

Abstract
TANK-binding kinase 1 (TBK1) is central to multiple biological processes that promote tumorigenesis including cell division, autophagy, innate immune response and AKT-pro survival signaling. TBK1 is well studied and most known for its function in innate immunity. However, the serine threonine protein kinase received significant attention as a synthetic lethal partner and effector of the major oncogene, RAS. This review summarizes newly identified cancer promoting functions of TBK1 and evaluates the therapeutic potential of targeting TBK1 in cancer.
Keywords: Autophagy, Cancer therapeutic target, Pancreatic cancer, RAS, TBK1
Introduction
As a member of the inhibitor of nuclear factor-κB (IκB) kinase (IKK) family, TANK-binding kinase 1 (TBK1) plays a central role in driving inflammation and innate immunity. Upon receptor-mediated pathogen detection, TBK1 can mobilize the interferon response pathway or activate the NF-kB pathway in host cell defense (Helgason et al. 2013). However, there is now a growing body of evidence implicating aberrant TBK1 activity in numerous cancer types.
Initial studies linking TBK1 to cancer involved the RAS effector, RALB (Chien et al. 2006, Ou et al. 2011). In NSCLC cells, RALB activated TBK1, leading to restriction of apoptosis, while having little effect on survival of non-tumorigenic epithelial cells. Moreover, expression of an oncogenic K-Ras allele in TBK1-deficient murine embryonic fibroblasts (MEFs) induced cell death, suggesting that TBK1 is integral for cells to tolerate transforming levels of oncogenic RAS. The critical contribution of RALB and TBK1 to RAS-induced lung cancer growth was corroborated in an RNAi screen of synthetic lethal partners of oncogenic K-RAS, where RALB and TBK1 were identified as top hits (Barbie et al. 2009). While RAS signaling through RALB and TBK1 is well established, the mechanism by which TBK1 promotes tumorigenesis remains unclear. Dozens of publications related to TBK1 signaling in cancer have resulted since these initial discoveries were made several years ago. In this review, we highlight some of these impactful discoveries, discuss the viability of TBK1 as a therapeutic target and evaluate the efficacy of TBK1 inhibition from recent clinical trials.
Cellular mechanisms of TBK1-mediated cancer growth
Cell division
TBK1 was originally linked to cell division in a phosphoproteomics screen performed in A549 lung adenocarcinoma cells (Kim et al. 2013). The stable isotope labeling by amino acids in cell culture (SILAC) mass spectrometry technique was used in control shRNA and shTBK1 knock-down A549 cells to define TBK1-regulated signaling networks based on quantitative differences in phosphoproteins. Pathway analyses and subsequent experimental validation revealed that TBK1 is induced at mitosis and directly phosphorylates the mitotic kinase, Polo-like kinase 1 (PLK1). In 2015, Pillai and colleagues (Pillai et al. 2015) confirmed TBK1 induction during mitosis in non-small cell lung cancer (NSCLC) cell lines. However, PLK1 overexpression did not rescue mitotic progression in cells treated with siRNA targeting TBK. This led to a search for and ultimately identification of novel mitotic TBK1 substrates including CEP170 and NUMA, proteins that promote microtubule stability and mitosis.
Given the relatively high frequency of KRAS mutations in lung cancer, one consideration is whether TBK1 facilitates mitotic spindle formation independent of mutant KRAS. The discovery of CEP170 and NUMA as mitotic TBK1 substrates was made in both mutant and wild-type (WT) KRAS NSCLC lines, suggesting that mutant KRAS may not be initiating this function of TBK1. Thus the identity of factors upstream that direct TBK1 to the centrosome during mitosis and induce TBK1 expression is of great interest. Evidence from recent studies indicates that TBK1 activation is dependent upon its subcellular localization as well as local TBK1 concentration (Ma et al. 2012; Helgason et al. 2013). Numerous adaptor proteins have been shown to escort TBK1 to various signaling complexes for distinctive cellular responses. Additionally, TBK1 can autophosphorylate itself through inter-dimer interactions between locally concentrated TBK1 molecules. Moving forward, it will be important to test these findings in vivo to understand the clinical significance of targeting TBK1 in NSCLC. Mitotic defects resulting from TBK1 inhibition in vitro indicate that therapeutically targeting TBK1 would likely have a cytotoxic effect by preventing tumor cell division. Inhibiting cancer cell proliferation by blocking TBK1 activity could be especially beneficial in combination with drugs that function independently of the cell cycle.
Autophagy
TBK1 has been shown to promote the intracellular degradation pathway, autophagy that is often deregulated in human cancers (Newman et al. 2012; Yang et al. 2016). Autophagy is a fundamental biological process of self-digestion, whereby a cell degrades various intracellular components, including damaged or excessive proteins and organelles, as a reactive survival mechanism or as a strategy to maintain cellular energy production. Autophagy is induced by various physiological stressors including hypoxia, nutrient deprivation, high temperatures and innate immune signals (Levine and Klionsky 2004). Deregulation of autophagy is implicated in various disease states, including cancer. However, the function of autophagy in cancer cells is complex with reports indicating it has oncogenic and tumor-suppressive roles. Depending on the tumor source and/or stage, autophagy can function as a tumor suppressor pathway that prevents tumor formation. For example, in the initial stages of pancreatic cancer, autophagy can limit inflammation and cell injury, processes that are critical for tumor development and progression (Gukovsky et al. 2013). Autophagy also functions as a pro-survival pathway in pancreas cancer, allowing tumor cells to tolerate metabolic stress and resist cell death induced by chemotherapy. In fact, several studies have reported elevated basal autophagy levels in human pancreatic ductal adenocarcinoma (PDA) cell lines and primary tumor tissues. Inhibition of autophagy in culture and in PDA mouse models caused marked growth suppression, indicating a large subset of PDA cells depend on autophagy (Yang et al. 2011; Rosenfeldt et al. 2013).
Originally, autophagy was thought to be a nonselective bulk degradation pathway in response to cellular stress. Recent studies have revealed that through the use of autophagic receptors and adaptors, the antibacterial form of autophagy, referred to as xenophagy, can selectively degrade intracellular pathogens (Reggiori et al. 2012; Fimia et al. 2013). In xenophagy, TBK1 activates the adaptor proteins OPTN and p62 that bind to and escort invading pathogens for rapid autophagic clearance (Wild et al. 2011; Pilli et al. 2012; Jo et al. 2013). We, along with another group (Pilli et al. 2012), observed an increase in the autophagic marker, LC3, in Tbk1-deficient mouse embryonic fibroblasts (MEFs) without any pathogenic stimulus (Fig. 1a). Additionally we detected an increase in autophagic markers, LC3 and p62, in tumor tissues collected from Tbk1 mutant mice (Tbk1 ∆/∆) that were crossed into a genetically engineered mouse model of PDA (Kras LSL-G12D ; Cdkn2a lox/lox ; Ptf1a Cre/+) compared to Tbk1 +/+ : PDA tumors (Fig. 1b). Tbk1 ∆/∆ mice have a truncated form of TBK1 that lacks the catalytic domain and is expressed at very low levels globally (Marchlik et al. 2010). These findings demonstrate that autophagy is altered in the absence of Tbk1 and suggest that TBK1 may regulate autophagy induced by stimuli other than bacteria. Furthermore, it is plausible that TBK1 contributes to the pro-survival effect of autophagy in tumors, including PDA.
Fig. 1.
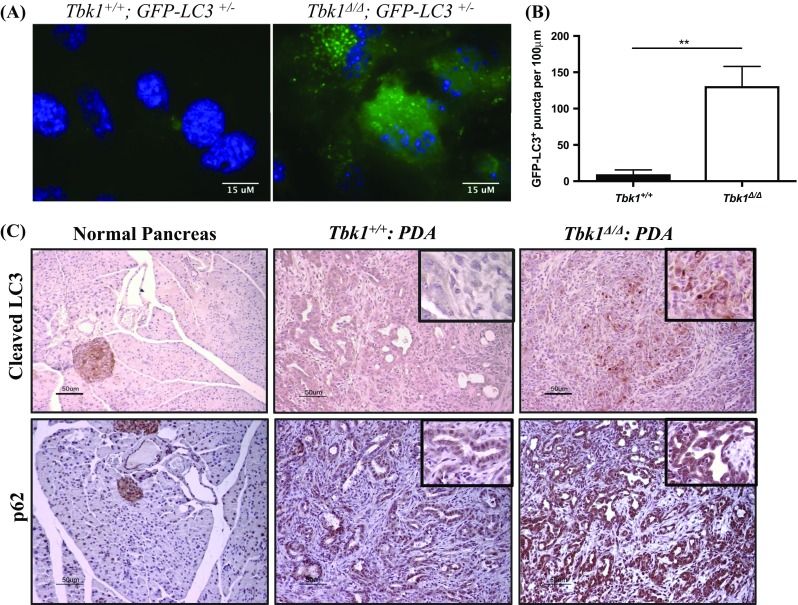
Autophagy is dysregulated in the absence of Tbk1. a MEFs isolated at E13.5 days were cultured in DMEM-10% serum. b GFP-LC3+ puncta were quantified in a minimum of four microscopic fields per cell line (3 cell lines/genotype). Results are presented as mean +/- SEM. **p<0.01, student’s t-test. c Tumor tissues from Tbk1 +/+ : PDA and Tbk1 Δ/Δ : PDA mice were stained for autophagy markers LC3 and p62 to analyze autophagic flux. Images are representative of 4 images/tumor, 5 tumors/genotype
Similarly, a recent study by Yang et al. (2016) demonstrated a link between autophagy inhibition and TBK1 signaling in pancreas cancer. They claim that a negative feedback mechanism exists between TBK1 and autophagy, whereby active TBK1 promotes basal autophagy in PDA cells and is then degraded in the autophagic process to limit over-activation of autophagy and TBK1-induced cytokine production, both of which fuel neoplasia. Accordingly, utilization of the multitarget JAK/TBK1/IKKε inhibitor CYT387 (momelotinib) proved advantageous in blocking autophagy and cytokine production in a PDA cell line with high basal autophagy and in a PDA mouse model, resulting in more intact pancreatic acinar tissue and limited pre-neoplastic lesions relative to vehicle control. While the authors mention that autophagy restricts TBK1 and cytokine activity, it isn’t clear how cell lines with low basal levels of autophagy would respond to the multi-target inhibitor CYT387. It is intriguing to consider but currently unclear whether baseline autophagy levels could serve as a predictor of CYT387 sensitivity. Or whether inflammation could function as a biomarker for this TBK1 inhibitor. It is also unclear if mutant active KRAS is necessary for the negative feedback between TBK1 and autophagy in PDA cells. This is especially relevant considering that oncogenic KRAS can induce autophagy and is commonly mutated in PDA (Guo et al. 2011). It is worth pointing out that CYT387 has undergone testing in multiple clinical trials including in combination with chemotherapy in PDA and in combination with the MEK inhibitor, Trametinib, in NCSLC. Results from these trials will be very informative from multiple interest points, including that of KRAS-driven cancers.
Immune response
Innate immune sensing is a critical step in promoting T-cell priming and infiltration in tumors. Better understanding of the cross talk between innate and adaptive immune systems is needed to pharmacologically facilitate enduring anti-tumor immunity. TBK1 is best known as an innate immune kinase that is downstream of the transmembrane protein stimulator of interferon genes (STING) in the type I interferon (IFN-I) response pathway. STING senses the presence of nucleic acids from intracellular pathogen infection and in turn initiates a downstream signaling cascade that includes TBK1-mediated activation of IRF3 and Stat6, resulting in IFN-I and cytokine production (Ishikawa and Barber 2008; Chen et al. 2011). Multiple STING agonists have been shown to generate anti-tumor immunity through interferon β (IFN-β) production in the tumor microenvironment (Woo et al. 2014; Corrales et al. 2015). Intratumoral injection of synthetic derived cyclic di-nucleotides to activate STING resulted in primary tumor regression in three different murine tumor models, including those of B16 melanoma, as well as 4T1 mammary and CT26 colon carcinomas. In each of these models, a systemic immune response involving IFN-β and cytokine production, priming of CD8+ T cells and immunologic memory was elicited that prevented primary tumor growth and the development of distal lesions. This work supports the development of innate immune modulatory strategies for anti-tumor efficacy and implicates TBK1 as a potential anti-tumor protein.
Contrary to these findings, a recent study of dendritic cell (DC) conditional Tbk1 knockout mice (Tbk1-DKO) found that Tbk1-DKO mice injected subcutaneously with tumor cells lived longer and had smaller tumors compared to WT control mice (Xiao et al. 2017). Bone marrow and spleen from Tbk1-DKO and WT control mice showed similar peripheral immune profiles, implying that TBK1 is not critical to myeloid cell development. Yet the assessment of anti-tumor immunity in Tbk1-DKO animals containing B16 melanoma tumors revealed greater T-effector cell infiltration into tumors and lymph nodes as well as synergy with anti-PD-1 treatment. These results were corroborated with two additional tumor cell lines (EG7-OVA and EL4 lymphoma cells). Gene expression profiling of DCs isolated from Tbk1-DKO spleens and tumors showed enhanced interferon-responsive gene expression compared to WT control DCs. To see if the Tbk1-DKO tumor suppressive phenotype was dependent upon interferon signaling, DCs harvested from Tbk1-DKO mice crossed into interferon alpha/beta receptor 1 (Ifnar1) KO mice were utilized in DC-based tumor immunotherapy in WT mice with established B16 melanoma tumors. Treatment with double TBK1 DKO IFNAR KO DCs allowed for tumor development unlike in the TBK1 DKO IFNAR WT DC therapy group where tumor growth was restrained. Collectively, these observations support a pro-tumor function for TBK1 in DCs that suppresses IFNAR1 signaling to mediate immune tolerance and enable tumor growth.
While the interpretation of the function of TBK1 in tumor immunity from these two publications (Corrales et al. 2015; Xiao et al. 2017) are conflicting, we can glean that the function of TBK1 is likely variable between different stromal cell types within a tumor and even within cancer types. Further work is needed to understand whether there are distinct tumor types that would respond better to modulation of TBK1 activity. In the previous publication (Xiao et al. 2017) for example, synergy was observed in Tbk1-DKO animals containing B16 melanoma tumors treated with anti-PD-1. Thus, it is plausible that PD-L1 expressing tumors may show a stronger response to TBK1 inhibition. Xiao and colleagues (Xiao et al. 2017) also showed greater T-effector cell tumor infiltration in tumor bearing Tbk1-DKO animals compared to tumor bearing WT control animals. Therefore, it may also be therapeutically beneficial to pretreat immune therapy non-responder patients with a TBK1 inhibitor in an effort to augment T-cell infiltration into tumors. These papers highlight the importance in understanding the unique function of TBK1 in each relevant cell type within a tumor as well as different tumor types and mouse models. This is especially pertinent when considering the therapeutic benefit of pharmacological modulation of TBK1 in cancer patients. If TBK1 does function differently between immune cell types, then going forward it would be important to evaluate whether one cell type is more abundant in a particular tumor or has more of a dominant effect on tumor growth.
AKT pro-survival signaling
One of the original reports linking TBK1 with RAS signaling identified AKT as a direct substrate of TBK1 in NSCLC (Ou et al. 2011). Follow up from these results has led to the characterization of TBK1 as a molecular vulnerability in subtypes of melanoma, in addition to NSCLC, in two recent studies (Cooper et al. 2017; Eskiocak et al. 2017). In the melanoma study (Eskiocak et al. 2017), a chemical compound library was employed in screening a panel of melanoma cell lines annotated with drug resistance status to identify molecular liabilities unique to BRAF inhibitor (BRAFi) resistant tumors. Bx795, a TBK1/PDK1 (3-phosphoinositide-dependent protein kinase-1) dual inhibitor was the top hit from the screen with selective toxicity in BRAFi resistant cell lines. Since Bx795 is a dual TBK1/PDK1 inhibitor, the mechanism of action causing toxicity in the drug resistant melanoma cell lines was unclear. Additional scaffolds targeting TBK1 were tested, including compound II, MRT6737 and momelotinib, and these showed similar toxicity profiles across the cell panel, incriminating TBK1 inhibition as the cause for cell death. Of note, TBK1 inhibitor (TBK1i) sensitivity positively correlated with MEK inhibitor resistance. TBK1 inhibitor compound II was selected for further testing in vivo in NSG (Cg-Prkdc scid Il2rg tm1Wjl /SzJ) mice subcutaneously implanted with three melanoma cell lines that were already proven to be sensitive to compound II in vitro. As expected, tumor growth in these immunodeficient animals was reduced by compound II. Though compound II does show therapeutic benefit in this model, the growth curves of the treated tumors have a positive slope indicating that tumor growth is slowed but not inhibited. In these TBK1-dependent cell lines, compound II treatment reduced AKT activity, yet chemical inhibition of AKT was not sufficient to recapitulate the level of toxicity associated with TBK1 inhibition. Interestingly, two members of the hippo tumor suppressor pathway, LATS1 and YAP1, co-immunoprecipitated with TBK1. Inhibition of TBK1 activated the hippo tumor suppressor pathway while combined siRNA-mediated knock down of YAP1 with AKT inhibition resulted in apoptosis at a level comparable to compound II-mediated apoptosis. Thus, the authors concluded that the basis of TBK1 addiction in drug resistant melanoma cells is through the combined activation of AKT survival signaling and suppression of hippo tumor suppressor pathway activity.
In the NSCLC study (Cooper et al. 2017), sensitivity to TBK1 inhibitors Bx795 and compound II along with gene expression data for 100 NSCLC cell lines were pooled to distinguish biological features of TBK1-dependent cell lines. This data included a screen of chemical compounds in pursuit of scaffolds with similar activity profiles. As in the melanoma study (Eskiocak et al. 2017), the TBK1i sensitivity profiles correlated well with one another. They also were highly correlative with the profiles of multiple AKT/mTOR pathway inhibitors, particularly in the mutant KRAS NSCLC lines, suggesting a mechanistic relationship between TBK1 and the mTOR pathway. Subsequent experiments in fibroblasts with genetic modulation or pharmacological inhibition of TBK1 uncovered a physical interaction between TBK1 and multiple components of the mTOR pathway. TBK1 promoted mTOR activation through direct phosphorylation of upstream activator AKT and downstream substrate S6K predominantly in the transition from the amino acid-starved to fed state. Further analysis of the TBK1i sensitive NSCLC lines revealed mutations in RAS family members and greater mesenchymal gene expression compared to the resistant lines that had a more epithelial gene expression profile. Intriguingly, TGFβ treatment in a TBK1i resistant cell line induced epithelial-to-mesenchymal transition (EMT) and sensitized the cell line to TBK1i in a reversible manner. All in all, this report characterized a TBK1-dependent subset of NSCLC lines that contain mesenchymal gene expression, RAS class member mutations and are liable to mTOR pathway inhibitors. A limitation of this study is that the mechanism of TBK1 dependency in the subset of NSCLC lines is attributed to TBK1-mediated activation of the mTOR/AKT pro-survival pathway that was observed primarily in fibroblasts. This investigation sheds light on the effect of TBK1 inhibition in a relevant stromal cell type which is informative considering small molecule inhibitors do not target cancer cells exclusively. However, additional studies to validate these interactions in lung cancer cell lines would substantiate the proposed mechanism in the NSCLC context. Additionally, it would be interesting to examine the non-tumor cell autonomous consequences of inhibiting TBK1-mediated mTOR activation in cancer associated fibroblasts or other stromal cells on overall tumor growth.
These two studies (Cooper et al. 2017; Eskiocak et al. 2017) highlight the potential of TBK1 as a therapeutic target in BRAF/MEKi resistant melanoma together with mutant RAS NSCLC. Moving forward, it will be essential to evaluate the therapeutic benefit of TBK1 inhibition in preclinical cancer models, particularly in immune-competent animals. The tumor xenograft models from the melanoma study were performed in immunodeficient animals so there is a gap in understanding how well TBK1 inhibition will affect the immune-competent tumor microenvironment. Also, the TBK1-dependent melanoma cell lines were BRAF/MEKi resistant and displayed an “innate immune” expression profile. This specific combination of drug resistance and gene expression is associated with a decreased response to immune therapy (Hugo et al. 2015). Therefore, TBK1 inhibition may be a viable alternative to the subset of melanoma patients who don’t respond to immune therapy. It is also possible that pretreatment of these patients with TBK1i could enhance immune therapy response. An unexplored question stemming from the melanoma study is whether NRAS mutation status, which can be found in up to 30% of melanoma patients, had any correlation with TBK1i sensitivity. One group observed that TBK1 is active in mutant NRAS melanoma and promoted migration and invasion of these cells (Vu and Aplin 2014). The group also demonstrated cooperation between TBK1 and MEK inhibition to promote apoptosis in mutant NRAS MEKi resistant melanomas, suggesting clinical utility for combined TBK1/MEK inhibition. While mutant NRAS is common in melanoma, KRAS activating mutations dominate PDA and to a lesser extent colon and lung cancers, respectively. Given the high sensitivity to TBK1i seen in NSCLCs with RAS class mutations it would also be appropriate to investigate TBK1 dependency in these other RAS-driven cancers. In our own analysis, data from a cohort of PDA patients within The Cancer Genome Atlas (TCGA) revealed a correlation between high TBK1 RNA expression and poorer overall survival (Fig. 2a). Expression of TBK1 homolog, IKBKE, on the other hand showed no correlation with overall survival (Fig. 2b). Though not causative, this data strengthens the relevance of TBK1 in RAS-driven PDA.
Fig. 2.

High TBK1 expression is associated with poor prognosis. Kaplan Meier curves of a cohort of PDA patients from TCGA showing the overall survival of a the TBK1-high (upper 25 %) and TBK1-low (lower 75 %) expression subgroups and b the IKBKE-high (upper 25 %) and IKBKE-low (lower 75 %) expression subgroups. Cox regression was used to calculate hazard ratios and Kaplan Meier survival analyses
TBK1 inhibitors
Over the past decade, research and interest in TBK1 have expanded along with the identification and development of small molecules targeting TBK1. There are at least six distinct small molecules that are known to inhibit TBK1 including Bx795, compound II, CYT387, MRT67307, GSK2292978A and amlexanox. Though most of these compounds are quite potent towards TBK1 and homolog IKKε, not all of them are highly selective. Bx795 inhibits the serine/threonine kinase, PDK1, in addition to TBK1 and IKKe. CYT387 or momelotinib was originally described as a selective JAK inhibitor but was recently discovered to inhibit TBK1 with low nanomolar potency (Zhu et al. 2014). Three years ago, momelotinib began testing in combination with either chemotherapy or Trametinib (MEKi) in multiple clinical trials for metastatic KRAS mutant NSCLC and PDA (NCT02258607, NCT02244489, NCT02101021). Unfortunately all three trials were terminated early by the sponsor, Gilead Sciences without the release of further information.
The only other TBK1i known to enter clinical trial testing in human patients is amelxanox in a phase 2 study for the treatment of type 2 diabetes, nonalcoholic fatty liver disease or obesity (NCT01975935, NCT01842282). Amlexanox was identified in a compound library screen for inhibitors of TBK1/IKKε, which had previously been shown to promote metabolic syndrome in an obese rodent model (Chiang et al. 2009; Reilly et al. 2013). Takeda developed amlexanox in the 1980s for the treatment of asthma and conjuctivitus in Japan. Though the mechanism of action was unknown until recently, amlexanox is still sold there today for treating asthma. Despite the fact that TBK1 inhibition in patients with metabolic syndrome is a different biology than cancer, the side effects from systemic TBK1 inhibition in this trial are applicable to the evaluation of TBK1 as a viable therapeutic target in cancer patients. In the open label trial to ensure the safe administration of amlexanox, there were no serious adverse events reported (Oral et al. 2017). However, mild to moderate events included two rash incidents that resolved in the presence of continued amlexanox treatment. The rashes, confirmed as perivascular inflammation by biopsy, are reminiscent of the reported phenotype seen in the previously mentioned (Marchlik et al. 2010) kinase dead Tbk1 ∆/∆ mice. These mice exhibited mild levels of immune cell infiltrates in multiple organs and tissues including skin. It is likely that modulation of the innate immune kinase TBK1 will result in some form of an altered immune reactivity. The question then becomes in what way and how will that affect a tumor? Aside from that, the mild side effects observed from the amlexanox trial are promising in terms of tolerability of systemic TBK1 inhibition, at least in the context of amelxanox.
Conclusion
The multifunctional kinase, TBK1, mediates numerous signaling pathways that drive malignant growth (Table 1). TBK1 facilitates cell division in lung cancer cells through its interaction with mitotic substrates PLK1, CEP170 and NUMA to promote microtubule stability and mitosis. TBK1 can also promote autophagy in pancreatic cancer cells to silence proinflammatory signals that elicit an immune response. Furthermore, the DC-specific function of TBK1 suppresses IFNAR1 signaling to enable tumor growth. In a subset of melanoma and NSCLC cancers, TBK1 has been implicated in the activation of AKT/mTOR signaling to promote cancer cell survival. To date, the majority of TBK1-related cancer research has conferred a pro-tumorigenic role for TBK1. Further investigations targeting TBK1 in immune-competent animals are needed to better predict response in human cancer patients and to analyze the effect on tumor metastases. It is estimated that nearly 90% of cancer mortalities are due to metastases, yet studies to date have evaluated the effect of TBK1 inhibition on primary tumor burden. Gene expression data from the melanoma and NSCLC studies (Cooper et al. 2017; Eskiocak et al. 2017) suggest TBK1 is involved in EMT, which is a process commonly associated with metastatic potential. Both subsets of TBK1i sensitive melanoma and NSCLC cell lines comprised a mesenchymal gene signature. Looking ahead, it will be critical to test the effect of targeting TBK1 on tumor cell migration, invasion and overall metastatic burden. These studies will aid in our overall understanding of the clinical benefit of targeted TBK1 inhibition.
Table 1.
Summary of TBK1-mediated malignant growth signaling
| Upstream/Predictor | Downstream/Substrate | Context | Reference |
|---|---|---|---|
| - | PLK1 | NSCLC | Kim et al. 2013 |
| - | CEP170, NUMA | NSCLC* | Pillai et al. 2015 |
| KRAS, IL1 | CCL5, IL6 | PDA | Zhu et al. 2014; Yang et al. 2016 |
| STING | IRF3 | Melanoma | Corrales et al. 2015 |
| - | IFNAR1, STAT3 | Melanoma, Lymphoma | Xiao et al. 2017 |
| BRAFi/MEKi resistance | AKT, YAP1, LATS1 | Melanoma | Eskiocak et al. 2017 |
| RAS class | S6K, AKT | NSCLC, Fibroblasts | Cooper et al. 2017 |
- Undefined/unmentioned by authors
*Experiments were carried out primarily in NSCLC cell lines but some experiments were validated in cell lines of other origins including myeloid leukemia, Daudi Burkitt lymphoma, HeLa, AALE cells
Acknowledgements
The authors thank Drs. Jonathan Cooper and Aubhishek Zaman for many helpful discussions and members of the Brekken lab for critical review of this manuscript. We also gratefully acknowledge Dr. Tae Hyun Hwang for his help with TCGA gene expression analysis and Dave Primm for editorial assistance.
Abbreviations
- AALE
Immortalized tracheobronchial epithelial cells
- AKT
Protein kinase B
- BRAFi
BRAF inhibitor
- CCL5
Chemokine ligand 5
- CEP1
Centrosomal protein 170
- DC
Dendritic cell
- DKO
Dendritic cell knockout
- EMT
Epithelial-to-mesenchymal transition
- GEMM
Genetically engineered mouse model
- GFP
Green fluorescent protein
- IFN-I
Type I interferon
- IFN-β
Interferon β
- IFNAR1
Interferon alpha/beta receptor 1
- IKB
Inhibitor of NFκβ
- IKK
Iκβ kinase
- IL1
Interleukin 1
- IL6
Interleukin 6
- IRF3
Interferon regulator factor 3
- LATS1
Large tumor suppressor kinase 1
- LC3
Microtubule-associated protein-1 light chain 3
- MEFs
Mouse embryonic fibroblasts
- MEKi
MEK inhibitor
- mTOR
Mammalian target of rapamycin
- NFκβ
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NSCLC
Non-small cell lung cancer
- NSG
NOD scid gamma
- NUMA
Nuclear mitotic apparatus protein
- OPTN
Optineurin
- PD-1
Programmed cell death protein 1
- PDA
Pancreatic ductal adenocarcinoma
- PDK1
3-phosphoinositide-dependent protein kinase-1
- PLK1
Polo-like kinase 1
- RAL
Ras-related protein
- RAS
Rat sarcoma virus
- S6K
p70 S6 kinase
- SILAC
Stable isotope labeling by amino acids in cell culture
- siRNA
Small interfering RNA
- STAT3
Signal transducer and activator of transcription 3
- STING
Stimulator of interferon genes
- TANK
TRAF family member associated NF-κβ activator
- TBK1
TANK -binding kinase 1
- TBK1i
TBK1 inhibitor
- TCGA
The cancer genome atlas
- TGFB
Transforming growth factor β
- WT
Wild-type
- YAP1
Yes associated protein 1
References
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, Frohling S, Chan EM, Sos ML, Michel K, Mermel C, Silver SJ, Weir BA, Reiling JH, Sheng Q, Gupta PB, Wadlow RC, Le H, Hoersch S, Wittner BS, Ramaswamy S, Livingston DM, Sabatini DM, Meyerson M, Thomas RK, Lander ES, Mesirov JP, Root DE, Gilliland DG, Jacks T. and Hahn WC (2009) "Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1." Nature 462(7269):108–112 [DOI] [PMC free article] [PubMed]
- Chen H, Sun H, You F, Sun W, Zhou X, Chen L, Yang J, Wang Y, Tang H, Guan Y, Xia W, Gu J, Ishikawa H, Gutman D, Barber G, Qin Z, Jiang Z. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell. 2011;147(2):436–446. doi: 10.1016/j.cell.2011.09.022. [DOI] [PubMed] [Google Scholar]
- Chiang SH, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, Ma JT, Zhou J, Qi N, Westcott D, Delproposto JB, Blackwell TS, Yull FE, Saltiel AR. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell. 2009;138(5):961–975. doi: 10.1016/j.cell.2009.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, Balakireva MG, Romeo Y, Kopelovich L, Gale M, Yeaman C, Camonis JH, Zhao Y and White MA (2006) RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 127(1):157–170 [DOI] [PubMed]
- Cooper JM, Ou YH, McMillan EA, Vaden RM, Zaman A, Bodemann BO, Makkar G, Posner BA, White MA. TBK1 provides context-selective support of the activated AKT/mTOR pathway in lung cancer. Cancer Res. 2017;77(18):5077–5094. doi: 10.1158/0008-5472.CAN-17-0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, Metchette K, Dubensky TW, Jr, Gajewski TF. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11(7):1018–1030. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskiocak B, McMillan EA, Mendiratta S, Kollipara RK, Zhang H, Humphries CG, Wang C, Garcia-Rodriguez J, Ding M, Zaman A, Rosales TI, Eskiocak U, Smith MP, Sudderth J, Komurov K, Deberardinis RJ, Wellbrock C, Davies MA, Wargo JA, Yu Y, De Brabander JK, Williams NS, Chin L, Rizos H, Long GV, Kittler R, White MA. Biomarker accessible and chemically addressable mechanistic subtypes of braf melanoma. Cancer Discov. 2017;7(8):832–851. doi: 10.1158/2159-8290.CD-16-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimia GM, Kroemer G, Piacentini M. Molecular mechanisms of selective autophagy. Cell Death Differ. 2013;20(1):1–2. doi: 10.1038/cdd.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gukovsky I, Li N, Todoric J, Gukovskaya A, Karin M. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144(6):1199.e1194–1209.e1194. doi: 10.1053/j.gastro.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, Coller HA, Dipaola RS, Gelinas C, Rabinowitz JD, White E. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25(5):460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgason E, Phung QT, Dueber EC. Recent insights into the complexity of Tank-binding kinase 1 signaling networks: the emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett. 2013;587(8):1230–1237. doi: 10.1016/j.febslet.2013.01.059. [DOI] [PubMed] [Google Scholar]
- Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman KB, Johnson DB, Sosman JA, Ribas A, Lo RS. Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell. 2015;162(6):1271–1285. doi: 10.1016/j.cell.2015.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo EK, Yuk JM, Shin DM, Sasakawa C. Roles of autophagy in elimination of intracellular bacterial pathogens. Front Immunol. 2013;4:97. doi: 10.3389/fimmu.2013.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Welsh EA, Oguz U, Fang B, Bai Y, Kinose F, Bronk C, Remsing Rix LL, Beg AA, Rix U, Eschrich SA, Koomen JM, Haura EB. Dissection of TBK1 signaling via phosphoproteomics in lung cancer cells. Proc Natl Acad Sci U S A. 2013;110(30):12414–12419. doi: 10.1073/pnas.1220674110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6(4):463–477. doi: 10.1016/S1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, Bowman KK, Starovasnik MA, Dueber EC. Molecular basis of tank-binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci U S A. 2012;109(24):9378–9383. doi: 10.1073/pnas.1121552109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchlik E, Thakker P, Carlson T, Jiang Z, Ryan M, Marusic S, Goutagny N, Kuang W, Askew GR, Roberts V, Benoit S, Zhou T, Ling V, Pfeifer R, Stedman N, Fitzgerald KA, Lin LL, Hall JP. Mice lacking Tbk1 activity exhibit immune cell infiltrates in multiple tissues and increased susceptibility to LPS-induced lethality. J Leukoc Biol. 2010;88(6):1171–1180. doi: 10.1189/jlb.0210071. [DOI] [PubMed] [Google Scholar]
- Newman AC, Scholefield CL, Kemp AJ, Newman M, McIver EG, Kamal A, Wilkinson S. TBK1 kinase addiction in lung cancer cells is mediated via autophagy of Tax1bp1/Ndp52 and non-canonical NF-kappaB signalling. PLoS ONE. 2012;7(11):e50672. doi: 10.1371/journal.pone.0050672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oral EA, Reilly SM, Gomez AV, Meral R, Butz L, Ajluni N, Chenevert TL, Korytnaya E, Neidert AH, Hench R, Rus D, Horowitz JF, Poirier B, Zhao P, Lehmann K, Jain M, Yu R, Liddle C, Ahmadian M, Downes M, Evans RM, Saltiel AR. Inhibition of IKKvarepsilon and TBK1 Improves Glucose Control in a Subset of Patients with Type 2 Diabetes. Cell Metab. 2017;26(1):157.e157–170.e157. doi: 10.1016/j.cmet.2017.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou YH, Torres M, Ram R, Formstecher E, Roland C, Cheng T, Brekken R, Wurz R, Tasker A, Polverino T, Tan SL, White MA. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell. 2011;41(4):458–470. doi: 10.1016/j.molcel.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai S, Nguyen J, Johnson J, Haura E, Coppola D, Chellappan S. Tank binding kinase 1 is a centrosome-associated kinase necessary for microtubule dynamics and mitosis. Nat Commun. 2015;6:10072. doi: 10.1038/ncomms10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, Bruun JA, Hansen TE, Johansen T, Deretic V. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012;37(2):223–234. doi: 10.1016/j.immuni.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori F, Komatsu M, Finley K, Simonsen A. Autophagy: more than a nonselective pathway. Int J Cell Biol. 2012;2012:219625. doi: 10.1155/2012/219625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, Larsen MJ, Rubin JR, Mowers J, White NM, Hochberg I, Downes M, Yu RT, Liddle C, Evans RM, Oh D, Li P, Olefsky JM, Saltiel AR. An inhibitor of the protein kinases TBK1 and IKK-varepsilon improves obesity-related metabolic dysfunctions in mice. Nat Med. 2013;19(3):313–321. doi: 10.1038/nm.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, Adams PD, Anderson KI, Gottlieb E, Sansom OJ, Ryan KM. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504(7479):296–300. doi: 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- Vu HL, Aplin AE. Targeting TBK1 inhibits migration and resistance to MEK inhibitors in mutant NRAS melanoma. Mol Cancer Res. 2014;12(10):1509–1519. doi: 10.1158/1541-7786.MCR-14-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dotsch V, Bumann D, Dikic I. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333(6039):228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, Alegre ML, Gajewski TF. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Zou Q, Xie X, Liu T, Li HS, Jie Z, Jin J, Hu H, Manyam G, Zhang L, Cheng X, Wang H, Marie I, Levy DE, Watowich SS, Sun SC. The kinase TBK1 functions in dendritic cells to regulate T cell homeostasis, autoimmunity, and antitumor immunity. J Exp Med. 2017;214(5):1493–1507. doi: 10.1084/jem.20161524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, Mautner J, Tonon G, Haigis M, Shirihai OS, Doglioni C, Bardeesy N, Kimmelman AC. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25(7):717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Imamura Y, Jenkins RW, Canadas I, Kitajima S, Aref A, Brannon A, Oki E, Castoreno A, Zhu Z, Thai T, Reibel J, Qian Z, Ogino S, Wong KK, Baba H, Kimmelman AC, Pasca Di Magliano M, Barbie DA. Autophagy inhibition dysregulates TBK1 signaling and promotes pancreatic inflammation. Cancer Immunol Res. 2016;4(6):520–530. doi: 10.1158/2326-6066.CIR-15-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Aref AR, Cohoon TJ, Barbie TU, Imamura Y, Yang S, Moody SE, Shen RR, Schinzel AC, Thai TC, Reibel JB, Tamayo P, Godfrey JT, Qian ZR, Page AN, Maciag K, Chan EM, Silkworth W, Labowsky MT, Rozhansky L, Mesirov JP, Gillanders WE, Ogino S, Hacohen N, Gaudet S, Eck MJ, Engelman JA, Corcoran RB, Wong KK, Hahn WC, Barbie DA. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014;4(4):452–465. doi: 10.1158/2159-8290.CD-13-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]