

Abstract
Loss of major histocompatibility Class II expression (MHCII) in diffuse large B-cell lymphoma (DLBCL) correlates with decreased survival. MHCII transcription is in part regulated by histone acetylation. We tested the hypothesis that combination of histone deacetylase inhibitor (HDACI) with standard chemotherapy would improve outcomes in DLBCL in part through increased MHCII expression. S0806 was a single arm phase I/II trial of vorinostat given at 400 mg po daily on days 1-9 (subsequently amended to days 1-5 due to toxicity), combined with R-CHOP given on day 3 of a 21-day cycle for 8 cycles, with primary phase II endpoint of 2-year progression free survival (PFS). With 72 evaluable patients, at median follow up of 3 years, 2-year PFS estimate was 73%, and OS estimate was 86%. Considering that the regimen fell short of pre-defined efficacy improvement and was associated with high rates of febrile neutropenia (38%) and sepsis (19%), it cannot be recommended for general use. Consistent with our hypothesis, patients with low MCHII expression on S0806 had numerically superior outcomes compared to those from trial S0433 which did not use an HDACI, but the difference was not statistically significant. Current studies are focused on finding biomarkers of response to HDACI.
Keywords: diffuse large B-cell lymphoma, vorinostat, histone deacetylase inhibitor
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common lymphoma diagnosed in the US, representing 30-40% of all non-Hodgkin lymphomas.1 While 55-60% of patients are cured with R-CHOP, a substantial number of patients relapse and die of their disease. Loss of major histocompatibility Class II antigen (MHCII) expression from the cell surface membrane in DLBCL is associated with a decreased CD8+ tumor-infiltrating T-lymphocyte (TIL) response and poor patient survival, independent of international prognostic index (IPI) score or histologic subtype.2 The negative correlation with outcome is presumably due to the loss of antigen presentation capability on the B cells. In addition, aberrant localization of MHC II protein away from the surface membrane into the cytoplasm, is also associated with shorter progression free survival,3,4 MHCII internalization has been associated with altered ubiquinization, which enhances MHCII endocytosis and localization in late endosomes, and a results in a phenotype similar to immature dendritic cells.5 Further investigations indicate that deletion of the MHCII gene is infrequent in MHCII surface negative cases, and that altered transcription of the MHCII gene complex is under the control of the master transactivator, CIITA.6 Activation of CIITA and upregulation of MHCII expression may therefore be a potential therapeutic strategy in DLBCL. The cause of internalized MHCII in B cell lymphomas is unknown, but alteration of the ubiquinization, proteasome, and/or trafficking processes are likely involved.
Epigenetic mechanisms such as DNA hypermethylation or histone deacetylation normally silence gene expression to regulate numerous cellular activities. CIITA is both itself regulated by histone acetylation and, in turn, modifies H3 and H4 histone acetylation at the HLA-DRA promoter region in human B cell lines.7,8 Importantly, both H3 and H4 acetylation is enforced by vorinostat (suberoylanilide hydroxamic acid, or SAHA), a first in class pan-histone deacetylase inhibitor (HDACI).9 Therefore, HDACI could potentially enhance MHCII expression. Indeed, in SWOG trial S0520, where another pan-HDACI belinostat was used in the treatment of relapsed/refractory DLBCL, of 2 on-treatment biopsies the one in the responding patient had increased levels of MHCII expression and increased numbers of CD8+ T cells, while the one in the non-responding patient lacked these changes.10 HDACI have been shown to affect other aspects of the immune system, including regulatory T-cells and cytokines,11,12 which may be of clinical relevance.
The phase II study of vorinostat-R-CHOP was thus designed with the hypothesis that the combination of vorinostat with standard chemotherapy would enhance MHCII expression and improve patient outcome in DLBCL, possibly through improving CD8+ T cell infiltration and immunosurveillance.
Methods
SWOG S0806 was a single arm open label phase I/II trial of vorinostat in combination with R-CHOP. Patients must have biopsy-proven CD20-positive newly diagnosed diffuse large B-cell ymphoma (DLBCL) with Stage II bulky, Stage III or Stage IV disease, with an International Prognostic Index (IPI) or revised IPI (R-IPI) score greater than 0. All patients must have bidimensionally measurable disease. Patients with evidence of central nervous system or known to be HIV positive were not eligible. No prior chemotherapy, radiation, or antibody therapy for lymphoma was allowed. Patients must have had a Zubrod performance status of 0-2, be at least age 18, and have adequate cardiac and hematologic function. All patients gave written informed consent to participate in the study. The trial was approved by the Institutional Review Board at each participating center, and was registered with Clinicaltrials.gov (NCT00972478).
The primary endpoint of phase I portion of the trial was to find a safe dose of vorinostat to be used in combination with standard dose of R-CHOP (vincristine capped at 2 mg, prednisone 100 mg flat dose). Vorinostat starting dose (dose 0) was 400 mg po daily days 1-9, with two preplanned dose de-escalation levels. Dose-limiting toxicity was defined as occurrence of the following in the first cycle of treatment: grade 3-4 neutropenia or thrombocytopenia lasting more than 7 days; grade 3-4 febrile neutropenia or hemorrhage; grade 3-4 non-hematologic toxicity that persists until cycle 2 day 1; grade 3-4 nausea, vomiting, diarrhea, hypomagnesemia, hypokalemia, hypophosphatemia, or hyperglycemia that persisted despite appropriate management. Ten patients were to be evaluated at the recommended dose prior to proceeding with the Phase II portion of the trial. A temporary closure was to occur prior to opening this study for the Phase II portion in order to assess dose, and to evaluate the safety profile more fully. If 3/10 or fewer patients were have DLT at Dose Level 0, this would be the dose for the Phase II trial. If more than 3/10 patients had DLT at Dose Level 0, enrollment was to stop at this dose after the 4th patient develops DLT and 10 patients were to be enrolled at Dose Level -1.
The primary endpoint of phase II portion was to estimate the two-year progression-free survival (PFS), with secondary endpoints of estimating the response rate (complete and partial) and two-year overall survival rate, and of evaluating the toxicity of the vorinostat-R-CHOP combination.
Vorinostat was initially administered at 400 mg po daily days 1-9 of 21-day cycle, while R-CHOP was given on day 3 of each 21-day cycle, for 8 cycles. Restaging was performed after 4 cycles and then after completion of all 8 cycles. PET/CT scans were not mandatory pre-treatment but were required after treatment completion. Thereafter, disease was assessed every 6 months for 2 years, and annually for a maximum of 5 years or until disease progression. Clinical responses were determined using revised International Workshop Group criteria.13
Laboratory Methods
Diagnosis of DLBCL was confirmed centrally by an expert hematopathologist (L.R.). The cell of origin designation was determined by the Hans algorithm, using antibodies for CD10, BCL6, and MUM1.14 MYC and BCL2 expression was evaluated by immunohistochemistry, using cutoffs of 40% and 50%, respectively.
Translational goals of the study included assessing correlation of the following factors with PFS or OS: 1) Pre-treatment histone acetylation status and MHCII expression on the tumor cells; 2) pre-treatment percentage of CD8+ tumor infiltrating lymphocytes (TIL) and other lymphocyte subsets; and 3) baseline or changes in serum cytokine levels.
Since S0806 did not have a randomized control arm, accessible tissues from patients who participated in SWOG S0433 trial were obtained to compare the impact of translational endpoints on S0806 to that in non-HDACI-containing standard therapy. S0433 sequentially administered R-CHOP × 6, CHOP × 2 and then iodine-131 tositumomab to patients with newly diagnosed advanced stage DLBCL, with outcomes similar to those obtained with R-CHOP.15
Immunohistochemistry
To assess if histone acetylation, MHCII expression, and percentage of CD8+ tumor infiltrating lymphocytes (TIL) and other lymphocyte subsets correlate with PFS (or OS), we performed immunohistochemistry on pre-treatment paraffin samples.
Immunohistochemistry was performed on Ventana Benchmark XT™ instrument utilizing antibodies to HLA-DR, FoxP3, CD3, CD4, CD8, CD68, and acetylated H3 and H4. Methods are described in supplementary table I. The primary antibody was added by hand (in a so-called “titre” run) for those antibodies not purchased from Ventana Medical systems. Following staining, slides were rinsed, washed in detergent, dehydrated, and cover slipped. Each antibody staining run included a reactive tonsil as the positive control. The negative control was the same tonsil but without addition of primary antibody.
Scoring and Image Analysis
We used an automated image analysis program from Definiens (Cambridge, Massachusetts) version 2.4 for evaluation of staining results of all IHC, with the exception of HLA-DR, which was scored manually since the pattern of staining was of interest (supplementary figure 1). Each slide was evaluated for percentage of HLA-DR positive cells, intensity, and pattern of staining (no staining, surface staining, cytoplasmic punctate staining, or mixed surface/punctate), to come up with an HLA-DR score as previously published.10 Cytoplasmic markers were CD3, CD4, CD8, CD68, and FoxP3; while nuclear markers were acetylated H3 and H4. Detailed methods for automated image analyses are described in Supplementary Tables II and III. All microscopic evaluations were performed by one of the authors, a hematopathologist (L.R.).
Serum cytokine analysis
To assess if baseline or changes in serum cytokine levels correlate with PFS (or OS), we collected2 ml of whole blood pre-treatment and on day 9 of cycle 1. We used the Human Cytokine Magnetic 30-plex panel from Luminex™. This panel of 30 measures include human IL-1beta, IL-1RA, Il-2, IL-sR, IL-4, IL-5, IL-6, IL-7,IL-8, IL-10, IL-12p40/p70, IL-13, IL-15, IL-17, TNF-alpha, IFN-alpha, IFN-gamma, GM-CSF, MIP-1alpha, MIP-1beta, IP-10, MIG, Eotaxin, RANTES, MCP-1, VEGF, G-CSF, EGF, FGF-basic and HGF. Previously stored serum samples were thawed on ice, spun to remove any aggregates, allowed to come to room temperature, then run immediately. If samples were cloudy, hemolyzed or had precipitate this was noted. Regardless, all samples were centrifuged, otherwise processed and analyzed, in duplicate, per manufacturer’s explicit instructions, without any modifications. Values were plotted by patient and time point (pre-treatment and cycle 1 day 9). Because of the skewed distribution of the serum cytokine level measurements, log 10 transformation was used to normalize the distribution. Standardization of the data was also performed to remove variability of measurements between plates by subtracting the mean of measurements. Measurements that were below the lower threshold or above the higher threshold were set as the value of lower threshold or higher threshold respectively.
Statistical Considerations
For Phase I portion of the study, the goal was to establish maximum tolerated dose of vorinostat with standard R-CHOP, with expected accrual of 10-30 patients. A temporary closure would occur prior to opening this study for the Phase II portion in order to assess dose, and to evaluate the safety profile more fully.
For the Phase II portion of study, the primary goal of this portion of the study was to assess the 2-year PFS rate, with secondary endpoints including toxicity, overall survival, and response probability. Sixty-five eligible patients accrued over 18 months with 24 months of additional follow-up would be sufficient to estimate the 2-year PFS within this group to ± 0.13. Sehn et al,16 found 2-year PFS to be approximately 80% and 60% for patients with intermediate (1-2) and high (3-5) IPI scores, respectively. Given these historical data and assuming that 50% of accrued patients will be in each IPI risk category, we expected 2-year PFS for R-CHOP to be 70%. Under these assumptions, an estimated 2-year PFS rate of 80% or greater for vorinostat-R-CHOP would be sufficient to warrant further investigation. The target value was to be adjusted based on the observed frequency of prognostic groups. This design had a power of 0.90 and a significance level of 0.05. Assuming 65 eligible patients were accrued to the Phase II portion of this trial, we would be able to estimate the probability of any particular toxicity to within ± 0.13. Any adverse event occurring with at least a 5% probability was likely to be seen at least once (96% chance).
PFS was defined as the time from date of registration to date of first observation of progressive disease or death due to any cause. Patients last known to be alive and without report of progression are censored at date of last contact. OS was defined as date of registration to date of death due to any cause. Patients last known to be alive are censored at date of last contact. OS and PFS estimates were calculated using the Kaplan-Meier method.
Translational studies included exploratory analyses to assess the association of baseline histone acetylation and T-cell subsets with PFS using univariate Cox regression model. For cytokine analyses, both nominal and Bonferroni adjustments were considered to address multiple comparisons. Additional analyses included patients from a prior SWOG study S0433, and studied the impact of vorinostat-R-CHOP on relative to outcomes on S0433. Comparisons between studies for PFS and OS were assessed by the stratified log-rank test at two-sided alpha level of 0.05.
Results
This study was closed to accrual on October 1, 2013, after meeting its enrollment goal. Data as of July 12, 2016, were included in analyses. Eighty-three patients were registered.
Study course
The Phase I portion of this study was activated in four SWOG institutions and was temporarily closed for pre-specified analysis with 11 patients registered. One patient was ineligible due to incorrect histology upon hematopathology review, and one patient did not receive any protocol treatment was not analyzable for any endpoint. Of 9 evaluable (10 toxicity-evaluable) phase I patients, two experienced DLTs – grade 3 febrile neutropenia and grade 4 hypokalemia, both in the second week of treatment. Therefore phase II study opened at all centers with vorinostat original starting dose at 400 mg po days 1-9.
As phase I patients completed all treatment and phase II patients started enrollment, excess rates of febrile neutropenia and sepsis were noted, occurring throughout the treatment, from both gram positive and gram negative organisms. Two of phase I patients developed sepsis and one died of it one week after completing all 8 cycles of treatment. The study was again placed on hold and amended to reduce the duration of vorinostat to 400 mg po days 1-5, to mandate neutrophil growth factor prophylaxis, and to recommend prophylactic antibiotics.
Seventy-two patients were registered on phase II part of the study, of which 2 withdrew consent and did not receive any protocol treatment, and 7 were ineligible (incorrect histology (3), inadequate tissue for diagnosis confirmation (1), non-bulky stage II disease (2), and low platelet count (1)). Sixty-three patients were thus evaluable in phase II part of the trial, for a total of 72 evaluable patients including 9 from the phase I part of the trial. Patient baseline characteristics are outlined in Table I. Median age was 64, 92% had stage III/IV disease, 63% had elevated LDH, and 33% had more than one extranodal site involvement, with 58% of patients having IPI 3-5.
Table I.
Patient characteristics.
| S0806 | |||
|---|---|---|---|
| Phase I (n=9) | Phase II (n=63) | Total (n=72) | |
| Clinical Variable | N (%) | N (%) | N (%) |
| Age, Years | |||
| Median | 67 | 64 | 64 |
| Range | 46 - 84 | 20 - 81 | 20-84 |
| >60 | 5 (56%) | 40 (63%) | 45 (63%) |
| Male sex | 6 (67%) | 36 (57%) | 42 (58%) |
| Hispanic race | 3 (33%) | 4 (6%) | 7 (10%) |
| White race | 9 (100%) | 54 (86%) | 63 (88%) |
| B Symptoms | 5 (56%) | 25 (40%) | 30 (42%) |
| Stage II | 1 (11%) | 5 (8%) | 6 (8%) |
| Performance Status | |||
| 0 | 0 (0%) | 26 (41%) | 26 (36%) |
| 1 | 6 (67%) | 32 (51%) | 38 (53%) |
| 2 | 3 (33%) | 5 (8%) | 8 (11%) |
| Bulky Disease | 3 (33%) | 14 (22%) | 17 (24%) |
| Bone Marrow Involvement | 1 (11%) | 8 (13%) | 9 (13%) |
| LDH > ULN | 7 (78%) | 38 (60%) | 45 (63%) |
| > 1 Extranodal Sites | 0 (0%) | 24 (38%) | 24 (33%) |
| IPI Score | |||
| 1 | 1 (11%) | 9 (14%) | 10 (14%) |
| 2 | 4 (44%) | 16 (25%) | 20 (28%) |
| 3 | 2 (22%) | 29 (46%) | 31 (43%) |
| 4 | 2 (22%) | 8 (13%) | 10 (14%) |
| 5 | 0 (0%) | 1 (2%) | 1 (1%) |
Safety
Five patients in phase I and 29 patients in phase II did not complete all 8 cycles of treatment. Patients received a median of 7 cycles of treatment on protocol. Toxicities are listed in table II. Notable grade 3-4 non-hematologic toxicities included: febrile neutropenia (38%), sepsis (19%), fatigue (17%), hypokalemia (14%), hyponatremia (8%), and small bowel perforation (3%). Grade 3-4 hematologic toxicities included: neutropenia (63%), anemia (36%), and thrombocytopenia (36%). While there was one death from sepsis and multi-organ failure on phase I as mentioned above, there were no deaths from toxicity in phase II part of the trial.
Table II.
Toxicities occurring in at least 15% of patients, listed by individual toxicity type in order of decreasing frequency of toxicity of any grade in combined phase I/II data.
| Phase I | Phase II | Total | ||||
|---|---|---|---|---|---|---|
| n = 9 | n = 63 | n = 72 | ||||
| Any Grade | Grade ≥ 3 | Any Grade | Grade ≥ 3 | Any Grade | Grade ≥ 3 | |
| By Individual Toxicity Type | No (%) | No (%) | No (%) | No (%) | No (%) | No (%) |
| Anemia | 9 (100%) | 4 (44%) | 51 (81%) | 22 (35%) | 60 (83%) | 26 (36%) |
| Fatigue | 8 (89%) | 3 (33%) | 46 (73%) | 9 (14%) | 54 (75%) | 12 (17%) |
| Neutrophil count decreased | 8 (89%) | 8 (89%) | 43 (68%) | 37 (59%) | 51 (71%) | 45 (63%) |
| White blood cell decreased | 8 (89%) | 7 (78%) | 43 (68%) | 32 (51%) | 51 (71%) | 39 (54%) |
| Nausea | 8 (89%) | 1 (11%) | 41 (65%) | 3 (5%) | 49 (68%) | 4 (6%) |
| Platelet count decreased | 8 (89%) | 4 (44%) | 37 (59%) | 22 (35%) | 45 (63%) | 26 (36%) |
| Lymphocyte count decreased | 7 (78%) | 4 (44%) | 29 (46%) | 20 (32%) | 36 (50%) | 24 (33%) |
| Diarrhea | 7 (78%) | 2 (22%) | 27 (43%) | 2 (3%) | 34 (47%) | 4 (6%) |
| Anorexia | 5 (56%) | 1 (11%) | 24 (38%) | 2 (3%) | 29 (40%) | 3 (4%) |
| Constipation | 4 (44%) | 0 (0) | 23 (37%) | 0 (0) | 27 (38%) | 0 (0) |
| Febrile neutropenia | 3 (33%) | 3 (33%) | 24 (38%) | 24 (38%) | 27 (38%) | 27 (38%) |
| Alopecia | 5 (56%) | 0 (0) | 21 (33%) | 0 (0) | 26 (36%) | 0 (0) |
| Vomiting | 4 (44%) | 0 (0) | 20 (32%) | 2 (3%) | 24 (33%) | 2 (3%) |
| Mucositis oral | 1 (11%) | 1 (11%) | 22 (35%) | 3 (5%) | 23 (32%) | 4 (6%) |
| Hypoalbuminemia | 3 (33%) | 1 (11%) | 19 (30%) | 3 (5%) | 22 (31%) | 4 (6%) |
| Hyponatremia | 3 (33%) | 0 (0) | 19 (30%) | 6 (10%) | 22 (31%) | 6 (8%) |
| Fever | 1 (11%) | 0 (0) | 20 (32%) | 0 (0) | 21 (29%) | 0 (0) |
| Hypokalemia | 3 (33%) | 2 (22%) | 18 (29%) | 8 (13%) | 21 (29%) | 10 (14%) |
| Peripheral sensory neuropathy | 3 (33%) | 0 (0) | 18 (29%) | 0 (0) | 21 (29%) | 0 (0) |
| Hypocalcemia | 2 (22%) | 1 (11%) | 18 (29%) | 0 (0) | 20 (28%) | 1 (1%) |
| Dizziness | 3 (33%) | 1 (11%) | 16 (25%) | 0 (0) | 19 (26%) | 1 (1%) |
| Abdominal pain | 2 (22%) | 1 (11%) | 15 (24%) | 3 (5%) | 17 (24%) | 4 (6%) |
| Headache | 2 (22%) | 0 (0) | 15 (24%) | 0 (0) | 17 (24%) | 0 (0) |
| Edema limbs | 1 (11%) | 0 (0) | 15 (24%) | 0 (0) | 16 (22%) | 0 (0) |
| Generalized muscle weakness | 5 (56%) | 2 (22%) | 11 (17%) | 2 (3%) | 16 (22%) | 4 (6%) |
| ALT increased | 1 (11%) | 0 (0) | 14 (22%) | 1 (2%) | 15 (21%) | 1 (1%) |
| Sepsis | 3 (33%) | 3 (33%) | 11 (17%) | 11 (17%) | 14 (19%) | 14 (19%) |
| AST increased | 2 (22%) | 0 (0) | 11 (17%) | 1 (2%) | 13 (18%) | 1 (1%) |
| Cough | 3 (33%) | 0 (0) | 10 (16%) | 0 (0) | 13 (18%) | 0 (0) |
| Weight loss | 2 (22%) | 0 (0) | 11 (17%) | 3 (5%) | 13 (18%) | 3 (4%) |
| Dyspnea | 0 (0) | 0 (0) | 12 (19%) | 1 (2%) | 12 (17%) | 1 (1%) |
| Dysgeusia | 3 (33%) | 0 (0) | 8 (13%) | 0 (0) | 11 (15%) | 0 (0) |
Efficacy
As specified in the protocol, clinical primary efficacy analysis was to be based on patients registered to the Phase II component of the study. Of 63 evaluable phase II patients, 33 had complete response and 18 had partial response, for an overall estimated response rate of 81% (95% CI: 69.1%, 89.8%). Two patients died prior to the first disease assessment and 10 patients could not have their response determined due to lack of radiographic scans within 30 days of coming off protocol as required by study. These twelve patients were counted as non-responders for the purpose of response estimation.
The median follow-up among Phase II patients last known alive is 3 years (range 1.7 - 4.3 years). Twenty-two patients have either progressed or died, with median PFS of 51.2 months. The estimate of 2-year PFS is 73% (95%CI: 60.2%, 82.3%) (figure 1A). There have been 14 deaths with median OS of 51.2 months. The estimate of 2-year OS is 86% (95%CI: 74.3%, 92.3%) (figure 1B). While it was protocol specified to evaluate efficacy on the Phase II portion of the study, the addition of the Phase I patients led to very similar results with a 2 year PFS of 75% and 2 year OS of 86%.
Figure 1.
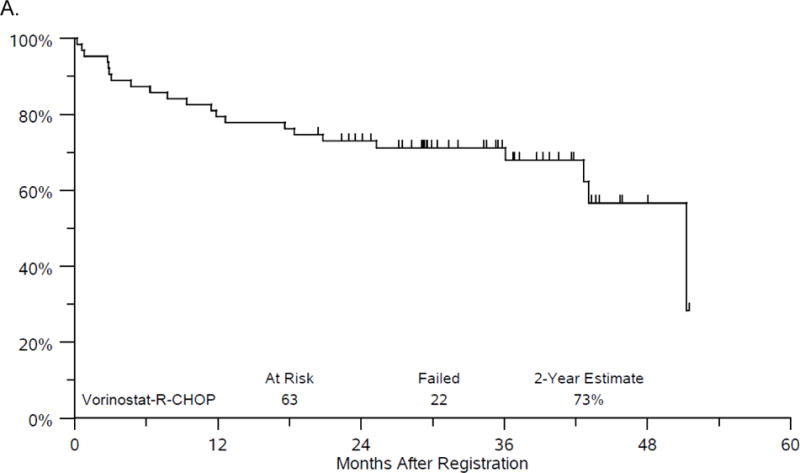
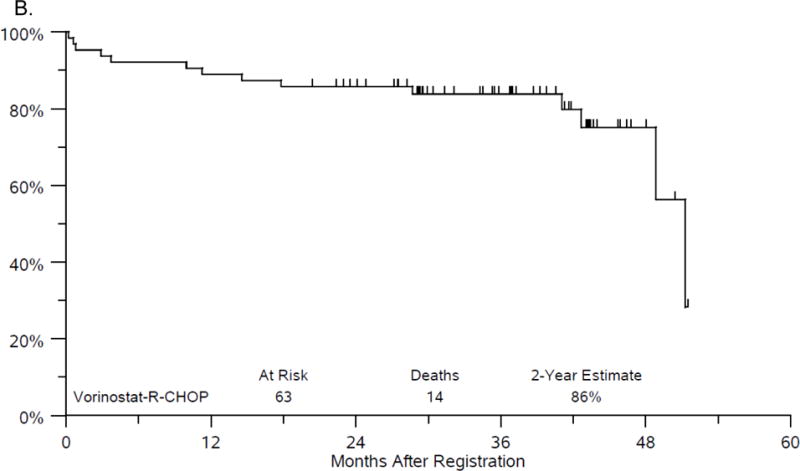
A. Progression-free survival.
B. Overall Survival.
Outcomes according to standard prognostic factors
Of 72 evaluable patients, 58% had high risk (3-5) R-IPI. While high risk R-IPI had numerically inferior outcomes compared to intermediate risk (1-2) R-IPI (2-year PFS 69% vs. 83%, 2-year OS 83% vs. 90%), it did not reach statistical significance. Ki67 of at least 80%, which correlated with unfavorable outcome in many DLBCL studies, was available in 47 phase I/II patients, and had even less significance: 2-year PFS 73% vs. 76%, and 2-year OS 81% vs. 90%.
Cell of origin (COO) analysis was assigned using the Hans algorithm in 57 eligible phase I/II patients, with 42 patients (74%) having germinal center B-cell- derived (GCB) DLBCL and 15 patients having non-GCB DLBCL. Fifteen patients who did not have COO assessments had numerically inferior but statistically similar outcomes compared to those who did have COO determination. Patients with GCB DLBCL had numerically superior 2-year PFS (81% vs. 67%) and OS (90% vs. 87%), but this did not reach statistical significance. Comparison of outcomes according to COO in S0806 vs. S0433 revealed numerically superior outcomes in GCB but similar outcomes in non-GCB, again without any statistically significant differences.
Fifty-three evaluable phase I/II patients had MYC expression assessed by IHC, of which 23 were positive; 62 evaluable patients had BCL2 assessment by IHC, of which 40 were positive; of 49 evaluable patients who had both MYC and BCL2 assessment by IHC, 11 (22%) were “double expressor.” Patients with “double expressor” DLBCL had similar PFS and OS to patients without MYC/BCL2 overexpression. Despite numerically superior outcomes for “double expressor” lymphoma in S0806 compared to S0433 (2-year PFS 73% vs. 58%, 2-year OS 91% vs. 75%), the differences were not statistically significant. Too few patients had FISH performed locally for MYC and BCL2 to make any conclusions about proportion and outcome of “double-hit lymphoma” on this study.
Translational studies
HLA-DR was assessed by immunohistochemistry for percent positivity (n=58), intensity (n=58) and pattern of staining (n=52). Forty-six patients (79%) had at least 10% HLA-DR staining, with those below 10% cutoff showing unfavorable outcomes in prior work.2 In keeping with our original rationale about enhancing immunosurveillance, in S0806, low pre-treatment HLA-DR expression did not carry an unfavorable outcome, with 2-year PFS of 75% (vs. 74% in high HLA-DR expressors) and OS of 92% (vs. 83%), p=0.9066 and 0.3916, respectively. By comparison, patients on S0433 with low pre-treatment HLA-DR expression did not fare as well – their 2-year PFS was only 58% and OS was 67%. However, this was not statistically significant (p=0.4843 for PFS and p=0.2459 for OS). High HLA-DR expressors on S0433 had more similar 2-year PFS and OS to S0806, both at 75%.
HLA-DR score, which takes into account staining positivity by quartile and intensity, as previously published,10 was not prognostic in S0806 when dichotomized at the median, highest vs. lowest quartile, or 0 vs. more than 0. S0433 by comparison showed numerically inferior outcomes for low HLA-DR score of 0, and more similar outcomes for higher HLA-DR scores. This result was in keeping with our original rationale for the study that vorinostat would increase surface HLA-DR expression and improve immunosurveillance, but again this was not statistically significant.
In addition to the poor outcome associated with complete loss of HLA-DR in B cell lymphomas, we recently reported on the unfavorable impact of loss from the cell surface with retained HLA-DR in cytoplasmic punctate staining pattern in DLBCL.3 In the current study, 5 patients had no staining, 12 had cytoplasmic punctate staining, 13 had mixed punctate and surface staining, and 22 had surface only staining. As expected, patients with no staining (truly negative) had the worst 2-year PFS at 40%, while punctate had 83%, mixed punctate/surface 85%, and surface only had 64% 2-year PFS (p=0.0627). However, 2-year OS was 80-85% for all 4 staining patterns. Of interest, patients with negative or punctate HLA-DR pattern were 2.7 times more likely to be GCB than those with any surface HLA-DR pattern (surface/punctate or surface), but this was not statistically significant (p=0.29). There were no associations between HLA-DR expression and IPI, MYC expression, BCL2 expression, double (MYC and BCL2) expression, Ki67, or COO.
Immunohistochemical stains for T-cell subsets and macrophages were available on 39 to 44 of phase I/II patients, depending on the individual stain. Logarithms of total counts for CD8, CD3, CD4, CD68, and FOXP3, dichotomized at the median, were not associated with PFS when used in a univariate Cox regression model.
Paired cytokine assessments were available on 5 phase I and 36 phase II patients. With adjusting for multiple (30) cytokine measurements using Bonferroni method, baseline elevated logarithm (log) of MIP-1beta concentration correlated with inferior OS (adj. p= 0.0156). No baseline measurements correlated with PFS. No changes in log cytokine concentrations correlated to outcomes after adjusting for multiple measurements.
Discussion
SWOG S0806 was a phase I/II study of 8 cycles of vorinostat-R-CHOP that achieved 2-year PFS estimate of 73%, which did not meet its pre-specified target. Taking into account unexpected and significant rates of febrile neutropenia (38%) and sepsis (19%), the regimen of vorinostat-R-CHOP cannot be recommended for broad use.
Febrile neutropenia and sepsis occurred predominantly during days of 9-13 of the cycles, which would be the time of blood count nadirs during R-CHOP, suggesting that addition of vorinostat lowered the neutrophil nadir in a very clinically meaningful way. Both gram negative and gram positive infections were seen, and febrile neutropenia and sepsis occurred throughout all cycles. There was no factor that predicted toxicity that we could identify.
This study is the largest trial combining an HDACI with chemotherapy in B-cell malignancy, where the role of this drug class remains to be defined. Straus et al17 reported a trial of 29 patients with relapsed/refractory DLBCL treated with vorinostat and R-CEPP without procarbazine, with vorinostat MTD established at 300 mg po days 1-10. Febrile neutropenia rate was 17%, with median PFS of 9.2 months and OS of 17.5 months. Our study used doxorubicin instead of etoposide, but was frontline and in younger patients, and therefore toxicity was more than expected. Oki at al18 combined vorinostat with CHOP in 14 patients with newly diagnosed T-cell lymphomas. Three patients discontinued due to toxicity, including one with repeat neutropenic fever, one with grade 3 mucositis, and one with grade 3 anemia. We used 8 cycles of R-CHOP in our trial, which could have contributed to toxicity, and the multicenter nature of a cooperative study probably reflects the true toxicity of a regimen to a greater extent than single center studies such as mentioned above.
Studies published since the design of this trial showed only modest activity of single agent HDACI in DLBCL.19,20 The rationale for this study, however, was to see if HDACI could reverse the unfavorable impact of the loss of MHCII expression. We previously published the results of SWOG phase II trial with belinostat in relapsed/refractory DLBCL, where of 2 on-treatment biopsies one responding patient had increase in MHCII expression and numbers of TILs, while a patient who did not respond did not.10 Unlike in that prior experience we could not find an association between MHCII (HLA-DR) and T-cells in this study. Comparison to our prior frontline DLBCL study, S0433, which did not use an HDACI,15 showed that low MCHII expressing patients have numerically inferior outcomes on that study compared to this trial. Thus, it may indicate that addition of vorinostat, as hypothesized, did address unfavorable features of low MCHII expression. However, the difference was not statistically significant, and bore all the limitations of cross-study comparisons. Complete lack of MHCII staining on S0806 was borderline statistically significant (p=0.0627), however there were only 5 patients in that cohort.
Cytokine profiling revealed that baseline elevated MIP-1beta concentration correlated with worse OS. MIP-1beta, also known as CCL4, is an autocrine chemokine that has been shown to a biomarker of B-cell receptor pathway and to correlate with worse outcomes in DLBCL.21 In this study we looked at FOXP3, a marker of T-reg lineage, by immunohistochemistry, but it did not correlate with PFS.
The future of HDACI in B-cell lymphoma is hampered by lack of biomarkers predicting response and outcomes. Inactivating mutations in the genes encoding histone acetyltransferases CREBBP and EP300 were found in up to one third of DLBCL.22,23 In vitro data indicated greater sensitivity of CREBBP and EP300 mutants to vorinostat.24 Assouline et al25 recently reported on 40 patients with relapsed/refractory/transformed DLBCL treated with panobinostat with or without rituximab. They found 64% of patients having mutations in histone-modifying enzymes. They also found that mutations in MEF2B gene, which cooperates with CREBBP and EP300 to acetylate histones, were significantly associated with higher response. Future study will look at S0806 to identify if there is a subset, perhaps DLBCL harboring CREBBP and EP300 mutations that may benefit from addition of vorinostat.
In summary, SWOG S0806 trial tested the addition of HDACI vorinostat to R-CHOP in advanced stage DLBCL. The regimen resulted in outcomes similar to R-CHOP, but caused increased toxicity, particularly febrile neutropenia and sepsis. As a result, this regimen cannot be recommended for DLBCL patients. Future studies will investigate if a subset of patients with DLBCL harboring CREBBP and EP300 may have benefitted from the addition of HDACI.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Numbers CA180888, CA180819, CA189957, CA189821, CA180830, CA189808, CA189860, CA189853, CA189953, CA189858, CA12644, CA189873, CA189822, CA189804, CA180826, CA189954, CA 11083, CA13612, CA46368, CA46282, CA35119, and in part by Merck & Co., Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or Merck & Co., Inc.
Footnotes
Author Contribution:
Study conception and/or design: DOP, RIF, JWF, HL, ML, LMR
Statistical Analysis: HL, ML
Data collection and/or assembly: All authors
Data acquisition and/or interpretation: All authors
Provision of study materials or patients: CLB, PMB, JWF, DOP, LLP, AVG
Reviewed and revised the manuscript for important intellectual content, and approved the final version: All authors
Author Conflict of Interest Disclosure:
DOP: Consultancy – Genentech, Sandoz
RIF: Consultancy – Pharmacyclics, F. Hoffmann-LaRoche, Kite, Seattle Genetics, Sandoz, Celgene, Genentech, Astra Zeneca, Bayer, Adaptive Biotechnologies
References
- 1.Morton LM, Wang SS, Devesa SS, Hartge P, Weisenburger DD, Linet MS. Lymphoma incidence patterns by WHO subtype in the United States, 1992–2001. Blood. 2006;107(1):265–276. doi: 10.1182/blood-2005-06-2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rimsza LM, Roberts RA, Miller TP, et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood. 2004;103(11):4251–4258. doi: 10.1182/blood-2003-07-2365. [DOI] [PubMed] [Google Scholar]
- 3.Kendrick S, Rimsza LM, Scott DW, et al. Aberrant cytoplasmic expression of MHCII confers worse progression free survival in diffuse large B-cell lymphoma. Virchows Arch. 2017;470(1):113–117. doi: 10.1007/s00428-016-2041-7. [DOI] [PubMed] [Google Scholar]
- 4.Wilkinson ST, Vanpatten KA, Fernandez DR, et al. Partial plasma cell differentiation as a mechanism of lost major histocompatibility complex class II expression in diffuse large B-cell lymphoma. Blood. 2012;119(6):1459–1467. doi: 10.1182/blood-2011-07-363820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma JK, Platt MY, Eastham-Anderson J, Shin JS, Mellman I. MHC class II distribution in dendritic cells and B cells is determined by ubiquitin chain length. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(23):8820–8827. doi: 10.1073/pnas.1202977109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rimsza LM, Roberts RA, Campo E, et al. Loss of major histocompatibility class II expression in non-immune-privileged site diffuse large B-cell lymphoma is highly coordinated and not due to chromosomal deletions. Blood. 2006;107(3):1101–1107. doi: 10.1182/blood-2005-04-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beresford GW, Boss JM. CIITA coordinates multiple histone acetylation modifications at the HLA-DRA promoter. Nature immunology. 2001;2(7):652–657. doi: 10.1038/89810. [DOI] [PubMed] [Google Scholar]
- 8.Lee SC, Bottaro A, Insel RA. Activation of terminal B cell differentiation by inhibition of histone deacetylation. Molecular immunology. 2003;39(15):923–932. doi: 10.1016/s0161-5890(03)00029-4. [DOI] [PubMed] [Google Scholar]
- 9.Richon VM, Emiliani S, Verdin E, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(6):3003–3007. doi: 10.1073/pnas.95.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puvvada SD, Li H, Rimsza LM, et al. A phase II study of belinostat (PXD101) in relapsed and refractory aggressive B-cell lymphomas: SWOG S0520. Leuk Lymphoma. 2016;57(10):2359–2369. doi: 10.3109/10428194.2015.1135431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reddy P, Maeda Y, Hotary K, et al. Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(11):3921–3926. doi: 10.1073/pnas.0400380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tao R, de Zoeten EF, Ozkaynak E, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13(11):1299–1307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 13.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579–586. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 14.Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103(1):275–282. doi: 10.1182/blood-2003-05-1545. [DOI] [PubMed] [Google Scholar]
- 15.Friedberg JW, Unger JM, Burack WR, et al. R-CHOP with iodine-131 tositumomab consolidation for advanced stage diffuse large B-cell lymphoma (DLBCL): SWOG S0433. Br J Haematol. 2014;166(3):382–389. doi: 10.1111/bjh.12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sehn LH, Berry B, Chhanabhai M, et al. The revised International Prognostic Index (R-IPI) is a better predictor of outcome than the standard IPI for patients with diffuse large B-cell lymphoma treated with R-CHOP. Blood. 2007;109(5):1857–1861. doi: 10.1182/blood-2006-08-038257. [DOI] [PubMed] [Google Scholar]
- 17.Straus DJ, Hamlin PA, Matasar MJ, et al. Phase I/II trial of vorinostat with rituximab, cyclophosphamide, etoposide and prednisone as palliative treatment for elderly patients with relapsed or refractory diffuse large B-cell lymphoma not eligible for autologous stem cell transplantation. Br J Haematol. 2015;168(5):663–670. doi: 10.1111/bjh.13195. [DOI] [PubMed] [Google Scholar]
- 18.Oki Y, Younes A, Copeland A, et al. Phase I study of vorinostat in combination with standard CHOP in patients with newly diagnosed peripheral T-cell lymphoma. Br J Haematol. 2013;162(1):138–141. doi: 10.1111/bjh.12326. [DOI] [PubMed] [Google Scholar]
- 19.Amengual JE, Clark-Garvey S, Kalac M, et al. Sirtuin and pan-class I/II deacetylase (DAC) inhibition is synergistic in preclinical models and clinical studies of lymphoma. Blood. 2013;122(12):2104–2113. doi: 10.1182/blood-2013-02-485441. [DOI] [PubMed] [Google Scholar]
- 20.Crump M, Coiffier B, Jacobsen ED, et al. Phase II trial of oral vorinostat (suberoylanilide hydroxamic acid) in relapsed diffuse large-B-cell lymphoma. Ann Oncol. 2008;19(5):964–969. doi: 10.1093/annonc/mdn031. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi K, Sivina M, Hoellenriegel J, et al. CCL3 and CCL4 are biomarkers for B cell receptor pathway activation and prognostic serum markers in diffuse large B cell lymphoma. Br J Haematol. 2015;171(5):726–735. doi: 10.1111/bjh.13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morin RD, Mendez-Lago M, Mungall AJ, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476(7360):298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pasqualucci L, Dominguez-Sola D, Chiarenza A, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471(7337):189–195. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andersen CL, Asmar F, Klausen T, Hasselbalch H, Gronbaek K. Somatic mutations of the CREBBP and EP300 genes affect response to histone deacetylase inhibition in malignant DLBCL clones. Leuk Res Rep. 2012;2(1):1–3. doi: 10.1016/j.lrr.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Assouline SE, Nielsen TH, Yu S, et al. Phase 2 study of panobinostat with or without rituximab in relapsed diffuse large B-cell lymphoma. Blood. 2016;128(2):185–194. doi: 10.1182/blood-2016-02-699520. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.