

Abstract
Transforming growth factor (TGF)-β is a multifunctional growth factor with potent pro-fibrotic effects. Endoglin is a TGF-β co-receptor that strongly regulates TGF-β signaling in a variety of cell types. Although aberrant regulation of TGF-β signaling is known to play a key role in fibrotic diseases such as scleroderma and impaired cartilage repair, the significance of endoglin function in regulating these processes is poorly understood. Here we examined whether endoglin haploinsufficiency regulates extracellular (ECM) protein expression and fibrotic responses during bleomycin induced skin fibrosis and surgically induced osteoarthritis, using endoglin-heterozygous (Eng+/−) mice and wild-type (Eng+/+) littermates. Skin fibrosis was induced by injecting mice intradermally with bleomycin or vehicle. Osteoarthritis was induced surgically by destabilization of medial meniscus. Dermal thickness, cartilage integrity and ECM protein expression were then determined. Eng+/− mice subjected to bleomycin challenge show a marked decrease in dermal thickness (P < 0.005) and reduced collagen content and decreased collagen I, fibronectin, alpha-smooth muscle actin levels as compared to Eng+/+ mice, both under basal and bleomycin treated conditions. Eng+/− mice undergoing surgically induced osteoarthritis show no differences in the degree of cartilage degradation, as compared to Eng+/+ mice, although chondrocytes isolated from Eng+/− display markedly enhanced collagen II levels. Our findings suggest that endoglin haploinsufficiency in mice ameliorates bleomycin-induced skin fibrosis suggesting that endoglin represents a pro-fibrotic factor in the mouse skin. However, endoglin haploinsufficiency does not protect these mice from surgically indiced cartilage degradation, demonstrating differential regulation of endoglin action during skin and cartilage repair.
Keywords: Fibrosis, skin, cartilage, osteoarthritis; Endoglin; TGF-beta; Scleroderma; Systemic sclerosis; Animal model
Introduction
Transforming growth factor (TGF)-β is a multifunctional growth factor that is important for tissue homeostasis, by regulating cell proliferation, differentiation and wound healing. However, excessive action of TGF-β leads to impaired wound repair as it can act as a potent fibrogenic factor by inducing the synthesis of extracellular matrix (ECM) proteins such as collagens and fibronectin, by inhibiting ECM degradation by repressing metalloproteinase and by stimulating tissue inhibitor of metalloproteinase synthesis (Morikawa et al. 2016). ECM deposition initially intensifies as a reparative response to insult or injury, but becomes pathogenic fibrosis when prolonged or excessive (Rittie 2015). Several studies indicate that aberrant TGF-β signaling plays a key role in the pathogenesis of fibrotic diseases such as scleroderma (Lafyatis 2014), and in impaired cartilage repair and fibrosis associated with osteoarthritis (Blaney Davidson et al. 2007).
TGF-β signaling is transduced by a pair of transmembrane serine/threonine kinases known as TGF-β receptor type I (TGFβRI, also known as activin receptor-like kinase-5 or ALK5) and type II (TGFβRII) receptors (Heldin and Moustakas 2016). Activation of TGF-β receptors induces phosphorylation of intracellular Smad2 and Smad3 proteins, which then complex with Smad4 and translocate to the nucleus, where they regulate the expression of TGF-β target genes such as type I collagen and fibronectin (Morikawa et al. 2016).
The central role of TGF-β in multiple cellular functions implicates tight regulation of its signaling. TGF-β co-receptors have been shown to be potent regulators of TGF-β receptor activity (Finnson et al. 2012). Endoglin is a TGF-β co-receptor that strongly modulates TGF-β signaling in a variety of cell types including endothelial cells (Goumans et al. 2009), skin fibroblasts (Maring et al. 2012) and chondrocytes (Finnson et al. 2010). Although regulation of TGF-β signaling is known to be important in skin and cartilage function, and aberrant TGF-β signaling is implicated in skin fibrosis (Maring et al. 2012) and impaired cartilage repair (Blaney Davidson et al. 2007; Finnson et al. 2013), the role of endoglin in regulating TGF-β signaling and fibrotic process in these tissues is poorly understood. Furthermore, it is not known whether endoglin differentially regulates ECM synthesis in these two tissues.
Scleroderma or systemic sclerosis is a rare autoimmune disorder characterized by inflammation, vasculopathy and fibrosis of skin and internal organs (Denton 2015; Desbois and Cacoub 2016). Endoglin levels have been reported be upregulated in scleroderma patient fibroblasts in vitro (Leask et al. 2002), but whether endoglin regulates TGF-β signaling or ECM production in these cells was not established. In addition, it has been demonstrated that endoglin promotes TGF-β/Smad1 signaling and fibrotic gene expression in scleroderma fibroblasts in vitro (Morris et al. 2011). However, whether endoglin plays a pro-fibrotic role in vivo in scleroderma remains to be determined.
We have previously reported that endoglin is expressed in chondrocytes (Parker et al. 2003) and that it is upregulated in osteoarthritis (Finnson et al. 2010). We have also shown that endoglin inhibits the canonical TGF-β/ALK5 pathway and decreases ECM synthesis in human chondrocytes in vitro (Finnson et al. 2010). Whether endoglin regulates cartilage repair in vivo is not known.
In the current study, we examined whether endoglin haploinsufficiency regulates ECM protein expression and fibrotic responses in a mouse model of scleroderma (bleomycin-induced skin fibrosis), using heterozygote (Eng+/−) mice. We also determined whether endoglin haploinsufficiency modulates ECM synthesis and cartilage repair after surgical induction of osteoarthritis, by destabilization of the medial meniscus (DMM), in these mice.
Methods and materials
Endoglin heterozygote mice (Eng+/−)
Endoglin heterozygote (Eng+/−) mice containing only one allele of endoglin were generated as previously described (Bourdeau et al. 1999) (obtained from Dr. Letarte from University of Toronto, Toronto, Ontario). (Endoglin knockout mice die due to cardiovascular malformation by embryonic day (E) 10.5 of gestation (Nomura-Kitabayashi et al. 2009) and therefore could not be used). Congenic Eng+/− and their respective littermate controls were successfully generated by backcrosses to C57BL/6 mice (Jackson Laboratory). The routine genotyping was performed to differentiate between Eng+/− and their WT littermates. Briefly, the tail clips of each mouse were collected at the time of weaning. DNA was extracted using a HotSHOT method (hot sodium hydroxide and tris) as previously described (Truett et al. 2000) followed by PCR reaction. PCR products were resolved by agarose gel electrophoresis and visualized by SYBR safe DNA gel stain (Life Technologies). The Facility Animal Care Committees (FACCs) and other animal ethics subcomittees of the McGill University and the McGill University Health Care (MUHC) Research Institute approved all animal procedures and protocols.
Bleomycin treatment
Endoglin heterozygote (Eng+/−) and wild-type (Eng+/+) littermate male mice (ages 6–8 weeks) were anesthetized with isoflurane, and their backs shaved and depilated using Nair (Church & Dwight). Mice were injected with 50 μl (5 μg) of filter-sterilized bleomycin sulfate (Sigma-Aldrich) in phosphate-buffered saline (PBS) or with PBS alone, intradermally into a single site on their shaved dorsal surface every other day for 28 days (8 groups of mice; n = 6 in each group). Mice were anesthetized with isoflurane and then sacrificed by CO2 asphyxiation followed by cervical dislocation. The injected skin tissue was harvested, bisected, and either snap-frozen in liquid nitrogen or fixed in 10% neutral buffered formalin (Sigma-Aldrich) for histologic analysis.
Histology and immunohistochemistry of skin
Formalin-fixed skin tissue was embedded in paraffin, and 7-μm sections were stained with picrosirius red for microscopic evaluation using ImageProPlus6 Software (Media Cybernetics). Dermal thickness was determined as the distance from the basement membrane to the hypodermis in 5 different high-power fields (hpf) per section, in 2 different sections from 6 different animals per group. Immunohistochemistry to assess levels of type I collagen, fibronectin and alpha-smooth muscle actin was performed by incubation with specific antibodies or negative control IgG at 4 °C overnight and incubation with a biotin-conjugated secondary antibody as previously described (Vorstenbosch et al. 2013a, 2013b).
Mouse fibroblast isolation and culture
Mouse skin dermal fibroblasts were prepared from 2 to 3 day-old neonates of Eng+/− and Eng+/+ mice. Mice were anesthetized and sacrificed by CO2 asphixiation, and whole skin was washed in PBS, minced into small pieces and digested with collagenase solution (Sigma-Aldrich) overnight at 37 °C. Dissociated cells were collected by centrifugation at 300 x g for 5 min, and suspended in Dulbecco’s modified Eagle’s medium (D-MEM; Life Technologies) supplemented with 10% (v/v) fetal bovine serum (FBS), 100 U/ml penicillin, and 50 μg/ml streptomycin as described previously (Finnson et al. 2006). Cells were maintained in 25 cm2 flasks and expanded in 75 cm2 flasks in a humidified atmosphere containing 5% CO2 at 37 °C. Experiments were performed using fibroblasts between passage 1 and 2.
For proliferation experiments, the cells were washed twice with PBS and then incubated with 0.25% trypsin-EDTA (Life Technologies) at 37 °C for 10 min. Dissociated cells were collected by centrifugation at 300 x g for 5 min and resuspended in D-MEM supplemented with FBS and antibiotics as described above. Cells were counted using a hemocytometer and seeded in triplicate in 6-well plates at a density of 3 × 105 cells/well. At the indicated time points, cells were detached from the plates by trypsin-ETDA treatment and counted using a hemocytometer. Cell numbers are represented as mean ± standard error of the mean (SEM).
Western blot analysis
Primary mouse Eng+/− and Eng+/+ dermal fibroblasts were left untreated or treated with TGF-β1 for 45 min. Cell lysates were prepared, separated by electrophoresis on SDS-polyacrylamide gels and transferred to nitrocellulose membranes (Fisher Scientific). The membranes were blocked with Tris-buffered saline-Tween20 (TBST) containing 5% milk for 1 h at room temperature and incubated with anti-phosphoSmad2 or anti-phosphoSmad1 antibodies (Cell Signaling Technologies) at 4 °C overnight. The membranes were washed 3 times for 10 min each and were incubated with horseradish peroxidase (HRP) conjugated secondary antibodies (Cell Signaling) for 1 h at room temperature. The signals on the membranes were detected using enhanced chemiluminescence (ECL) system (GE Healthcare, Canada). The membranes were stripped and then reprobed for total Smad2 (Cell Signaling Technologies) or α-tubulin (Santa Cruz, TX) as a loading control.
Destabilization of medial meniscus (DMM) surgery
Eng+/− mice and WT littermates at the age of 14-weeks were subjected to the DMM surgery to induced OA as described previously (Glasson et al. 2007). Briefly, after isoflurane anaesthesia, the medial meniscotibial ligament was transected, displacing medial meniscus resulting in the free movement of medial meniscus medially. A sham operation (without the transection of the medial meniscotibial ligament) was performed in the right knee joint of the control group (Zhang et al. 2015). At 14 weeks post-surgery, the mice were sacrificed and right knee joints of each mouse were collected for histological assessment and biochemical studies.
Histology of mouse knee joint
Mouse knee joints were dissected and fixed overnight in Tissue-Fix (Chaptec, Montreal, Quebec, Canada), decalcified for 1.5–2 h with RDO Rapid Decalcifier (Apex Engineering, Plainfield, Illinois, USA). The decalcified knee joints were washed with PBS twice followed by embedding in paraffin and sectioning. For Safranin-O/Fast Green staining, the sections were stained according to the manufacturer’s protocol (Sigma–Aldrich, Oakville, Ontario, Canada). Osteoarthritis Research Society International (OARSI) histological scoring was used to assess cartilage integrity of the samples (Glasson et al. 2010). The stained slides were evaluated blindly and independently by two individuals with expertise in evaluating slides using OARSI scoring system.
Immunohistochemistry for mouse cartilage
For mouse cartilage IHC studies, we used the Universal Dako Labelled Streptavidin-Biotin-2 System, Horseradish Peroxidase (LSAB2 System, HRP; DAKO, Burlington, ON, Canada). The samples were incubated with 3% hydrogen peroxide for 5 min at RT to block endogenous peroxide followed by blocking with bovine serum albumin (0.1%) in PBS for 1 h. Incubation with the anti-type II collagen antibody (Millipore, 1:500) was performed overnight at 4 °C. Next day, biotinylated link incubation for 30 min followed by streptavidin for 1 h was performed at RT. The DAB-substrate chromogen solution was added and the slides were counterstained with hematoxylin. For negative control, the primary antibody was omitted.
Mouse primary chondrocyte cell culture
Mouse primary chondrocytes from Eng+/− and Eng+/+ mice were isolated from femur head and knee joints as previously described (Monemdjou et al. 2012). Dissected articular cartilage was rinsed with PBS, then pronase digestion for 30 min at 37 °C followed by collagenase digestion (1 mg/ml in DMEM/F12) for 2 h at 37 °C. Supernatant was collected, centrifuged (4000 RPM for 5 min at RT) and the pellet was collected. The pellet was washed, re-suspended and plated into T25 flask with 10% FBS containing DMEM/F12 with antibiotics and grown at 37 °C until confluence. 1–2 passaged chondrocytes were used for the experiments. Conditioned media (proteins precipitated with cold ethanol) and cell lysates were analyzed using anti-endoglin (Santa Cruz) and type II collagen (Chemicon) levels by Western blot (described above). Membranes were stripped and reprobed with an anti-β-actin antibody (Santa Cruz Biotechnology) (loading control).
Statistical analysis
Numerical results are represented as means of n ≥ 4 independent experiments ± standard error of the mean (SEM). For statistical tests where only two data sets were being compared, a Student’s t test (two-tailed) was used where P < 0.05 was deemed statistically significant.
Results
Endoglin heterozygote mice are resistant to bleomycin-induced skin fibrosis
In the current study, we examined the role of endoglin in skin fibrosis using Eng+/− and Eng+/+ mice subjected to bleomycin-induced skin fibrosis. Figure 1 shows the genotyping result of Eng+/− mice where the primers amplified the recombinant product (476 bp) that was inserted to disrupt exon 1 of endoglin gene. This product was not detected in WT littermate control mice, as previously reported (Bourdeau et al. 1999). Figure 2a and b shows that Eng+/− mice treated with bleomycin display a marked decrease in dermal thickness (P < 0.005) as compared to Eng+/+ mice. Also, Eng+/− mice showed a thinner dermis under basal (PBS control injection) conditions as compared to wild-type littermates (P < 0.05). In addition, polarized light assessment of picrosirius red stained skin sections demonstrates that bleomycin-treated Eng+/− mice display reduced birefringence of polarized light suggesting decreased cross-linking (maturation) of collagen as compared to Eng+/+ mice (Fig. 2c). These data suggest that Eng+/− mice are more resilient to bleomycin-induced skin fibrosis than Eng+/+ mice.
Fig. 1.

Tail clips from each mouse were collected and DNA was extracted using a HotSHOT method (hot sodium hydroxide and tris) followed by PCR reaction, as described in Methods section. PCR products were resolved by agarose gel electrophoresis and visualized by SYBR safe DNA gel stain. Genotyping confirmed the presence of recombinant product (476 bp) confirming the presence of endoglin in heterozygote mice and its absence in the wild type littermates
Fig. 2.
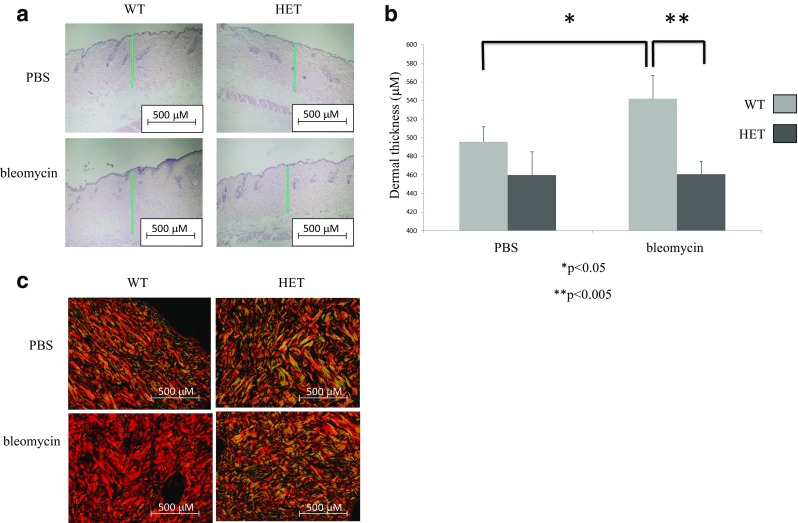
Endoglin heterozygote mice are resistant to bleomycin-induced skin fibrosis. a Hematoxylin and Eosin (H & E) staining of skin sections from endoglin heterozygote mice and wild-type littermates injected with bleomycin or PBS for 28 days. Vertical green line indicates a representative measurement of dermal thickness which was determined as the distance from the superficial aspect of the hypodermis to the epidermal–dermal junction at the edge of the wound. Scale bar = 500 μM. b Quantitative analysis indicates that endoglin heterozygote mice display a resistance to bleomycin-induced skin fibrosis (dermal thickening) as compared to wild-type littermates. *P < 0.05; **P < 0.005. c Picrosirius red staining of skin sections from endoglin heterozygote mice and wild-type littermates injected with bleomycin or PBS for 28 days. The bleomycin-induced increase in birefringence was markedly reduced in endoglin heterozygote mice as compared to their WT littermates
Endoglin heterozygote mice display diminished basal and bleomycin-induced ECM protein expression
The bleomycin-induced skin fibrosis mouse model is characterized by increased levels of ECM proteins (type I collagen and fibronectin) and myofibroblast differentiation (alpha smooth muscle actin expression) in the dermis (Vorstenbosch et al. 2013a). We therefore examined levels of these factors by IHC in skin sections from bleomycin- and PBS-injected Eng+/− and Eng+/+ mice. Figure 3 shows that Eng+/− mice treated with bleomycin show reduced levels of type I collagen, fibronectin and alpha-smooth muscle actin as compared to Eng+/+ mice. In addition, even in the absence of bleomycin challenge, Eng+/− mice showed a decrease in the ECM protein levels as compared to Eng+/+ mice. These results are consistent with the above findings that Eng+/− mice are resistant to bleomycin-induced skin fibrosis.
Fig. 3.

Endoglin heterozygote mice display diminished basal and bleomycin-induced ECM protein expression. Immunohistochemistry staining of skin sections from endoglin heterozygote mice and wild-type littermates injected with bleomycin or PBS for 28 days. Immunohistochemistry was performed using anti-type I collagen a anti-fibronectin b and anti-alpha smooth muscle actin c antibodies
Previous studies have shown that bleomycin-induced skin fibrosis is mediated by the stimulatory effects of TGF-β on ECM production by dermal fibroblasts (Lakos et al. 2004; Yamamoto and Nishioka 2002). We therefore examined whether endoglin haploinsufficiency affected TGF-β signaling and ECM production in dermal fibroblasts isolated from Eng+/− and Eng+/+ mice. The results indicate that primary dermal fibroblasts from Eng+/− and Eng+/+ mice show no difference in TGF-β-induced Smad2/3 or Smad1/5 phosphorylation, although type I collagen production is increased compared to Eng+/+ fibroblasts.
Endoglin haploinsufficiency does not alter proliferation rate or Smad phosphorylation but enhances plating efficiency of primary mouse dermal fibroblasts in vitro
We next examined proliferation of dermal fibroblasts isolated from Eng+/− and Eng+/+ mice. Although Eng+/− fibroblasts appeared to proliferate more quickly than Eng+/+ fibroblasts (Fig. 4a), the proliferation rates are similar for Eng+/− and Eng+/+ fibroblasts, as evidenced by fold-increase in cell number over time, when the cell count data are replotted as “proliferation rate” (Fig. 4b). We therefore reasoned that the observed differences between cell counts shown in Fig. 4a might be due to differences in plating efficiency (cell survival/apotosis or adhesion). To address this question, equivalent numbers of Eng+/− and Eng+/+ fibroblasts were seeded in 6-well plates and allowed to attach to the wells for 3 h. Cells were then detached by trypsin treatment and counted on a hemocytometer. Results shown in Fig. 4c indicate that Eng+/− fibroblasts display increased number of attached cells as compared to Eng+/+ fibroblasts. Results shown in Fig. 4d indicates that Eng+/− and Eng+/+ fibroblasts display similar levels of TGF-β-induced phosphorylation of Smad2 and Smad1.
Fig. 4.
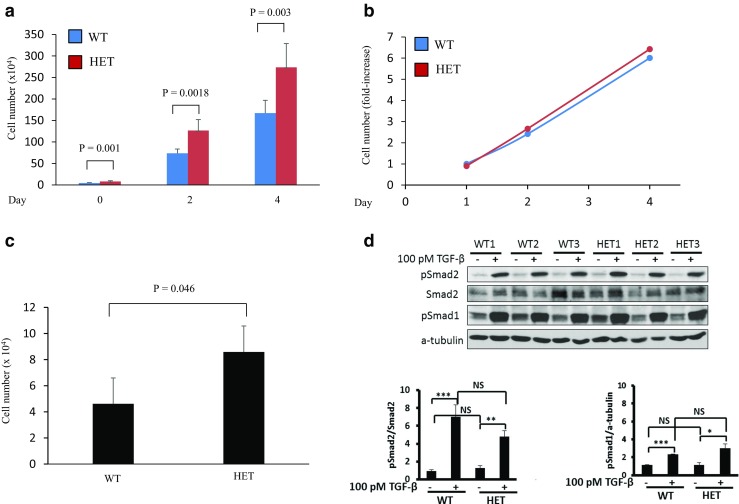
Endoglin haploinsufficiency does not alter proliferation rate but enhances plating efficiency of primary mouse dermal fibroblasts in vitro. a Primary dermal fibroblasts from endoglin heterozygote (Eng+/−) and wild-type (Eng+/+) littermates were seeded at 1 × 105 cells/well in 6-well plates. Cells were collected by trypsinization at the indicated time points (days) after plating and cell numbers counted using a haemocytometer. Results from three independent experiments were pooled and data are presented as cell number (mean ± SEM). b Data shown in (A) are presented as fold-increase (mean ± SEM). c Primary dermal fibroblasts from endoglin heterozygote (Eng+/−) and wild-type (Eng+/+) littermates were seeded at 1 × 105 cells/well in 6-well plates. Cells were harvested 3 h later and counted using a haemocytometer. Results from three independent experiments were pooled and data are presented as mean (±SEM) in cell number. (P = 0.046). d Western blot analysis shows that Eng+/− and Eng+/+ fibroblasts display similar levels of TGF-β-induced phosphorylation of Smad2 and Smad1 (NS = not significant), while TGF-β significantly induces Smad2 and Smad1 phosphorylation (mean ± SD) in both cell types (P < 0.05)
Eng+/− mice do not display any structural alterations in knee joints at 14 weeks after DMM surgery when compared to WT mice
Having determined the effect of endoglin haploinsufficiency on ECM synthesis and fibrotic responses during skin fibrosis, we examined whether endoglin haploinsufficiency alters ECM synthesis and cartilage repair during surgically-induced (by destabilization of medial meniscus) osteoarthritis. We subjected the 14-week old male Eng+/− and WT mice to DMM surgery to induce OA or were sham operated (as a control group) as described in the Experimental Design (Fig. 5a). Histological analysis was performed at 14 weeks post-DMM surgery. Our data demonstrate that there is no difference in proteoglycan content (as indicated in the Safranin-O-staining, red color), roughness of the articular cartilage, and articular chondrocyte cellularity between the knee joints of WT and Eng+/− mice in the control (sham operated) group (Fig. 5a). The knee joints of the WT mice from the DMM group (Fig. 5a, bottom panel) show the decreased proteoglycan content, decreased chondrocyte cellularity and increased roughness of the articular cartilage compared to the WT mice from the control group indicating that the mice subjected to the DMM surgery developed OA. The graphical representation of the evaluated sections using OARSI scoring system is represented in the Fig. 5c. We also evaluated sections for synovial fibrosis and both Eng+/− and WT mice show similar levels of synovial fibrosis (Fig. 5c).
Fig. 5.
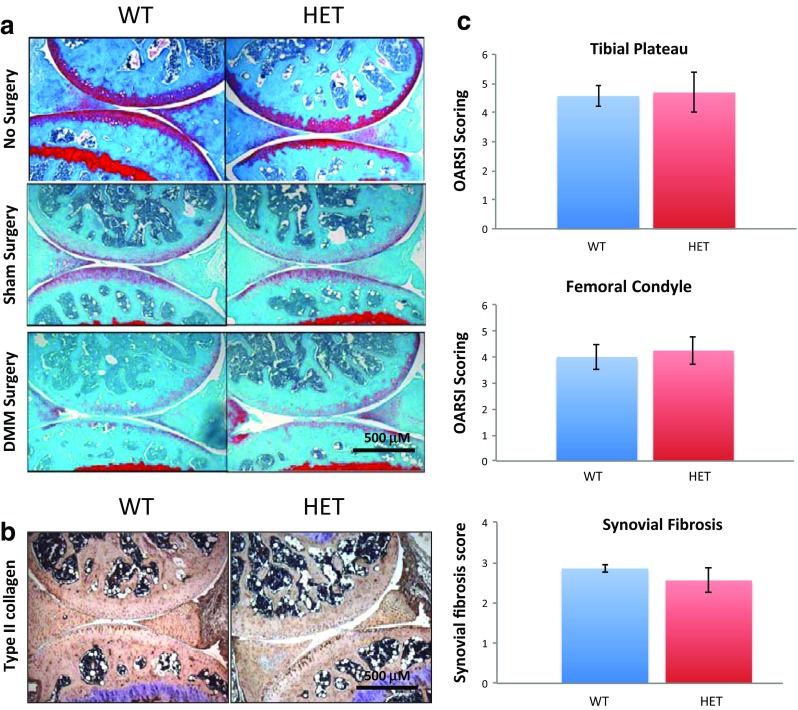
Endoglin haploinsufficiency has no effect on surgically-induced (DMM) osteoarthritis in mice. a Histological analysis using Safranin O/fast green staining of 14 weeks post-OA surgery knee joint sections demonstrate that Eng+/− mice and WT mice show similar degree of cartilage degradation when surgically challenged; b IHC results demonstrate that Eng+/− mice and WT mice show similar levels of type II collagen when surgically challenged; c Osteoarthritis Research Society International (OARSI) scoring of medial tibial plateau and medial femoral condyle from the surgically challenged WT and Eng+/− mice are performed by two individuals independently. Synovial fibrosis was evaluated and compared between WT OA and Eng+/− OA. Results shown are representative of minimum five mice
Endoglin heterozygote (Eng+/−) mice show the similar levels of type II collagen in knee joints as compared to their WT counterparts
Although the safranin-O-staining data do not show any apparent difference between Eng+/− mice and WT mice knee joints after the DMM surgery, we examined whether levels of type II collagen is altered in Eng+/− mice. 14-weeks old male mice knee joint were dissected followed by histological staining and assessment. Our data show that there are no differences in the levels of type II collagen between Eng+/− and WT knee joints (Fig. 5b).
Chondrocytes isolated from endoglin heterozygote (Eng+/−) mice show decreased endoglin levels as expected while showing increased type II collagen levels
As our results showed that Eng+/− mice display similar levels of type II collagen in knee joints as compared to their WT counterparts, we examined whether isolated chondrocytes show similar results in vitro. Chondrocytes were isolated from knee joint of both Eng+/− and WT mice and cultured, as described in the Method section. At confluence, both conditioned media and cell lysates were prepared and western blot analysis was performed using anti-endoglin and anti-type II collagen antibodies. Here, we observe that endoglin levels are decreased in Eng+/− compared to WT chondrocytes, as expected (Fig. 6). Importantly, both cell lysates and conditioned media from Eng+/− chondrocytes show increased levels of type II collagen, as compared to WT mouse chondrocytes while β-actin levels remain unchanged.
Fig. 6.

Endoglin heterozygote (Eng+/−) chondrocytes show increased type II collagen level as compared to wild-type chondrocytes in vitro. Primary mouse chondrocytes were isolated from articular cartilage of endoglin heterozygote and wild-type mice. Western blot analysis indicates that type II collagen protein levels are higher in cell lysate and conditioned media of chondrocytes from endoglin heterozygote mice as compared to WT littermates
Discussion
Endoglin is a TGF-β co-receptor that strongly modulates TGF-β signaling in a variety of cell types (Finnson et al. 2010; Lebrin et al. 2004; Morris et al. 2011). Whether endoglin plays a role in the fibrotic process in scleroderma or in the impaired cartilage repair in osteoarthritis has not been established. Also, it is not known whether endoglin differentially regulates ECM synthesis in the skin and cartilage. Determining this is important, as current evidence indicates that endoglin may enhance or inhibit the effects of TGF-β in a context dependent manner (Maring et al. 2012). Because endoglin knockout mice display an embryonic lethal phenotype, endoglin heterozygote (Eng+/−) mice have been used as a model system to study the effect of endoglin loss of function in vivo. In the current study, we show that Eng+/− mice subjected to bleomycin challenge show a marked decrease in dermal thickness (P < 0.005) and reduced ECM protein production as compared to Eng+/+ mice, both under basal and bleomycin treated conditions. Eng+/− mice undergoing surgically induced osteoarthritis show no differences in the degree of cartilage degradation, as compared to Eng+/+ mice, although chondrocytes isolated from Eng+/− display markedly enhanced collagen II levels. Our findings suggest that endoglin haploinsufficiency in mice ameliorates bleomycin-induced skin fibrosis suggesting that endoglin represents a pro-fibrotic factor in the mouse skin. However, endoglin haploinsufficiency does not protect these mice from cartilage degradation, demonstrating differential regulation of endoglin action during skin and cartilage repair.
That Eng+/− mice are resistant to bleomycin-induced skin fibrosis is evidenced by a decrease in dermal thickness, reduced collagen cross-linking (maturation) and lower ECM (type I collagen and fibronectin) and myofibroblast marker (alpha-smooth muscle actin) levels as compared to Eng+/+ mice subjected to bleomycin treatment. Whether the reduced collagen maturation in bleomycin treated Eng+/− mice reflects decreased activity of factors such as lysyl oxidases (LOXs), enzymes that catalyze the cross-linking of collagens and elastin remains to be determined (Trackman 2017). Interestingly, under basal (PBS treatment) conditions, Eng+/− mice display reduced ECM (type I collagen and fibronectin) protein levels in the dermis as compared to wild-type littermate, and appear to have a thinner dermis. Taken together, these results using the bleomycin-induced skin fibrosis model of scleroderma, suggest that endoglin acts as a pro-fibrotic factor and that decreasing its expression or function may represent a new strategy to reduce skin fibrosis in scleroderma. The marked decrease in the levels of ECM proteins observed in Eng+/− mice may represent decreased ECM protein synthesis and/or increased ECM protein degradation in the fibroblasts. Also, indirect effects of endoglin haploinsufficiency on other cell types such as endothelial cells or keratinocytes may contribute to the decreased fibrotic responses observed.
The mechanism by which endoglin decreases fibrotic responses during skin fibrosis remains to be delineated. Our in vitro studies using primary dermal fibroblasts from Eng+/− and Eng+/+ mice failed to show any differences in TGF-β-induced Smad2 or Smad1 phosphorylation. This is consistent with a previous report showing that dermal fibroblasts from Eng+/− and Eng+/+ mice show similar levels of Smad2/3 and Smad1/5 phosphorylation (Pericacho et al. 2013). Thus, it is possible that the profibrotic effects of endoglin may be mediated via TGF-β-dependent non-Smad pathways or TGF-β-independent mechanism(s) in the dermal fibroblasts in mice. In addition, the possibility that the results obtained in vitro, may not reflect the in vivo situation, cannot be ruled out.
A possible explanation for our results that primary fibroblasts isolated from Eng+/− mice display a higher plating efficiency as compared to Eng+/+ mice, is that Eng+/− fibroblasts may survive better than wild-type fibroblasts during the plating process. Along these lines, one study has shown that Eng+/− fibroblasts are more resistant to apoptosis induced by a combination of extrinsic (anti-Fas antibody) and intrinsic pathway (cycloheximide) activators and have enhanced Akt activation (survival pathway) as compared to wild-type fibroblasts (Pericacho et al. 2013). A second possibility for the increased plating efficiency of the Eng+/− fibroblasts is altered cell adhesion for which a role for endoglin in mouse fibroblasts has been well documented (Guerrero-Esteo et al. 1999).
Several studies have implicated endoglin in the pathogenesis of fibrosis in scleroderma patients but whether endoglin plays a causative role in the disease has not been established. Endoglin was shown to be upregulated on the endothelium of lesional skin of scleroderma patients as compared to normal skin with higher expression in the diffuse scleroderma subgroup (Dharmapatni et al. 2001). In addition, endoglin levels have been shown to be increased in scleroderma patient fibroblasts in vitro with its expression increasing with disease progression (Leask et al. 2002). More recently, Trojanowska et al. showed that endoglin promotes TGF-β/Smad1 signaling (a response not observed in Eng+/− mouse fibroblasts in the current study or Pericacho et al. 2013) and type I collagen and CCN2/CTGF expression in scleroderma patient fibroblasts revealing a pro-fibrotic role for endoglin in the human (Morris et al. 2011). Our results showing that endoglin haploinsufficiency confers resistance to bleomycin-induced skin fibrosis in mice is consistent with a role of endoglin as a pro-fibrotic factor.
TGF-β is an important anabolic factor in cartilage homeostasis (Blaney Davidson et al. 2007) and deregulation of TGF-β signaling is implicated in joint tissue diseases such as OA (Blaney Davidson et al. 2007). To determine whether endoglin plays a role in ECM synthesis and cartilage repair in vivo, we examined type II collagen and proteoglycan content in surgically-induced (DMM surgery) osteoarthritis in Eng+/− versus Eng+/+ mice. Our in vivo data showing a similar degree of cartilage degradation in the knee joints of the Eng+/− mice and Eng+/+ mice during surgically induced osteoarthritis, indicate that endoglin haploinsufficiency does not protect from cartilage degradation in mice. In addition, the immunohistochemistry data of type II collagen in Eng+/− versus WT knee joints suggest that endoglin does not alter the levels of type II collagen in mouse cartilage. This is in contrast to the results obtained with primary chondrocytes isolated from Eng+/− and Eng+/− mice and maintained in culture, which showed markedly enhanced type II collagen production in Eng+/− chondrocytes and Smad1/5 phosphorylation. This is consistent with our results in human chondrocytes in vitro which show that endoglin strongly inhibits type II collagen production while enhancing Smad 1/5 phosphorylation (Finnson et al. 2010). One possible explanation for the discrepancy between the effects of endoglin haploinsufficiency in vitro and in vivo, in the Eng+/− mice is that the mice may have developed compensatory mechanisms. Previous studies using other types of heterozygous mice have noted that such mice often develop compensatory mechanisms (Minamisawa et al. 1999; Wang et al. 2001). Development of a cartilage specific endoglin knockout mice is warranted to avoid this limitation.
Previously, our group has shown that in human chondrocytes in vitro, endoglin associates with TGF-β signaling receptors and enhances TGF-β1-induced Smad1/5 phosphorylation while decreasing TGF-β1-mediated Smad2 phosphorylation and ECM protein production (Parker et al. 2003). We have also shown that endoglin expression is linked to chondrocyte phenotype with dedifferentiated chondrocytes and osteoarthritic cartilage expressing higher levels of endoglin (Finnson et al. 2012, 2010). However, the mechanisms by which endoglin regulates TGF-β signaling pathway are highly cell-type specific and context dependent as evidenced by previous studies (Santibanez et al. 2007; Velasco et al. 2008) and thus may explain the discrepancy between the results obtained with chondrocytes in vitro and in vivo in Eng+/− mice.
In summary, we show that endoglin is a profibrotic factor during skin fibrosis in vivo in a mouse model of scleroderma where Eng+/− mice were compared to Eng+/+ mice. Our findings suggest that endoglin is a potetnenial target for anit-fibrotic theray in the skin. Our results on the role of endoglin in cartilage repair using a surgical model of OA where Eng+/− versus Eng+/+ mice were compared, demonstrate that endoglin haploinsufficiency does not protect mice from osteoarthritis. Unraveling the precise molecular mechanisms governing endoglin action in the skin and cartilage and elucidation of the discrepancy between the in vitro versus in vivo results will provide insight into the cellular processes underlying aberrant repair and fibrotic disease progression in those organs.
Acknowledgements
This study was supported by a Canadian Institutes of Health Research (CIHR) operating grant (FRN13732) to AP, a Research Award to AA from the King AbdulAziz University, Jeddah, Saudi Arabia, and a PhD studentship award to YC from FRQS, Quebec.
Compliance with Ethical Standards
Conflicts of Interest
The authors have no conflicts of interest.
Footnotes
Anas Alzahrani and Yoon Chi contributed equally to this work.
References
- Blaney Davidson E, van der Kraan P, van den Berg W. TGF-β and osteoarthritis. Osteoarthr Cartil. 2007;15:597–604. doi: 10.1016/j.joca.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. 1999;104:1343–1351. doi: 10.1172/JCI8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton CP (2015) Systemic sclerosis: from pathogenesis to targeted therapy. Clin Exp Rheumatol 33(4 Suppl 92):S3–S7 [PubMed]
- Desbois AC, Cacoub P. Systemic sclerosis: An update in 2016. Autoimmun Rev. 2016;15:417–426. doi: 10.1016/j.autrev.2016.01.007. [DOI] [PubMed] [Google Scholar]
- Dharmapatni AA, Smith MD, Ahern MJ, Simpson A, Li C, Kumar S, Roberts-Thomson PJ. The TGF-β receptor endoglin in systemic sclerosis. Asian Pac J Allergy Immunol. 2001;19:275–282. [PubMed] [Google Scholar]
- Finnson K, Tam B, Liu K, Marcoux A, Lepage P, Roy S, Bizet A, Philip A. Identification of CD109 as part of the TGF-β receptor system in human keratinocytes. FASEB J. 2006;20:E780–E795. doi: 10.1096/fj.05-5229fje. [DOI] [PubMed] [Google Scholar]
- Finnson KW, Parker WL, Chi Y, Hoemann C, Goldring MB, Antoniou J, Philip A. Endoglin differentially regulates TGF-β-induced Smad2/3 and Smad1/5 signalling and its expression correlates with extracellular matrix production and cellular differentiation state in human chondrocytes. Osteoarthr Cartil. 2010;18:1518–1527. doi: 10.1016/j.joca.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Finnson K, Chi Y, Bou-Gharios G, Leask A, Philip A. TGF-β signaling in cartilage homeostasis and osteoarthritis. Front Biosci. 2012;4:251–268. doi: 10.2741/s266. [DOI] [PubMed] [Google Scholar]
- Finnson K, McLean S, Di Guglielmo GM, Philip A. Dynamics of transforming growth factor-β signaling in wound healing and scarring. Advances in wound care. 2013;2:195–214. doi: 10.1089/wound.2013.0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthr Cartil. 2007;15:1061–1069. doi: 10.1016/j.joca.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthr Cartil. 2010;18(Suppl 3):S17–S23. doi: 10.1016/j.joca.2010.05.025. [DOI] [PubMed] [Google Scholar]
- Goumans MJ, Liu Z, Ten Dijke P. TGF-β signaling in vascular biology and dysfunction. Cell Res. 2009;19:116–127. doi: 10.1038/cr.2008.326. [DOI] [PubMed] [Google Scholar]
- Guerrero-Esteo M, Lastres P, Letamendia A, Perez-Alvarez MJ, Langa C, Lopez LA, Fabra A, Garcia-Pardo A, Vera S, Letarte M, Bernabeu C. Endoglin overexpression modulates cellular morphology, migration, and adhesion of mouse fibroblasts. Eur J Cell Biol. 1999;78:614–623. doi: 10.1016/S0171-9335(99)80046-6. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Moustakas A (2016) Signaling receptors for TGF-β family members. Cold Spring Harb Perspect Biol 8. 10.1101/cshperspect.a022053 [DOI] [PMC free article] [PubMed]
- Lafyatis R. Transforming growth factor β-at the centre of systemic sclerosis. Nat Rev Rheumatol. 2014;10:706–719. doi: 10.1038/nrrheum.2014.137. [DOI] [PubMed] [Google Scholar]
- Lakos G, Takagawa S, Chen S-J, Ferreira AM, Han G, Masuda K, Wang X-J, DiPietro LA, Varga J. Targeted disruption of TGF-β/Smad3 signaling modulates skin fibrosis in a mouse model of scleroderma. Am J Pathol. 2004;165:203–217. doi: 10.1016/S0002-9440(10)63289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A, Abraham DJ, Finlay DR, Holmes A, Pennington D, Shi-Wen X, Chen Y, Venstrom K, Dou X, Ponticos M, Black C, Bernabeu C, Jackman JK, Findell PR, Connolly MK. Dysregulation of transforming growth factor β signaling in scleroderma: overexpression of endoglin in cutaneous scleroderma fibroblasts. Arthritis Rheum. 2002;46:1857–1865. doi: 10.1002/art.10333. [DOI] [PubMed] [Google Scholar]
- Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M, Mummery C, Arthur HM, ten Dijke P. Endoglin promotes endothelial cell proliferation and TGF-β/ALK1 signal transduction. EMBO J. 2004;23:4018–4028. doi: 10.1038/sj.emboj.7600386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maring JA, Trojanowska M, Ten Dijke P. Role of endoglin in fibrosis and scleroderma. Int Rev Cell Mol Biol. 2012;297:295–308. doi: 10.1016/B978-0-12-394308-8.00008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamisawa S, Gu Y, Ross J, Jr, Chien KR, Chen J. A post-transcriptional compensatory pathway in heterozygous ventricular myosin light chain 2-deficient mice results in lack of gene dosage effect during normal cardiac growth or hypertrophy. J Biol Chem. 1999;274:10066–10070. doi: 10.1074/jbc.274.15.10066. [DOI] [PubMed] [Google Scholar]
- Monemdjou R, Vasheghani F, Fahmi H, Perez G, Blati M, Taniguchi N, Lotz M, St-Arnaud R, Pelletier JP, Martel-Pelletier J, Beier F, Kapoor M. Association of cartilage-specific deletion of peroxisome proliferator-activated receptor gamma with abnormal endochondral ossification and impaired cartilage growth and development in a murine model. Arthritis Rheum. 2012;64:1551–1561. doi: 10.1002/art.33490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa M, Derynck R, Miyazono K (2016) TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb Perspect Biol 8 [DOI] [PMC free article] [PubMed]
- Morris E, Chrobak I, Bujor A, Hant F, Mummery C, Ten Dijke P, Trojanowska M. Endoglin promotes TGF-β/Smad1 signaling in scleroderma fibroblasts. J Cell Physiol. 2011;226:3340–3348. doi: 10.1002/jcp.22690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura-Kitabayashi A, Anderson GA, Sleep G, Mena J, Karabegovic A, Karamath S, Letarte M, Puri MC. Endoglin is dispensable for angiogenesis, but required for endocardial cushion formation in the midgestation mouse embryo. Dev Biol. 2009;335:66–77. doi: 10.1016/j.ydbio.2009.08.016. [DOI] [PubMed] [Google Scholar]
- Parker WL, Goldring MB, Philip A. Endoglin is expressed on human chondrocytes and forms a heteromeric complex with betaglycan in a ligand and type II TGFβ receptor independent manner. J Bone Miner Res. 2003;18:289–302. doi: 10.1359/jbmr.2003.18.2.289. [DOI] [PubMed] [Google Scholar]
- Pericacho M, Velasco S, Prieto M, Llano E, Lopez-Novoa JM, Rodriguez-Barbero A. Endoglin haploinsufficiency promotes fibroblast accumulation during wound healing through Akt activation. PLoS One. 2013;8:e54687. doi: 10.1371/journal.pone.0054687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittie L. Another dimension to the importance of the extracellular matrix in fibrosis. J Cell Commun Signal. 2015;9:99–100. doi: 10.1007/s12079-015-0282-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santibanez J, Letamendia A, Perez-Barriocanal F, Silvestri C, Saura M, Vary C, Lopez-Novoa J, Liliana A, Bernabeu C. Endoglin increases eNOS expression by modulating Smad2 protein levels and Smad2-dependent TGF-β signaling. J Cell Physiol. 2007;210:456–468. doi: 10.1002/jcp.20878. [DOI] [PubMed] [Google Scholar]
- Trackman PC (2017) Functional importance of lysyl oxidase family propeptide regions. J Cell Commun Signal. 10.1007/s12079-017-0424-4 [DOI] [PMC free article] [PubMed]
- Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT) BioTechniques. 2000;29(52):54. doi: 10.2144/00291bm09. [DOI] [PubMed] [Google Scholar]
- Velasco S, Alvarez-Munoz P, Pericacho M, Ten Dijke P, Bernabeu C, Lopez-Novoa JM, Rodriguez-Barbero A. L- and S-endoglin differentially modulate TGF-β1 signaling mediated by ALK1 and ALK5 in L6E9 myoblasts. J Cell Sci. 2008;121:913–919. doi: 10.1242/jcs.023283. [DOI] [PubMed] [Google Scholar]
- Vorstenbosch J, Al-Ajmi H, Winocour S, Trzeciak A, Lessard L, Philip A. CD109 overexpression ameliorates skin fibrosis in mouse model of bleomycin-induced scleroderma. Arthritis Rheum. 2013;65:1378–1383. doi: 10.1002/art.37907. [DOI] [PubMed] [Google Scholar]
- Vorstenbosch J, Gallant-Behm C, Trzeciak A, Roy S, Mustoe T, Philip A. Transgenic mice overexpressing CD109 in the epidermis display decreased inflammation and granulation tissue and improved collagen architecture during wound healing. Wound Repair Regen. 2013;21:235–246. doi: 10.1111/wrr.12023. [DOI] [PubMed] [Google Scholar]
- Wang Q, Hummler E, Maillard M, Nussberger J, Rossier BC, Brunner HR, Burnier M. Compensatory up-regulation of angiotensin II subtype 1 receptors in alpha ENaC knockout heterozygous mice. Kidney Int. 2001;59:2216–2221. doi: 10.1046/j.1523-1755.2001.00739.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Nishioka K. Animal model of sclerotic skin. V: Increased expression of alpha-smooth muscle actin in fibroblastic cells in bleomycin-induced scleroderma. Clin Immunol. 2002;102:77–83. doi: 10.1006/clim.2001.5138. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Vasheghani F, Li YH, Blati M, Simeone K, Fahmi H, Lussier B, Roughley P, Lagares D, Pelletier JP, Martel-Pelletier J, Kapoor M. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann Rheum Dis. 2015;74:1432–1440. doi: 10.1136/annrheumdis-2013-204599. [DOI] [PubMed] [Google Scholar]