

Abstract
Non-alcoholic fatty liver disease (NAFLD) is a condition in which fat accumulates in the liver of patients without a prior history of alcohol abuse. The most severe form, nonalcoholic steatohepatitis (NASH), often leads to hepatic fibrosis and cirrhosis with ensuing complications. To date, there is no pharmacologic treatment for NASH. The biological effects of CCN3, specifically its role in the regulation of inflammation, reactive oxygen species production and angiogenesis, have been recently established. Additional data suggests that CCN3 is associated with the development of tumors in the liver yet may be protective of liver fibrogenesis. Currently, the role of CCN3 in NAFLD/NASH remains unexplored. Therefore, the objective of our investigation was to decipher the role of myeloid-deficient CCN3 in the pathogenesis of NASH and the underlying mechanisms of CCN3 in modulation of hepatic function. Wild type and myeloid CCN3-deficient mice were fed a methionine- and choline-deficient diet to induced NASH. Increased lipid, cholesterol, and cholesterol ester accumulation was observed in myeloid CCN3-deficient mice when compared to the control group. This disease state was associated with alterations of key genes involved in lipid synthesis, β-oxidation and lipid uptake. Additionally, the levels of important molecules critical for inflammation, ROS generation, ER stress and liver injury were significantly elevated; as was the observed severity of hepatic apoptosis and necroptosis. Therefore, CCN3 is critical for protection from hepatic apoptosis and necroptosis in our induced NASH model and our findings suggest that CCN3 can be exploited as a therapeutic target for the treatment of NASH.
Keywords: CCN3, ER stress, NAFLD, NASH, Programmed cell death, SREBP-1
Introduction
Nonalcoholic fatty liver disease (NAFLD) is regarded as the most common chronic liver disease in the Western world and incidence have increased worldwide over recent decades. NAFLD is characterized by fat accumulation within the liver and can be classified into two types: 1) simple steatosis and 2) nonalcoholic steatohepatitis (NASH). Both types are associated with obesity, insulin resistance, metabolic syndrome and type II diabetes. While simple steatosis is considered a benign condition, NASH is associated with inflammation and hepatocyte damage and can often lead to fibrosis, cirrhosis, and liver cancer if not diagnosed and treated at early stages. It is currently unknown why some people develop one form over the other. To date, there are no approved medicines to treat either condition, and diet modification and weight control are the current recommended treatments.
The CCN family of matricellular proteins consists of six members (CCN1-CCN6) (Yeger and Perbal 2007). The family name originates from the common names of the first three proteins characterized (cysteine-rich 61 (CYR61) – CCN1 / connective tissue growth factor (CTGF) – CCN2 / nephroblastoma overexpressed (NOV) – CCN3). CCN3 is associated with many cardiovascular processes; ranging from neovascular development in utero and angiogenesis (Klenotic et al. 2016), to mitigation of aortic aneurysm formation and atherosclerotic plaque development (Liu et al. 2014; Zhang et al. 2016; Shi et al. 2017). Interestingly, it was found that CCN3 originated within the myeloid compartment was critical for mitigation of vascular disease progression. CCN3 also plays a role in osteoblast differentiation (Ouellet and Siegel 2012), obesity, insulin resistance and diabetes (Martinerie et al. 2016; Escote et al. 2017), and tumor development (Barreto et al. 2016; Yeger and Perbal 2016). Less is known about the role of CCN3 within the liver. Borkham-Kamphorst et al., has shown that CCN3 expression increased significantly at the mRNA and protein levels in models of experimental hepatic fibrosis (Borkham-Kamphorst et al. 2012b), yet overexpression of CCN3 fails to mitigate liver fibrogenesis, in part, due to enhanced reaction oxygen species production and cytokine signaling, which lead to the induction of hepatocyte injury and apoptosis (Borkham-Kamphorst et al. 2016). Since the absence of myeloid-derived CCN3 has previously been shown to exacerbate cardiovascular disease, it was of great interest to determine whether CCN3’s protective effects also translate to the liver in the context of NASH progression.
Materials and methods
LysMCre/Cre mice were commercially available from Jackson Laboratory (Farmington, CT). CCN3flox/flox mice were generated by Ozgene (Cambridge, MA) by insertion of LoxP sites that flank exon 2 of the CCN3 gene. The result of the crossing yielded LysMCre/CreCCN3flox/flox mice which were used in the experiments described below, with LysMCre/Cre mice used as control animals. All animal genotypes were verified by PCR. Mice were fed either a methionine- and choline-deficient (MCD) diet (MP Biomedicals, Santa Ana, CA) or normal chow (CHOW) for 8 weeks. All animal protocols were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University, which is certified by the American Association of Accreditation for Laboratory Animal Care.
Plasma ALT and AST and liver triglycerides
Plasma alanine aminotransferase (ALT) and plasma aspartate aminotransferase (AST) assay kits were purchased from Sekisui Diagnostics (Lexington, MA) and the protocol provided by the manufacturer was followed.
Lipid analysis
Measurements of liver tissue triglyceride, cholesterol and cholesterol ester levels were performed by the Vanderbilt NIH Mouse Metabolic Phenotyping Core.
RNA isolation and qPCR
Total RNA from mouse liver tissue was isolated using Aurum™ Total RNA Fatty and Fibrous Tissue Kit (Bio-Rad, Hercules, CA). cDNA was synthesized from 1 μg mRNA template using iScript Reverse Transcriptase (Bio-Rad) and subjected to qPCR with SYBR green Real-Time PCR Master Mix (Applied Biosystems, Waltham, MA) on a QuantStudio 6 Flex real-time PCR System (Applied Biosystems). Gene expression was normalized to β-actin using the ΔΔCt method of calculation.
Protein isolation and western blot analysis
Protein was extracted from tissues using RIPA buffer (Sigma-Aldrich, St. Louis, MO) supplemented with proteinase and phosphatase inhibitors (Roche Diagnostics, Indianapolis, IN). Protein concentrations were determined using a bicinchoninic acid (BCA) protein assay kit (Pierce Biotechnology, Waltham, MA). Western blots of liver were performed following standard protocols. SREBP-1 antibody was from BD Biosciences (San Jose, CA). CHOP, XBP-1 and mouse β-actin antibodies were from Santa Cruz Biotechnology (Dallas, TX). Phospho-RIP3 antibody was from Abcam (Cambridge, MA).
Liver histological and immunohistochemical analysis
Livers were fixed in 10% phosphate-buffered formalin acetate at 4 °C overnight and embedded in paraffin wax. Paraffin-embedded serial sections (7 μm) were cut and mounted onto glass slides for hematoxylin and eosin staining and for assessment of F4/80 (Santa Cruz), MCP-1 (Santa Cruz) and CD45 (Santa Cruz). Paraffin-embedded sections were also used for Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick-end labeling (TUNEL) using the DNA Fragmentation Assay Kit from Clontech Laboratories, (Mountain View, CA). Staining for Oil Red O (Alfa Aesar, Haverhill, MA) and Dihydroethidium (DHE) (Invitrogen, Carlsbad, CA) was performed on sections cut from frozen liver embedded in OCT. Nuclei were labeled using Vectashield with DAPI (Vector Laboratories, Burlingame, CA) or hematoxylin.
Statistics
All values are reported as means ± standard error of the mean (SEM), n = 4 chow-fed and n = 7 for MCD-fed mice. To analyze the difference between 2 groups, the Student’s t test was used. A P value less than 0.05 was considered significant.
Results
CCN3 protects against NASH progression
We employed the Cre-lox recombination system to delete CCN3 from myeloid cells to test its importance in the progression of NASH. LysMcre/cre mice were crossed with CCN3flox/flox mice to generate LysMcre/creCCN3flox/flox progeny, which lack CCN3 expression within circulating myeloid cells. When fed a diet lacking both methionine and choline (MCD) to induce NASH, LysMcre/creCCN3flox/flox mice (grey bars) showed significant increases in triglycerides (TG), total cholesterol (T Chol) and cholesterol ester (CE) concentrations, all markers of fatty acid disease, when compared to the LysMcre/cre control group (black bars) (Fig. 1a). No difference between the two groups was observed when fed a normal CHOW diet (Fig. 1a). Enhanced lipid accumulation was observed in liver tissue harvested from LysMcre/creCCN3flox/flox mice, with increased lipid droplet formation (compare bottom two panels, MCD diet, Fig. 1b) as well as greater lipid and triglyceride accumulation, as seen in Oil Red O-stained sections in Fig. 1c (compare bottom two panels, MCD diet).
Fig. 1.

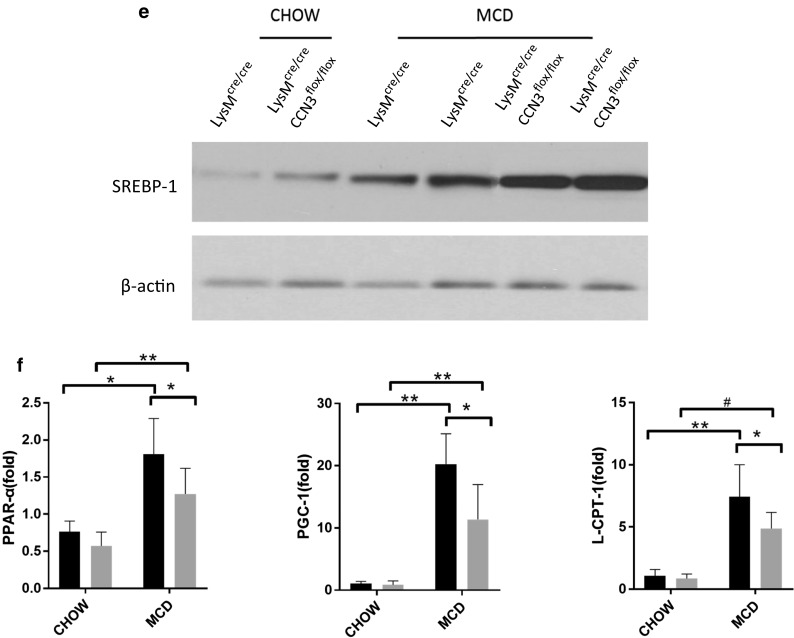
CCN3 protects against NASH progression. a Tissue from LysMcre/cre and LysMcre/creCCN3flox/flox mice were analyzed for triglyceride (TG), total cholesterol (T Chol) and cholesterol ester (CE) concentrations. Tissue were also stained with hematoxylin and eosin (b), and Oil Red O (c). d qPCR analysis of Sterol regulatory element binding protein-1 (SREBP-1), Fatty acid synthase (FASN), elongation of very long chain fatty acid protein-6 (ELOVL-6), acetyl-CoA carboxylase (ACC) and stearoyl-Coenzyme A desaturase-1 (SCD-1) levels. Samples tested were derived from liver tissue. e Western blot of liver tissue from LysMcre/cre and LysMcre/creCCN3flox/flox mice probed for SREBP-1. f qPCR analysis of Peroxisome proliferator-activated receptor alpha (PPAR-α), Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1), and carnitine palmitoyltransferase I (L-CPT-1) levels. g qPCR analysis of Peroxisome proliferator-activated receptor gamma (PPAR-γ) and cluster of differentiation 36 (CD36) levels. Scale bars = 200 μm. In all graphs, LysMcre/cre is depicted with black bars and LysMcre/creCCN3flox/flox is depicted with grey bars. *P < 0.05, **P < 0.01, #P < 0.001
To gain insight into how the loss of myeloid CCN3 results in increased lipid accumulation within the liver, qPCR was performed to assess the levels of key genes involved in lipid synthesis (Fig. 1d). SREBP-1, FASN, ELOVL-6, ACC, and SCD-1 all were significantly increased at the messenger level in LysMcre/creCCN3flox/flox mice on the MCD diet (Fig. 1d, grey bars) when compared to the LysMcre/cre control group (Fig. 1d, black bars). SREBP-1 is also shown to be significantly increased at the protein level (Fig. 1e). Concurrently, LysMcre/creCCN3flox/flox mice on the MCD diet, when compared to control, had decreased levels of PPAR-α, PGC-1 and L-CPT-1, genes important for lipid oxidation (Fig. 1f), while both PPAR-γ and CD36, genes critical for oxidized lipoprotein uptake into cells, were significantly increased (Fig. 1g). Taken together, these data show that CCN3 affects several pathways involved in lipid regulation, and the loss of myeloid CCN3 results in increased lipid accumulation within the liver.
Loss of myeloid-derived CCN3 leads to an increased inflammatory state
To further assess the increase in NASH severity due to myeloid CCN3 ablation, LysMcre/cre and LysMcre/creCCN3flox/flox mice were tested for alanine transaminase (ALT) and aspartate transaminase (AST) levels in plasma. These two transaminases are well known biomarkers for overall liver health, and increased concentrations of one or both are indicative of liver disease. Plasma samples from mice on normal chow showed very low ALT and AST levels (Fig. 2a). On the MCD diet, both control LysMcre/cre mice, and myeloid-deficient CCN3 LysMcre/creCCN3flox/flox mice showed elevated ALT and AST amounts as expected. Without myeloid-derived CCN3 present, however, the ALT and AST levels are markedly, and significantly increased, approximately double that of the control group (compare LysMcre/cre, MCD, with that of LysMcre/creCCN3flox/flox, MCD, Fig. 2a). Concomitantly, increased inflammatory cytokines were elevated in the MCD diet groups (Fig. 2b). While both IL-6 and IL-10 were unchanged between LysMcre/cre, and LysMcre/creCCN3flox/flox groups (Fig. 2b, right two panels), TNFα and IL-1β were both elevated in LysMcre/creCCN3flox/flox tissue Fig. 2b, left two panels), approximately 2-fold over LysMcre/cre alone. Further evidence of increased NASH in LysMcre/creCCN3flox/flox mice is shown in liver tissue stained for macrophages, F4/80 (Fig. 2c), hematopoietic marker CD45 (Fig. 2d), and cytokine MCP-1 (Fig. 2e). Collectively, increases in biomarkers, tissue cytokines and inflammatory cells suggest an increased inflammatory state in liver from mice lacking myeloid-derived CCN3.
Fig. 2.
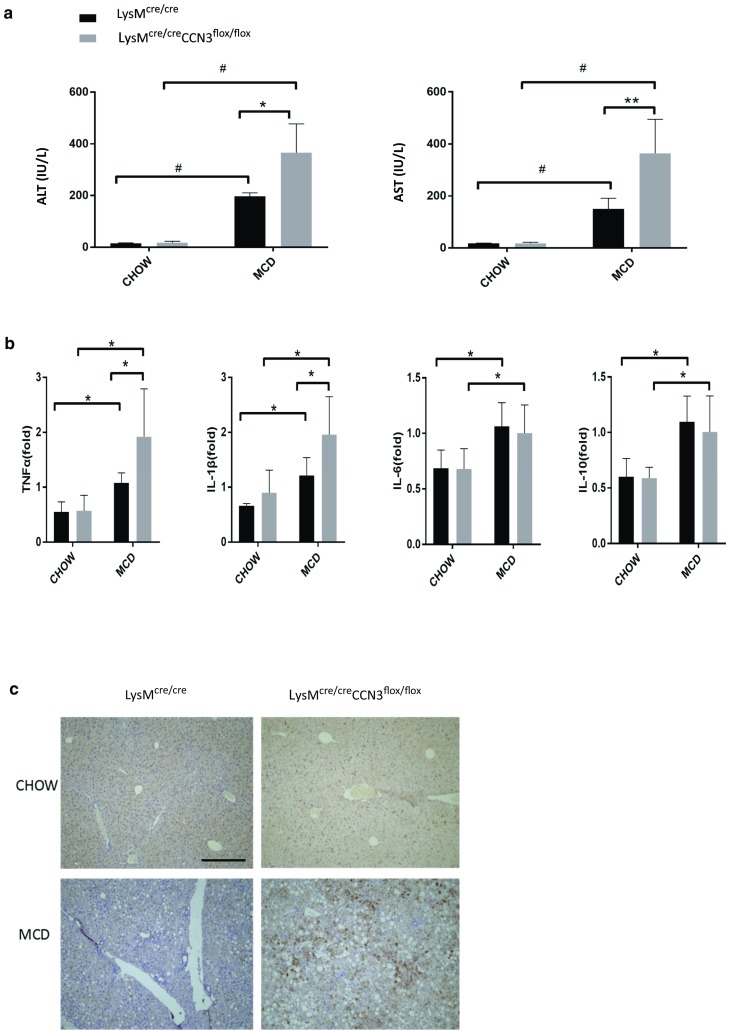

Loss of CCN3 is associated with an increased inflammatory state in the liver. a Plasma samples from LysMcre/cre and LysMcre/creCCN3flox/flox mice on normal chow and MCD were tested for alanine transaminase (ALT) and aspartate transaminase (AST). b qPCR analysis of tumor necrosis factor alpha (TNFα), interleukin-1 beta (IL-1β), interleukin-6 (IL-6) and interleukin-10 (IL-10) levels from these same groups. Samples derived from liver tissue. c,d,e Immunohistochemistry of paraffin-embedded liver tissue stained for F4/80 (c), CD45 (d) and MCP-1 (e). Scale bars = 200 μm. In all graphs, LysMcre/cre is depicted with black bars and LysMcre/creCCN3flox/flox is depicted with grey bars. *P < 0.05, **P < 0.01, #P < 0.001
Loss of myeloid-derived CCN3 contributes to increased stress signals within the liver
Both endoplasmic reticulum (ER) and oxidative stress contribute to overall tissue health and homeostasis. During onset of NASH, an influx of cytokines and inflammatory cells can severely increase stress signals within the liver. To assess the effect of the myeloid deficiency of CCN3 on ER stress, protein levels of two transcription factors important in the ER stress response, C/EBP homologous protein (CHOP) and X-box binding protein 1 (XBP-1) were analyzed. On the MCD diet, LysMcre/creCCN3flox/flox mice had from moderately (XBP-1) to significantly (CHOP) higher protein levels in liver tissue when compared to the LysMcre/cre mice control group (Fig. 3a). This was recapitulated in qPCR results (Fig. 3b) along with an increase in nuclear factor (erythroid-derived 2)-like 2 (Nrf2), a regulator of proteins that protect the tissue from oxidative damage (Fig. 3b, right panel). Additional evidence of increased oxidative stress in liver tissue from LysMcre/creCCN3flox/flox mice is shown in Fig. 3c. When treated with dihydroethidium (DHE), LysMcre/creCCN3flox/flox tissue shows an intense staining when compared to controls (Fig. 3c, lower right panel), suggesting the presence of increased reactive oxygen species (ROS). These data show that the absence of myeloid-derived CCN3 contributes to increased severity of NASH, in part, by increased ER and oxidative stress within the liver.
Fig. 3.

ER and oxidative stress levels are CCN3 dependent. a Western blot of C/EBP homologous protein (CHOP) and X-box binding protein 1 (XBP-1) from liver tissue of LysMcre/cre and LysMcre/creCCN3flox/flox mice, either on chow or MCD. b qPCR analysis of CHOP, XBP-1 and nuclear factor (erythroid-derived 2)-like 2 (Nrf2) levels from the same conditions as in (a). c Frozen sections of liver tissue stained for dihydroethidium (DHE), a marker of reactive oxygen species (ROS) generation. Scale bar = 100 μm. In all graphs, LysMcre/cre is depicted with black bars and LysMcre/creCCN3flox/flox is depicted with grey bars. *P < 0.05, **P < 0.01, #P < 0.001
Myeloid CCN3-deficiency causes increased programmed cell death in NASH-induced liver disease
A downstream effect in tissue with aberrant inflammatory cell deposition and oxidative stress signals is an increase in programmed cell death. A TUNEL assay in Fig. 4a shows an increase in apoptosis within LysMcre/creCCN3flox/flox tissue from mice that were on a MCD diet when compared to the LysMcre/cre mice control group. Necroptosis is also induced in LysMcre/creCCN3flox/flox mice as evidenced by increased receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and receptor-interacting serine/threonine-protein kinase 3 (RIPK3) expression (Fig. 4b), with increases in p-RIPK3 verified at the protein level (Fig. 4c). Therefore, myeloid CCN3 deficiency leads to increased NASH severity, in part, due to increased apoptosis and necroptosis within the liver.
Fig. 4.
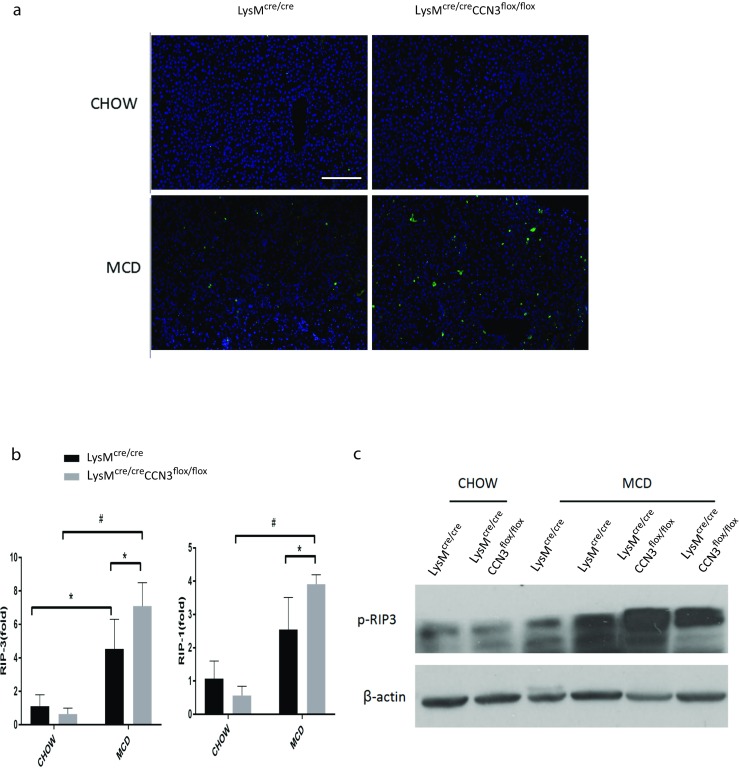
Myeloid CCN3 deficiency contributes to increased programmed cell death within the liver. a TUNEL assay on liver tissue from LysMcre/cre and LysMcre/creCCN3flox/flox mice fed either normal chow or MCD diet. b qPCR analysis of receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and receptor-interacting serine/threonine-protein kinase 3 (RIPK3) levels from liver tissue of the same groups as described in A. c Western blot of p-RIPK3 from liver tissue as described in A. Scale bar = 200 μm. In all graphs, LysMcre/cre is depicted with black bars and LysMcre/creCCN3flox/flox is depicted with grey bars. *P < 0.05, **P < 0.01, #P < 0.001
Discussion
Tissue-specific and inducible Cre systems have been developed to drive or delete a gene of interest in a time- and/or tissue-specific manner. Previously, we have taken advantage of the Cre-Lox recombination system to study the effects of the loss of CCN3 within myeloid cells upon aortic aneurysm development (Zhang et al. 2016) and atherosclerosis (Shi et al. 2017). Bone marrow (BM) transplantation of ApoE −/− ; CCN3 −/− BM into lethally irradiated ApoE −/− recipient mice significantly exacerbated atherosclerotic plaque development while the reciprocal experiment greatly reduced the severity of the induced phenotype. This highlighted the importance of non-tissue-derived CCN3 (instead of resident CCN3 produced in endothelial cells and smooth muscle cells for example) in the development of vascular diseases. Since myeloid-derived CCN3 can be ubiquitously transported to other tissue through the circulatory system, and CCN3 has been shown to effect liver homeostasis, it was of great interest to determine whether its protective effects also translate to the liver, where fat deposition and ensuing disease progression and complications in many ways mimic those seen in the vasculature.
Mice, deficient in CCN3 within myeloid cells, were fed a methionine- and choline-deficient diet to induce NASH. Indeed, in mice lacking myeloid-derived CCN3, there was increased fat deposition within the liver, concomitant with increased immune cell infiltration, reactive oxygen species generation and programmed cell death, all physiological events associated with NASH (Fig. 5). This is consistent with the results seen in aortic tissue upon aneurysm development (Zhang et al. 2016) and in our atherosclerotic mouse model (Shi et al. 2017).
Fig. 5.

Myeloid-derived CCN3 protects the liver from nonalcoholic steatohepatitis. On mice fed a methionine- and choline-deficient diet to induce NASH, loss of myeloid-derived CCN3 enhances the accumulation of hepatic lipid, in part, by regulation of key factors involved in lipid synthesis, oxidation and uptake. Additionally, there is increased inflammatory cell infiltration into the tissue as well as increased ER stress and ROS generation within these cells, leading to subsequent hepatocytic death
At the molecular level, loss of myeloid-derived CCN3 resulted in increases in genes involved in lipid synthesis and oxidized lipoprotein uptake. In fact, CD36, a well-known transmembrane protein involved in oxidized LDL uptake into macrophages, was upregulated in both of our models of atherosclerosis and NASH. Loss of myeloid-derived CCN3 also increased expression of transcription factors involved in the ER stress response (Fig. 5). Recent work has highlighted the effects of CCN proteins on induction of the unfolded protein response (UPR) due to ER stress (Borkham-Kamphorst et al. 2016). Borkham-Kamphorst et al. suggest that elevated levels of CCNs aid in tissue repair after hepatic injury, but there is a plateau where additional CCN protein beyond that results in increased apoptosis. CCN3 is of special interest because it has no endogenous expression in hepatocytes (Borkham-Kamphorst et al. 2012a, b; 2016) and its targeted expression has shown therapeutic properties via inhibition of CCN2 gene expression (Borkham-Kamphorst et al. 2012b, van Roeyen et al. 2012, Abd El Kader et al. 2013). Our results suggest that myeloid-derived CCN3 may play a protective role in NASH, in part, by limiting ER stress and ROS production, but also by affecting the UPR and CCN2 gene expression. Further experiments to test this will be of great interest.
Acknowledgements
This work was supported by NIH grants AA021390 and HL117759 (ZL) and National Natural Science Foundation of China grants NSFC-81400290 (XH) and NSFC-81070342 (QZ).
Abbreviations
- ACC
acetyl-CoA carboxylase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CCN
Cyr61/CTGF/NOV
- CD36
cluster of differentiation 36
- CHOP
C/EBP homologous protein
- Cyr61
cysteine-rich 61
- CTGF
connective tissue growth factor
- DHE
dihydroethidium
- ELOVL-6
elongation of very long chain fatty acid protein-6
- ER
endoplasmic reticulum
- FASN
fatty acid synthase
- IL
interleukin
- L-CPT-1
carnitine palmitoyltransferase I (L-CPT-1)
- MCD
methionine- choline- deficient
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NOV
nephroblastoma overexpressed
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- PCR
polymerase chain reaction
- PPAR
peroxisome proliferator-activated receptor
- PCG-1
peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- qPCR
quantitative polymerase chain reaction
- RIPK
receptor-interacting serine/threonine-protein kinase
- ROS
reactive oxygen species
- SCD-1
stearoyl-Coenzyme A desaturase-1
- SREBP-1
sterol regulatory element binding protein-1
- TNFα
tumor necrosis factor alpha
- TUNEL
terminal deoxynucleotidyl transferase nick end labeling
- dUTP
deoxyuridine triphosphate
- XBP-1
X-box binding protein 1
Contributor Information
Qi Zhou, Phone: (86)27-83662658, Email: zhouqi1973@hotmail.com.
Zhiyong Lin, Phone: 404 712 0974, Email: zhiyong.lin@emory.edu.
References
- Abd El Kader T, Kubota S, Janune D, Nishida T, Hattori T, Aoyama E, Perbal B, Kuboki T, Takigawa M. Anti-fibrotic effect of CCN3 accompanied by altered gene expression profile of the CCN family. J Cell Commun Signal. 2013;7(1):11–18. doi: 10.1007/s12079-012-0180-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto SC, Ray A, Ag Edgar P. Biological characteristics of CCN proteins in tumor development. J BUON. 2016;21(6):1359–1367. [PubMed] [Google Scholar]
- Borkham-Kamphorst E, Huss S, Van de Leur E, Haas U, Weiskirchen R. Adenoviral CCN3/NOV gene transfer fails to mitigate liver fibrosis in an experimental bile duct ligation model because of hepatocyte apoptosis. Liver Int. 2012;32(9):1342–1353. doi: 10.1111/j.1478-3231.2012.02837.x. [DOI] [PubMed] [Google Scholar]
- Borkham-Kamphorst E, van Roeyen CR, Van de Leur E, Floege J, Weiskirchen R. CCN3/NOV small interfering RNA enhances fibrogenic gene expression in primary hepatic stellate cells and cirrhotic fat storing cell line CFSC. J Cell Commun Signal. 2012;6(1):11–25. doi: 10.1007/s12079-011-0141-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkham-Kamphorst E, Steffen BT, Van de Leur E, Tihaa L, Haas U, Woitok MM, Meurer SK, Weiskirchen R. Adenoviral CCN gene transfers induce in vitro and in vivo endoplasmic reticulum stress and unfolded protein response. Biochim Biophys Acta. 2016;1863(11):2604–2612. doi: 10.1016/j.bbamcr.2016.07.006. [DOI] [PubMed] [Google Scholar]
- Escote X, Gomez-Zorita S, Lopez-Yoldi M, Milton-Laskibar I, Fernandez-Quintela A, Martinez JA, Moreno-Aliaga MJ, Portillo MP. Role of Omentin, Vaspin, Cardiotrophin-1, TWEAK and NOV/CCN3 in obesity and diabetes development. Int J Mol Sci. 2017;18(8):1770. doi: 10.3390/ijms18081770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenotic PA, Zhang C, Lin Z. Emerging roles of CCN proteins in vascular development and pathology. J Cell Commun Signal. 2016;10(3):251–257. doi: 10.1007/s12079-016-0332-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ren Y, Kang L, Zhang L. Overexpression of CCN3 inhibits inflammation and progression of atherosclerosis in apolipoprotein E-deficient mice. PLoS One. 2014;9(4):e94912. doi: 10.1371/journal.pone.0094912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinerie C, Garcia M, Do TT, Antoine B, Moldes M, Dorothee G, Kazazian C, Auclair M, Buyse M, Ledent T, Marchal PO, Fesatidou M, Beisseiche A, Koseki H, Hiraoka S, Chadjichristos CE, Blondeau B, Denis RG, Luquet S, Feve B. NOV/CCN3: a new Adipocytokine involved in obesity-associated insulin resistance. Diabetes. 2016;65(9):2502–2515. doi: 10.2337/db15-0617. [DOI] [PubMed] [Google Scholar]
- Ouellet V, Siegel PM. CCN3 modulates bone turnover and is a novel regulator of skeletal metastasis. J Cell Commun Signal. 2012;6(2):73–85. doi: 10.1007/s12079-012-0161-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Zhang C, Pasupuleti V, Hu X, Prosdocimo DA, Wu W, Qing Y, Wu S, Mohammad H, Gerson SL, Perbal B, Klenotic PA, Dong N, Lin Z. CCN3 regulates macrophage foam cell formation and atherosclerosis. Am J Pathol. 2017;187(6):1230–1237. doi: 10.1016/j.ajpath.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Roeyen CR, Boor P, Borkham-Kamphorst E, Rong S, Kunter U, Martin IV, Kaitovic A, Fleckenstein S, Perbal B, Trautwein C, Weiskirchen R, Ostendorf T, Floege J. A novel, dual role of CCN3 in experimental glomerulonephritis: pro-angiogenic and antimesangioproliferative effects. Am J Pathol. 2012;180(5):1979–1990. doi: 10.1016/j.ajpath.2012.01.031. [DOI] [PubMed] [Google Scholar]
- Yeger H, Perbal B. The CCN family of genes: a perspective on CCN biology and therapeutic potential. J Cell Commun Signal. 2007;1(3–4):159–164. doi: 10.1007/s12079-008-0022-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeger H, Perbal B. CCN family of proteins: critical modulators of the tumor cell microenvironment. J Cell Commun Signal. 2016;10(3):229–240. doi: 10.1007/s12079-016-0346-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, van der Voort D, Shi H, Zhang R, Qing Y, Hiraoka S, Takemoto M, Yokote K, Moxon JV, Norman P, Rittie L, Kuivaniemi H, Atkins GB, Gerson SL, Shi GP, Golledge J, Dong N, Perbal B, Prosdocimo DA, Lin Z. Matricellular protein CCN3 mitigates abdominal aortic aneurysm. J Clin Invest. 2016;126(4):1282–1299. doi: 10.1172/JCI82337. [DOI] [PMC free article] [PubMed] [Google Scholar]