

Abstract
Background
Airway hyperresponsiveness (AHR) is a major feature of asthma attributed predominantly to an extrinsic immune/inflammatory response increasing airway smooth muscle (ASM) contractility.
Objective
We investigated whether increased ASM expression of ORMDL3, a gene on chromosome 17q21 highly linked to asthma, induced increased ASM proliferation and contractility in vitro, as well as influenced airway contractility and calcium flux in ASM in precision cut lung slices from WT and hORMDL3Zp3-Cre mice (which express increased levels of human ORMDL3).
Methods
Levels of ASM proliferation and contraction were assessed in ASM cells transfected with ORMDL3 in vitro. In addition, airway contractility and calcium oscillations were quantitated in ASM cells in precision cut lung slices derived from naïve WT and naïve hORMDL3Zp3-Cre mice which do not have a blood supply.
Results
Increased ASM expression of ORMDL3 in vitro resulted in increased ASM proliferation and contractility. Precision cut lung slices derived from naïve hORMDL3Zp3-Cre mice which do not have airway inflammation exhibit increased airway contractility with increased calcium oscillations in ASM cells. Increased ASM ORMDL3 increases ASM sarcoplasmic reticulum Ca2+ ATPase 2b (SERCA2b) which increases ASM proliferation and contractility.
Conclusion
Overall, these studies provide evidence that an intrinsic increase in ORMDL3 in ASM can induce increased ASM proliferation and contractility which may contribute to increased AHR in the absence of airway inflammation in asthma.
Keywords: ORMDL3, Asthma, Airway smooth muscle, SERCA2b, Airway hyperresponsiveness
INTRODUCTION
Airway hyperresponsiveness (AHR) is a major feature of asthma1. Conceptually AHR may either be due to an intrinsic abnormality in airway smooth muscle (ASM) resulting in increased contractility, or to an extrinsic effect of inflammatory cells and their mediators on ASM to increase contractility. The effects of extrinsic airway inflammation on ASM is considered to be the primary driving force for increased AHR mediated by ASM in asthma. Thus, the presence of airway inflammation in asthma forms the basis for NIH expert panel recommendations regarding early institution of anti-inflammatory therapy to reduce persistent asthma symptoms, exacerbations, and improve lung function and AHR1. The episodes of airway inflammation in asthma are commonly triggered by either viruses or allergen and are associated with release of inflammatory mediators from a variety of lung resident (mast cell, epithelium, macrophage) and recruited cells (eosinophils, T cell, neutrophil) that may contribute to increased ASM contractility, AHR, airway remodelling, and decline in lung function1.
In contrast to this well established role of extrinsic inflammation contributing to increased ASM contractility and AHR, the role of an intrinsic abnormality in ASM contributing to increased ASM contractility and AHR are less well established. Theoretically the contribution of an intrinsic abnormality in ASM to AHR may be mediated by either increased amounts of ASM, increased expression of contractile proteins in ASM cells, and/or increased activation of the contractile apparatus2–4. ASM contraction is initiated by the binding of calcium and calmodulin to activate myosin light chain (MLC) kinase, which phosphorylates MLC to form cross-bridge cycling of MLC with actin filaments4. Thus, the regulation of intracellular calcium concentration ([Ca2+]i) in ASM is essential for airway contraction. Mathematical models suggest that an increased amount of ASM in the airway submucosa is sufficient to cause AHR through mechanical effects alone5–7. The presence of increased airway smooth muscle mass in large and small airways in asthma is well described3,8–13 and is considered to be a major contributor to bronchoconstriction of airways, and persistent airflow obstruction6,11. The increase in ASM in asthma can be due to hyperplasia3 and/or hypertrophy11. There are studies demonstrating ASM hypertrophy in severe asthma10,11 but not in mild asthma3. There are several studies demonstrating that ex vivo asthmatic ASM proliferation (i.e. hyperplasia) is increased compared to non-asthmatics12,13. Studies of ASM obtained by laser capture from mild asthmatics and controls have shown that ASM cell number was nearly twofold higher in subjects with asthma, and the amount of ASM in the submucosa was increased 50–83%3. As with studies of hypertrophy of ASM in asthma, there are some studies that have not demonstrated increased proliferation of asthmatic ASM ex vivo14. Thus, while there is extensive evidence from multiple laboratories that there is increased ASM in asthma3,8–13, the mechanism mediating the increased ASM is incompletely understood. Current paradigms consider ASM as a target of mediators released from inflammatory cells in the airway that result in ASM changes (hypertrophy, hyperplasia) and increased AHR. Thus, the mechanisms behind these changes in ASM are considered to be mediated by repeated episodes of airway inflammation in asthma, rather than an intrinsic abnormality in ASM1.
Our previous studies of ORMDL3 (oroscomucoid 3), a gene on chromosome 17q21 which is highly linked to asthma in genome wide association studies (GWAS)15, demonstrated that these mice generated to express universal increased levels of human ORMDL3 (hORMDL3Zp3-Cre mice) spontaneously developed increased ASM, and increased AHR in the absence of airway inflammation16. These results suggest that ORMDL3 may be a key gene regulating ASM hypertrophy and hyperplasia, and more importantly, that lung structural cells (such as ASM), rather than inflammatory cells, expressing increased ORMDL3, may be the cells contributing to the increased AHR as observed in hORMDL3Zp3-Cre mice. In this study, we make the novel observation that ORMDL3 upregulates cardinal features of asthma including ASM proliferation, contractility, and Ca2+ oscillations in ASM cells. We also provide evidence that ORMDL3 (localized to the endoplasmic reticulum)17 upregulates sarcoendoplasmic reticulum calcium ATPase 2b (SERCA2b) (localized in proximity to ORMDL3 in the sarcoplasmic endoplasmic reticulum)18 which regulates Ca2+ flux and contractility in ASM cells. Therefore, this ORMDL3, SERCA2b, Ca2+ oscillation, and ASM contractility pathway in ASM may be an important genetic pathway intrinsic to ASM leading to increased AHR in the 62% of asthmatics harboring the SNP on chromosome 17q21 linked to increased levels of ORMDL3 expression15. These studies support the paradigm that ASM changes and AHR in asthma are not only driven by extrinsic inflammation, but can also be mediated by an intrinsic abnormality in lung structural cells such as ASM.
METHODS
Generation of hORMDL3zp3-Cre chimeric mice
hORMDL3zp3-Cre chimeric mice were generated as described in the online supplement16.
ASM studies in vitro
The methods for mASM or hASM culture, transfection with ORMDL3 or SERCA2b, siRNA knockdown with SERCA2b, quantitation of ASM proliferation and contraction in vitro, and qRT-PCR have been described in the online supplement16,19.
Mouse precision-cut lung slices (PCLSs)
The methods for generation of PCLSs, measurement of mouse airway contraction and relaxation, and measurement of Ca2+ signaling have been described in the online supplement20–23.
Statistical Analysis
All data are reported as the mean ± SEM. A P<0.05 was considered significant. Detailed methods for statistical analysis have been described in the online supplement.
RESULTS
Chimeric hORMDL3Zp3-Cre mice have increased ASM without airway inflammation
As our previous studies had demonstrated that naïve hORMDL3Zp3-Cre mice have increased ASM without airway inflammation16, we generated bone marrow chimeric mice in which the hORMDL3Zp3-Cre chimeric mouse had wild type (WT) bone marrow (with no expression of hORMDL3) and the mouse lung structural cells expressed increased levels of hORMDL3. Establishment of chimerism (WT bone marrow into irradiated hORMDL3Zp3-Cre recipient) was confirmed by RT-qPCR demonstrating hORMDL3 expressed in the lungs but not the blood or bone marrow leukocytes of hORMDL3Zp3-Cre chimeric mouse (Fig 1A). These studies demonstrated that 12 week old naïve chimeric hORMDL3Zp3-Cre mice had increased levels of peribronchial smooth muscle (p<0.05 vs WT control) as assessed by image analysis quantitation of the area of α-smooth muscle actin immunostaining (Fig 1 B–E). The naïve chimeric hORMDL3Zp3-Cre mice with increased ASM did not have evidence of airway inflammation (Fig 1F). These studies provide evidence that lung structural cell expression of hORMDL3 is sufficient to induce increased ASM independent of airway inflammation. However, these bone marrow chimera studies are unable to identify which lung structural cell expressing ORMDL3 (ASM, fibroblast, epithelium) is able to induce increased ASM mass independent of airway inflammation.
Figure 1. hORMDL3zp3-Cre chimeric mice and increased airway smooth muscle.
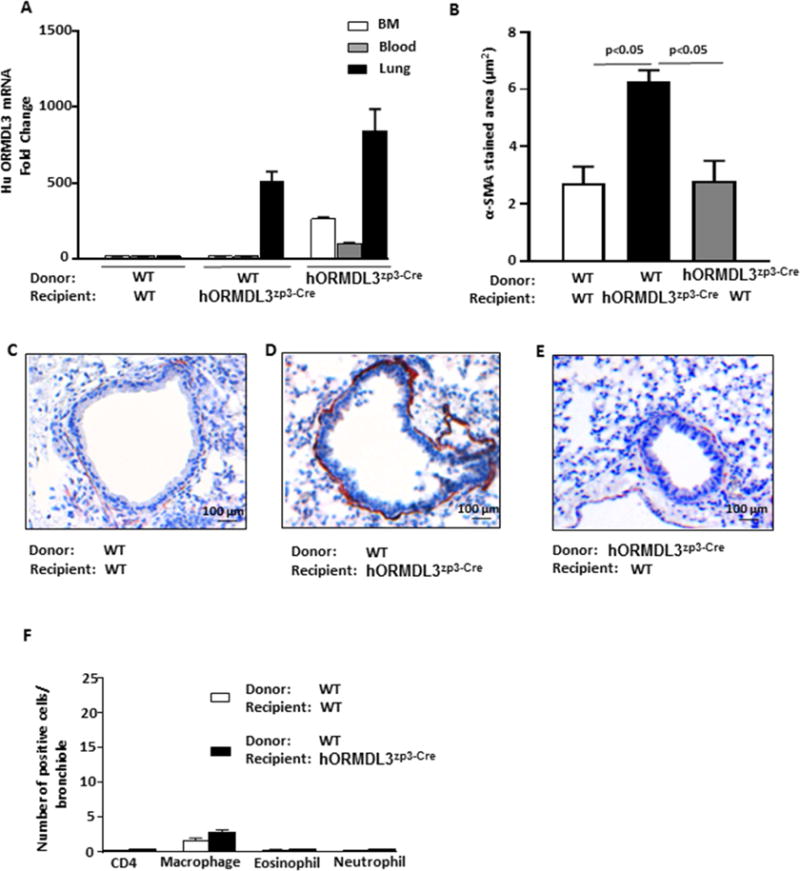
Wild type (WT) and hORMDL3zp3-Cre mice (n= 8 mice/group) were irradiated and each injected i.v. with WT bone marrow and sacrificed 12 weeks later. (A) Efficiency of chimerism was assessed by RT-qPCR to detect expression of human ORMDL3 in blood and lungs. (B–E). Lungs of chimeric mice (n= 8 mice/group) were immunostained with an anti-α smooth muscle actin antibody and the area of peribronchial anti-α smooth muscle actin immunostaining quantitated by image analysis. (F). Lungs of chimeric mice (n= 8 mice/group) were immunostained with either an anti-CD4, anti-F4/80, anti-MBP, or anti-neutrophil elastase antibody and the number of peribronchial cells immunostaining positive quantitated by image analysis.
Human ASM cells expressing increased levels of ORMDL3 have increased proliferation, contraction, and expression of SERCA2b
To determine whether ASM expression of ORMDL3 itself could contribute to the increased ASM observed in naïve chimeric hORMDL3Zp3-Cre mice, we transfected normal human ASM cells (hASM) with hORMDL3 (Fig 2A, 2B). These studies demonstrated that Ad-ORMDL3-GFP transfected hASM cells had significantly increased proliferation assessed by Ki67 mRNA (Fig 2C) and hASM cell counts (Fig 2D) as compared to empty vector control. In addition, Ad-ORMDL3-GFP transfected hASM had significantly increased contractility in response to acetylcholine (Ach) (Fig 2E, Fig 2F). These studies demonstrate that increased ORMDL3 in hASM can induce increased proliferation and increased hASM contractility a major feature of asthma.
Figure 2. ORMDL3, SERCA2b, and airway smooth muscle proliferation and contraction.
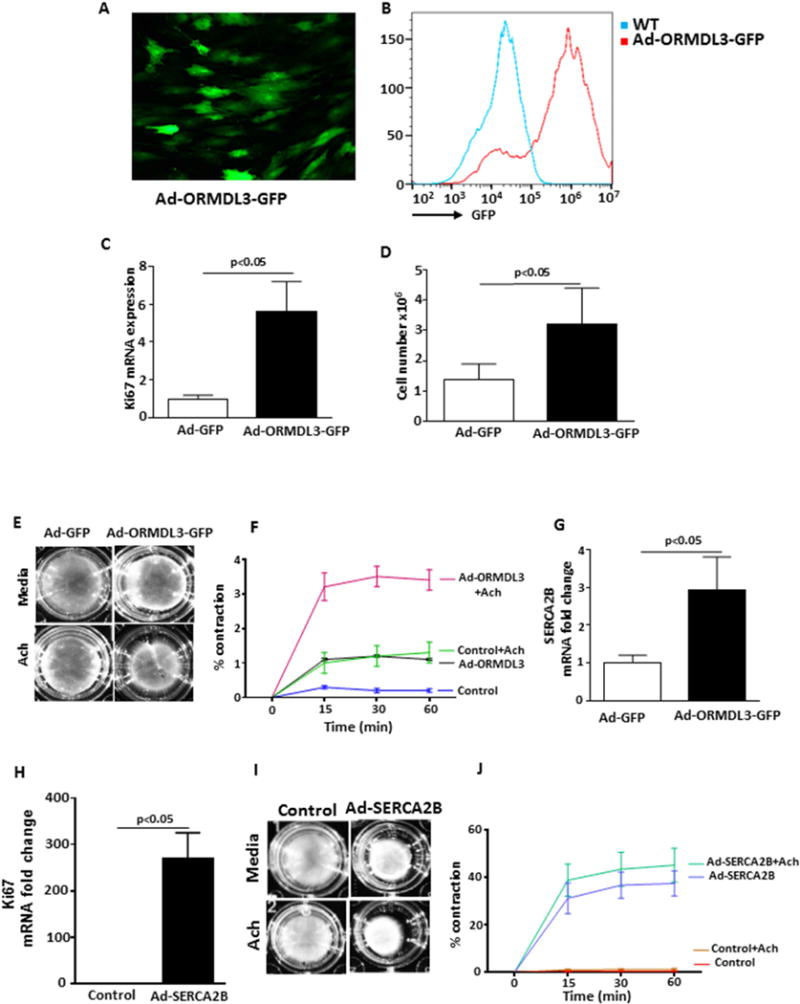
Primary human airway smooth muscle cells (hASM) were transfected with Ad-ORMDL3-GFP or as a control the empty vector Ad-GFP. Efficiency of transfection was assessed by immunofluorescence microscopy (A) and FACS (B). hASM proliferation was assessed 72 hours later either by (C) RT-qPCR of Ki67 mRNA levels (n= 3/group), or (D) by ASM cell counts by light microscopy (n= 6/group). (E–F). hASM contraction (n= 3/group) was assessed in a gel contraction assay in hASM cells transfected with either Ad-ORMDL3-GFP or Ad-GFP in the presence or absence of acetylcholine (Ach). Ad-ORMDL3 vs Control, p<0.05; Ad-ORMDL3 vs Ad-ORMDL3+Ach, p<0.05; Ad-ORMDL3+Ach vs Control +Ach, p<0.05; Ad-ORMDL3 vs Control + Ach, p= 0.20; Control vs Control + Ach, p<0.05. (G). Levels of SERCA2b (n= 3/group) were quantitated by RT-qPCR in hASM cells transfected with either Ad-ORMDL3-GFP or Ad-GFP. (H) Levels of hASM proliferation (n= 3/group) were assessed by RT-qPCR of Ki67 mRNA levels in hASM cells transfected with either Ad-SERCA2b or control. (I–J). hASM contraction (n= 3/group) was assessed in a gel contraction assay in hASM cells transfected with either Ad-SERCA2b or Ad empty vector control in the presence or absence of Ach. Ad-SERCA2b vs Control, p<0.05; Ad-SERCA2b vs Ad-SERCA2b+Ach, p=0.13; Ad-SERCA2b3+Ach vs Control +Ach, p<0.05; Ad-SERCA2b + vs Control + Ach, P<0.05.
We have previously demonstrated that hORMDL3Zp3-Cre mice have increased expression of the calcium pump SERCA2b in their lungs16, but have not previously investigated whether ORMDL3 regulates SERCA2b in ASM, and whether this influences ASM contractility. Thus, we transfected Ad-ORMDL3-GFP into hASM cells which resulted in significantly increased levels of expression of SERCA2b in hASM (Fig 2G). The importance of SERCA2b induction by ORMDL3 is suggested from subsequent studies in which we transfected normal hASM with Ad-SERCA2b. These studies demonstrated that Ad-SERCA2b transfected hASM cells had significantly increased proliferation as assessed by Ki67 mRNA (Fig 2H) as compared to empty vector control. In addition, Ad-SERCA2b transfected hASM cells had significantly increased contractility compared to control transfected hASM (p<0.05) (Fig 2I, Fig 2J), consistent with in vivo studies demonstrating increased cardiac smooth muscle contractility in mice expressing increased levels of SERCa2b in the heart24,25. Incubation of Ad-SERCA2b transfected hASM cells with Ach in vitro resulted in a trend for increased ASM contractility which was not statistically significant (Ad-SERCA2b vs Ad-SERCA2b + Ach) (p=0.13) (Fig 2J). Thus, the majority of the increased ASM contractility in Ad-SERCA2b transfected ASM incubated with Ach was due to Ad-SERCA2b transfection rather than Ach. These studies demonstrate that in hASM cells, increased SERCA2b (a downstream pathway from ORMDL3)16,17 can induce increased proliferation and increased hASM contractility providing evidence for an intrinsic pathway in hASM in which ORMDL3 upregulates SERCA2b resulting in increased hASM and increased contractility.
Increased airway contraction in hORMDL3Zp3-Cre mouse precision cut lung slices (PCLSs)
To determine whether ORMDL3 expression in the lung influences airway contraction, we visualized (Figure 3A, and Supplemental Video 1) and quantitated (Figure 3B–E) airway contractility in naïve hORMDL3Zp3-Cre mouse precision cut lung slices (PCLSs) which reflect the in situ physiology of mASM of relatively smaller airways devoid of a blood supply and thus unable to recruit inflammatory or immune cells from the circulation. To quantitate airway contraction in PCLSs, we used both MCh (a cholinergic agonist used to assess AHR in asthma) and 5-hydroxytryptamine (5-HT) which each trigger Ca2+ oscillation and contraction in ASM26 by binding to their respective ASM cell surface receptors, which as a consequence activate different downstream G-protein coupled pathways each generating inositol triphosphate (IP3)4,27–28. IP3 then binds to intracellular IP3 receptors on the SR to release Ca2+ from the SR into the cytoplasm4,27–28.
Figure 3. hORMDL3zp3-Cre mice have increased airway contraction compared to WT mice.
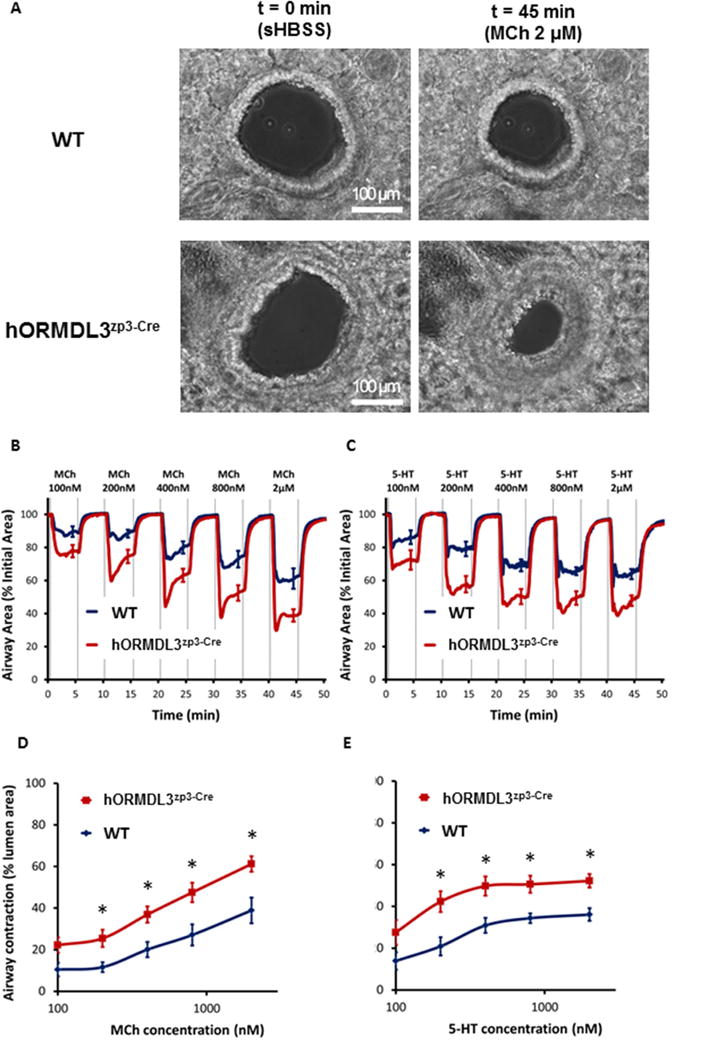
(A) Representative phase-contrast images of two airways in a precision cut lung slices (PCLSs) derived from WT and hORMDL3zp3-Cre mouse, respectively, under resting condition (left panel, scale bar = 100 μm), exposed to 2000 nM MCh (right panel) at 37°C. The response of airways in PCLSs derived from WT (blue line) and hORMDL3zp3-Cre (red line) mouse, respectively, to increased dose of MCh (B) and 5-HT (C) at 37°C, followed by a comparison of dose response curve to MCh (D) and 5-HT (E) between WT (blue line) and hORMDL3zp3-Cre (red line) mouse. hORMDL3zp3-Cre vs WT, *P<0.05. Each line in (B) and (C) represents the mean and each point represents the mean ± SEM at selected time points of the airway lumen area normalized to the initial size at t = 0 s. Data in (B) and (D) “WT” represent n = 9 airways, “hORMDL3zp3-Cre” represent n = 11 airways from 5 mice, respectively; data in (C) and (E) “WT” represent n = 9 airways, “hORMDL3zp3-Cre” represent n = 13 airways from 5 mice, respectively.
There was no significant difference in the baseline size of airways chosen for testing between hORMDL3zp3-Cre and WT group (supplemental Figure S1A–D). In addition, no pre-contraction of the airways was observed in either hORMDL3zp3-Cre or WT mice, as none of the airways showed relaxation to (R,R)-formoterol, a long-acting β2-receptor agonist that relaxes ASM by inactivating MLC kinase and activating MLC phosphatase (supplemental Figure S1E and S1F). Both MCh (Figure 3B) and 5-HT (Figure 3C) induced a dose-dependent increase in airway contraction in WT mouse PCLSs at 37°C. As compared to WT mice, the extent of airway contraction in hORMDL3zp3-Cre mice were significantly increased for both MCh (Figure 3B, 3D, and Supplemental Video 1) and 5-HT (Figure 3C, 3E) at all concentrations tested (100 to 2000 nM) suggesting an overall increased airway contractility in hORMDL3zp3-Cre mice. A similar pattern of increased airway contraction in hORMDL3zp3-Cre mice were noted at room temperature (supplemental Figure S2). These studies demonstrate that increased ORMDL3 expression in naïve lung PCLSs without a blood supply results in increased airway reactivity to agonists including MCh, a major feature of asthma.
Increased Ca2+ oscillation frequency in ASM cells in hORMDL3Zp3-Cre mouse PCLSs
As ORMDL3 induced increased ASM expression of SERCA2b (Fig 2G), which regulates [Ca2+]i that control ASM contraction, we further examined whether ASM cells with increased ORMDL3 expression in PCLSs from hORMDL3zp3-Cre mice could exhibit increased frequency of Ca2+ oscillations in response to either MCh or 5-HT, as Ca2+ oscillation frequency has been implicated in ASM contractile responses in vivo26–28. No spontaneous Ca2+ signaling was observed in mASM cells derived from either WT or hORMDL3zp3-Cre mice. In response to both MCh and 5-HT at 370C, mASM cells in WT PCLSs showed a rapid onset within 15 seconds of oscillatory increases of [Ca2+]i, which occurred in the form of Ca2+ waves propagating along the length of the mASM cells (Figure 4A–4E, and Supplemental Video 2). These Ca2+ oscillations were accompanied by contraction and shortening in mASM cells resulting in airway contraction (Figure 3A). In the mASM cell in PCLSs of WT mice (Figure 4F), MCh induced a concentration dependent increase in Ca2+ oscillation frequency in a similar way as observed in airway contraction. As compared to WT mice, the Ca2+ oscillation frequency in mASM cells in PCLSs of hORMDL3zp3-Cre mice were significantly increased for both MCh (Figure 4G and 4H) and 5-HT (Figure 4I) at all concentrations tested (100 to 2000 nM). A similar pattern of increased Ca2+ oscillations in mASM cells of hORMDL3zp3-Cre mice were noted at room temperature (supplemental Figure S3). These results demonstrate that in the absence of airway inflammation, mASM cells with increased ORMDL3 expression exhibit increased Ca2+ oscillation frequency response to agonists. Thus, mASM with increased Ca2+ oscillations is associated with increased airway contractility in hORMDL3zp3-Cre mice.
Figure 4. hORMDL3zp3-Cre mice have increased Ca2+ oscillations in airway smooth muscle cells compared to WT mice.
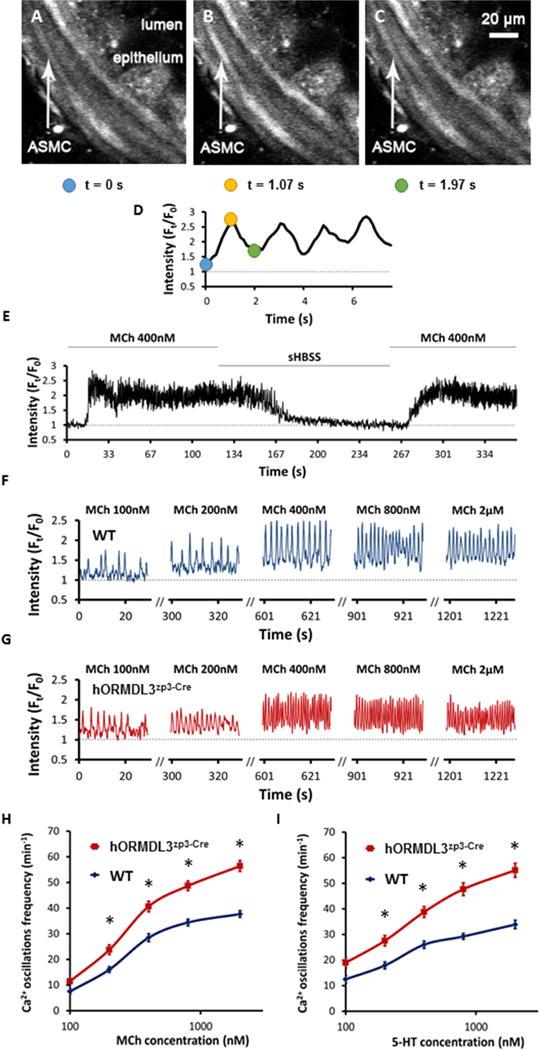
(A–D) Representative 2-photon images of MCh-induced Ca2+ oscillations in the same airway smooth muscle cell (labeled ASMC with white arrow) in mouse precision cut lung slices (PCLSs). A cycle of Ca2+ oscillations in the same ASMC over time can be visualized in A–D as a change in fluorescence intensity of Oregon Green (white color change in ASMC in figure A–C, and normalized intensity change in figure D) recorded at baseline pre-MCh at 0 s (A), and at two time points after MCh, 1.07 s (B), and 1.97 s (C) (scale bar = 20μm). (E) Representative trace shows that 400 nM MCh induced Ca2+ oscillations in an ASMC at 37°C, which sustained until MCh was washed off by sHBSS, adding back MCh would induce Ca2+ oscillations again. Representative traces show Ca2+ oscillations of ASMC in PCLSs derived from WT (F) and hORMDL3zp3-Cre mouse (G), respectively, response to increased dose of MCh at 37°C, followed by a comparison of dose response curve to MCh (H) and 5-HT (I) between WT (blue line) and hORMDL3zp3-Cre (red line) mouse. hORMDL3zp3-Cre vs WT, *P<0.05. Representative traces are expressed as intensity (Ft) normalized to the initial intensity at t = 0 s (F0), measured from a region of interest with size of 8 × 10 pixel within a single ASMC. Data in (H) “WT” represent n = 26 ASMCs, “hORMDL3zp3-Cre” represent n = 31 ASMCs from 5 mice, respectively; data in (I) “WT” represent n = 17 ASMCs, “hORMDL3zp3-Cre” represent n = 26 ASMCs from 5 mice, respectively.
Knockdown or inhibition of SERCA2b inhibits ASM contractility and Ca2+ oscillations
To determine the role of SERCA2b expression induced by ORMDL3 in mediating increased contractility and Ca2+ oscillations in hASM, we initially demonstrated that the levels of SERCA2b in mASM cells derived from hORMDL3zp3-Cre mice were increased compared to WT mice (Fig 5A) as quantitated by RT-qPCR. In hASM, SERCA2b but not SERCA2a was detected (Fig 5B). We then used siRNA to knockdown SERCA2b expression in hASM cells. siRNA knockdown of SERCA2b was efficient in hASM cells (Fig 5C). siRNA knockdown of SERCA2b significantly inhibited hASM contraction in vitro in response to histamine starting within 15 minutes of exposure to histamine (Fig 5D). Control scrambled siRNA did not inhibit histamine induced hASM contraction. These results demonstrated an important role of SERCA2b in ASM contractility in vitro.
Figure 5. Knockdown or inhibition of SERCA2b inhibits airway smooth muscle contractility and Ca2+ oscillations.
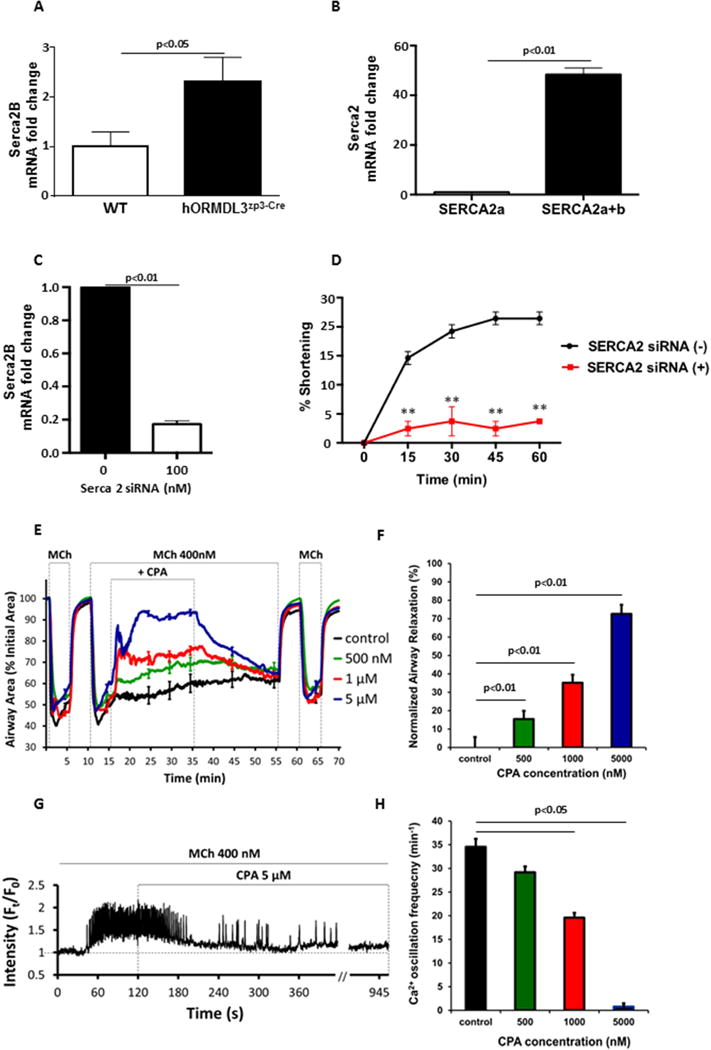
(A). RT-qPCR was used to detect levels of SERCA2b mRNA in mouse airway smooth muscle (mASM) derived from either hORMDL3zp3-Cre mice or WT control mice (n=6/group). (B) Levels of SERCA2a and SERCA2a and 2b mRNA were quantitated by RT-qPCR in human ASM (hASM) cells (n= 6/group). (C). Levels of SERCA2b mRNA were quantitated in hASM cells in which SERCA2 siRNA was used to knockdown SERCA. Scrambled siRNA was used as a control (n= 4/group). (D). hASM contraction was assessed in a gel contraction assay for 0–60 minutes in the presence of histamine in hASM cells in which siRNA (or scrambled control siRNA) was used to knockdown SERCA2 (n=8/group). SERCA2 siRNA vs Scrambled siRNA. **P<0.01. (E) The effect of cyclopiazonic acid (CPA) (500 to 5000 nM) on airways contracted with 400 nM MCh in precision cut lung slices (PCLSs) derived from WT mice, followed by (F) a summary of relaxation effect normalized to control. Controls were treated with vehicle of 0.5% DMSO in sHBSS, P<0.01. Each line in (E) represents the mean and each point represents the mean ± SEM at selected time points of the airway lumen area normalized to the initial size at t = 0 s. Data in (F) represent n = 8 – 10 airways for each bar from 5 mice. (G) Representative trace shows 5 μM CPA gradually stopped Ca2+ oscillations induced by 400 nM MCh in a mASM cell in mouse PCLSs. (H) A summary for the effect of CPA on Ca2+ oscillation frequency induced by 400 nM MCh in mASM cells in PCLSs, P <0.05. Data in (H) represent n = 17 – 19 mASM cells for each bar from 5 mice.
To further confirm the role of SERCA2b in airway contraction and underlied Ca2+ oscillations in ASM cells, we investigate the effect of cyclopiazonic acid (CPA), a specific SERCA pump inhibitor29, on the MCh-induced airway contraction, as well as the Ca2+ oscillations in ASM cells in mouse PCLSs (Fig 5E–H; supplemental Figure S4). Treatment of MCh-contracted airways with CPA (from 500 nM to 5 μM) relaxed airway contraction in a concentration-dependent manner (Fig 5E and 5F), 5 μM CPA could induce nearly 73% relaxation as normalized to the control (Fig 5F). Consistently, CPA also inhibited MCh-induced Ca2+ oscillations in a concentration-dependent manner (Fig. 5G and 5H), while 5 μM CPA gradually stopped Ca2+ oscillations in 15 min (Fig. 5G). Taken together, these results suggest that SERCA2b expression or activity is important to sustain ASM contractility via regulating Ca2+ oscillation frequency in ASM cells. Thus, increased SERCA2b expression induced by ORMDL3 in ASM cells could contribute to the increased reactivity of ASM in response to agonists such as MCh and 5-HT as observed in the PCLSs of hORMDL3zp3-Cre mice.
DISCUSSION
This study makes the novel observation that ORMDL3 expression in ASM may explain the strong genetic linkage of ORMDL3 on chromosome 17q21 to asthma15. In addition, this study provides evidence that intrinsic expression of a gene highly linked to asthma in ASM can lead to increased ASM proliferation and contractility independent of extrinsic inflammation. Thus, asthma may be a disease of both airway inflammation as well as an intrinsic change in ASM. In support of this, we demonstrate that ORMDL3 expression in ASM cells in vitro induces increased ASM proliferation and increased ASM contractility to Ach. In addition, in vivo studies demonstrate that naïve hORMDL3Zp3-Cre chimeric mice with ORMDL3 expressed in lung structural cells (but not in bone marrow derived inflammatory and immune cells) have increased ASM in the absence of lung inflammation. Importantly, in vivo studies demonstrate that ASM in naïve hORMDL3Zp3-Cre mouse PCLSs without a blood supply have increased contractility and increased Ca2+ oscillation frequency, a fundamental signaling mechanism mediating ASM contraction, in response to MCh and 5-HT. Taken together, these studies underscore the important role that ORMDL3 expressed in ASM plays in inducing increased ASM proliferation and increased ASM contractility to MCh, a major feature of asthma. However, this study is unable to determine whether ORMDL3 induces increased AHR by effects on increasing ASM proliferation and/or by increasing ASM contractility. As ORMDL3 is highly linked in genetic epidemiology studies to childhood asthma15, ORMDL3 expression in ASM may help to explain why individuals with the SNP linked to chromosome 17q21 and increased ORMDL3 expression have increased AHR, a major feature of asthma.
The concept that Th2 inflammation is important to the development of allergic disease in the upper airway and lower airway is well accepted. However, there is limited insight into why certain individuals with the same level of Th2 inflammation will either only have allergic rhinitis (with no asthma), while others will have allergic rhinitis and asthma. In this study we provide evidence that ORMDL3, a gene most highly linked to asthma15 (but not linked to allergic rhinitis)30, when expressed in a lung structural ASM cells in vitro or in vivo spontaneously induced increased airway contractility to MCh without an additional inflammatory stimulus. This may help to explain how Th2 inflammation when combined with an ASM expressed asthma associated gene such as ORMDL3 can result in allergic rhinitis and asthma, whereas Th2 inflammation alone in the absence of an abnormality in ASM may result in allergic rhinitis but not asthma. ORMDL3 has been linked asthma15 but not to allergic rhinitis30, underscoring differences between asthma and allergic rhinitis. In addition, the SNP linked to increased ORMDL3 expression in asthma is common affecting approximately 62% of childhood asthmatics15. Another example of a structural gene being important to allergic disease is the loss of function of the filaggrin gene in epithelial cells in the skin in atopic dermatitis which influences barrier function in Th2 inflammation in the skin31.
Previous studies have also provided evidence that AHR can be independent from airway inflammation. For example, a study of allergic asthmatics did not find a correlation between AHR to MCh and levels of inflammatory cells in sputum, BAL, or bronchial biopsy32. In addition, inhaled corticosteroids can effectively reduce levels of airway inflammation, but only attenuate AHR33. Patients with eosinophilic bronchitis can also have severe inflammation but no AHR33. Population studies have also demonstrated that AHR is a heritable trait, with an estimated heritability of 30 to 66%34,35. Thus, while airway inflammation plays an important role in the pathogenesis of asthma, there are likely genetic, as well as airway components (airway inflammation; airway remodeling) that all contribute to levels of AHR in asthma36.
Our study also provides evidence that SERCA2b may be the downstream pathway by which ORMDL3 influences ASM contractility. Our prior studies have demonstrated that in human lung epithelial cells increased expression of hORMDL3 activates several downstream pathways including ATF6α, SERCA2b, sphingolipids, chemokines, and remodeling pathways16,17. As ORMDL3 regulates several downstream pathways, some downstream pathways may be more important in ASM compared to other cell types expressing ORMDL3. For example, the importance of increased SERCA2b to ASM hypertrophy and increased contractility is suggested from studies of the heart, in which mice expressing increased levels of SERCA2b in the heart had increased cardiac contractility with increased Ca2+ flux in cardiomyocytes, and increased cardiac muscle hypertrophy24,25. SERCA are intracellular ATP driven Ca2+ pumps that transport Ca2+ from the cytoplasm into the sarco-endoplasmic reticulum18,37 (the domain of the endoplasmic reticulum that lacks ribosomes). SERCA proteins are encoded by three genes Serca1, Serca2, and Serca3, of which SERCA2 is the only SERCA expressed in ASM18,37. Alternative splicing of the Serca2 gene encodes two major protein isoforms, SERCA2a which is expressed predominantly in cardiomyocytes and slow twitch skeletal muscle18,37, while SERCA2b, the focus of this study is expressed in ASM and most tissues18,37. Much of the Ca2+ entering the ASM cell and released from stores may be sequestered by the superficial SR through SERCA. Cycling between SR Ca2+ uptake and release mechanisms leads to Ca2+ oscillations27,28. Force generation by ASM is dependent on actin–myosin cross-bridge formation4,28. When cytosolic Ca2+ levels are increased, the combination of Ca2+ and calmodulin activate myosin light chain (MLC) kinase, which phosphorylates MLC, leading to enhanced contraction4,38. Therefore, ASM cell contraction is regulated by the frequency of oscillations in [Ca2+]i27,28, which correspond to the speed of cyclic release of Ca2+ from the SR and re-uptake of Ca2+ from the SR by SERCA38,39. In this study we demonstrate that ORMDL3 increases levels of SERCA2b in ASM which is associated with increased frequency of Ca2+ oscillations in ASM. In addition, increased levels of SERCA2b in ASM increases ASM proliferation, while siRNA knockdown of SERCA2b reduces ASM contractility. Furthermore, inhibition of SERCA by giving CPA in PCLSs reduced Ca2+ oscillation frequency in ASM cells and thus airway contraction in a dose-dependent way. These results together suggest that SERCA2b, regulated by ORMDL3 in ASM, plays an important role in sustaining agonists-induced Ca2+ oscillations in ASM cells and is associated with increased airway contractility as shown in PCLSs of hORMDL3Zp3-Cre mice. Transfection of SERCA2b into ASM resulted in greater enhancement of contraction than that noted with transfection of ORMDL3. One possible explanation for these results is that transfection of ORMDL3 not only increases SERCA2b (which increases ASM contractility), but also inhibits the function of the enzyme serine palmitoyltransferase or SPT40 (which could reduce ASM contractility). For example, increased levels of ORMDL3 inhibit the enzyme SPT, the first and rate limiting step in sphingolipid synthesis resulting in lower levels of sphingosine 1 phosphate (S1P)40. As S1P induces ASM contraction41, lower levels of S1P induced by increased levels of ORMDL3 could reduce ASM contractility. Thus, ORMDL3 induction of SERCA2b could increase ASM contractility, while its concomitant induction of the enzyme SPT could reduce S1P and reduce ASM contractility. Thus, the net effect of ORMDL3 on ASM contractility may be less than that of SERCA2b, because of the induction not only of SERCA2b, but also inhibition of S1P by ORMDL3. Interestingly, therapeutically targeting Ca2+ signaling pathways in smooth muscle in asthma to influence smooth muscle contractility and AHR is currently an active area of investigation42. However, there are also studies which question whether Ca2+ signals change in asthmatic ASM while stimulated with agonists43–45.
Interestingly, some studies have reported that decreased expression of SERCA in ASM from asthmatics contributes to airway remodeling43. It should be noted that our study differs from the prior study43 in examining the effect of increased ORMDL3 (simulating asthmatics with the SNP on chromosome 17q21 linked to increased ORMDL3 expression) on SERCA2b in ASM, whereas the prior study did not genotype asthmatics to determine whether they had the SNP associated with increased ORMDL3 expression which we demonstrate is associated with increased SERCA2b expression. Thus, we hypothesize that based on genotype not all asthmatics will have increased SERCA2b expression. In addition, prior studies have demonstrated that suppressed SERCA by cytokines might slow [Ca2+]i response to agonists, and thus maintain a relatively high [Ca2+]i leading to increased ASM contractility46. In agreement with this, our results showed that treatment of PCLSs with high dose (10 M) of CPA stopped MCh-induced Ca2+ oscillations and lead to a rise of [Ca2+]i baseline in ASM cells with partially contracted airway (supplemental Fig S4). However, lower dose of CPA (from 500 to 5000 nM) reduced Ca2+ oscillations and airway contraction in a dose-dependent manner, suggesting that under a certain range, increased/decreased SERCA is proportional to increased/decreased Ca2+ oscillations and therefore increased/decreased airway contraction, while fully block or knockdown of SERCA would result in an [Ca2+]i plateau that mediates only impaired ASM contraction response to agonists47.
ORMDL3 is expressed in multiple cell types including lung structural cells (epithelium, smooth muscle) and immune and inflammatory cells (T cells, eosinophils, and macrophages)16,17. As the SNP linking chromosome 17q21 to asthma is associated with increased ORMDL3 expression15 we have previously used cre-lox techniques to generate mice selectively deficient in ORMDL3 in airway epithelium (Ormdl3Δ2-3/Δ2-3CC10)41 to simulate an inhaled therapy that effectively inhibited ORMDL3 expression in the airway. In contrast to the anticipated reduction in AHR, allergen challenged Ormdl3Δ2-3/Δ2-3CC10 mice had a significant increase in AHR compared to WT mice41. Levels of sphingosine-1-phosphate (S1P) were significantly increased in Ormdl3Δ2-3/Δ2-3CC10 mice as well as in airway epithelial cells in which ORMDL3 was inhibited with siRNA and incubation of S1P with ASM cells significantly increased contractility41. Overall, Ormdl3Δ2-3/Δ2-3CC10 mice exhibit increased allergen induced AHR independent of inflammation and associated with increased S1P generation41. These studies suggest that therapies that selectively and effectively inhibit ORMDL3 in airway epithelium in asthma, may not be the best cell to target. Further studies are needed to determine whether targeting reduction of ORMDL3 in airway ASM would reduce AHR.
In summary, in this study we provide evidence that an intrinsic gain of function in ORMDL3 in ASM can influence ASM contractility in asthma, by demonstrating that increased ORMDL3 expression in ASM cells induces increased expression of SERCA2b with consequent increased Ca2+ oscillation in ASM cells, and increased ASM contractility. Overall, these studies provide evidence that ORMDL3 (a gene on chromosome 17q21 highly linked to asthma in GWAS)15 can induce increased ASM contractility in the absence of inflammation. As AHR may conceptually be driven by either chronic inflammation inducing increased ASM contractility, and/or an intrinsic change in ASM inducing increased ASM hyperplasia and contractility, this study provides evidence that targeting a gene inducing a fundamental abnormality in ASM such as ORMDL3, may result in a new therapeutic approach to the treatment of asthma.
Supplementary Material
A representative comparison of airway contraction in mouse precision-cut lung slices (PCLS) induced by increasing doses of MCh in hORMDL3zp3-Cre and WT mouse.
Representative Ca2+ oscillations in airway smooth muscle cells (ASMCs) induced by 400 nM MCh in precision-cut lung slices (PCLS) derived from WT mouse. Note that Ca2+ oscillations occurred in the form of Ca2+ waves propagating along the length of ASMCs.
KEY MESSAGES.
Increased ASM expression of ORMDL3 induces increased ASM proliferation and contractility in vitro.
Precision cut lung slices derived from naïve hORMDL3Zp3-Cre mice (which express increased levels of human ORMDL3) and do not have airway inflammation exhibit increased airway contractility with increased calcium oscillations in ASM cells.
In addition to extrinsic inflammation, intrinsic ASM expression of ORMDL3, can increase ASM contractility and proliferation and might contribute to increased AHR in asthma.
CAPSULE SUMMARY.
This study demonstrates that an intrinsic increase in ORMDL3 in ASM can induce increased ASM proliferation and contractility which might contribute to increased AHR in the absence of airway inflammation in asthma.
Acknowledgments
Research reported in this publication was supported by NIH grants AI 107779, AI 38425, AI 070535, AI 72115, AI 242236 to D.H.B
Funding: This work was supported by NIH grants AI 107779, AI 38425, AI 070535, AI 72115, AI 242236 to D.H.B
Abbreviations
- AHR
airway hyperresponsiveness
- ASM
airway smooth muscle
- [Ca2+]i
intracellular Ca2+ concentration
- CPA
cyclopiazonic acid
- 5-HT
5-hydroxytryptamine
- GWAS
genome wide association study
- MCh
methacholine
- MLC
myosin light-chain
- OG
Oregon Green
- ORMDL3
oroscomucoid like 3
- PCLS
precision-cut lung slices
- SERCA
sarcoplasmic reticulum Ca2+ transport ATPases
- SNP
single nucleotide polymorphism
- SR
sarcoplasmic reticulum
- TG
transgenic
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Expert Panel Report 3: Guidelines for the diagnosis and management of asthma. NHLBI; 2007. https://www.nhlbi.nih.gov/health-pro/guidelines/current/asthma-guidelines. [DOI] [PubMed] [Google Scholar]
- 2.Bentley JK, Hershenson MB. Airway smooth muscle growth in asthma: proliferation, hypertrophy, and migration. Proc Am Thorac Soc. 2008;5:89–96. doi: 10.1513/pats.200705-063VS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woodruff PG, Dolganov GM, Ferrando RE, Donnelly S, Hays SR, Solberg OD, et al. Hyperplasia of smooth muscle in mild to moderate asthma without changes in cell size or gene expression. Am J Respir Crit Care Med. 2004;169:1001–1006. doi: 10.1164/rccm.200311-1529OC. [DOI] [PubMed] [Google Scholar]
- 4.Erle DJ, Sheppard D. The cell biology of asthma. J Cell Biol. 2014;205:621–631. doi: 10.1083/jcb.201401050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.James AL, Pare PD, Hogg JC. The mechanics of airway narrowing in asthma. Am Rev Respir Dis. 1989;139:242–246. doi: 10.1164/ajrccm/139.1.242. [DOI] [PubMed] [Google Scholar]
- 6.Lambert RK, Wiggs BR, Kuwano K, Hogg JC, Paré PD. Functional significance of increased airway smooth muscle in asthma and COPD. J Appl Physiol. 1993;74:2771–2781. doi: 10.1152/jappl.1993.74.6.2771. [DOI] [PubMed] [Google Scholar]
- 7.Macklem PT. A theoretical analysis of the effect of airway smooth muscle load on airway narrowing. Am J Respir Crit Care Med. 1996;153:83–89. doi: 10.1164/ajrccm.153.1.8542167. [DOI] [PubMed] [Google Scholar]
- 8.Ebina M, Yaegashi H, Chiba R, Takahashi T, Motomiya M, Tanemura M. Hyperreactive site in the airway tree of asthmatic patients revealed by thickening of bronchial muscles: a morphometric study. Am Rev Respir Dis. 1990;141:1327–1332. doi: 10.1164/ajrccm/141.5_Pt_1.1327. [DOI] [PubMed] [Google Scholar]
- 9.Saetta M, Di Stefano A, Rosina C, Thiene G, Fabbri LM. Quantitative structural analysis of peripheral airways and arteries in sudden fatal asthma. Am Rev Respir Dis. 1991;143:138–143. doi: 10.1164/ajrccm/143.1.138. [DOI] [PubMed] [Google Scholar]
- 10.Ebina M, Takahashi T, Chiba T, Motomiya M. Cellular hypertrophy and hyperplasia of airway smooth muscles underlying bronchial asthma: a 3-D morphometric study. Am Rev Respir Dis. 1993;148:720–726. doi: 10.1164/ajrccm/148.3.720. [DOI] [PubMed] [Google Scholar]
- 11.Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway structural alterations selectively associated with severe asthma. Am J Respir Crit Care Med. 2003;167:1360–1368. doi: 10.1164/rccm.200209-1030OC. [DOI] [PubMed] [Google Scholar]
- 12.Johnson PR, Roth M, Tamm M, Hughes M, Ge Q, King G, et al. Airway smooth muscle cell proliferation is increased in asthma. Am J Respir Crit Care Med. 2001;164:474–477. doi: 10.1164/ajrccm.164.3.2010109. [DOI] [PubMed] [Google Scholar]
- 13.Trian T, Benard G, Begueret H, Rossignol R, Girodet PO, Ghosh D, et al. Bronchial smooth muscle remodeling involves calcium-dependent enhanced mitochondrial biogenesis in asthma. J Exp Med. 2007;204:3173–3181. doi: 10.1084/jem.20070956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaur D, Hollins F, Saunders R, Woodman L, Sutcliffe A, Cruse G, et al. Airway smooth muscle proliferation and survival is not modulated by mast cells. Clin Exp Allergy. 2010;40:279–288. doi: 10.1111/j.1365-2222.2009.03423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moffatt MF, Kabesch M, Liang L, Dixon AL, Strachan D, Heath S, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 16.Miller M, Rosenthal P, Beppu A, Mueller JL, Hoffman HM, Tam AB, et al. ORMDL3 transgenic mice have increased airway remodeling and airway responsiveness characteristic of asthma. J Immunol. 2014;192:3475–3487. doi: 10.4049/jimmunol.1303047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller M, Tam AB, Cho JY, Doherty TA, Pham A, Khorram N, et al. ORMDL3 is an inducible lung epithelial gene regulating metalloproteases, chemokines, OAS, and ATF6. Proc Natl Acad Sci USA. 2012;109:16648–16653. doi: 10.1073/pnas.1204151109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wuytack F, Raeymaekers L, Missiaen L. Molecular physiology of the SERCA and SPCA pumps. Cell Calcium. 2002;32:279–305. doi: 10.1016/s0143416002001847. [DOI] [PubMed] [Google Scholar]
- 19.Miller M, Beppu A1, Rosenthal P1, Pham A1, Das S1, Karta M, et al. Fstl1 promotes asthmatic airway remodeling by inducing oncostatin M. J Immunol. 2015;195:3546–56. doi: 10.4049/jimmunol.1501105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez JF, Sanderson MJ. The frequency of calcium oscillations induced by 5-HT, ACH, and KCl determine the contraction of smooth muscle cells of intrapulmonary bronchioles. J Gen Physiol. 2005;125:535–553. doi: 10.1085/jgp.200409216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bai Y, Sanderson MJ. The contribution of Ca2+ signaling and Ca2+ sensitivity to the regulation of airway smooth muscle contraction is different in rats and mice. Am J Physiol-Lung Cell Mol Physiol. 2009;296:L947–L958. doi: 10.1152/ajplung.90288.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan X, Sanderson MJ. Bitter tasting compounds dilate airways by inhibiting airway smooth muscle calcium oscillations and calcium sensitivity. Br J Pharmacol. 2014;171:646–662. doi: 10.1111/bph.12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bai Y, Sanderson MJ. Modulation of the Ca2+ sensitivity of airway smooth muscle cells in murine lung slices. American J Physiol-Lung Cellular Mol Physiol. 2006;291:L208–221. doi: 10.1152/ajplung.00494.2005. [DOI] [PubMed] [Google Scholar]
- 24.Greene AL, Lalli MJ, Ji Y, Babu GJ, Grupp I, Sussman M, et al. Overexpression of SERCA2b in the heart leads to an increase in sarcoplasmic reticulum calcium transport function and increased cardiac contractility. J Biol Chem. 2000;275:24722–24727. doi: 10.1074/jbc.M001783200. [DOI] [PubMed] [Google Scholar]
- 25.Ver Heyen M, Heymans S, Antoons G, Reed T, Periasamy M, Awede B, et al. Replacement of the muscle-specific sarcoplasmic reticulum Ca(2+)-ATPase isoform SERCA2a by the nonmuscle SERCA2b homologue causes mild concentric hypertrophy and impairs contraction-relaxation of the heart. Circ Res. 2001;89:838–846. doi: 10.1161/hh2101.098466. [DOI] [PubMed] [Google Scholar]
- 26.Leybaert L, Sanderson MJ. Intercellular Ca (2+) waves: mechanisms and function. Physiol Rev. 2012;92:1359–1392. doi: 10.1152/physrev.00029.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lauzon AM, Bates JH, Donovan G, Tawhai M, Sneyd J, Sanderson MJ. A multi-scale approach to airway hyperresponsiveness: from molecule to organ. Front Physiol. 2012;3:191, 1–25. doi: 10.3389/fphys.2012.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanderson MJ, Bai Y, Perez-Zoghbi J. Ca (2+) oscillations regulate contraction of intrapulmonary smooth muscle cells. Adv Exp Med Biol. 2010;661:77–96. doi: 10.1007/978-1-60761-500-2_5. [DOI] [PubMed] [Google Scholar]
- 29.Michelangeli F, East JM. A diversity of SERCA Ca2+ pump inhibitors. Biochem Soc Trans. 2011;39:789–797. doi: 10.1042/BST0390789. [DOI] [PubMed] [Google Scholar]
- 30.Crimi E, Spanevell A, Neri M, Ind PW, Rossi GA, Brusaasco V. Dissociation between airway inflammation and airway responsiveness in asthma. Am J Resp Crit Care Med. 1998;157:4–9. doi: 10.1164/ajrccm.157.1.9703002. [DOI] [PubMed] [Google Scholar]
- 31.Sandilands A, Terron-Kwiatkowski A, Hull PR, O’Regan GM, Clayton TH, Watson RM, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet. 2007;39:650–654. doi: 10.1038/ng2020. [DOI] [PubMed] [Google Scholar]
- 32.Sont JK, Willems LN, Bel EH, Van Krieken JH, Vandenbroucke JP, Sterk PJ. Clinical control and histopathologic outcome of asthma when using airway hyperresponsiveness as an additional guide to long-term treatment. The AMPUL Study Group. Am J Respir Crit Care Med. 1999;159:1043–1051. doi: 10.1164/ajrccm.159.4.9806052. [DOI] [PubMed] [Google Scholar]
- 33.Brightling CE, Ward R, Goh KL, Wardlaw AJ, Pavord ID. Eosinophilic bronchitis is an important cause of chronic cough. Am J Respir Crit Care Med. 1999;160:406–410. doi: 10.1164/ajrccm.160.2.9810100. [DOI] [PubMed] [Google Scholar]
- 34.Hopp RJ, Bewtra AK, Watt GD, Nair NM, Townley RG. Genetic analysis of allergic disease in twins. J Allergy Clin Immunol. 1984;73:265–270. doi: 10.1016/s0091-6749(84)80018-4. [DOI] [PubMed] [Google Scholar]
- 35.Palmer LJ, Burton PR, Faux JA, James AL, Musk AW, Cookson WO. Independent inheritance of serum immunoglobulin E concentrations and airway responsiveness. Am J Respir Crit Care Med. 2000;161:1836–1843. doi: 10.1164/ajrccm.161.6.9805104. [DOI] [PubMed] [Google Scholar]
- 36.Busse WW. The relationship of airway hyperresponsiveness and airway inflammation: Airway hyperresponsiveness in asthma: its measurement and clinical significance. Chest. 2010;138:4S–10S. doi: 10.1378/chest.10-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vandecaetsbeek I, Vangheluwe P, Raeymaekers L, Wuytack F, Vanoevelen J. The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb Perspect Biol. 2011;3:1–24. doi: 10.1101/cshperspect.a004184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanderson MJ, Madison JM. Bitter treats for better breathing. Nat Med. 2010;16:1190–1191. doi: 10.1038/nm1110-1190. [DOI] [PubMed] [Google Scholar]
- 39.Nivala M, de Lange E, Rovetti R, Qu Z. Computational modeling and numerical methods for spatiotemporal calcium cycling in ventricular myocytes. Front Physiol. 2012;3:114, 1–12. doi: 10.3389/fphys.2012.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Breslow DK, Collins SR, Bodenmiller B, Aebersold R, Simons K, Shevchenko A, et al. Orm family proteins mediate sphingolipid homeostasis. Nature. 2010;463:1048–1053. doi: 10.1038/nature08787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller M, Tam AB, Mueller JL, Rosenthal P, Beppu A, Gordillo R, et al. Cutting Edge: Targeting Epithelial ORMDL3 Increases, Rather than Reduces, Airway Responsiveness and Is Associated with Increased Sphingosine-1-Phosphate. J Immunol. 2017;198:3017–3022. doi: 10.4049/jimmunol.1601848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yarova PL, Stewart AL, Sathish V, Britt RD, Jr, Thompson MA, P Lowe AP, et al. Calcium-sensing receptor antagonists abrogate airway hyperresponsiveness and inflammation in allergic asthma. Sci Transl Med. 2015;7:1–26. doi: 10.1126/scitranslmed.aaa0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mahn K, Hirst SJ, Ying S, Holt MR, Lavender P, Ojo OO, et al. Diminished sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) expression contributes to airway remodeling in bronchial asthma. Proc Natl Acad Sci USA. 2009;106:10775–10780. doi: 10.1073/pnas.0902295106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahn K, Ojo OO, Chadwick G, Aaronson PI, Ward JP, Lee TH. Ca2+ homeostasis and structural and functional remodelling of airway smooth muscle in asthma. Thorax. 2010;65:547–52. doi: 10.1136/thx.2009.129296. [DOI] [PubMed] [Google Scholar]
- 45.Trian T, Benard G, Begueret H, Rossignol R, Girodet PO, Ghosh D, et al. Bronchial smooth muscle remodeling involves calcium-dependent enhanced mitochondrial biogenesis in asthma. J Exp Med. 2007;204:3173–81. doi: 10.1084/jem.20070956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sathish V, Thompson MA, Bailey JP, Pabelick CM, Prakash YS, Sieck GC. Effect of proinflammatory cytokines on regulation of sarcoplasmic reticulum Ca2+ reuptake in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2009;297:L26–34. doi: 10.1152/ajplung.00026.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Croisier H, Tan X, Perez-Zoghbi JF, Sanderson MJ, Sneyd J, Brook BS. Activation of store-operated calcium entry in airway smooth muscle cells: insight from a mathematical model. PLoS One. 2013;8:e69598. doi: 10.1371/journal.pone.0069598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A representative comparison of airway contraction in mouse precision-cut lung slices (PCLS) induced by increasing doses of MCh in hORMDL3zp3-Cre and WT mouse.
Representative Ca2+ oscillations in airway smooth muscle cells (ASMCs) induced by 400 nM MCh in precision-cut lung slices (PCLS) derived from WT mouse. Note that Ca2+ oscillations occurred in the form of Ca2+ waves propagating along the length of ASMCs.