μg of EVs (fig. S1H). To determine whether this was physiologically relevant, we determined the amount of circulating EVs in 24 glioblastoma patients and found that they contained an average of 2 × 1012 EVs/ml of blood (fig. S1I). Because 5 × 107 EVs is equal to approximately 5 μg of protein, these results indicate that the dose-response curve was well within the physiological range.
EVs fluorescently labeled with palmitoylated tdTomato (palmtdT) and palmitoylated green fluorescent protein (GFP) (palmGFP) (20) could be visualized to localize to the surface of PBMCs and CD3+-sorted cells (fig. S2A and movie S1). To investigate whether there was in vivo colocalization of labeled EVs to infiltrating lymphocytes in mouse glioblastomas, we intracranially injected palmGFP-labeled murine CT2A glioma cells that constantly produce palmGFP-EVs in mice. Figure S2C shows visual evidence of colocalization of these EVs to CD3+ cells in vivo in mouse glioblastomas. In summary, our observations suggest that GSC EVs can directly inhibit TCR-mediated T cell activation and that these effects were mediated by both PD-L1high and PD-L1low EVs.
Because both PD-L1high and PD-L1low EVs inhibited T cell activation, we wanted to explore whether PD-L1 expression was superfluous to the observed EV inhibition. We thus proceeded to add a PD1 blocking antibody to the inhibition assays. PD1 blockade did partially restore the anti-CD3–mediated activation of CD4+ and CD8+ T cells exposed to the PD-L1high GSC EVs, as measured by CD69 (Fig. 3, A and B) and CD25 (fig. S3, A and B). Similar data were observed in isolated CD3 cells (Fig. 3, C and D). However, this did not occur in cells exposed to the PD-L1low GSC EVs, whereas PD1 levels increased through TCR activation (fig. S3C). Neither did we observe an activation of T cells by the PD1 blocking antibody itself (fig. S3D). EVs from PD-L1high GSCs fluorescently labeled with palmtdT (21) bound specifically to plates coated with recombinant PD1, and this binding could be reversed by adding soluble anti-PD1 (Fig. 3E). Furthermore, binding of fluorescently labeled PD-L1high EVs to PD1 was dose-dependent (fig. S3E). We then asked whether PD-L1 on PD-L1high GSC EVs could directly bind to PD1 on lymphocytes. GSCs were transduced with a vector encoding a PD-L1/red fluorescent protein (RFP) fusion protein to visualize PD-L1 expression. Purified PD-L1/RFP EVs from GSCs could be observed bound to the outer surface of PBMCs (fig. S3F). Addition of anti-PD1 reduced binding of PD-L1/RFP EVs to the surface of PBMCs (fig. S3G). The EVs shown to bind the surface of lymphocytes (presumably to PD1) and displaced by the PD1 antibody were seen to localize both to the surface and inside the cell. Quantification of high-power photomicrographs, where there were only a few lymphocytes per field, showed that there were 30 EVs internalized versus 18 EVs surface-bound on 18 randomly selected cells (table S1). Together, these results thus showed that PD-L1 in the PD-L1high GSC EVs interacted with PD1 on PBMCs, and this interaction partially blocks suppression of T cell activation in PD-L1high EVs.
Fig. 3. Glioblastoma EVs contain PD-L1 that directly interacts with PD1, and the interaction is displaced by anti-PD1 treatment.
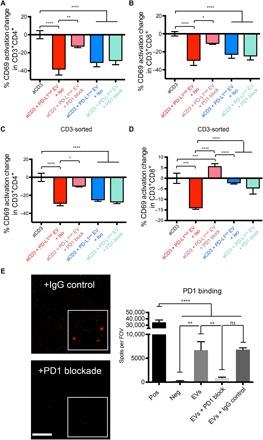
(A and B) PD1 blockade prevents the inhibition of PD-L1high GSC EVs on PBMCs. Percent change in CD69 expression for CD4+ (A) and CD8+ (B) T cells. PD1 blocking antibody (10 μg/ml) or isotype control (10 μg/ml) was added at day 0 (n = 7 PBMC donors, means ± SD). (C and D) PD1 blockade furthermore prevents the inhibition of PD-L1high GSC EVs on CD3+ isolated cells. CD3+CD4+ (C) and CD3+CD8+ (D) cells (n = 3) after treatment. (E) PD-L1–carrying, palmtdT-labeled PD-L1high GSC EVs can bind to wells coated with recombinant PD1, whereas PD1 antibody blockade inhibits EV binding. Representative confocal images are shown on the left, whereas quantification is provided on the right. Spots per field of view (FOV) on the y axis represent palmtdT-positive dots. Scale bar, 50 μm; ×500 magnification inserts; quadruplicates as means ± SD. One-way ANOVA, with post hoc Bonferroni’s correction, was used to differentiate multiple groups (****P < 0.0001, ***P < 0.001, **P < 0.01, and *P < 0.05).
As shown in Fig. 1, there was detectable PD-L1 expression in PD-L1high GSCs in contrast to PD-L1low GSCs. Because up-regulation of PD-L1 is linked to infiltrating lymphocytes in some solid tumors, we investigated whether activated lymphocytes induce PD-L1 expression on GSCs. Supernatants of activated PBMCs significantly up-regulated PD-L1 on GSCs that were initially PD-L1low, and this up-regulation was inhibited when an anti–IFN-γ neutralizing antibody was added to the supernatants, confirming a likely role for IFN-γ in mediating the observed effect (Fig. 4A). IFN-γ treatment itself, which has previously been shown to increase PD-L1 expression in some glioblastoma cells (22), resulted in a profound induction in PD-L1 levels in PD-L1low GSCs but minimally increased PD-L1 in the PD-L1high GSCs (Fig. 4B). IFN-γ also increased PD-L1 levels in GSCs differentiated in serum (fig. S4A). Significantly, this up-regulation was also observed in EVs released by the PD-L1low GSCs (Fig. 4B and fig. S4B). The observation of IFN-γ–mediated induction of PD-L1 in PD-L1low cells allowed us to further investigate its role in inhibition of T cell activation, and thus, we sought to determine whether EVs from IFN-γ–treated PD-L1low GSCs also inhibited T cells. These EVs inhibited T cell activation (Fig. 4C and fig. S4C), and blockade of PD1/PD-L1 interaction did significantly reverse the inhibition of T cell activation (Fig. 4C and fig. S4C). As expected from the Western blot analysis in Fig. 4B, there was no effect of IFN-γ treatment of PD-L1high EVs on further inhibition of T cell activation or reversal by PD1 blockade (fig. S4D). These results thus showed that even PD-L1low EVs had the potential to inhibit T cell activation if PD-L1 was up-regulated by IFN-γ. We also show that PD-L1low EVs increase the levels of immunosuppressive molecules IDO and IL-10, which primarily derive from the CD3-negative cell population (Fig. 4, E to H). This demonstrates the presence of multiple layers of immunosuppression that can be mediated by GSC EVs.
Fig. 4. PD-L1 is up-regulated in response to IFN-γ in PD-L1low GSCs and their EVs that can now suppress activated T cells in a PD-L1–dependent manner.
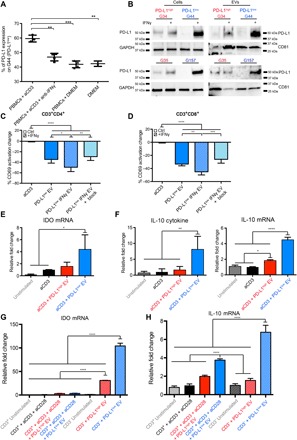
(A) Glioma GSCs up-regulate PD-L1 in vitro in response to activated PBMC supernatants. PBMCs were stimulated with anti-CD3, and supernatants were collected and co-incubated with GSCs (G44, a PD-L1low GSC) in the presence or absence of anti–IFN-γ. PD-L1 expression was measured by flow cytometry. DMEM, Dulbecco’s modified Eagle’s medium. (B) IFN-γ–mediated increase of PD-L1 expression levels in PD-L1High and PD-L1low GSCs as shown by Western blots of four different GSCs. (C and D) EVs derived from IFN-γ–treated PD-L1low GSCs inhibit anti-CD3–stimulated T cell activation, and this can be partially reversed by PD1 blockade. Inhibition potential was measured by the percentage change of CD69+ levels on anti-CD3–stimulated CD3+CD4+ (C) or CD3+CD8+ (D) cells, isolated from five human volunteers (means ± SD). Representative dot plots for (C) and (D) can be found in fig. S4C. (E) PD-L1low EVs up-regulated indoleamine 2,3-dioxygenase (IDO) mRNA in PBMCs treated with PD-L1low EVs. Quantitative polymerase chain reaction (qPCR) expression levels are shown (n = 3). (F) PD-L1low EVs cause interleukin-10 (IL-10) up-regulation in PBMCs. IL-10 cytokine (left) and qPCR expression levels (right) are shown (n = 3). (G and H) Immunosuppressive molecules IDO and IL-10 primarily derive from the CD3-negative population. IDO (G) and IL-10 (H) mRNA levels are shown after CD3+ magnetic-activated cell sorting (n = 3). Data sets consist of EVs from four different glioblastoma cell lines with means ± SD. One-way ANOVA, with post hoc Bonferroni’s correction, was used to differentiate multiple groups (****P < 0.0001, ***P < 0.001, **P < 0.01, and *P < 0.05). Student’s t test was used to differentiate between two groups, and one-way ANOVA with post hoc Bonferroni’s correction was used for multiple groups (****P < 0.0001, ***P < 0.001, **P < 0.01, and *P < 0.05).
We next asked whether GSCs that were PD-L1high versus PD-L1low correlated with any glioblastoma transcriptome classes. We analyzed RNA sequencing (RNA-seq) data sets from The Cancer Genome Atlas (TCGA) (23) and Ivy Glioblastoma Atlas Project (Ivy GAP) (24) and found that M glioblastoma showed elevated PD-L1 expression compared with other subtypes (Fig. 5A). The higher levels of PD-L1 in M glioblastoma were very pronounced in a subset of the samples. In the whole glioblastoma data set, the expression of IFN-γ response genes (25), such as interferon regulatory factor 1 (IRF1), HLA-A, and ICAM1, positively correlated with levels of PD-L1 (Fig. 5B), in agreement with a model where glioblastoma evades immune recognition by expression of PD-L1 and engagement of PD1 (26). Similar results were obtained from two independent glioblastoma RNA-seq data sets (fig. S5). Additional RNA-seq analysis of laser-captured glioblastomas showed anatomical clustering of PD-L1 with known IFN-γ–responsive genes in different glioblastoma regions (infiltrating edge versus cellular tumor versus perinecrotic areas) (Fig. 5C). PD-L1 gene expression anatomically localized within glioblastoma niches with high expression of other known mesenchymal genes, such as perinecrotic zones and pseudopalisading cells (Fig. 5D). The expression of PD-L1 in these areas did not correlate with the expression of known T cell markers, such as CD25, CD69, CD3, CD4, and CD8 (Fig. 5E). However, it did correlate with tumor cell markers, such as CD44, but not with astrocytic or neuronal markers, such as GFAP (glial fibrillary acidic protein), MAP2 (microtubule-associated protein 2), NCAM1 (neural cell adhesion molecule 1), or PECAM1 (platelet endothelial cell adhesion molecule 1) (Fig. 5F). This in vivo association of PD-L1 expression predominantly with M glioblastoma genes was valid for the cells that were studied in Figs. 1 to 3, where PD-L1high cells turned out to be mesenchymal, whereas the PD-L1low cells were proneural (13, 27). These data thus showed that PD-L1 gene expression was localized to glioblastoma niches characterized by relatively high expression of mesenchymal and IFN-γ–responsive genes and relatively low expression of activated T cell, astrocytic, and neural genes.
Fig. 5. PD-L1 expression correlates with M glioblastoma and IFN-γ response genes.
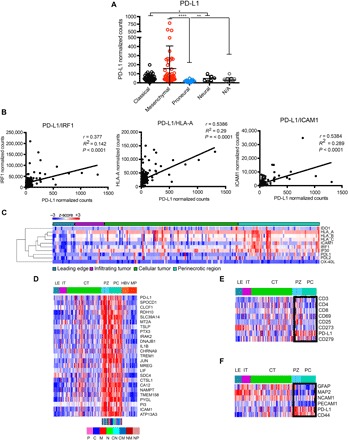
(A) Analysis of TCGA RNA-seq data for PD-L1 expression shows mesenchymal enrichment. The dots represent individual patients. (B) PD-L1 expression correlates with IFN-γ response factor (IRF1), human leukocyte antigen A (HLA-A), and intercellular adhesion molecule 1 (ICAM1) levels. (C) Ivy Glioma database RNA-seq analysis of laser-captured specimens from different regions of glioblastomas (leading edge, infiltrating tumor, cellular tumor, and perinecrotic areas) shows anatomical clustering of PD-L1 with known IFN-γ response genes. (D) The expression of PD-L1 correlates with immune response genes in perinecrotic and pseudopalisading niches of glioblastoma tumors. The correlation of PD-L1 expression (R > 0.6) with Ivy GAP database–based expression signatures in different anatomic areas of glioblastoma was analyzed (LE, leading edge; IT, infiltrating tumor; CT, cellular tumor; PZ, perinecrotic zone; PC, pseudopalisading cells around necrosis; HBV, hyperplastic blood vessels; MP, microvascular proliferation; P, proneural; C, classical; M, mesenchymal; N, neural). (E) The expression of PD-L1 inversely correlates with T cell markers in the tumor anatomic niche. Selected genes were queried with Ivy GAP database–based expression signatures in different anatomic areas of glioblastomas. The box denotes PZ and PC areas of tumor. (F) The expression of PD-L1 positively correlates with mesenchymal tumor markers in the tumor anatomic niche. Selected genes were queried with Ivy GAP database–based expression signatures in different anatomic areas of glioblastoma. The boxes denote PZ and PC areas of tumor. The Kruskal-Wallis test with Dunn’s correction was used to differentiate TCGA groups (****P < 0.0001, **P < 0.01, and *P < 0.05).
Tumor EVs may have systemic effects, are important potential biomarkers of disease, and may represent a useful tool to analyze tumor progression and responses. Therefore, we analyzed circulating patient EVs in serum and plasma from glioblastoma patients before surgery for their PD-L1 protein expression and compared it to healthy donor controls (fig. S6, A and B). There was no significant difference measured by Western blotting and enzyme-linked immunosorbent assay (ELISA)–type sandwich analysis. Because EVs contain DNA and RNA, we measured the amount of PD-L1 DNA by droplet PCR and found that 14 of 21 glioblastoma patient–derived samples showed enrichment of PD-L1 DNA in isolated EVs (Fig. 6A). Next, we correlated EV PD-L1 DNA abundance with glioblastoma tumor volume, as measured by magnetic resonance (MR) imaging and specifically by contrast-enhancing tumor volume (CET) (fig. S6, C and D). The full patient cohort showed no significant correlation between PD-L1 DNA abundance and tumor volume. However, this was due to two patients with CET > 60 cm3, whose PD-L1 DNA levels were very low (fig. S6E). When we set the cutoff at 60 cm3 to remove these outliers, there was a highly significant correlation between PD-L1 DNA abundance in circulating EVs and CET (Fig. 6B). There was also a positive correlation between PD-L1 expression in glioblastomas and PD-L1 DNA in patient EVs (Fig. 6C), whereas the PD-L1 DNA did not correlate with the necrotic volume of the tumor (fig. S6F). The sum of these findings suggests that EV PD-L1 DNA may serve as a biomarker of glioblastoma in humans.
Fig. 6. Glioblastoma patient–derived serum and plasma EVs contain PD-L1 DNA whose levels correlate with tumor volume.
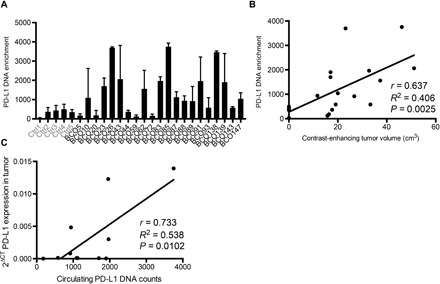
(A) PD-L1 DNA enrichment is found specifically in patient-derived EVs (healthy controls, n = 5; glioblastoma patients, n = 21). (B) PD-L1 DNA enrichment highly correlates with CET of <60 cm3 (Pearson’s correlation with nonlinear regression, r = 0.637, R2 = 0.406, P = 0.0025). (C) PD-L1 expression in tumors correlates with PD-L1 DNA in patient blood (Pearson’s correlation with nonlinear regression, r = 0.733, R2 = 0.538, P = 0.0102).
DISCUSSION
Here, we hypothesized that PD-L1 found on glioblastoma EVs could mediate inhibition of T cell activation, thus providing a novel mechanism for the capacity of glioblastoma to evade the immune system. We could show that (i) EVs from glioblastomas that are PD-L1high significantly inhibit the magnitude of T cell activation and that this is partially reversed by PD1 blockade; (ii) EVs from glioblastomas that are PD-L1low also inhibit T cell activation but not through PD-L1, unless they are stimulated by IFN-γ (IFN-γ up-regulates PD-L1 in EVs from PD-L1low glioblastomas, and now, T cell inhibition can be partially reversed by PD1 blockade); (iii) PD-L1 on EVs binds to purified PD1 and to lymphocytes in vitro and localizes to lymphocytes in mice in vivo; (iv) PD-L1 gene expression clusters with mesenchymal and IFN-γ–responsive genes in glioblastoma niches that are anatomically localized to the perinecrotic and pseudopalisading cells but does not cluster with activated T cell markers and astrocytic or neural genes; and (v) circulating EV PD-L1 DNA correlates with glioblastoma tumor volumes of up to 60 cm3 in patients. Together, these findings suggest that PD-L1 on EVs may be another possible mechanism used by glioblastomas, particularly of the mesenchymal subtype, to evade T cell surveillance.
To measure the effects of EVs on T cells, we used activation markers, such as CD69, CD25, and PD1 (an activation/“exhaustion” marker), and the functional assay of T cell proliferation. Although CD69 is classically described as an “early” activation marker and CD25 as a “late” activation marker, in our hands, we found that both were significantly elevated at the 48-hour time point when our assay was performed. This is in agreement with other studies (18, 19). The regulation of additional T cell functional changes in these assays is currently under detailed investigation over a series of time points. Although the observed inhibition of T cell activation by EVs was dose-dependent, it was not complete, suggesting that additional mechanisms of T cell inhibition are operative. PD-L1low EVs caused an increase in mRNA levels of the immunosuppressive molecules IDO1 and IL-10 (Fig. 4), which primarily derive from the non–T cell fraction of the PBMCs used in the assay. Our novel findings should be placed in the context of other published papers. In a series of elegant papers, the Whiteside laboratory has shown that tumor microvesicles expanded T regulatory cells and caused apoptosis of effector cytotoxic T cells through FasL (Fas ligand) and other immunosuppressive molecules (28–31). In addition, others have shown that tumor exosomes inhibited natural killer (NK) cells (16). Novel findings in our publication relate to the immunosuppressive role of PD-L1 in EVs, showing that it directly binds to PD1, that only EVs from PD-L1high glioblastoma cells have such a function constitutively, and that PD-L1low glioblastoma cells must be stimulated with IFN-γ to observe some level of PD-L1–dependent T cell inhibition.
Although our experiments were performed with activated and presumably immunologically functional T cells from healthy human volunteers, it is not known whether this can also be reproduced with dysfunctional and immunologically “exhausted” T cells from human glioblastoma patients. Although these experiments are in progress, we argue that these T cells may not activate well to begin with, thus rendering it difficult to measure inhibition by EVs. Physiologically, we did find that EVs injected in mice colocalized with lymphocytes in mouse brain glioblastomas. In addition, by immunoelectron microscopy, we estimate that 16% of EVs from PD-L1high GSCs expressed PD-L1. Because an IC50 of T cell activation was provided by 25 μg of EV, this would translate to a concentration of 4 μg of PD-L1–containing EVs. These EV amounts in blood are within physiological ranges of what we measured in glioblastoma patients, thus suggesting validation of the inhibitory action of PD-L1 EVs.
We found that in vitro EV PD-L1 bound to immobilized purified PD1 and that this can be displaced by anti-PD1 in a dose-dependent manner. We also visualized EVs that contain PD-L1 binding to the surface of lymphocytes and localizing in vivo to mouse tumor-infiltrating lymphocytes. However, it is evident that the EV PD-L1/PD1 interaction is only one of many possible immunosuppressive mechanisms mediated by EVs, as shown not only in this paper but also by others (16, 28–31). In this context, we showed that PD-L1low EVs inhibited T cell activation in a PD-L1–independent manner. However, when PD-L1low GSCs were exposed to IFN-γ, there was an up-regulation of PD-L1 in cells and their EVs. PD1 blockade experiments now suggested that this PD-L1 also led to a significant inhibition of T cell activation. Our data do not rule out a potential role for the other PD-L1 ligand, PD-L2, which may also be functional in our system, and is the subject of ongoing studies.
Although our experiments were conducted with GSCs, our data show that PD-L1 expression in cells and EVs is also found in glioblastoma cells from patients (Fig. 2B), in differentiated GSCs (fig. S4A), and in vivo in tumor niches that colocalize with IFN-γ response genes (Fig. 5C). These findings suggest that expression is independent of GSC differentiation status. In the tumor microenvironment, PD-L1 is expressed by other cell types including microglia, macrophages, and myeloid-derived suppressor cells, and these may also be sources of PD-L1 EVs in glioblastoma (32). PD-L1 expression from these other cells has also been shown to be important for glioblastoma immunosuppression (32).
The TCGA and Ivy data would seem to show a correlation in humans with glioblastomas between PD-L1 expression, CET, and colocalization with IFN-γ response genes. However, the elevated PD-L1 levels in M glioblastoma were observed in a subset of M samples. At present, it is not clear what features of these specific samples were involved in PD-L1 elevation. However, our data suggest that the expression of PD-L1 is dependent on glioblastoma subtype. Here, we used four independent GSC lines: Two were PD-L1high and were in the M subclass, and the two PD-L1low were in the P subclass. Together with gene expression data, this suggests that M glioblastoma has elevated PD-L1 compared with P. However, there are P and M regions within the same tumor; thus, PD-L1 expression (along with subtype) may be dependent on the local microenvironment.
The in vitro dose-response analysis of PD-L1 EVs shows that the amount needed to achieve 50% inhibition of T cell activation would be achievable in humans. We thus argue that this is strong suggestive evidence of an important physiological role for EV PD-L1 in glioblastoma. EVs have been reported not only to express tumor-associated antigens that can prime antitumor immune responses (33) but also to have inhibitory functions (34). Our data provide evidence that EVs hinder immune activation against glioblastoma by multiple mechanisms including the EV PD-L1/PD1 interaction described here. Together, we show that glioblastoma EVs hinder T cell activation in a subtype-specific manner and support studies of others who have shown immunosuppressive effects of glioblastoma EVs on other arms of the immune system (35), including microglia (26). Our findings represent one of the likely many mechanisms involved in EV-mediated immunosuppression. Emerging data from other tumor types reveal effects on macrophages, T cells, and NK cells and show that effects may vary depending on the tumor type (36, 37).
Circulating patient EVs also exhibited significantly elevated levels of PD-L1 DNA when compared to serum from five control subjects, and this correlated with tumor volumes of up to approximately 60 cm3. At this juncture, we do not have an explanation for the lack of correlation of serum PD-L1 DNA with tumor volumes above 60 cm3. If these findings can be further validated, they would imply that serum EV PD-L1 DNA could provide a surrogate marker of tumor volume and possibly help in real-time monitoring of disease progression. This should be validated in a prospective fashion.
MATERIALS AND METHODS
Human specimens
Tumor samples were obtained as approved by the Dana-Farber Cancer Institute (DFCI) Institutional Review Board (IRB). GSCs were obtained by dissociation of tumor samples and cultivated under stem cell–enriching conditions using glutamine-, B27-, fibroblast growth factor–, and epidermal growth factor–supplemented Neurobasal medium. Serum-free conditions were used to isolate and culture cells from patient specimens with no stem cell enrichment because this allows selection of cells with diverse transcriptomic characteristics. The unique identity of cultured patient-derived cells was confirmed by short tandem repeat analysis (table S2). Mycoplasma testing was routinely carried out by PCR. Glioblastoma subtype classification was carried out by gene expression profiling, as previously described (13). PBMCs were obtained from healthy donors approved by the IRB at Brigham and Women’s Hospital and by the Medical Ethics Committee of the Chamber of Physicians of Hamburg. Informed consent was obtained from all patients. All experiments were performed in accordance with local guidelines and regulations. PBMCs were isolated from heparin-anticoagulated venous blood using Ficoll Paque Plus. Isolation was performed according to the manufacturer’s recommendations.
Cell culture
Primary human GSCs (G34, G35, G44, and G157) were obtained by dissociation of gross tumor samples and cultivated in neurosphere-specific medium, as previously described (13). Cells were grown in neurosphere stem cell medium, consisting of Neurobasal (Invitrogen) supplemented with 1% glutamine (Invitrogen), 2% B27 (Invitrogen), epidermal growth factor (20 ng/ml; PeproTech), and fibroblast growth factor–2 (20 ng/ml; PeproTech). Human NSCs were purchased from EMD Millipore. For IFN-γ stimulation, cells were treated with increasing doses of IFN-γ (10, 100, and 1000 IU/ml) for 48 hours. GSCs stably transduced with PALM-Tomato (M GSC) and PALM-GFP (P GSC) were sorted using fluorescence-activated cell sorting by the Harvard Medical School Flow Cytometry Core Facility. GSCs stably transfected with PDL1/RFP were cultured in neurosphere stem cell medium containing hygromycin (250 μg/ml; Thermo Fisher Scientific). Lymphocytes were cultured in RPMI (Invitrogen) containing 10% exosome-depleted fetal bovine serum (FBS; Invitrogen), 1% glutamine (Invitrogen), and 1% penicillin/streptomycin (Invitrogen). Exosome depletion was achieved by ultracentrifugation.
Isolation of EVs
Conditioned medium, serum, and plasma were collected, and EVs were isolated by differential centrifugation and analyzed using NanoSight, as previously described (13). Briefly, conditioned media were centrifuged at 200g for 5 min to eliminate cells, followed by filtration through a 0.22-μm filter (Millipore), and, in the case of human serum and plasma, were centrifuged at 15,000g for 10 min. EVs were pelleted by ultracentrifugation (Thermo Fisher Scientific) at 100,000g for 70 min and subsequently washed with phosphate-buffered saline (PBS). The number and size of EVs were determined as previously described using NanoSight analysis (13). Briefly, EVs were analyzed using a NanoSight LM10 Nanoparticle Characterization system. All nanoparticle tracking analyses were carried out with identical experiment settings. Particles were measured for 60 s, and for optimal results, microvesicle concentrations were adjusted to obtain ~50 microvesicles per field of view.
Plasmid constructs
Palmitoylation sequences of growth cone–associated protein (GAP43) were genetically fused to the NH2 terminus of GFP (palmGFP) and tdTomato (palmtdT) and cloned into lentiviral constructs, and in combination with lentiviral packaging, constructs were used to establish stable lines (13). The PD-L1/RFP fusion protein vector was purchased from Sino Biological Inc. Lipofectamine-transfected cell clones were positively selected by hygromycin resistance.
Flow cytometry
Flow cytometry was performed on a FACS LSR II or Fortessa (BD Biosciences). For lymphocyte activation studies, soluble anti-CD3 (500 ng/ml; clone OKT3, Bio X Cell) ± anti-CD28 (1 μg/ml; clone CD28.2, BD Biosciences) or IL-2 (PeproTech) was added to media for 2 days. Proliferation was measured by CFSE (eFluor 670, BD Biosciences) after 3 days. EVs were added every 24 hours (5 μg/ml). PD1 blockade was achieved by adding anti-PD1 (10 μg/ml; EH12) or immunoglobulin G control (clone MOPC-21, Bio X Cell). Cells were gated by FSC (forward scatter)/SCC (side scatter) while excluding duplets by FSC-A/FSC-H. Subsequently, CD3+CD56− T cells were gated with further classification of CD4+ and CD8+ T cells. Finally, CD4+ and CD8+ T cells were measured for their CD69, CD25, PD1, and TIM3 expression (seen as a downloadable figure). Cells were stained with the following antibodies: CD3/PE-Cy5.5 (1:100; clone SK7, eBioscience), CD4/FITC (1:100; clone RPA-T4, BD Biosciences), CD8/AmCyan (1:10; clone SK1, BD Biosciences), CD16/APC-Cy7 (1:100, BD Biosciences), CD25/PE-Cy7 (1:10; clone BC96, eBioscience), CD56/V450 (1:30; clone B159, BD Biosciences), CD69/APC (1:10; clone FN50, BD Biosciences), TIM3/APC-Cy7 (1:100, clone F38-2E2, BD Biosciences), and PD1/PE (1:100; clone J105, eBioscience).
Cell sorting
CD3+ magnetic-activated cell sorting was performed using CD3 MicroBeads (#130-050-101, Miltenyi Biotec), as described by the manufacturer’s protocol. CD3+ purified cells were stimulated with soluble anti-CD3 and anti-CD28 for sufficient TCR cross-linking.
EV suppression assay in mice
Bone marrow DCs (BMDCs) were collected from femora and tibiae of naïve 6- to 8-week-old C57BL/6J mice and cultured in RPMI (Gibco), supplemented with 10% FBS, l-glutamine, penicillin/streptomycin, 50 μM β-mercaptoethanol, granulocyte-macrophage colony-stimulating factor (20 ng/ml; PeproTech), and IL-4 (10 ng/ml; PeproTech) for 7 days. Loosely adherent BMDCs were harvested thereafter and pulsed with LCMV gp33-41 peptide (10 μg/ml; GeneScript) for 2 hours at 37°C, followed by lipopolysaccharide (LPS) (10 μg/ml; Escherichia coli serotype 026:B6, Sigma-Aldrich) activation for 24 hours at 37°C. Gp33-41 antigen–specific P14 cells were isolated from the spleens of naïve 6- to 8-week-old P14 mice (bred in-house; a gift from P. Penaloza, Northwestern University) by CD8-negative selection (Miltenyi Biotec). For EV suppression assay, LPS-activated, gp33-41 peptide–pulsed BMDCs were cocultured with CellTrace Violet–labeled P14 cells at a ratio of 1:10 in a 96-well flat-bottom plate for 72 hours at 37°C, in the presence or absence of CT2A-derived EVs at a dose of 1 μg per well, supplemented every 24 hours. Fluorochrome-conjugated anti-mouse antibodies [anti-CD3 (17A2), anti-CD8α (53-6.7), anti-CD69 (H1.2F3), and anti-CD44 (IM7)] and the LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Invitrogen) for live cells were used for FACS (fluorescence-activated cell sorting) analysis (BD LSR II flow cytometer).
Electron microscopy
EVs or cryo-sections were labeled with anti–PD-L1 (1:20; 9A11) and 5-nm gold particles. Grids were examined in a JEOL 1200EX transmission electron microscope or a Tecnai G2 Spirit BioTWIN, and images were recorded with an AMT 2k charge-coupled device (CCD) camera.
PD-L1/PD1 binding assay
Human PD1 Fc fusion protein (2 μg/ml; BPS Bioscience) was coated on high protein-binding 96-well plates (#3590, Costar). After 1-hour incubation with blocking buffer (BPS Bioscience), PD-L1–carrying, palmtdT-labeled EVs were incubated for 2 hours at room temperature (RT). Finally, plates were washed and analyzed by confocal microscopy.
Immunohistochemistry
Human brain tumor tissue specimens were obtained following the guidelines approved by the IRB at DFCI. Immunohistochemistry (IHC) staining was performed as previously described (21). Briefly, tissue specimens were incubated with antibodies against PD-L1 (E1L3N, Cell Signaling Technology) and CD3 (Dako), followed by hematoxylin and eosin counterstaining.
ELISA-type sandwich fluorescent measurement
Purified exosomes were captured on a glass slide precoated with CD81 antibodies (JS-81, BD Biosciences). Subsequently, exosomes were labeled with fluorescent antibodies, and the fluorescence signal was measured. A commercially available kit (A20186, Thermo Fisher Scientific) was used for antibody-fluorophore conjugation, as recommended by the manufacturer’s protocol.
In vivo studies
All animal work was approved by the authorities for health and consumer protection in Hamburg, Germany (protocol number 41/17). Six-week-old female C57BL/6 mice were purchased from the Jackson Laboratory. GL261-palmGFP (100,000 cells) or CT2A-palmGFP (100,000 cells) in 2 μl of PBS was injected intracranially to establish mouse brain tumors (2 mm right lateral, 1 mm frontal to the bregma, and 3 mm deep). For fluorescence microscopy, brains were perfused, fixed, and paraffin-embedded before making 10-μm sections.
Human EVs and droplet PCR
Two milliliters of serum or plasma from each patient was centrifuged at 300g for 5 min to remove cell debris. Supernatants were centrifuged at 15,000g for 15 min, and EVs were pelleted subsequently at 100,000g for 90 min. The pellet was resuspended in 30 μl of PBS. One microliter was used as template for Evergreen Droplet PCR (Bio-Rad). Primers can be found in table S3. RNA from human tumor samples was isolated, and the expression of PD-L1 was measured by qPCR.
Imaging methods
MR images were corrected for gradient nonlinearity, and intensity was corrected using N4 bias field correction. Images were subsequently registered to the Montreal Neurological Institute 152 1-mm3 template using methods from ANTS (Advanced Normalization Tools) (22). Four subjects were excluded because of the lack of reliable images. A total of 19 subjects with MR imaging were segmented for CET using the iterative probabilistic voxel labeling (IPVL) algorithm (38). Visual inspections were performed by two independent reviewers (T.S. and J.T.) to ensure accurate preprocessing and segmentation. Necrosis volumes were segmented using custom software. Briefly, voxels within tumor-associated volumes determined from IPVL automatic segmentation were further segmented using T1 intrinsic signal intensity. Necrosis, on T1, is generally hyperintense to cerebrospinal fluid and hypointense to edematous brain. The resulting segmentations were reviewed manually to ensure accuracy.
Immunoblotting
Cells were lysed using radioimmunoprecipitation assay buffer containing 50 mM tris-HCl (pH 7.5), 150 mM NaCl, aprotinin (10 μg/ml), 1 mM phenylmethylsulfonyl fluoride, leupeptin (10 μg/ml), 2 mM Na3VO4, 4 mM EDTA, 10 mM NaF, 10 mM sodium pyrophosphate, 1% NP-40, 0.1% sodium deoxycholate, 1% protease inhibitor (Calbiochem), and 5% phosphatase inhibitor cocktail (Roche). Total protein concentration was measured using the Bradford protein assay. The PD-L1 antibody 9A11 (1:1000) was obtained from Gordon Freeman (DFCI). CD81 (1:1000; sc-7635), and GAPDH (glyceraldehyde phosphate dehydrogenase; 1:10,000; Abcam #105430) were used according to the manufacturer’s protocols.
Immunohistochemistry
All slides were baked for 60 min in an oven set to 60°C. They were then loaded into the Bond III staining platform with appropriate labels. Antigen retrieval was performed using Bond Epitope Retrieval 2 (Leica Biosystems) for 30 min. They were then incubated with PD-L1 (1:200; E1L3N, Cell Signaling Technology) for two applications of 60 min at RT. Primary antibody was detected using the Bond Polymer Refine Detection Kit (Leica Biosystems) and detected using DAB (3,3′-diaminobenzidine). Slides were then incubated in the second primary antibody CD3 (polyclonal) (Dako) at 1:250 for 30 min. Secondary antibody was detected using the Bond Polymer Refine Red Detection Kit (Leica Biosystems). Secondary antibody was developed using Fast Red, part of the detection kit. Slides were then dehydrated and coverslipped.
Microscopy
All fluorescent and bright-field microscopy–based assays were observed using a Nikon Eclipse Ti microscope. High-resolution confocal fluorescent microscopy was performed using a Zeiss LSM 710 confocal microscope system (Carl Zeiss Inc.) and visualized using the ZEN Zeiss Imaging software. Electron microscopy was carried out as previously described (24310399) using transmission electron microscopy Tecnai G2 Spirit BioTWIN with an AMT 2k CCD camera.
Electron microscopy
For negative staining, 5 μl of the sample was adsorbed for 1 min to a formvar/carbon-coated grid. The grids were floated on a drop of water and then stained with 0.75% uranyl formate for 30 s. PBMCs were incubated with EVs for 3 to 4 hours before fixation (4% paraformaldehyde in 0.1 M sodium phosphate buffer for 2 hours at RT). Pellets were infiltrated with 2.3 M sucrose in PBS containing 0.2 M glycine for 15 min. Samples were snap-frozen in liquid nitrogen and sectioned at −120°C to 60- to 80-nm sections. Sections were transferred to formvar/carbon-coated copper grids. Grids were labeled with anti–PD-L1 (1:20; 9A11) in PBS containing 1% bovine serum albumin for 30 min, followed by rabbit anti-mouse bridging for 20 min. Protein A–gold (5 nm) was added for 20 min. Grids were examined in a JEOL 1200 EX transmission electron microscope or a Tecnai G2 Spirit BioTWIN, and images were recorded with an AMT 2k CCD camera.
Quantitative PCR
qPCR was carried out as previously described (1). Total RNA was extracted using TRIzol (Invitrogen) and treated with ribonuclease-free deoxyribonuclease (Qiagen). mRNA expression analysis was carried out using Power SYBR Green (Applied Biosystems). RNA concentration was quantified using NanoDrop RNA 6000 nano-assays and analyzed using the StepOnePlus Applied Biosystems PCR machine. See table S3 for the primer sequences.
Bioinformatic analysis
Functional bioinformatic analyses were performed using Qiagen Ingenuity Pathway Analysis (IPA; Qiagen, www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/). The IPA analysis was used to select immune response module in GSC EV. The collection of the data from TCGA (23) was compliant with all applicable laws, regulations, and policies for the protection of human subjects, and necessary ethical approvals were obtained. Experimental and clinical data were analyzed using the Glioblastoma Bio Discovery Portal (https://gbm-biodp.nci.nih.gov/) (24), as previously described (13). Clinical data were downloaded from the TCGA data portal (https://tcga-data.nci.nih.gov/docs/publications/tcga/), as described in the TCGA research network. The two individual RNA-seq data sets (Bao and Ivy GAP) were obtained from the GlioVis portal (http://gliovis.bioinfo.cnio.es). Gene expression in the various anatomical regions of glioblastoma was analyzed using data from the Ivy GAP (http://glioblastoma.alleninstitute.org/).
Glioblastoma samples with RNA-seq data were downloaded from TCGA (December 2015), and samples with a 1p19q codeletion were removed in accordance with the classification of glioblastoma in the World Health Organization 2016 guidelines. These data were merged with the supplementary data from Ceccarelli et al. (39) and those samples without a purity score obtained from ABSOLUTE (determined on the basis of mutant allele frequencies from exome data and therefore takes into account normal brain cells as well as immune infiltrates) were filtered out. Of the remaining samples (n = 134), the expression values were normalized to ABSOLUTE purity score, and each gene was correlated with the normalized expression score of PD-L1. Bonferroni’s correction was used for statistical analysis of multiple testing.
In silico analysis of immune response modules in GSC EVs
Functional bioinformatics analyses were performed using Qiagen IPA using casual network (25). Analysis was performed using EV proteome analysis from subtype-determined GSCs (13, 27). Lists of proteins differentially secreted between GSCs were analyzed on the basis of the IPA library of canonical pathways (content date, 8 May 2012). This approach did not take into account individual expression levels but instead assumed that transcriptionally altered genes were determined using a suitable cutoff applied to the measured expression change, which allowed list curation where each gene in the data set could be either up- or down-regulated. The significance of the association between each list and a canonical pathway was measured by Fisher’s exact test. As a result, a P value was obtained, determining whether the probability that the association between the genes in experimental data set and a canonical pathway could be explained by chance alone.
Gene expression in the various anatomical regions of glioblastoma tumors was analyzed using the Ivy GAP (http://glioblastoma.alleninstitute.org/). Gene expression in five major anatomic structures of glioblastoma [leading edge (LE), infiltrating tumor (IT), cellular tumor (CT), microvascular proliferation (MVP), and pseudopalisading cells around necrosis (PAN)] was quantified by RNA-seq in the anatomic structure RNA-seq study. The Ivy GAP cohort was composed of 42 tumors. Samples from the anatomic structures were collected by laser microdissection and validated by in situ hybridization. A curated list of genes expressed in GSC based on IPA analysis was queried with the Ivy GAP data set, and expression was visualized as a z-score heat map.
Data and statistical analysis
All microscopy-based assays were edited/quantified using ImageJ (https://imagej.nih.gov/ij/index.html), including the Analyze Particles function of binary images with automatic threshold. RNA-seq data from TCGA and the Ivy GAP were downloaded from https://tcga-data.nci.nih.gov/docs/publications/tcga/ and http://glioblastoma.alleninstitute.org, respectively (40). Data are means ± SD. The unpaired two-tailed Student’s t test was used for comparison between two groups. Each group was tested for Gaussian distribution, if one-way ANOVA was passed, followed by Bonferroni’s test. If this failed, the Kruskal-Wallis test, followed by Dunn’s correction, was conducted to test for significance among multiple groups. Pearson’s correlation with nonlinear regression analysis was performed to compute Pearson r, R2, and P values. Statistical analyses were performed using Microsoft Office Excel 2011 or GraphPad Prism 6 software. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank M. Ericsson from the Harvard Electron Microscopy Core Facility for technical support. Funding: This work was funded, in part, by the following: NIH [National Cancer Institute (NCI) P01 CA69246 (to E.A.C. and B.S.C.), NCI 1R01 CA176203-01A1 (to J.G.), and NIH UH3 TR000931 (to B.S.C.)], Deutsche Forschungsgemeinschaft (scholarship to F.L.R.; RI 2616/1-1 and RI 2616/2-1), 1RO1NS097649-01, the Doris Duke Charitable Foundation Clinical Scientist Development Award, the Sontag Foundation Distinguished Scientist Award, the Kimmel Scholar Award, and BWF 1006774.01 (to C.C.C.). U19 CA179563 was supported by the NIH Common Fund through the Office of Strategic Coordination/Office of the NIH Director (to X.O.B.) and NIH/NCI P01 CA069246 (to X.O.B.) and by an American-Italian Cancer Foundation Postdoctoral Research Fellowship (to C.P.). S.R. acknowledges support from the Center for Immuno-Oncology, Dana-Farber Cancer Institute. Author contributions: F.L.R. designed, acquired, and analyzed overall assays; designed the study; and wrote the paper. Q.A. performed in vivo assays. H.K. acquired data and designed the study. A.B.M. acquired data and interpreted the results. M.C.S. acquired data and designed the study. J.L.H. performed bioinformatic analysis. K.L. and K.Y. performed ELISA-type sandwich fluorescent measurements. L.B. performed and analyzed droplet PCR. C.P. designed and performed the PD-L1/PD1 binding assay. A.K.R. performed short tandem repeat analysis and interpreted the data. S.K. performed confocal microscopy and interpreted the results. C.C.C. and I.N. obtained and controlled patient specimens. T.S. and J.T. analyzed MR data. S.R. performed and interpreted IHC staining. S.C., H.L., X.O.B., R.W., J.G., M.W., K.L., A.B., and G.J.F. assisted with the writing of the manuscript and with analysis and interpretation of the data. S.E.L. designed the study, analyzed the data, and wrote the manuscript. E.A.C. acquired patient specimens, designed the study, analyzed the data, and wrote the manuscript. All authors discussed the results and commented on the manuscript. Competing interest: G.J.F. has patents/pending royalties on the PD1 pathway from Roche, Merck, Bristol-Myers Squibb, EMD Serono, Boehringer Ingelheim, AstraZeneca, Dako, and Novartis. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Correspondence and requests for materials should be addressed to S.E.L. and E.A.C.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/3/eaar2766/DC1
fig. S1. Glioblastoma EVs contain PD-L1.
fig. S2. Glioblastoma EVs are bound to the plasma membrane.
fig. S3. Glioblastoma EVs that contain PD-L1 bind to PD1.
fig. S4. IFN-γ increases PD-L1–positive EVs in PD-L1low GSCs.
fig. S5. PD-L1 expression correlates with IFN-γ response genes.
fig. S6. EV PD-L1 is found in serum from both glioblastoma patients and healthy controls.
table S1. EV number and their localization: Fluorescently labeled EVs were added to lymphocytes (5 μg/ml) for 3 hours.
table S2. Short tandem repeat profile of cell lines used in the study.
table S3. Primers used in the study.
movie S1. PalmtdT EV bound to CD3+ T cell.
REFERENCES AND NOTES
- 1.Stupp R., Mason W. P., van den Bent M. J., Weller M., Fisher B., Taphoorn M. J. B., Belanger K., Brandes A. A., Marosi C., Bogdahn U., Curschmann J., Janzer R. C., Ludwin S. K., Gorlia T., Allgeier A., Lacombe D., Cairncross J. G., Eisenhauer E., Mirimanoff R. O. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group , Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Reardon D. A., Freeman G., Wu C., Chiocca E. A., Wucherpfennig K. W., Wen P. Y., Fritsch E. F., Curry W. T. Jr, Sampson J. H., Dranoff G., Immunotherapy advances for glioblastoma. Neuro Oncol. 16, 1441–1458 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pardoll D. M., The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahoney K. M., Rennert P. D., Freeman G. J., Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 14, 561–584 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Kmiecik J., Poli A., Brons N. H. C., Waha A., Eide G. E., Enger P. Ø., Zimmer J., Chekenya M., Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 264, 71–83 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Hodi F. S., O’Day S. J., McDermott D. F., Weber R. W., Sosman J. A., Haanen J. B., Gonzalez R., Robert C., Schadendorf D., Hassel J. C., Akerley W., van den Eertwegh A. J. M., Lutzky J., Lorigan P., Vaubel J. M., Linette G. P., Hogg D., Ottensmeier C. H., Lebbé C., Peschel C., Quirt I., Clark J. I., Wolchok J. D., Weber J. S., Tian J., Yellin M. J., Nichol G. M., Hoos A., Urba W. J., Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larkin J., Hodi F. S., Wolchok J. D., Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 1270–1271 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Motzer R. J., Escudier B., McDermott D. F., George S., Hammers H. J., Srinivas S., Tykodi S. S., Sosman J. A., Procopio G., Plimack E. R., Castellano D., Choueiri T. K., Gurney H., Donskov F., Bono P., Wagstaff J., Gauler T. C., Ueda T., Tomita Y., Schutz F. A., Kollmannsberger C., Larkin J., Ravaud A., Simon J. S., Xu L.-A., Waxman I. M., Sharma P.; CheckMate 025 Investigators , Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 373, 1803–1813 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freeman G. J., Long A. J., Iwai Y., Bourque K., Chernova T., Nishimura H., Fitz L. J., Malenkovich N., Okazaki T., Byrne M. C., Horton H. F., Fouser L., Carter L., Ling V., Bowman M. R., Carreno B. M., Collins M., Wood C. R., Honjo T., Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keir M. E., Liang S. C., Guleria I., Latchman Y. E., Qipo A., Albacker L. A., Koulmanda M., Freeman G. J., Sayegh M. H., Sharpe A. H., Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 203, 883–895 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel A. P., Tirosh I., Trombetta J. J., Shalek A. K., Gillespie S. M., Wakimoto H., Cahill D. P., Nahed B. V., Curry W. T., Martuza R. L., Louis D. N., Rozenblatt-Rosen O., Suvà M. L., Regev A., Bernstein B. E., Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doucette T., Rao G., Rao A., Shen L., Aldape K., Wei J., Dziurzynski K., Gilbert M., Heimberger A. B., Immune heterogeneity of glioblastoma subtypes: Extrapolation from the cancer genome atlas. Cancer Immunol. Res. 1, 112–122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ricklefs F., Mineo M., Rooj A. K., Nakano I., Charest A., Weissleder R., Breakefield X. O., Chiocca E. A., Godlewski J., Bronisz A., Extracellular vesicles from high-grade glioma exchange diverse pro-oncogenic signals that maintain intratumoral heterogeneity. Cancer Res. 76, 2876–2881 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bronisz A., Wang Y., Nowicki M. O., Peruzzi P., Ansari K. I., Ogawa D., Balaj L., De Rienzo G., Mineo M., Nakano I., Ostrowski M. C., Hochberg F., Weissleder R., Lawler S. E., Chiocca E. A., Godlewski J., Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer Res. 74, 738–750 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skog J., Würdinger T., van Rijn S., Meijer D. H., Gainche L., Curry W. T. Jr, Carter B. S., Krichevsky A. M., Breakefield X. O., Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 10, 1470–1476 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clayton A., Mitchell J. P., Court J., Mason M. D., Tabi Z., Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res. 67, 7458–7466 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Nduom E. K., Wei J., Yaghi N. K., Huang N., Kong L.-Y., Gabrusiewicz K., Ling X., Zhou S., Ivan C., Chen J. Q., Burks J. K., Fuller G. N., Calin G. A., Conrad C. A., Creasy C., Ritthipichai K., Radvanyi L., Heimberger A. B., PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 18, 195–205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simms P. E., Ellis T. M., Utility of flow cytometric detection of CD69 expression as a rapid method for determining poly- and oligoclonal lymphocyte activation. Clin. Diagn. Lab. Immunol. 3, 301–304 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Motamedi M., Xu L., Elahi S., Correlation of transferrin receptor (CD71) with Ki67 expression on stimulated human and mouse T cells: The kinetics of expression of T cell activation markers. J. Immunol. Methods 437, 43–52 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Lai C. P., Kim E. Y., Badr C. E., Weissleder R., Mempel T. R., Tannous B. A., Breakefield X. O., Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat. Commun. 6, 7029 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi M., Roemer M. G., Chapuy B., Liao X., Sun H., Pinkus G. S., Shipp M. A., Freeman G. J., Rodig S. J., Expression of programmed cell death 1 ligand 2 (PD-L2) is a distinguishing feature of primary mediastinal (thymic) large B-cell lymphoma and associated with PDCD1LG2 copy gain. Am. J. Surg. Pathol. 38, 1715–1723 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Avants B. B., Tustison N. J., Stauffer M., Song G., Wu B., Gee J. C., The Insight ToolKit image registration framework. Front. Neuroinform. 8, 44 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cancer Genome Atlas Research Network , Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Celiku O., Johnson S., Zhao S., Camphausen K., Shankavaram U., Visualizing molecular profiles of glioblastoma with GBM-BioDP. PLOS ONE 9, e101239 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krämer A., Green J., Pollard J. Jr, Tugendreich S., Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Vos K. E., Abels E. R., Zhang X., Lai C., Carrizosa E., Oakley D., Prabhakar S., Mardini O., Crommentuijn M. H. W., Skog J., Krichevsky A. M., Stemmer-Rachamimov A., Mempel T. R., El Khoury J., Hickman S. E., Breakefield X. O., Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro Oncol. 18, 58–69 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Godlewski J., Ferrer-Luna R., Rooj A. K., Mineo M., Ricklefs F., Takeda Y. S., Nowicki M. O., Salińska E., Nakano I., Lee H., Weissleder R., Beroukhim R., Chiocca E. A., Bronisz A., MicroRNA signatures and molecular subtypes of glioblastoma: The role of extracellular transfer. Stem Cell Rep. 8, 1497–1505 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szajnik M., Czystowska M., Szczepanski M. J., Mandapathil M., Whiteside T. L., Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLOS ONE 5, e11469 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wieckowski E. U., Visus C., Szajnik M., Szczepanski M. J., Storkus W. J., Whiteside T. L., Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J. Immunol. 183, 3720–3730 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J. W., Wieckowski E., Taylor D. D., Reichert T. E., Watkins S., Whiteside T. L., Fas ligand–positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin. Cancer Res. 11, 1010–1020 (2005). [PubMed] [Google Scholar]
- 31.Muller L., Mitsuhashi M., Simms P., Gooding W. E., Whiteside T. L., Tumor-derived exosomes regulate expression of immune function-related genes in human T cell subsets. Sci. Rep. 6, 20254 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bloch O., Crane C. A., Kaur R., Safaee M., Rutkowski M. J., Parsa A. T., Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 19, 3165–3175 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho J.-A., Yeo D.-J., Son H.-Y., Kim H.-W., Jung D.-S., Ko J.-K., Koh J. S., Kim Y.-N., Kim C.-W., Exosomes: A new delivery system for tumor antigens in cancer immunotherapy. Int. J. Cancer 114, 613–622 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Iorgulescu J. B., Ivan M. E., Safaee M., Parsa A. T., The limited capacity of malignant glioma-derived exosomes to suppress peripheral immune effectors. J. Neuroimmunol. 290, 103–108 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Hellwinkel J. E., Redzic J. S., Harland T. A., Gunaydin D., Anchordoquy T. J., Graner M. W., Glioma-derived extracellular vesicles selectively suppress immune responses. Neuro Oncol. 18, 497–506 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berchem G., Noman M. Z., Bosseler M., Paggetti J., Baconnais S., Le Cam E., Nanbakhsh A., Moussay E., Mami-Chouaib F., Janji B., Chouaib S., Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-β and miR23a transfer. Oncoimmunology 5, e1062968 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lundholm M., Schröder M., Nagaeva O., Baranov V., Widmark A., Mincheva-Nilsson L., Wikström P., Prostate tumor-derived exosomes down-regulate NKG2D expression on natural killer cells and CD8+ T cells: Mechanism of immune evasion. PLOS ONE 9, e108925 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steed T. C., Treiber J. M., Patel K. S., Taich Z., White N. S., Treiber M. L., Farid N., Carter B. S., Dale A. M., Chen C. C., Iterative probabilistic voxel labeling: Automated segmentation for analysis of The Cancer Imaging Archive glioblastoma images. AJNR Am. J. Neuroradiol. 36, 678–685 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ceccarelli M., Barthel F. P., Malta T. M., Sabedot T. S., Salama S. R., Murray B. A., Morozova O., Newton Y., Radenbaugh A., Pagnotta S. M., Anjum S., Wang J., Zoppoli P., Ling S., Rao A. A., Grifford M., Cherniack A. D., Zhang H., Poisson L., Carlotti C. G. Jr, da Cunha Tirapelli D. P., Rao A., Mikkelsen T., Lau C. C., Yung W. K. A., Rabadan R., Huse J., Brat D. J., Lehman N. L., Barnholtz-Sloan J. S., Zheng S., Hess K., Rao G., Meyerson M., Beroukhim R., Cooper L., Akbani R., Wrensch M., Haussler D., Aldape K. D., Laird P. W., Gutmann D. H., TCGA Research Network, Noushmehr H., Iavarone A., Verhaak R. G. W., Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164, 550–563 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hawrylycz M. J., Lein E. S., Guillozet-Bongaarts A. L., Shen E. H., Ng L., Miller J. A., van de Lagemaat L. N., Smith K. A., Ebbert A., Riley Z. L., Abajian C., Beckmann C. F., Bernard A., Bertagnolli D., Boe A. F., Cartagena P. M., Chakravarty M. M., Chapin M., Chong J., Dalley R. A., Daly B. D., Dang C., Datta S., Dee N., Dolbeare T. A., Faber V., Feng D., Fowler D. R., Goldy J., Gregor B. W., Haradon Z., Haynor D. R., Hohmann J. G., Horvath S., Howard R. E., Jeromin A., Jochim J. M., Kinnunen M., Lau C., Lazarz E. T., Lee C., Lemon T. A., Li L., Li Y., Morris J. A., Overly C. C., Parker P. D., Parry S. E., Reding M., Royall J. J., Schulkin J., Sequeira P. A., Slaughterbeck C. R., Smith S. C., Sodt A. J., Sunkin S. M., Swanson B. E., Vawter M. P., Williams D., Wohnoutka P., Zielke H. R., Geschwind D. H., Hof P. R., Smith S. M., Koch C., Grant S. G. N., Jones A. R., An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/3/eaar2766/DC1
fig. S1. Glioblastoma EVs contain PD-L1.
fig. S2. Glioblastoma EVs are bound to the plasma membrane.
fig. S3. Glioblastoma EVs that contain PD-L1 bind to PD1.
fig. S4. IFN-γ increases PD-L1–positive EVs in PD-L1low GSCs.
fig. S5. PD-L1 expression correlates with IFN-γ response genes.
fig. S6. EV PD-L1 is found in serum from both glioblastoma patients and healthy controls.
table S1. EV number and their localization: Fluorescently labeled EVs were added to lymphocytes (5 μg/ml) for 3 hours.
table S2. Short tandem repeat profile of cell lines used in the study.
table S3. Primers used in the study.
movie S1. PalmtdT EV bound to CD3+ T cell.