

Abstract
Background
Olfactory inflammation in CRS is associated with cytokines that may result in the death of olfactory sensory neurons. Principal signaling molecules involved in the apoptotic pathway are c-Jun N-terminal kinases (JNK). While the JNK pathway has emerged as a key player in programmed cell death in neuroinflammation, its specific role in CRS-associated olfactory loss has not been thoroughly investigated.
Methods
JNK activation was studied in human tissue samples from 9 control and 11 CRS patients by immunohistochemical staining for phosphorylated c-Jun. A mouse model of inducible olfactory cytokine expression was used to experimentally control inflammation and assess JNK activation over time.
Results
In patients with CRS, activation of c-Jun is significantly increased relative to non-CRS control subjects, and there is an associated loss of sensory neurons. In the olfactory inflammation mouse model, prolonged induction of inflammation results in elevation of c-Jun expression and neuronal apoptosis.
Conclusions
Activation of neuronal JNK is a feature of chronic olfactory inflammation that is associated with neuronal apoptosis. Given that inhibition of JNK activity is neuroprotective in other settings, antagonism of this pathway may have therapeutic potential in the management of inflammatory olfactory loss or other disorders linked to olfactory neuronal apoptosis.
Keywords: Olfactory disorders, Chronic rhinosinusitis, Olfaction, Inflammation, JNK, Olfactory sensory neurons
Introduction
Olfactory dysfunction is an important symptom frequently reported by patients with chronic rhinosinusitis (CRS). Many CRS patients have some degree of nasal obstruction that can impede airflow to the olfactory cleft. However, impaired airflow alone cannot account for the olfactory loss, and, in fact, olfactory dysfunction in CRS often occurs with olfactory cleft patency1. It is likely that CRS-associated olfactory loss is caused in part by damage to the olfactory epithelium (OE) with loss of sensory neurons2. That being said, olfactory loss due solely to OE destruction is not completely consistent with clinical observations of fluctuating impairment in CRS that can often be reversed transiently with administration of systemic corticosteroids3,4. Corticosteroids potently inhibit production of inflammatory mediators in the sinonasal mucosa, leading in turn to reduction of the inflammatory cell infiltrate that is the histologic hallmark of CRS. Because inflammatory processes affecting the sinonasal mucosa also directly involve the adjacent OE, it is hypothesized that neuroepithelial inflammation itself causes a sensory deficit, perhaps mediated by CRS-associated inflammatory cytokines2,5,6.
A wide array of cytokines have been identified within the sinonasal mucosa of patients with CRS, including within the olfactory cleft7. A number of these cytokines have been reported to be associated with olfactory loss7–10. The signal transduction mechanisms of key CRS-associated inflammatory mediators bifurcate into two separate pathways with opposing physiologic roles11,12. The NF-κB-dependent pathway is both pro-inflammatory and anti-apoptotic. In contrast, the other pathway is apoptotic and leads directly to activation of a caspase cascade. The principal signaling molecules involved in the TNF-α apoptotic pathway are c-Jun N-terminal kinases (JNK)13,14. The TNF-α/JNK pathway has emerged as a key player in programmed cell death in neuroinflammation15. Activation of JNK leads to formation of the AP-1 transcription factor and initiation of apoptosis. Multiple reports have demonstrated that inhibition of JNK activity is neuroprotective, including a study by Gangadhar et al. showing olfactory sensory neuron (OSN) death to be mediated by JNK in a bulbectomy model16. NF-κB, which is also expressed in neuronal tissue, exerts an anti-apoptotic effect in part through antagonism of the JNK pathway17,18. Both NF-κB and JNK pathways cross-talk extensively with diverse intracellular processes that impact non-apoptotic cell functions19.
In multiple cell types, including neurons, inflammatory cytokines activate signaling pathways involving NF-κB and JNK. JNK has been implicated as a mediator of both apoptotic and necrotic cell death20,21. In the olfactory system, treatment of olfactory bulbectomized mice with JNK inhibitors promotes survival of mature OSNs16 and, in cultured OSNs, suppresses caspase activation and promotes survival. It is intriguing to consider JNK inhibition as a strategy for olfactory neuroprotection in CRS. JNK inhibitors are in human trials for visual and auditory system conditions associated with sensory neuronal death. In the setting of inflammation, JNK activity is closely linked to the transcription factor NF-κB. Many inflammatory cytokines, including TNF-α, activate NF-κB, which increases expression of pro-inflammatory and anti-apoptotic genes. NF-κB-stimulated genes promoting survival act largely through suppression of JNK activation and anti-oxidation18.
Because detailed study of CRS in human OE is limited by relative difficulty in obtaining tissue and the infeasibility of experimental manipulation, there is a clear need for animal models. In rodent models of acute infectious or allergic rhinosinusitis, olfactory dysfunction and increased neuronal cell death occur within the OE, suggesting a role for neuroepithelial damage in this setting.22–24 Acute loss of neurons is followed by a wave of progenitor cell proliferation and OE regeneration. To investigate the effects of chronic inflammation, we developed a transgenic mouse model in which cytokine expression is used to initiate olfactory inflammation.8 In brief, this IOI (inducible olfactory inflammation) mouse model employs the tet-on expression system to drive temporally-controlled gene expression by olfactory sustentacular cells. The IOI mouse model chronically express the cytokine TNF-α when induced with doxycycline (DOX). The effect of chronic TNF-α exposure was an initial desensitization of odorant responses, followed by a progressive reorganization of olfactory architecture characterized by inflammatory cell infiltration, loss of axon bundles, and thinning of the OE above the basement membrane. During active inflammation, proliferation of olfactory progenitor cells is inhibited, preventing regeneration. Once the inflammation resolves, there is robust reconstitution of the normal olfactory architecture. This model mimics important aspects of CRS-associated olfactory loss and allows us to pursue important mechanistic questions relevant to the human disease.
In this study, we examine olfactory tissue from human subjects and from the IOI mouse model to determine whether neuronal JNK activation is associated with inflammation.
Methods
Animals
TNF-α receptor 1 (TNFR1) knockout mice were purchased from Jackson Laboratories. The IOI strain has been described previously8,25,26. Briefly, the reverse tetracycline transactivator gene (rtTA) was knocked into the olfactory sustentacular cell-specific cyp2g1 coding region to generate a cyp2g1-rtTA strain. The IOI strain was generated by crossing cyp2g1-rtTA mouse with a strain carrying a tet-response element-driven TNF-α transgene. 8 week old animals were given doxycycline impregnated food for 3–4 weeks to induce TNF-α expression by olfactory sustentacular cells. The animal handling procedures were approved by the Animal Care and Use Committee at the Johns Hopkins University.
Human olfactory tissue
Human olfactory epithelial samples were collected from 11 CRS patients and 9 control subjects undergoing endonasal surgical approaches for non-CRS disease processes. This tissue was collected from the medial surface of resected superior turbinate, and in some cases, from the upper septum. The characteristics of the patient cohort are detailed in Table 1. None of the control subjects reported decreased olfactory function. All of the CRS patients reported fluctuating olfactory loss with disease exacerbations. Objective olfactory identification testing was not performed. The removed tissue was maintained on ice until transported to the laboratory. After fixation in 4% PFA for 30 minutes on ice, the tissue was washed in PBS and immersed in 30% sucrose for cryoprotection. Any bone present was dissected out, and the tissue fragment was then embedded in OCT. Twelve μm sections were collected for immunohistochemistry analyses. Only patients with neuroepithelium-containing tissue samples were included in this study. The research protocol was approved by the Johns Hopkins Institutional Review Board, and all subjects gave signed informed consent.
Table 1.
Characteristics of study subjects
| Control (n = 9) | CRS (n = 11) | |
|---|---|---|
| Age (years)a | 56.9 ± 11.9 | 48.9 ± 11.8 |
| Gender (% male) | 55.6 | 54.5 |
| Polyposis (%) | 0.0 | 63.4 |
| Allergy (%) | 22.2 | 54.5 |
| Asthma (%) | 11.1 | 45.5 |
Data expressed as mean ± standard deviation. CRS = chronic rhinosinusitis; Polyposis = polyps visualized on nasal endoscopy; Allergy = tested positive on at least one allergen sample.
Immunohistochemistry
Mouse olfactory tissue was processed for histology as previously described. Briefly, anesthetized animals were transcardially perfused with PBS and 4% PFA. After removing the nasal dorsal bones, the nasal cavities with attached cribriform plate were dissected out and post-fixed in 4% PFA on ice for 20 minutes with gentle rotation. Frozen sections (12 micron) from mouse and human tissue were obtained. The human tissue fragments did not have a specific orientation. Mouse olfactory epithelial sections were obtained in a coronal plane from posterior to anterior. Immunostaining was performed on sections representing consistent anatomic levels in all mice. The following primary antibodies were used: Goat anti-OMP (1:1000, Wako 544-10001), which targets olfactory sensory neurons; Rabbit anti-Caspase-3 (1:400, Cell signaling 9661), which targets apoptotic cells; Rabbit anti-p-c-Jun (1:800, Cell signaling 3270), which targets phosphorylated c-Jun. Alexa Fluor 488 or 546 conjugated secondary antibodies were used to visualize the primary antibodies. Nuclei were stained with blue-fluorescent DAPI.
Confocal imaging and quantification
Immunostaining Images were obtained using a Zeiss LSM 780 Confocal Microscope. For each antibody, positive cells in 3 sections per sample were counted. For each section, ~2 mm neuroepithelium was imaged and counted. Statistical analysis was performed based on 3–4 independent animals.
Quantitative RT-PCR
Total RNA was extracted from isolated neuroepithelium using a RNeasy Mini Kit (QIAGEN). 500ng RNA was used to synthesize the first-strand cDNA by a Omniscript Reverse Transcription (RT) Kit (QIAGEN). 10 ng cDNA was added to a 20 μl reaction of SYBR Green Master Mix (Applied Biosystems). Realtime-PCR was performed on StepOne Plus System (Applied Biosystems). In each group, cDNA samples from 3 mice were examined. Primer sequences for mouse RT-PCR: c-Jun Forward-5′ GCACATCACCACTACACCGA, Reverse-5′ GGGAAGCGTGTTCTGGCTAT; IL-1β Forward-5′ CGACAAAATACCTGTGGCCT, IL-1β Reverse-5′ TCTTCTTTGGGTATTGCTTGG.
Statistical analysis
Quantified results of three independent experiments were expressed as the mean ± SD. The statistical significance of data was determined by Two-tailed Student’s t test. A value of P < 0.05 was considered statistically significant.
Results
Chronic rhinosinusitis is accompanied by JNK activation in olfactory neuroepithelium
To explore the importance of the JNK pathway in olfaction loss in the setting of CRS, we first investigated whether c-Jun is expressed and phosphorylated. The JNK pathway involves a sequential kinase-signaling pathway. Mixed lineage kinases (MLKs) phosphorylate and thereby activate c-Jun. The phosphorylated c-Jun then acts on cytoplasmic and nuclear targets. These targets have been associated with apoptosis. For this reason, antibodies to the phosphorylated c-Jun has served as a marker for apoptosis. To utilize this technique in olfaction, human olfactory mucosa was harvested from patients with and without CRS. Immunostaining was performed for phosphorylated c-Jun (Fig 1). In patients with CRS, human olfactory mucosa demonstrated expression of phosphorylated c-Jun. When compared with controls, olfactory neurons, identified by OMP staining, from CRS patients expressed significantly greater phosphorylated c-Jun (p = 0.001).
Figure 1. JNK activation in olfactory sensory neurons (OSNs) of CRS patients.
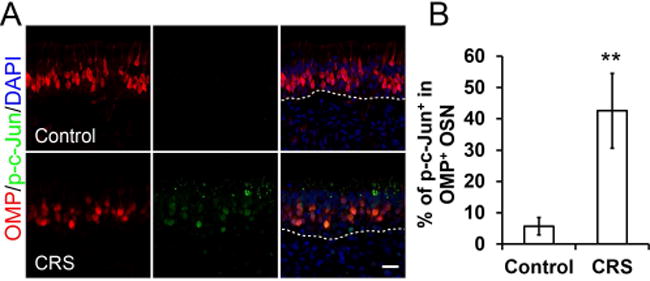
(A) Co-staining of OMP (olfactory marker protein) and p-c-Jun in human olfactory mucosa. p-c-Jun could be readily detected in OMP+ OSNs of CRS patient. Scale bar, 20 μm.
(B) Ratio of p-c-Jun+/ OMP+ population in OMP+ OSNs. Nine control samples and ten CRS patient samples were examined. *P = 0.001.
TNFR1 loss inhibits JNK activation in olfactory sensory neurons
We next sought to better understand the pathway of JNK activation in OSNs, using the IOI mouse model. Doxycycline (Dox) was given in the mouse food to induce tet-regulated activation of TNF-α, resulting in a uniform chronic olfactory inflammatory state. We collected the olfactory neuroepithelium after 3-weeks of Dox treatment and assayed for JNK activation using immunofluorescence. As evidenced by phosphorylated c-Jun staining, chronic inflammation caused evident JNK activation in the neuroepithelium and lamina propria (Fig 2A). Co-staining with OMP suggested that JNK was activated in ~30% of OMP+ olfactory neurons, which was similar to our observations in CRS patients (Fig 2B). Differences in the cellular pattern of JNK activation between the IOI mouse and CRS patients may relate to the high levels of sustentacular cell TNF-α expression in the mouse model, as well as different time courses of inflammation and medical treatment in humans. Interestingly, genetic ablation of the TNF-α receptor 1 (TNFR1) was accompanied by diminished phosphorylated c-Jun (p = 0.0012). Moreover, the elevated expression of JNK target gene c-Jun further supports that JNK is activated in chronically inflamed IOI mice. Loss of TNFR1 significantly inhibited this JNK activation (Fig 2C). The absence of TNF-α receptors 1 and 2 were previously shown not to affect normal immune homeostasis or stem cell proliferation in uninjured olfactory mucosa27 The present results demonstrate the critical role of TNFR1 in JNK activation in TNF-α-induced olfactory inflammation model.
Figure 2. Deficiency of TNFR1 receptors prevents JNK activation in olfactory sensory neurons.
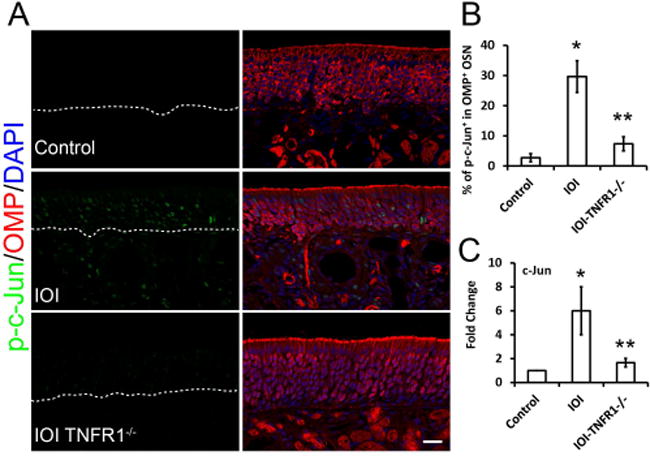
(A) Co-staining of phosphorylated c-Jun (p-c-Jun) with OMP in olfactory epithelium. Positive nuclei staining of p-c-Jun in an IOI (inducible olfactory inflammation) mouse model indicated JNK signaling activation. In IOI background, the knockout of the TNF-α receptor 1 (TNFR1) blocked JNK activation, suggesting the key role of TNFR1 in JNK activation in the TNF-α pathway.(B) Ratio of p-c-Jun+/ OMP+ population in OMP+ OSNs. *P = 0.0012, **P = 0.005.(C) Q-PCR analysis of JNK target gene c-Jun mRNA expression. IOI mice were treated with Dox for 3 weeks (n = 3), olfactory mucosa was collected for real-time PCR analysis. *P = 0.0002. **P = 0.011. Scale bar, 20 μm.
Olfactory sensory neuron apoptosis is induced by chronic inflammation
Although olfactory inflammation is initiated by TNF-α in the IOI mouse model, there is a resulting cascade of other cytokines that are upregulated, most prominently IL-1β (Fig 3A). This inflammatory mediator, which is present at low levels in control olfactory tissue (p = 0.0001), also induces JNK activity and may be associated with apoptosis. Caspase-3 immunostaining was used to identify apoptotic olfactory neurons and progenitors in the setting of chronic inflammation. We observed caspase-3+ cells distributed above the basal cell layer in IOI mice, consistent with apoptotic neurons. As compared to control mice, the number of caspase-3+ cells was significant elevated (Fig 3B and 3C). Caspase-3 staining in control or CRS human tissue demonstrated few scattered positive cells (data not shown).
Figure 3. Induced chronic inflammation cause OSN apoptosis.
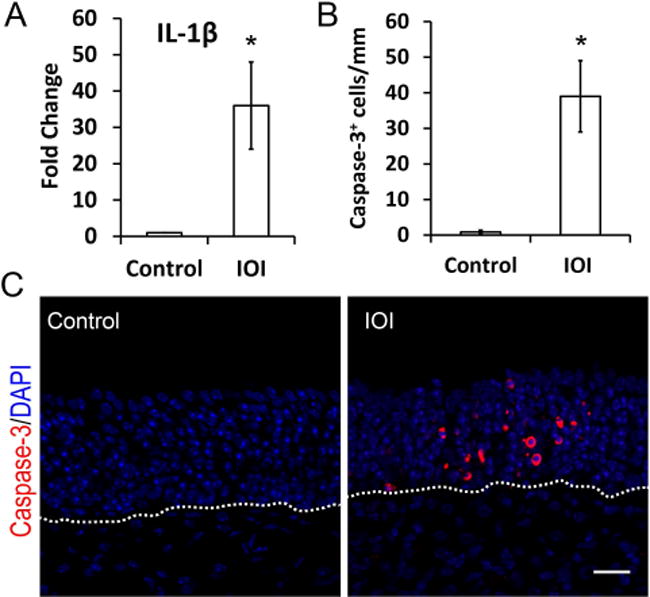
(A) Q-PCR analysis of IL-1β mRNA expression. Compared to control mice, the inflammatory cytokine IL-1β was upregulated 32-fold in olfactory mucosa derived from Dox treated IOI mice. IOI mice were treated with Dox for 3 weeks (n = 3). *P = 0.0001.
(B) Quantification of Caspase-3+ apoptosis in olfactory sensory neurons (OSNs).
(C) Representative confocal images of caspase-3 immunostaining. IOI mice were treated with Dox for 3 weeks (n = 3). Scale bar, 20 μm.
Discussion
In this study, we have demonstrated that olfactory activation of c-Jun is significantly increased in CRS patients relative to non-CRS control subjects and is associated with a loss of sensory neurons. In an inducible olfactory inflammation mouse model, prolonged induction of inflammation results similarly in elevation of c-Jun expression and neuronal apoptosis. Thus, activation of neuronal JNK is a feature of chronic olfactory inflammation in humans and mouse models, suggesting an apoptotic molecular pathway underlying CRS-associated olfactory loss.
The mechanisms through which CRS inflammation impacts olfactory function are incompletely understood. There is indirect evidence that suggests prolonged inflammation in CRS results in destruction and replacement of some OE with squamous or respiratory epithelium28–30. However, recent research using a mouse model of acute olfactory injury showed that a regulated and transient inflammatory response is necessary to begin the process of neural repair and regeneration27. The TNFR1/NF-κB pathway appears to serve a critical role in the cross talk between the immune system and olfactory progenitor cells. In addition, the NF-κB pathway is important for neuronal differentiation and survival. The TNF-α/TNFR1 pathway serves an important role in stimulating JNK activation in OSNs. Prior studies in the IOI mouse model demonstrate that ongoing chronic inflammation causes neuronal loss and impairs regeneration8. Although multiple cytokines are induced in the setting of chronic TNF-α induced inflammation, the present study reveals that loss of TNFR1 inhibits JNK activation, suggesting that TNF-α mediated inflammation can induce apoptosis in OSNs through the JNK pathway. This neuronal loss, together with inhibition of progenitor cell proliferation, may lead to persistent neuronal depletion and loss of olfaction in CRS. This would support the observed reversibility of dramatic histologic changes in the OE when inflammation is inhibited8.
The JNK pathway also plays a role in other neuronal tissues. JNK is involved in sensorineural hearing loss related to cochlear trauma and inflammation31–33. In the cochlea, stress-injured hair cells and spiral ganglia neurons undergo JNK mediated apoptosis34–36. Unlike the olfactory neuroepithelium, the cochlea does not have a repository of neural progenitor cells to restore hearing following an injury. JNK signaling also has a central role in Alzheimer’s disease. Alzheimer’s is an age-related neurodegenerative disease characterized by progressive dementia. Studies of brain tissue and cerebrospinal fluid of Alzheimer’s disease patients has demonstrated robust JNK activity37. Expression level of JNK has even been correlated with the degree of Alzheimer’s disease.
We hypothesize that cytokines act on mature olfactory sensory neurons and their progenitor cells through well-established intracellular pathways. A variety of cytokines such as TNF-α or IL-1β may act on these intracellular pathways7,38 Although it remains unclear which of these cytokines is most important, our previous work with the IOI mouse, as well as other published studies, suggests that JNK is a central participant in the apoptosis of mature OSNs via downstream death domain proteins and caspases12,39. Downstream signal transduction elements such as JNK are common to multiple inflammatory cytokines, and thus are also likely involved in human OSN dysfunction, even if the effect is not necessarily initiated by TNF-α as in the IOI model. In addition, the effects of TNF-α on proliferation and differentiation of neural progenitor cells in the CNS suggests a role of associated signaling pathways in altered regeneration of the OE in the setting of inflammation.40,41
While the IOI mouse is a useful model for studying chronic olfactory inflammation, it has limitations that must be recognized when interpreting findings and extrapolating to human CRS. The over-expression of TNF-α initiates a broad inflammatory infiltrate and cytokine cascade but still results in a higher level of TNF-α than would be present endogenously in CRS. This may impact the degree of JNK activation in the IOI mouse. In most cases, CRS patients have had their disease for years and have undergone medical and surgical interventions likely modulating their olfactory inflammation. The diminished caspase-3 staining in human tissue compared to the IOI mouse may reflect less apoptosis, or alternatively may be a technical consequence of the different processing of the tissue from humans and mice. Of note, increased human olfactory caspase-3 in the setting of sinus disease has been previously reported by Kern et al42. Despite the limitations of the model, the activation of JNK in both human CRS and IOI mouse olfactory epithelium strongly suggests that this is a feature of the neuronal response to chronic inflammatory cytokine exposure.
Olfactory dysfunction is a significant problem in patients with CRS with a prevalence ranging from 30–80% of the population43. Poor olfaction can be debilitating with significant effects on quality of life44. Although diminished nasal airflow in CRS can contribute to olfactory loss in this disease process, chronic inflammation clearly can also impair the functioning of olfactory epithelium. Our study suggests that persistent OSN apoptosis from JNK activation may contribute to CRS-related loss of olfaction. In the future, JNK may prove to be a therapeutic target allowing olfactory function to be maintained in the setting of acute or chronic sinonasal inflammation.
Acknowledgments
Research supported by NIH DC009026 (A.P.L.).
Footnotes
Presented at the Annual Meeting of the American Rhinologic Society, September 9, 2017, Chicago, IL
Conflict-of-interest disclosure: The authors declare no competing financial interests.
References
- 1.Zhao K, Jiang J, Pribitkin EA, et al. Conductive olfactory losses in chronic rhinosinusitis? A computational fluid dynamics study of 29 patients. Int Forum Allergy Rhinol. 2014;4(4):298–308. doi: 10.1002/alr.21272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kern RC. Chronic sinusitis and anosmia: pathologic changes in the olfactory mucosa. Laryngoscope. 2000;110(7):1071–1077. doi: 10.1097/00005537-200007000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Jafek BW, Moran DT, Eller PM, Rowley JC, 3rd, Jafek TB. Steroid-dependent anosmia. Arch Otolaryngol Head Neck Surg. 1987;113(5):547–549. doi: 10.1001/archotol.1987.01860050093023. [DOI] [PubMed] [Google Scholar]
- 4.Stevens MH. Steroid-dependent anosmia. Laryngoscope. 2001;111(2):200–203. doi: 10.1097/00005537-200102000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Mellert TK, Getchell ML, Sparks L, Getchell TV. Characterization of the immune barrier in human olfactory mucosa. Otolaryngol Head Neck Surg. 1992;106(2):181–188. [PubMed] [Google Scholar]
- 6.Haruna S, Otori N, Moriyama H, Nakanishi M. Olfactory dysfunction in sinusitis with infiltration of numerous activated eosinophils. Auris Nasus Larynx. 2006;33(1):23–30. doi: 10.1016/j.anl.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 7.Schlosser RJ, Mulligan JK, Hyer JM, Karnezis TT, Gudis DA, Soler ZM. Mucous Cytokine Levels in Chronic Rhinosinusitis-Associated Olfactory Loss. JAMA Otolaryngol Head Neck Surg. 2016;142(8):731–737. doi: 10.1001/jamaoto.2016.0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lane AP, Turner J, May L, Reed R. A genetic model of chronic rhinosinusitis-associated olfactory inflammation reveals reversible functional impairment and dramatic neuroepithelial reorganization. J Neurosci. 2010;30(6):2324–2329. doi: 10.1523/JNEUROSCI.4507-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henkin RI, Schmidt L, Velicu I. Interleukin 6 in hyposmia. JAMA Otolaryngol Head Neck Surg. 2013;139(7):728–734. doi: 10.1001/jamaoto.2013.3392. [DOI] [PubMed] [Google Scholar]
- 10.Oyer SL, Mulligan JK, Psaltis AJ, Henriquez OA, Schlosser RJ. Cytokine correlation between sinus tissue and nasal secretions among chronic rhinosinusitis and controls. Laryngoscope. 2013;123(12):E72–78. doi: 10.1002/lary.24305. [DOI] [PubMed] [Google Scholar]
- 11.Hehlgans T, Pfeffer K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology. 2005;115(1):1–20. doi: 10.1111/j.1365-2567.2005.02143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci. 2002;22(7):RC216. doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varfolomeev EE, Ashkenazi A. Tumor necrosis factor: an apoptosis JuNKie? Cell. 2004;116(4):491–497. doi: 10.1016/s0092-8674(04)00166-7. [DOI] [PubMed] [Google Scholar]
- 14.Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin Immunol. 2014;26(3):237–245. doi: 10.1016/j.smim.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang LW, Chang YC, Chen SJ, et al. TNFR1-JNK signaling is the shared pathway of neuroinflammation and neurovascular damage after LPS-sensitized hypoxic-ischemic injury in the immature brain. J Neuroinflammation. 2014;11(1):4. doi: 10.1186/s12974-014-0215-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gangadhar NM, Firestein SJ, Stockwell BR. A novel role for jun N-terminal kinase signaling in olfactory sensory neuronal death. Mol Cell Neurosci. 2008;38(4):518–525. doi: 10.1016/j.mcn.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennett J, Moretti M, Thotakura AK, Tornatore L, Franzoso G. The Regulation of the JNK Cascade and Programmed Cell Death by NF-κB: Mechanisms and Functions. In: Resende R, Ulrich H, editors. Trends in Stem Cell Proliferation and Cancer Research. 2013. pp. 297–336. [Google Scholar]
- 18.Papa S, Zazzeroni F, Pham CG, Bubici C, Franzoso G. Linking JNK signaling to NF-kappaB: a key to survival. J Cell Sci. 2004;117(Pt 22):5197–5208. doi: 10.1242/jcs.01483. [DOI] [PubMed] [Google Scholar]
- 19.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12(8):695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 20.Tournier C, Hess P, Yang DD, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288(5467):870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 21.Morgan MJ, Kim YS, Liu ZG. TNFalpha and reactive oxygen species in necrotic cell death. Cell Res. 2008;18(3):343–349. doi: 10.1038/cr.2008.31. [DOI] [PubMed] [Google Scholar]
- 22.Ge Y, Tsukatani T, Nishimura T, Furukawa M, Miwa T. Cell death of olfactory receptor neurons in a rat with nasosinusitis infected artificially with Staphylococcus. Chem Senses. 2002;27(6):521–527. doi: 10.1093/chemse/27.6.521. [DOI] [PubMed] [Google Scholar]
- 23.Epstein VA, Bryce PJ, Conley DB, Kern RC, Robinson AM. Intranasal Aspergillus fumigatus exposure induces eosinophilic inflammation and olfactory sensory neuron cell death in mice. Otolaryngol Head Neck Surg. 2008;138(3):334–339. doi: 10.1016/j.otohns.2007.11.029. [DOI] [PubMed] [Google Scholar]
- 24.Ozaki S, Toida K, Suzuki M, et al. Impaired olfactory function in mice with allergic rhinitis. Auris Nasus Larynx. 2010;37(5):575–583. doi: 10.1016/j.anl.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 25.Jung YG, Lane AP. Inhibition of Inflammation-Associated Olfactory Loss by Etanercept in an Inducible Olfactory Inflammation Mouse Model. Otolaryngol Head Neck Surg. 2016;154(6):1149–1154. doi: 10.1177/0194599816632177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner JH, May L, Reed RR, Lane AP. Reversible loss of neuronal marker protein expression in a transgenic mouse model for sinusitis-associated olfactory dysfunction. Am J Rhinol Allergy. 2010;24(3):192–196. doi: 10.2500/ajra.2010.24.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen M, Reed RR, Lane AP. Acute inflammation regulates neuroregeneration through the NF-kappaB pathway in olfactory epithelium. Proc Natl Acad Sci U S A. 2017 doi: 10.1073/pnas.1620664114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee SH, Lim HH, Lee HM, Park HJ, Choi JO. Olfactory mucosal findings in patients with persistent anosmia after endoscopic sinus surgery. Ann Otol Rhinol Laryngol. 2000;109(8 Pt 1):720–725. doi: 10.1177/000348940010900804. [DOI] [PubMed] [Google Scholar]
- 29.Yee KK, Pribitkin EA, Cowart BJ, et al. Neuropathology of the olfactory mucosa in chronic rhinosinusitis. American journal of rhinology & allergy. 2010;24(2):110–120. doi: 10.2500/ajra.2010.24.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aiba T, Nakai Y. Influence of experimental rhino-sinusitis on olfactory epithelium. Acta Otolaryngol Suppl. 1991;486:184–192. doi: 10.3109/00016489109134995. [DOI] [PubMed] [Google Scholar]
- 31.Hu BH, Henderson D, Nicotera TM. Involvement of apoptosis in progression of cochlear lesion following exposure to intense noise. Hear Res. 2002;166(1-2):62–71. doi: 10.1016/s0378-5955(02)00286-1. [DOI] [PubMed] [Google Scholar]
- 32.Ma C, Billings P, Harris JP, Keithley EM. Characterization of an experimentally induced inner ear immune response. Laryngoscope. 2000;110(3 Pt 1):451–456. doi: 10.1097/00005537-200003000-00024. [DOI] [PubMed] [Google Scholar]
- 33.Barkdull GC, Hondarrague Y, Meyer T, Harris JP, Keithley EM. AM-111 reduces hearing loss in a guinea pig model of acute labyrinthitis. Laryngoscope. 2007;117(12):2174–2182. doi: 10.1097/MLG.0b013e3181461f92. [DOI] [PubMed] [Google Scholar]
- 34.Zine A, van de Water TR. The MAPK/JNK signalling pathway offers potential therapeutic targets for the prevention of acquired deafness. Curr Drug Targets CNS Neurol Disord. 2004;3(4):325–332. doi: 10.2174/1568007043337166. [DOI] [PubMed] [Google Scholar]
- 35.Abi-Hachem RN, Zine A, Van De Water TR. The injured cochlea as a target for inflammatory processes, initiation of cell death pathways and application of related otoprotectives strategies. Recent Pat CNS Drug Discov. 2010;5(2):147–163. doi: 10.2174/157488910791213121. [DOI] [PubMed] [Google Scholar]
- 36.Suckfuell M, Lisowska G, Domka W, et al. Efficacy and safety of AM-111 in the treatment of acute sensorineural hearing loss: a double-blind, randomized, placebo-controlled phase II study. Otol Neurotol. 2014;35(8):1317–1326. doi: 10.1097/MAO.0000000000000466. [DOI] [PubMed] [Google Scholar]
- 37.Gourmaud S, Paquet C, Dumurgier J, et al. Increased levels of cerebrospinal fluid JNK3 associated with amyloid pathology: links to cognitive decline. J Psychiatry Neurosci. 2015;40(3):151–161. doi: 10.1503/jpn.140062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sultan B, May LA, Lane AP. The role of TNF-alpha in inflammatory olfactory loss. Laryngoscope. 2011;121(11):2481–2486. doi: 10.1002/lary.22190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki Y, Farbman AI. Tumor necrosis factor-alpha-induced apoptosis in olfactory epithelium in vitro: possible roles of caspase 1 (ICE), caspase 2 (ICH-1), and caspase 3 (CPP32) Exp Neurol. 2000;165(1):35–45. doi: 10.1006/exnr.2000.7465. [DOI] [PubMed] [Google Scholar]
- 40.Iosif RE, Ekdahl CT, Ahlenius H, et al. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci. 2006;26(38):9703–9712. doi: 10.1523/JNEUROSCI.2723-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Widera D, Mikenberg I, Elvers M, Kaltschmidt C, Kaltschmidt B. Tumor necrosis factor alpha triggers proliferation of adult neural stem cells via IKK/NF-kappaB signaling. BMC Neurosci. 2006;7:64. doi: 10.1186/1471-2202-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kern RC, Conley DB, Haines GK, 3rd, Robinson AM. Pathology of the olfactory mucosa: implications for the treatment of olfactory dysfunction. Laryngoscope. 2004;114(2):279–285. doi: 10.1097/00005537-200402000-00018. [DOI] [PubMed] [Google Scholar]
- 43.Kohli P, Naik AN, Harruff EE, Nguyen SA, Schlosser RJ, Soler ZM. The prevalence of olfactory dysfunction in chronic rhinosinusitis. Laryngoscope. 2017;127(2):309–320. doi: 10.1002/lary.26316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soler ZM, Smith TL, Alt JA, Ramakrishnan VR, Mace JC, Schlosser RJ. Olfactory-specific quality of life outcomes after endoscopic sinus surgery. Int Forum Allergy Rhinol. 2016;6(4):407–413. doi: 10.1002/alr.21679. [DOI] [PMC free article] [PubMed] [Google Scholar]