

Put a Ring on It
Merging the characteristics of transfer hydrogenation and carbonyl addition, a broad new class of ruthenium(0) catalyzed cycloadditions has been developed. Fused or bridged bicyclic ring systems are accessible in a redox-independent manner in C-C bond forming hydrogen transfer reactions of diols, α-ketols or 1,2-diones with diverse unsaturated reactants.
Keywords: Cycloaddition, Hydrogen Transfer, Ruthenium, Diol, Alcohol, C-C Bond Formation
1. Introduction
Since the advent of photocycloaddition (1908)[1a] and the Diels-Alder reaction (1928)[1b] many distinct and powerful classes of cycloadditions have been developed, including metal catalyzed processes.[2] As illustrated by ethylene glycol (23 × 106 tons in 2015) and propylene glycol (1.8 × 106 tons in 2015),[3c] diols are high-volume commodity chemicals that are of interest vis-a-vis biomass conversion.[3] Diols also appear ubiquitously as intermediate or terminal structural elements in target oriented synthesis, which has motivated the development of methods for their formation and elaboration, for example, the interconversion of diols and olefins.[4,5] In the course of developing catalytic hydrogen transfer reactions that convert lower alcohols to higher alcohols,[6] we recently found that phosphine-modified zero-valent ruthenium complexes derived from Ru3(CO)12 catalyze a diverse array of C-C couplings between vicinally dioxygenated hydrocarbons (diols, ketols, diones) and unsaturated reactants.[6e] These transformations occur through a common mechanistic motif in which oxidative formation of ruthenium(II) metalacycles,[7,8] which accompanies or triggers dione addition, is followed by diol- or ketol-mediated transfer hydrogenonlysis[9] of the ruthenacycle to release product, regenerate the dione and return ruthenium to its zero-valent form (Figure 1).
Figure 1.
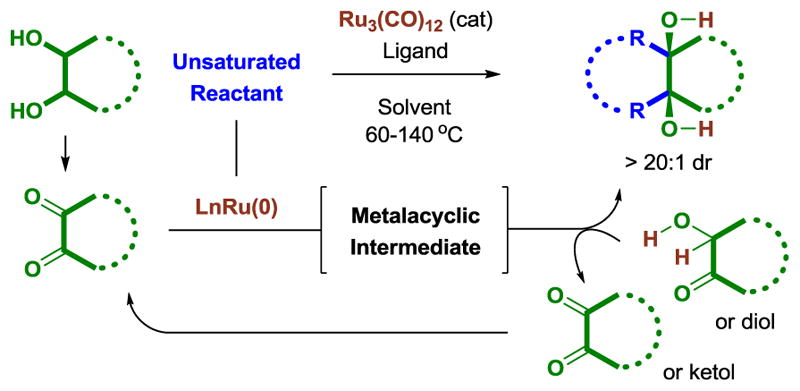
General catalytic mechanism for ruthenium(0) catalyzed cycloaddition of 1,2-diols
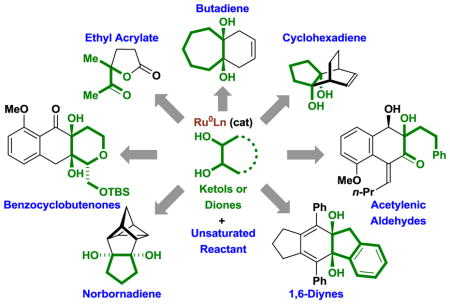
This pattern of reactivity served as the basis for the design of redox-independent cycloadditions between 1,2-diols, ketols or diones with unsaturated partners, including acyclic 1,3-dienes,[10] cyclic 1,3-dienes,[11] norbornadienes and trienes,[11] α,β-unsaturated esters,[12] benzannulated diynes[13a] and acetylenic aldehydes,[13b] 1,6-diynes[14] and benzocyclobutenones (Figure 2).[15] In this review, we provide an overview of transfer hydrogenative cycloadditions to form fused and bicyclic carbocyclic ring systems via metal catalyzed cycloadditions.[10–15] These processes, which occur through formal alcohol C-H functionalization, may be distinguished from related ring forming reactions that involve alcohol substitution via dehydrogenation-condensation-reduction (so-called “borrowing hydrogen” processes), which are largely restricted to heterocycle formation.[16]
Figure 2.
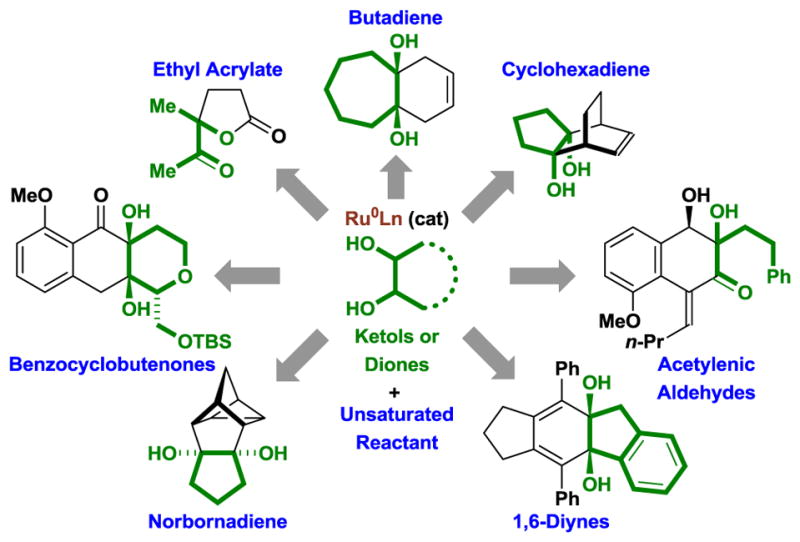
Ruthenium(0) catalyzed cycloaddition of diols, ketols or diones via alcohol-mediated hydrogen transfer.
2. 1,3-Dienes, Norbornadienes and Trienes
In ruthenium(0) catalyzed hydrohydroxyalkylations of isoprene or myrcene mediated by α-hydroxy esters,[17] mononuclear[18] oxaruthenacycles were postulated as reactive intermediates. These metalacycles were isolated, characterized and their reversible formation was demonstrated.[8] The allylruthenium moiety embedded in these metalacycles evoked the possibility of tandem C-C bond forming processes wherein oxidative coupling is followed by intramolecular allylation onto a tethered carbonyl partner to form products of cycloaddition. This possibility was initially realized in reactions of acyclic 1,3-dienes with diols.[10] Specifically, using the ruthenium(0) catalyst generated from Ru3(CO)12 and various chelating phosphine ligands, cycloalkane diols react with the feedstock dienes butadiene, isoprene and myrcene to form fused carbocycles bearing diols at the ring junction (Table 1). The cis- or trans-diols react with equal facility. Additionally, acyclic diols participate in cycloaddition to form monocyclic adducts.
Table 1.
Ruthenium(0) catalyzed [4+2] cycloaddition of diols with the feedstock dienes butadiene, isoprene and myrcene to form cyclohexene diols.a

|
Yields are of material isolated by silica gel chromatography. dppp = bis-(diphenylphosphino)propane. BINAP = 2,2′-bis-(diphenylphosphino)-1,1′-binaphthalene. BIPHEP = 2,2′-bis-(diphenylphosphino)-1,1′-biphenyl.
The cis-1,2-diol was employed.
3,5-Me2BzOH (10 mol%), 7 hrs.
3,5-Me2BzOH (10 mol%), myrcene (400 mol%).
The trans-1,2-diol was employed.
myrcene (300 mol%).
3,5-Me2BzOH (10 mol%), 18 hrs, myrcene (400 mol%).
4 hrs, 5:1 mixture of olefin regioisomers.
8 hrs, 14:1 mixture of olefin regioisomers.
Diol-diene cycloaddition is an oxidative process in which one equivalent of diene is required as hydrogen acceptor. The cycloaddition of α-ketols with dienes occurs under identical conditions and is a redox neutral process. Corresponding reductive cycloadditions of diones with dienes employing formic acid as terminal reductant have also been developed. Thus, ruthenium catalyzed [4+2] cycloaddition may be conducted from the diol, ketol or dione oxidation levels (Scheme 1).[10a]
Scheme 1.
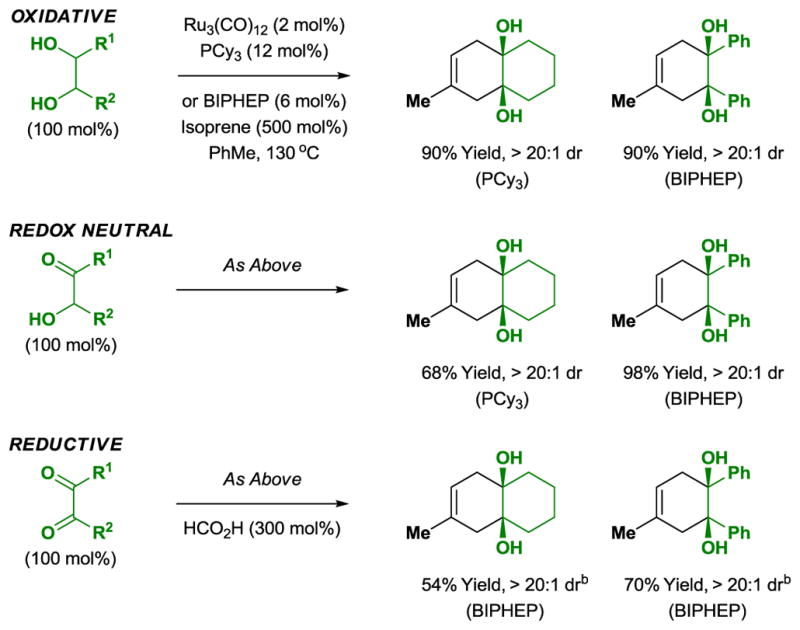
Redox independent [4+2] cycloaddition of diols, ketols and diones with isoprene.a
aYields are of material isolated by silica gel chromatography. BIPHEP = 2,2′-bis-(diphenylphosphino)-1,1′-biphenyl. bRuH2CO(PPh3)3 (6 mol%).
Tandem cycloaddition-dehydration provides a novel protocol for benzannulation.[10c] For example, the indicated acenaphthalene diol reacts with various butadienes to furnish cycloadducts that aromatize under the conditions of acid catalysis to form substituted fluoranthenes (Scheme 2). Substoichiometric quantities of 3,5-dimethylbenzoic acid improve conversion by catalyzing alcohol exchange and transfer hydrogenolysis of certain oxaruthenacycle intermediates.[19]
Scheme 2.

Ruthenium(0) catalyzed diol-diene benzannulation to form substituted fluoranthenes.a
aYields are of material isolated by silica gel chromatography. dppp = bis-(diphenylphosphino)propane. dppPh = bis-(1,2-diphenylphosphino)benzene. b120 °C. cdppPh, 3,5-Me2BzOH was omitted.
The diol-mediated cycloaddition of cyclohexadiene would enable access to bridged carbocycles. However, aromatization of cyclohexadiene to form benzene rendered the feasibility of this process uncertain. In the event, the reaction of cyclohexadiene to cyclic or acyclic diols in the presence of a dppe-modified ruthenium(0) catalyst, good to excellent isolated yields of the [4+2] cycloadducts were obtained with complete exo-selectivity as determined by 1H NMR analysis (Scheme 3).[11]
Scheme 3.

exo-Selective ruthenium(0) catalyzed [4+2] cycloaddition of cyclohexadiene.a
aYields are of material isolated by silica gel chromatography. dppe = bis-(diphenylphosphino)ethane. bThe cis-1,2-diol was employed. cA mixture of cis- and trans-1,2-diols was employed. drac-BINAP (6 mol%), BINAP = 2,2′-bis-(diphenylphosphino)-1,1′-binaphthalene. e3,5-Me2BzOH was omitted, 150 °C. fdCype (6 mol%), dCype = bis-(dicyclohexylphosphino)ethane. A 13% yield of the ketol cycloadduct also was obtained.
The ability of Ru3(CO)12 to catalyze olefin isomerization[20] enables recruitment of non-conjugated dienes in diol-mediated [4+2] cycloaddition with efficiencies roughly equivalent to that observed for the corresponding conjugated dienes (Scheme 4).[10a,11] A remarkable application of tandem olefin isomerization-cycloaddition is found in the reaction of 1,5,9-cyclododecatriene. A transient 1,3,5-triene is formed that exists in equilibrium with the corresponding diene-containing [6.4.0] bicycle, which is converted to the indicated bridged polycycle as a single diastereomer.
Scheme 4.
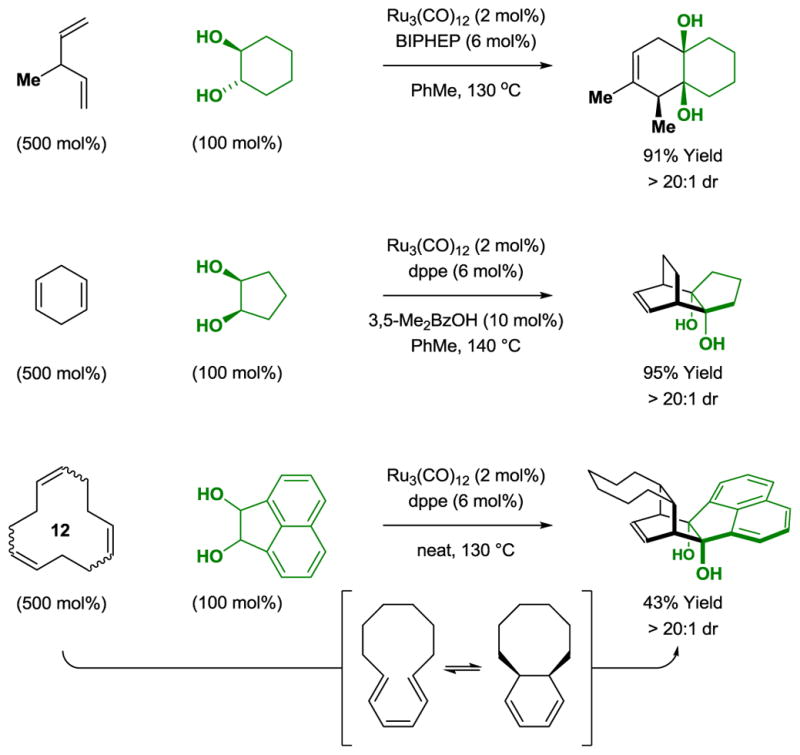
Tandem ruthenium(0) catalyzed olefin isomerization [4+2] cycloaddition of diols with non-conjugated dienes and 1,5,9-cyclododecatriene.a
aYields are of material isolated by silica gel chromatography. BIPHEP = 2,2′-bis-(diphenylphosphino)-1,1′-biphenyl. dppe = bis-(diphenylphosphino)ethane.
Mirroring the behavior of conjugated dienes, norbornadiene is known to participate in thermal homo-Diels-Alder reactions[21] and related metal catalyzed cycloadditions.[22] Based on this precedent, the diol-mediated cycloaddition of norbornadiene was explored.[11] Using the ruthenium(0) catalyst modified by dppe, cycloadducts were formed in good isolated yields with complete levels of exo-selectivity as determined by 1H NMR (Scheme 5).
Scheme 5.
exo-Selective ruthenium(0) catalyzed cycloaddition of norbornadiene.a
aYields are of material isolated by silica gel chromatography. dppe = bis-(diphenylphosphino)ethane. bThe cis-1,2-diol was employed. cA mixture of cis- and trans-1,2-diols was employed.
As demonstrated in the cycloaddition of diols with acyclic dienes, cyclohexadiene and norbornadiene also are capable of engaging in redox-independent cycloaddition (Scheme 6).[11] That is, cycloadditions may be conducted in oxidative, redox-neutral and reductive cycloaddition modes. In the latter case, formic acid is employed as terminal reductant.
Scheme 6.
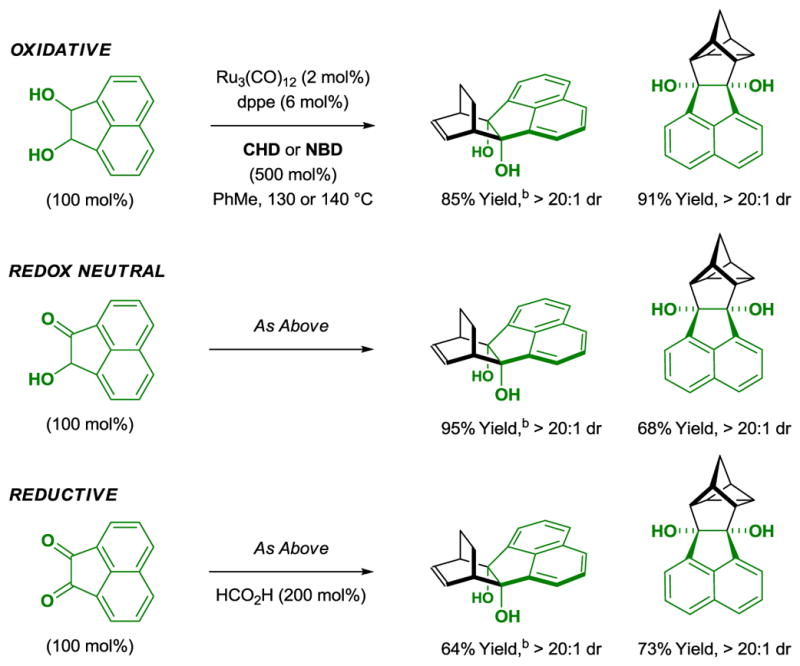
Redox independent cycloaddition of diols, ketols and diones with cyclohexadiene or norbornadiene.a
aYields are of material isolated by silica gel chromatography. dppe = bis-(diphenylphosphino)ethane. b3,5-Me2BzOH (10 mol%).
3. Acrylates and Unsaturated Esters
Spiro- and α-methylene-γ-butyrolactones are frequently encountered as structural motifs in natural products[23] and fragrance compounds.[24] It was posited that oxaruthenacycles obtained through ruthenium(0) mediated oxidative coupling of vicinal diones with α,β-unsaturated esters could undergo cyclization to form γ-lactones.[12] In the event, using the ruthenium(0) catalyst derived from Ru3(CO)12 and dppp, a variety of cyclic and acyclic diols react with methyl acrylate to form γ-lactones in good to excellent yields (Table 2).
Table 2.
Ruthenium(0) catalyzed [3+2] cycloaddition of methyl acrylate with cyclic and acyclic diols to form γ-lactones.a

|
Yields are of material isolated by silica gel chromatography. dppp = bis-(diphenylphosphino)propane. C10H15CO2H refers to 1-adamantanecarboxylic acid.
The cis-1,2-diol was employed.
The trans-1,2-diol was employed.
A mixture of cis- and trans-1,2-diols was employed.
Methyl acrylate (400 mol%).
Attempted ruthenium(0) catalyzed cycloadditions of β-substituted acrylic esters with cyclic and acyclic diols did not provide the desired lactone products. However, it was found that N-benzyl-3-hydroxy-2-oxindole reacts readily with β-substituted α,β-unsaturated esters due to the exceptional reactivity of the vicinal dicarbonyl moiety of the transient isatin. In these redox-neutral processes, spirolactones are formed in good to excellent isolated yields with complete control of diastereoselectivity as determined by 1H NMR analysis (Scheme 7).[12]
Scheme 7.

Ruthenium(0) catalyzed [3+2] cycloaddition of N-benzyl-3-hydroxy-2-oxindole with substituted α,β-unsaturated esters to form spirolactones.a
aYields are of material isolated by silica gel chromatography. dppp = bis-(diphenylphosphino)propane.
The lack of reactivity observed in attempted reactions of substituted acrylic esters with cyclic and acyclic diols may be attributed to unfavorable equilibria evident in reversible oxaruthenacycle formation.[8] For oxaruthenacycles bearing suitably disposed leaving groups, elimination might drive formation of the C-C coupling product. The veracity of this hypothesis is demonstrated in cycloadditions of cyclic and acyclic diols with the indicated α-hydroxymethyl-substituted acrylic ester, where elimination of water enables formation of α-exo-methylene γ-butyrolactones (Scheme 8).[12]
Scheme 8.
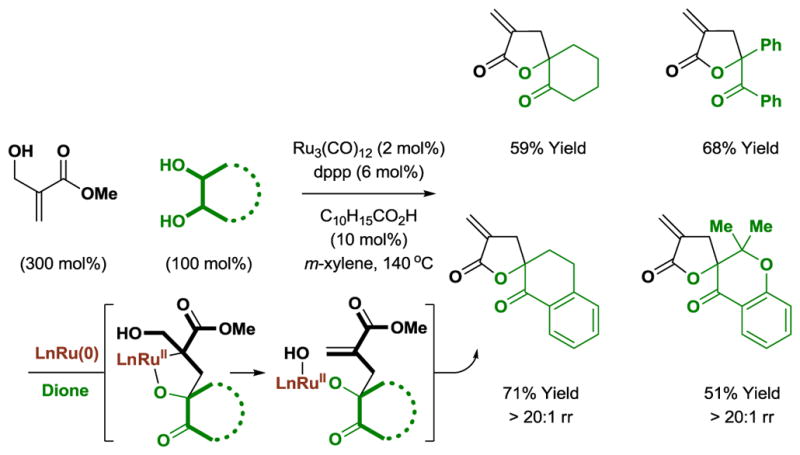
Ruthenium(0) catalyzed [3+2] cycloaddition with an α-hydroxymethyl-substituted α,β-unsaturated ester to form α-exo-methylene-γ-butyrolactones.a
aYields are of material isolated by silica gel chromatography. dppp = bis-(diphenylphosphino)propane. C10H15CO2H refers to 1-adamantanecarboxylic acid.
As illustrated in the cycloaddition of hydrobenzoin, benzoin and benzil with methyl acrylate, lactone formation may be accomplished in oxidative, redox-neutral or reductive modes from the diol, ketol or dione oxidation levels, respectively. The latter reaction of benzil employs 2-propanol (300 mol%) as terminal reductant (Scheme 9).[12]
Scheme 9.
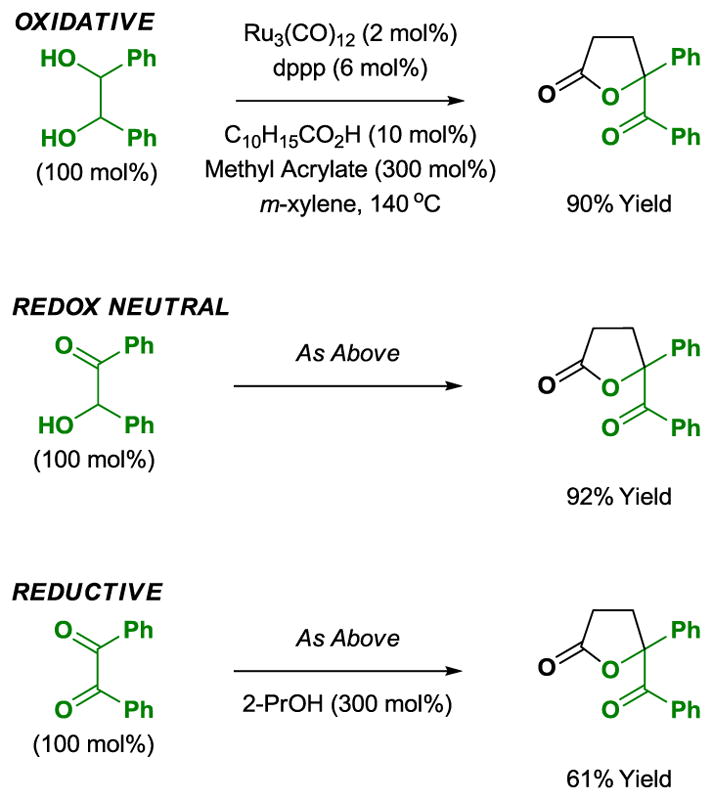
Redox independent [3+2] cycloaddition of hydrobenzoin, benzoin and benzil with methyl acrylate.a
aYields are of material isolated by silica gel chromatography. dppp = bis-(diphenylphosphino)propane. C10H15CO2H refers to 1-adamantanecarboxylic acid.
4. Benzannulated Diynes and ortho-Acetylenic Benzaldehydes
The ruthenium(0) catalyzed reaction of 3,4-benzannulated 1,5-diynes with α-ketols occurs through successive alkyne-carbonyl oxidative coupling events to form products of [4+2] cycloaddition.[13a] To suppress alkyne reduction, the reaction is conducted in a redox-neutral manner from the hydroxyketone oxidation level rather than using diol reactants, which requires a sacrificial hydrogen acceptor. The cycloadditions proceed at relatively low temperatures in aqueous organic media. As anticipated based on corresponding reactions of mono-alkynes,[19b] each alkyne-carbonyl oxidative coupling event occurs in a regioselective fashion at the aryl-substituted alkyne terminus (Scheme 10).
Scheme 10.

Ruthenium(0) catalyzed [4+2] cycloaddition of 3,4-benzannulated 1,5-diynes with α-ketols.a
aYields are of material isolated by silica gel chromatography. RuPhos = 2-dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl. bRu3(CO)12 (2 mol%), CyJohnPhos (12 mol%), CyJohnPhos = 2-(dicyclohexylphosphino)biphenyl.
The regioselective cycloaddition of non-symmetric diynes is achieved through differential substitution of the alkyne termini by n-propyl and t-butyl groups (Scheme 11). Ruthenium(0)-mediated alkyne-carbonyl oxidative coupling occurs initially at the less sterically demanding n-propyl substituted alkyne. After a second oxidative coupling at the t-butyl substituted alkyne and transfer hydrogenolysis of the resulting oxaruthenacycle, the cycloadduct is obtained as a single constitutional isomer.
Scheme 11.
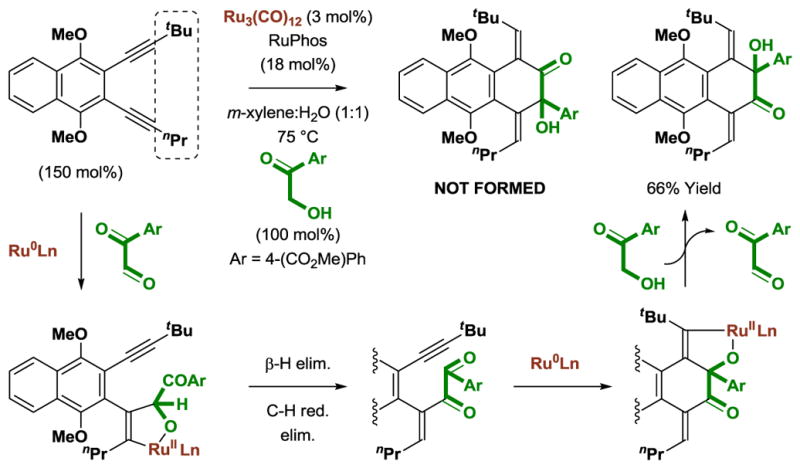
Regioselective ruthenium(0) catalyzed [4+2] cycloaddition of 3,4-benzannulated 1,5-diynes with α-ketols and general catalytic mechanism.a
aYields are of material isolated by silica gel chromatography. RuPhos = 2-dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl.
Through the use of ruthenium(0) complexes modified by CyJohnPhos or RuPhos, ortho-acetylenic benzaldehydes react with α-ketols to form products of [4+2] cycloaddition (Table 3).[13b] As in related reactions of 3,4-benzannulated 1,5-diynes,[13a] a catalytic mechanism involving successive oxidative coupling events is proposed. However, in the case of ortho-acetylenic benzaldehydes, alkyne-carbonyl oxidative coupling is followed by aldehyde-dione oxidative coupling. Notably, all C-C coupling events occurs in a highly regio- and stereoselective fashion to deliver cycloadducts as single diastereomers.
Table 3.
Ruthenium(0) catalyzed [4+2] cycloaddition of ortho-acetylenic benzaldehydes with α-ketols.a

|
Yields are of material isolated by silica gel chromatography. CyJohnPhos = 2-(dicyclohexylphosphino)biphenyl. RuPhos = 2-dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl.
Ru3(CO)12 (3 mol%), RuPhos (18 mol%).
5. 1,6-Diynes
Metal catalyzed [2+2+2] cycloadditions enable convergent construction of (poly)cyclic molecules from π-unsaturated reactants.[2c,2e,25] In the presence of ruthenium(0) catalysts, 1,6-diynes form ruthenacyclopentadienes that participate in successive carbonyl addition with transient diones derived from diol reactants to form products of [2+2+2] cycloaddition (Table 4).[14] Structurally diverse cyclic and acyclic diols deliver cycloadducts with complete levels of syn-diastereoselectivity as determined by 1H NMR analysis. A carboxylic acid cocatalyst was again required to enhance rate and conversion.[19] 1,6-Diynes that are unsubstituted at the acetylenic terminus and 1,7-diynes are not efficient partners for cycloaddition under these initially developed conditions.
Table 4.
Ruthenium(0) catalyzed [2+2+2] cycloaddition of diols with 1,6-diynes.a

|
Yields are of material isolated by silica gel chromatography. dppe = bis-(diphenylphosphino)ethane. C10H15CO2H refers to 1-adamantanecarboxylic acid. E = CO2Et.
The cis-1,2-diol was employed.
A mixture of cis- and trans-1,2-diols was employed.
As cycloaddition occurs through a catalytic mechanism involving successive dehydrogenation of the diol reactant to form a transient α-ketol and, ultimately, the vicinal dione required for C-C bond formation, diols, α-ketols and vicinal diones should all be competent partners for [2+2+2] cycloaddition. Indeed, beyond oxidative [2+2+2] cycloadditions of diol reactants, redox-neutral cycloadditions of α-ketols and reductive cycloadditions of diones are possible (Scheme 12). In the latter case, 2-propanol serves as terminal reductant.
Scheme 12.
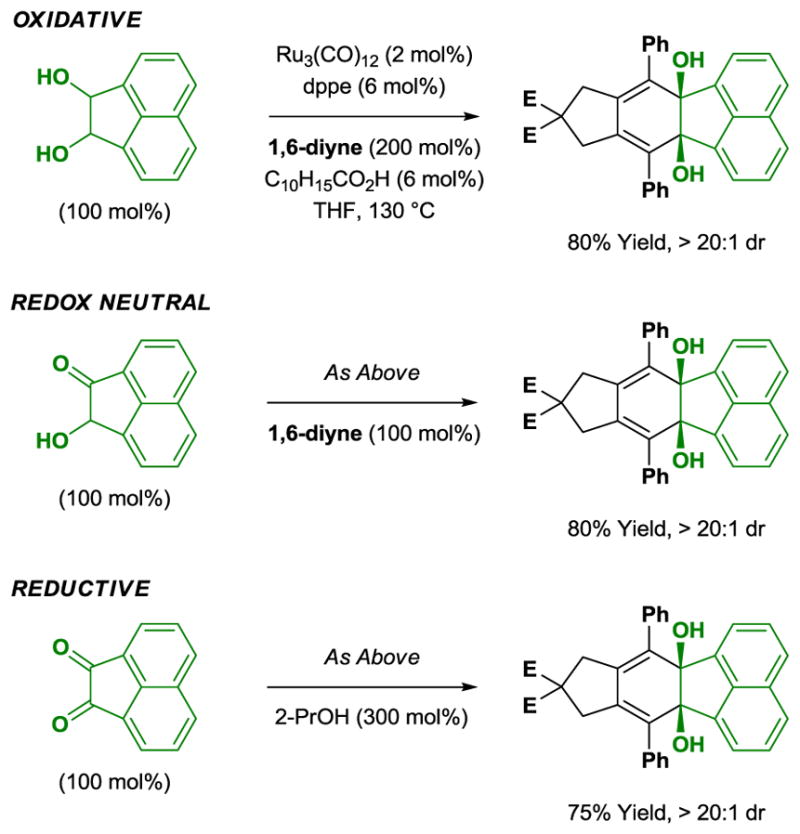
Redox independent [2+2+2] cycloadditions of diols, ketols and diones with 1,6-diynes.a
aYields are of material isolated by silica gel chromatography. dppe = bis-(diphenylphosphino)ethane. C10H15CO2H refers to 1-adamantanecarboxylic acid.
6. Benzocyclobutenones
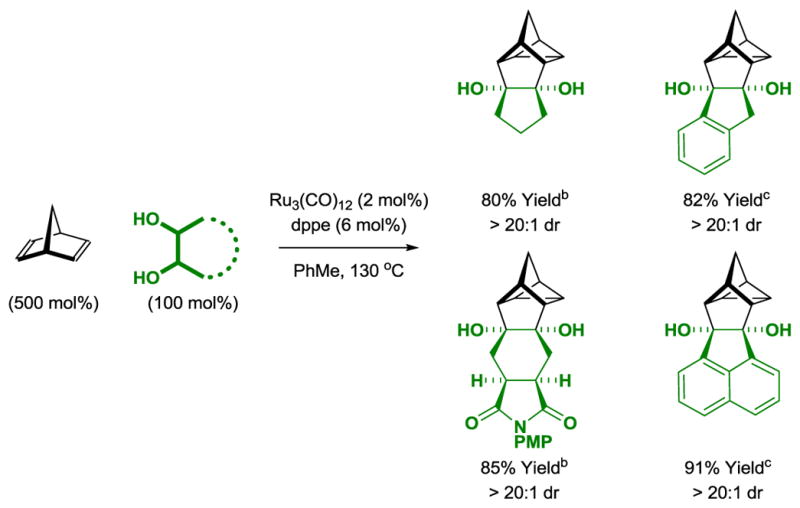
The preceding transformations suggested the feasibility of exploiting ruthenacycles derived upon C-C bond oxidative addition[26] as partners for cycloaddition. Given reports on the intermolecular cycloadditions of (benzo)cyclobutenone derivatives catalyzed by nickel,[27] palladium,[28] rhodium[29] and ruthenium,[30] the formal insertion of adjacent diol carbon atoms into C-C bonds appeared feasible. In the event, upon exposure to the ruthenium(0) catalyst derived from Ru3(CO)12 and dppp, benzocyclobutenones react with 1,2-diols to form cycloadducts with complete levels of diastereoselectivity as determined by 1H NMR analysis (Table 5).[15] Nonsymmetric diols delivered cycloadducts as single regioisomers. Whereas catalytic processes involving C-C bond activation rely on the insertion of π-unsaturated coupling partners, to our knowledge, the present cycloadditions involve formal insertion of saturated C-H bonds into C-C σ-bonds.
Table 5.
Ruthenium(0) catalyzed [4+2] cycloaddition of diols with benzocyclobutenones.a

|
Yields are of material isolated by silica gel chromatography. dppp = bis-(diphenylphosphino)propane.
The trans-1,2-diol was employed.
Reaction conducted from the ketol oxidation level.
130 °C.
A mixture of cis- and trans-1,2-diols was employed.
Cycloaddition is possible in oxidative, redox-neutral, and reductive modes (Scheme 13). As corroborated by the isolation of the ring-opened hydrogenolysis product (2-methoxy-6-methylphenyl)methanol, diol cycloadditions are oxidative processes in which excess benzocyclobutenone (50 mol%) accepts two equivalents of hydrogen. Reactions conducted from the ketol oxidation level exploit equimolar quantities of reactant, establishing the practicality of this method for the convergent union of complex fragments. Finally, for reductive cycloaddition from the dione oxidation level, 2-propanol is employed as terminal reductant.
Scheme 13.
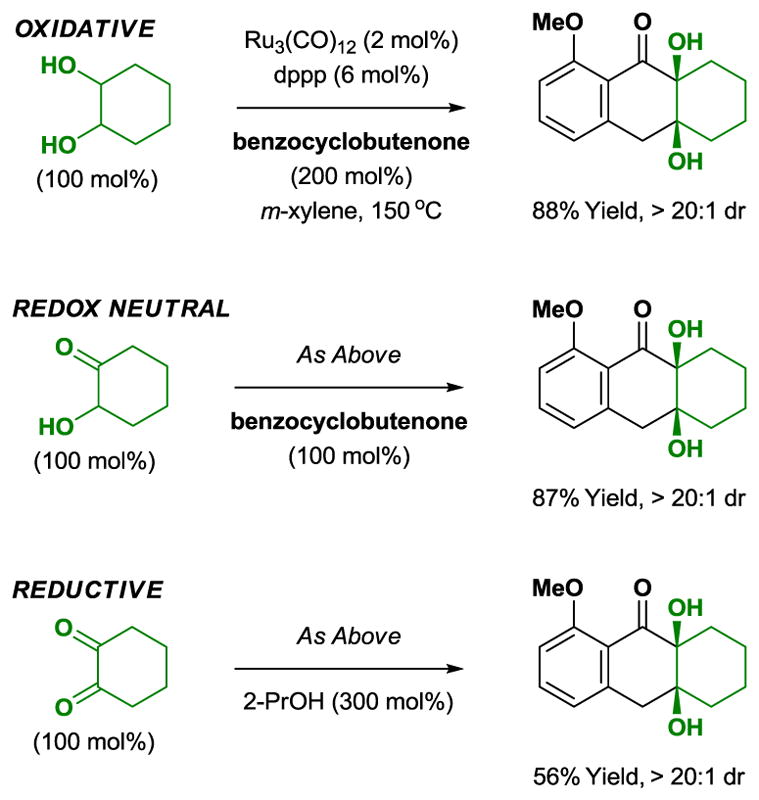
Redox independent [4+2] cycloaddition of cyclohexane diols, cyclohexane ketol and cyclohexane dione with a benzocyclobutenone.a
aYields are of material isolated by silica gel chromatography. dppp = bis-(diphenylphosphino)propane.
7. Conclusion and Outlook
New reactivity is the most fundamental and far-reaching basis for innovation in the field of chemical synthesis. By harnessing the native reducing ability of alcohols, we have developed a broad, new class of metal catalyzed carbonyl additions.[6] Beyond bypassing the use of discrete organometallic reagents in known carbonyl additions,[6g] the unique mechanisms embodied by these processes have availed transformations that have no counterpart in the current lexicon of synthetic methods. Indeed, the transfer hydrogenative cycloadditions described herein represent an outgrowth of this reactivity pattern that extends beyond the capabilities of classical carbonyl addition and cycloaddition chemistry. It is the authors hope that the present review will accelerate further expansion of this distinctive subset of metal catalyzed cycloadditions and, more broadly, the development of new alcohol-mediated C-C bond formations.
Acknowledgments
The Welch Foundation (F-0038), the NIH (R01 GM093905) and the NSF (CHE-1565688) are acknowledged for partial support of this research. The Japan Student Services Organization (JASSO) and the Japan Society for the Promotion of Science (JSPS) are acknowledged for graduate fellowship (HS) and research fellowship (KF) fellowship support, respectively.
References
- 1.a) Ciamician G, Silber P. Chem Ber. 1908;41:1928–1935. [Google Scholar]; b) Diels O, Alder K. Ann. 1928;460:98–122. [Google Scholar]
- 2.For selected reviews on metal catalyzed cycloadditions, see: Lautens M, Klute W, Tam W. Chem Rev. 1996;96:49–92. doi: 10.1021/cr950016l.Kondo T, Mitsudo T. Chem Lett. 2005;34:1462–1467.Chopade PR, Louie J. Adv Synth Catal. 2006;348:2307–2327.Gulías M, López F, Mascareñas JL. Pure Appl Chem. 2011;83:495–506.Shibata Y, Tanaka K. Synthesis. 2012;44:323–350.Pellissier H. Tetrahedron. 2015;71:8855–8869.Fructos MR, Prieto A. Tetrahedron. 2016;72:355–369.Kondo T. Eur J Org Chem. 2016:1232–1242.
- 3.For selected reviews on the conversion of biomass to diol feedstocks, see: Gallezot P. Chem Soc Rev. 2012;41:1538–1558. doi: 10.1039/c1cs15147a.Dapsens PY, Mondelli C, Pérez-Ramírez J. ACS Catal. 2012;2:1487–1499.Zheng M, Pang J, Sun R, Wang A, Zhang T. ACS Catal. 2017;7:1939–1954.
- 4.For selected reviews on olefin dihydroxylation, see: Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483–2547.Cha JK, Kim NS. Chem Rev. 1995;95:1761–1795.Donohoe TJ. Synlett. 2002:1223–1232.Zaitsev AB, Adolfsson H. Synthesis. 2006:1725–1756.Bataille CJR, Donohoe TJ. Chem Soc Rev. 2011;40:114–128. doi: 10.1039/b923880h.
- 5.For selected reviews on diol deoxydehydration, see: Boucher-Jacobs C, Nicholas KM. Top Curr Chem. 2014;353:163–184. doi: 10.1007/128_2014_537.Raju S, Moret M-E, Klein Gebbink RJM. ACS Catal. 2015;5:281–300.Dethlefsen JR, Fristrup P. ChemSusChem. 2015;8:767–775. doi: 10.1002/cssc.201402987.Petersen AR, Fristrup P. Chem Eur J. 2017;23:10235–10243. doi: 10.1002/chem.201701153.
- 6.For recent reviews on hydrogen transfer reactions that convert lower alcohols to higher alcohols, see: Ketcham JM, Shin I, Montgomery TP, Krische MJ. Angew Chem. 2014;126:9294–9302. doi: 10.1002/anie.201403873.Angew Chem Int Ed. 2014;53:9142–9150. doi: 10.1002/anie.201403873.Dechert-Schmitt AMR, Schmitt DC, Gao X, Itoh T, Krische MJ. Nat Prod Rep. 2014;31:504–513. doi: 10.1039/c3np70076c.Feng J, Kasun ZA, Krische MJ. J Am Chem Soc. 2016;138:5467–5478. doi: 10.1021/jacs.6b02019.Shin I, Krische MJ. Top Curr Chem. 2016;372:85–101. doi: 10.1007/128_2015_651.Perez F, Oda S, Geary LM, Krische MJ. Top Curr Chem. 2016;374:365–387. doi: 10.1007/s41061-016-0028-0.Nguyen KD, Park BY, Luong T, Sato H, Garza VJ, Krische MJ. Science. 2016;354:300. doi: 10.1126/science.aah5133. (aah5133-1–5) Kim SW, Zhang W, Krische MJ. Acc Chem Res. 2017;50:2371–2380. doi: 10.1021/acs.accounts.7b00308.
- 7.For use of zero-valent ruthenium complexes derived from Ru3(CO)12 in catalytic processes that involve oxidative coupling, see: Chatani N, Tobisu M, Asaumi T, Fukumoto Y, Murai S. J Am Chem Soc. 1999;121:7160–7161.Tobisu M, Chatani N, Asaumi T, Amako K, Ie Y, Fukumoto Y, Murai S. J Am Chem Soc. 2000;122:12663–12674.
- 8.For isolation, characterization and reversible formation of catalytically competent oxaruthenacycles formed via diene-carbonyl oxidative coupling, see: Park BY, Montgomery TP, Garza VJ, Krische MJ. J Am Chem Soc. 2013;135:16320–16323. doi: 10.1021/ja4087193.
- 9.For use of zero-valent ruthenium complexes derived from Ru3(CO)12 in catalytic processes that involve alcohol dehydrogenation, see: Blum Y, Reshef D, Shvo Y. Tetrahedron Lett. 1981;22:1541–1544.Shvo Y, Blum Y, Reshef D, Menzin M. J Organomet Chem. 1982;226:C21–C24.Meijer RH, Ligthart GBWL, Meuldijk J, Vekemans JAJM, Hulshof LA, Mills AM, Kooijmanc H, Spek AL. Tetrahedron. 2004;60:1065–1072.Johnson TC, Totty WG, Wills M. Org Lett. 2012;14:5230–5233. doi: 10.1021/ol302354z.Zhang M, Imm S, Bähn S, Neumann H, Beller M. Angew Chem. 2011;123:11393–11397. doi: 10.1002/anie.201104309.Angew Chem Int Ed. 2011;50:11197–11201. doi: 10.1002/anie.201104309.
- 10.a) Geary LM, Glasspoole BW, Kim MM, Krische MJ. J Am Chem Soc. 2013;135:3796–3799. doi: 10.1021/ja400691t. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kasun ZA, Geary LM, Krische MJ. Chem Comm. 2014;50:7545–7547. doi: 10.1039/c4cc03983a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Geary LM, Chen TY, Montgomery TP, Krische MJ. J Am Chem Soc. 2014;136:5920–5922. doi: 10.1021/ja502659t. [DOI] [PubMed] [Google Scholar]
- 11.Sato H, Fukaya K, Poudel BS, Krische MJ. Angew Chem. 2017;129 doi: 10.1002/ange.201708189;. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2017;56 doi: 10.1002/anie.201708189. [DOI] [Google Scholar]
- 12.McInturff EL, Mowat J, Waldeck AR, Krische MJ. J Am Chem Soc. 2013;135:17230–17235. doi: 10.1021/ja410533y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Saxena A, Perez F, Krische MJ. J Am Chem Soc. 2015;137:5883–5886. doi: 10.1021/jacs.5b02755. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Saxena A, Perez F, Krische MJ. Angew Chem. 2016;128:1515–1519. doi: 10.1002/anie.201509646. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2016;55:1493–1497. doi: 10.1002/anie.201509646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato H, Bender M, Chen W, Krische MJ. J Am Chem Soc. 2016;138:16244–16247. doi: 10.1021/jacs.6b11746. [DOI] [PubMed] [Google Scholar]
- 15.Bender M, Turnbull BWH, Ambler BR, Krische MJ. Science. 2017;357:779–781. doi: 10.1126/science.aao0453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For reviews on metal catalyzed alcohol amination including heterocycle formation, see: Guillena G, Ramón DJ, Yus M. Angew Chem. 2007;119:2410–2416. doi: 10.1002/anie.200603794.Angew Chem Int Ed. 2007;46:2358–2364. doi: 10.1002/anie.200603794.Hamid MHSA, Slatford PA, Williams JMJ. Adv Synth Catal. 2007;349:1555–1575.Nixon TD, Whittlesey MK, Williams JMJ. Dalton Trans. 2009:753–762. doi: 10.1039/b813383b.Dobereiner GE, Crabtree RH. Chem Rev. 2010;110:681–703. doi: 10.1021/cr900202j.Guillena G, Ramón DJ, Yus M. Chem Rev. 2010;110:1611–1641. doi: 10.1021/cr9002159.Yang Q, Wang Q, Yu Z. Chem Soc Rev. 2015;44:2305–2329. doi: 10.1039/c4cs00496e.Nandakumar A, Midya SP, Landge VG, Balaraman E. Angew Chem. 2015;127:11174–11186. doi: 10.1002/anie.201503247.Angew Chem Int Ed. 2015;54:11022–11034. doi: 10.1002/anie.201503247.Huang F, Liu Z, Yu Z. Angew Chem. 2016;128:872–885.Angew Chem Int Ed. 2016;55:862–875. doi: 10.1002/anie.201507521.Quintard A, Rodriguez J. Chem Comm. 2016;52:10456–10473. doi: 10.1039/c6cc03486a.Quintard A, Rodriguez J. ChemSusChem. 2016;9:28–30. doi: 10.1002/cssc.201501460.Chelucci G. Coord Chem Rev. 2017;331:37–53.
- 17.Leung JC, Geary LM, Chen T-Y, Zbieg JR, Krische MJ. J Am Chem Soc. 2012;134:15700–15703. doi: 10.1021/ja3075049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ru3(CO)12 reacts with dppe in benzene solvent to provide Ru(CO)3(dppe): Sanchez-Delgado RA, Bradley JS, Wilkinson G. J Chem Soc, Dalton Trans. 1976:399–404.
- 19.For carboxylic acid catalyzed hydrogenolysis and transfer hydrogenolysis of metalacycles, see: Ngai MY, Barchuk A, Krische MJ. J Am Chem Soc. 2007;129:280–281. doi: 10.1021/ja0670815.McInturff EL, Nguyen KD, Krische MJ. Angew Chem. 2014;126:3296–3299. doi: 10.1002/anie.201311130.Angew Chem Int Ed. 2014;53:3232–3235. doi: 10.1002/anie.201311130.
- 20.For selected examples of Ru3(CO)12 catalyzed olefin isomerization, see: Kašpar J, Spogliarich R, Graziani M. J Organomet Chem. 1985;281:299–304.Hilal HS, Khalaf S, Jondi W. J Organomet Chem. 1993;452:167–173.Jun CH, Lee H, Park JB, Lee DY. Org Lett. 1999;1:2161–2164.Alvila L, Pakkanen TA, Krause O. J Mol Catal. 1993;84:145–156.
- 21.For seminal reports on the thermal homo-Diels-Alder reactions of norbornadiene, see: Ullman EF. Chem Ind. 1958:1173–1174.Blomquist AT, Meinwald YC. J Am Chem Soc. 1959;81:667–672.Hall HK., Jr J Org Chem. 1960;25:42–44.
- 22.For selected examples on the metal catalyzed homo-Diels-Alder reactions of norbornadiene, see: Schrauzer GN, Eichler S. Chem Ber. 1962;95:2764–2768.Lyons JE, Myers HK, Schneider A. J Chem Soc, Chem Commun. 1978:638–639.Lautens M, Lautens JC, Smith AC. J Am Chem Soc. 1990;112:5627–5628.Lautens M, Edwards LG, Tam W, Lough AJ. J Am Chem Soc. 1995;117:10276–10291.Tenaglia A, Gaillard S. Org Lett. 2007;9:3607–3610. doi: 10.1021/ol701463r.Kettles TJ, Cockburn N, Tam W. J Org Chem. 2011;76:6951–6957. doi: 10.1021/jo2010928.
- 23.For selected reviews of γ-butyrolactones and α-methylene-γ-butyrolactones, respectively, see: Bartoli A, Rodier F, Commeiras L, Parrain JL, Chouraqui G. Nat Prod Rep. 2011;28:763–782. doi: 10.1039/c0np00053a.Kitson RRA, Millemaggi A, Taylor RJK. Angew Chem. 2009;121:9590–9615.Angew Chem Int Ed. 2009;48:9426–9451. doi: 10.1002/anie.200903108.
- 24.For selected reviews on fragrance chemistry, see: Fráter G, Bajgrowicz JA, Kraft P. Tetrahedron. 1998;54:7633–7703.Kraft P, Bajgrowicz JA, Denis C, Fráter G. Angew Chem. 2000;112:3106–3138. doi: 10.1002/1521-3773(20000901)39:17<2980::aid-anie2980>3.0.co;2-#.Angew Chem Int Ed. 2000;39:2980–3010. doi: 10.1002/1521-3773(20000901)39:17<2980::aid-anie2980>3.0.co;2-#.
- 25.For selected reviews on metal catalyzed[2+2+2]cycloaddition, see: Domínguez J. Períez-Castells, [2+2+2] cycloadditions. In: Knochel P, editor. Comprehensive Organic Synthesis II. 2. Vol. 5. Elsevier; Amsterdam: 2014. pp. 1537–1581;.Domínguez G, Pérez-Castells J. Chem Eur J. 2016;22:6720–6739. doi: 10.1002/chem.201504987.
- 26.For selected reviews on C-C bond activation, see: Bishop KC., III Chem Rev. 1976;76:461–486.Mitsudo T, Kondo T. Synlett. 2001:309–321.Kondo T, Mitsudo T. Chem Lett. 2005;34:1462–1467.Murakami M, Matsuda T. Chem Commun. 2011;47:1100–1105. doi: 10.1039/c0cc02566f.Kondo T. Bull Chem Soc Jpn. 2011;84:441–458.Nakao Y. Top Curr Chem. 2014;346:33–58. doi: 10.1007/128_2013_494.Dreis AM, Douglas CJ. Top Curr Chem. 2014;346:85–110. doi: 10.1007/128_2013_523.Souillart L, Cramer N. Chem Rev. 2015;115:9410–9464. doi: 10.1021/acs.chemrev.5b00138.Shaw MH, Bower JF. Chem Commun. 2016;52:10817–10829. doi: 10.1039/c6cc04359c.Kondo T. Eur J Org Chem. 2016:1232–1242.Chen P-h, Dong G. Chem Eur J. 2016;22:18290–18315. doi: 10.1002/chem.201603382.Murakami M, Ishida N. J Am Chem Soc. 2016;138:13759–13769. doi: 10.1021/jacs.6b01656.Chen P-h, Billet BA, Tsukamoto T, Dong G. ACS Catal. 2017;7:1340–1360. doi: 10.1021/acscatal.6b03210.Fumagalli G, Stanton S, Bower JF. Chem Rev. 2017;117:9404–9432. doi: 10.1021/acs.chemrev.6b00599.
- 27.For selected examples of intermolecular nickel catalyzed cycloadditions of cyclobutenone derivatives, see: Huffman MA, Liebeskind LS. J Am Chem Soc. 1991;113:2771–2772.Murakami M, Ashida S, Matsuda T. J Am Chem Soc. 2005;127:6932–6933. doi: 10.1021/ja050674f.Auvinet AL, Harrity JPA. Angew Chem. 2011;123:2821–2824.Angew Chem Int Ed. 2011;50:2769–2772. doi: 10.1002/anie.201007598.Ho KYT, Aïssa C. Chem Eur J. 2012;18:3486–3489. doi: 10.1002/chem.201200167.Ishida N, Yuhki T, Murakami M. Org Lett. 2012;14:3898–3901. doi: 10.1021/ol3016447.Kumar P, Louie J. Org Lett. 2012;14:2026–2029. doi: 10.1021/ol300534j.Thakur A, Evangelista JL, Kumar P, Louie J. J Org Chem. 2015;80:9951–9958. doi: 10.1021/acs.joc.5b01458.Stalling T, Harker WRR, Auvinet AL, Cornel EJ, Harrity JPA. Chem Eur J. 2015;21:2701–2704. doi: 10.1002/chem.201405863.
- 28.For an example of the intermolecular palladium catalyzed cycloaddition of cyclobutanone derivatives, see: Okumura S, Sun F, Ishida N, Murakami M. J Am Chem Soc. 2017;139:12414–12417. doi: 10.1021/jacs.7b07667.
- 29.For selected examples of intermolecular rhodium catalyzed cycloadditions of cyclobutenone derivatives, see: Kondo T, Taguchi Y, Kaneko Y, Niimi M, Mitsudo T. Angew Chem. 2004;116:5483–5486. doi: 10.1002/anie.200461002.Angew Chem Int Ed. 2004;43:5369–5372. doi: 10.1002/anie.200461002.Kondo T, Niimi M, Nomura M, Wada K, Mitsudo T-a. Tetrahedron Lett. 2007;48:2837–2839.
- 30.For an example of the intermolecular ruthenium catalyzed cycloaddition of cyclobutenone derivatives, see: Kondo T, Nakamura A, Okada T, Suzuki N, Wada K, Mitsudo T. J Am Chem Soc. 2000;122:6319–6320.