

Abstract
Single nucleotide polymorphism (SNP)-heritability estimation is an important topic in several research fields, including animal, plant and human genetics, as well as in ecology. Linear mixed model estimation of SNP-heritability uses the structures of genomic relationships between individuals, which is constructed from genome-wide sets of SNP-markers that are generally weighted equally in their contributions. Proposed methods to handle dependence between SNPs include, “thinning” the marker set by linkage disequilibrium (LD)-pruning, the use of haplotype-tagging of SNPs, and LD-weighting of the SNP-contributions. For improved estimation, we propose a new conceptual framework for genomic relationship matrix, in which Mahalanobis distance-based LD-correction is used in a linear mixed model estimation of SNP-heritability. The superiority of the presented method is illustrated and compared to mixed-model analyses using a VanRaden genomic relationship matrix, a matrix used by GCTA and a matrix employing LD-weighting (as implemented in the LDAK software) in simulated (using real human, rice and cattle genotypes) and real (maize, rice and mice) datasets. Despite of the computational difficulties, our results suggest that by using the proposed method one can improve the accuracy of SNP-heritability estimates in datasets with high LD.
Introduction
With the availability of genome-wide single nucleotide polymorphism (SNP) markers, researchers are now interested in estimating SNP-heritability/genomic heritability in animal, plant and human genetics, as well as in ecology (Visscher et al. 2006; Sillanpää 2011a; de los Campos et al. 2015). However, it is generally known that SNP-heritability estimation suffers from the missing heritability problem (Manolio et al. 2009; Eichler et al. 2010; Gibson 2012). That is, the quantitative traits that are known to have a substantial genetic component and a high heritability estimated in pedigree data sets show very low values of SNP-heritability: either when they are estimated using a few single SNPs showing most strong trait associations in genome-wide studies (Jakobsdottir et al. 2009), or estimated using genome-wide sets of SNP markers (Yang et al. 2010). However, recently, for some well studied human traits, partial concordance (60% of the pedigree value) has been reached (Yang et al. 2015; Kim et al. 2017). The origin of this missing heritability is currently the subject of heated debate. Possible explanations include loose associations between SNPs and causal variants (Shen 2013; de los Campos et al. 2013a), high fraction of causal effects may be due to rare variants (Zuk et al. 2014; Goldstein 2011), population admixture/structure (Zaitlen et al. 2014; Browning and Browning 2011), epistasis (Zuk et al. 2012; Hemani et al. 2013) and unaccounted haplotypes of common SNPs (Bhatia et al. 2015; Sun et al. 2016), all of which evidently have a role in the linkage disequilibrium (LD) pattern of the genome. Additionally, small sample sizes and gene-by-environment interactions may also be possible reasons.
SNP-heritability (the proportion of genetic factors explaining the total variance) is generally estimated under a linear mixed model (Henderson 1984), in which the random effect covariance structure between individuals is replaced with a sample covariance matrix estimated from genome-wide sets of SNP-markers. Here, we call this model the genomic best linear unbiased prediction (G-BLUP) model (Meuwissen et al. 2001; Habier et al. 2007; VanRaden 2008).
It is also possible to estimate SNP-heritability by fitting thousands of SNPs simultaneously to the whole-genome regression (WGR) model and applying variable selection for them (Meuwissen et al. 2001). This process is analogous to performing the G-BLUP analysis with a trait-specific relationship matrix having own variance component for each SNP in the diagonal (Zhang et al. 2010; Resende et al. 2012; Shen et al. 2013). In general, such G-BLUP model is equivalent to one WGR model called generalized ridge regression model (Piepho et al. 2012; Shen et al. 2013; Strandén and Garrick 2009). In simulations, WGR models following the Bayesian alphabet (BayesA, Bayes B, BayesC, etc.) have shown improved performance over G-BLUP (de los Campos et al. 2013a, b). Moreover, WGR models are more interpretable and so it is possible to develop better priors which leads to the improved performance. However, in practice, the G-BLUP model has been adopted more often than the WGR model, due to its robust performance in varying scenarios including different genetic architectures (Wimmer et al. 2013). This fact is true despite the strong assumption of equal weighting of loci in the G-BLUP model. Additionally, there are various WGR models proposed (Conti and Witte 2003; Sillanpää and Bhattacharjee 2005; Malo et al. 2008; Tsai et al. 2008; Fridley and Jenkins 2010; Yang and Tempelman 2012; Yi et al. 2015) in order to account for strong LD in the data.
Both G-BLUP and WGR models rely on the LD between SNPs and QTLs. While many methods seek to increase statistical power by better modeling of the LD, in some cases, strong LD is a problem. For instance, when QTLs are in strong LD, using the unweighted genomic relationship matrix in G-BLUP can cause upward bias in the heritability estimation (Speed et al. 2012; Fernando et al. 2017; Legarra 2016). Moreover when QTLs are in heterogeneous regions with varying degree of LD between SNPs and QTLs in each, the heritability estimate can be biased (Yang et al. 2015; Gusev et al. 2013; Yang et al. 2017). Therefore, there is a need of correcting for LD in some way in G-BLUP and WGR models.
To cope with strong LD in the G-BLUP context, the following approaches have been proposed: (1) pruning or “thinning” the SNP set (Purcell et al. 2007), (2) finding and using haplotype tagging SNPs (Lin and Altman 2004; Meng et al. 2003) and (3) using the LD weighting (Speed et al. 2012). Even if human geneticists and ecologists consider SNP selection to be an option, animal and plant breeders are more or less omitting it due to its minor influence on the genomic breeding value estimates (Ober et al. 2012). In this study, our main focus is on LD-correction and LD-weighting. To improve SNP-heritability estimation from genotype data, Speed et al. (2012) utilized LD information to calculate better weights for the contribution of each SNP to the genomic relationship matrix (GRM). This approach was used to correct for uneven LD distribution between SNPs in regions in which causal variants lie. Instead of individual SNP weighting, we present a novel conceptual framework that utilizes the linkage disequilibrium pattern between SNPs to calculate the genomic relationship matrix.
Materials and methods
Let us consider the basic G-BLUP model:
| 1 |
where y is an n × 1 vector of phenotypic observations, β is a n × 1 vector of fixed (environmental) effects with design matrix X, g is a n × 1 vector of random genetic effects with design matrix Z and is a n × 1 vector of error terms, , where is the error variance. Let M be n × m the marker matrix (n is the number of individuals and m is the number of loci), for which the elements are coded as −1, 0, and 1 for the homozygote, heterozygote and the other homozygote genotype, respectively. Let the column i of matrix P contains the allele frequencies as the difference from 0.5 and multiplied by 2 (i.e., 2 (pi − 0.5), pi is the frequency of the second allele). Then, the unscaled genomic relationship matrix G0 can be calculated as:
| 2 |
Here, the subtraction of P from M provide more credit to the rare alleles. Following VanRaden (2008), the scaled genomic relationship matrix (G), hereafter described as VanRaden genomic relationship matrix (VanRaden G matrix), can be calculated as: G = G0/k, where the scaling parameter k = . This scaling makes the scale of G comparable to that of the additive genetic relationship matrix calculated from the pedigree.
Here, the computation of the elements of the genomic relationship matrix (genomic relationships between individuals based on the marker information) can be accomplished in many ways (Speed and Balding 2015). Most of the current genomic relationship matrix computation methods assume equal weighting of the markers.
Let us consider the unscaled genomic relationship matrix (G0) from Eq. (2): G0 = ZZ′. Here, Z = M − P, then G0 = (M − P)(M − P)′. Furthermore, let I be an identity matrix of order m (the number of markers). Then, G0 can be represented as:
| 3 |
Thus, the construction of the unscaled genomic relationship matrix can be observed to include the same weighting (the ones on the diagonal) for all markers. However, in a breeding population, there exists correlation between the loci due to linkage and various other factors. It is thus important to consider this LD covariance structure in the computation of the genomic relationship matrix.
One of the easiest ways to account for the LD structure in genomic relationship matrix calculation is to use the squared Mahalanobis distance (Mahalanobis 1936; De Maesschalck et al. 2000; Mitchell and Krzanowski 1985), which can take the covariance structure of SNPs into account. The concept of the Mahalanobis distance has been used in various fields, including bioclimatic modeling (Farber and Kadmon 2003) and outlier detection (Hodge and Austin 2004). Given a matrix S which contains the covariance structure of linkage disequilibrium (covariance of SNPs), then the LD-corrected genomic relationship matrix Gld0 can be calculated as:
| 4 |
Here, the right hand side of Eq. (4) represents the squared multivariate Mahalanobis distance between individuals. The Mahalanobis distance has the property of projecting measurements to the space where independence holds, and it measures a distance between the individuals therein (see appendix for details). Note that a density function of the multivariate normal distribution uses Mahalanobis distance and thus it is very commonly applied procedure in practice. To simplify the example analyses, we estimated the LD structure for each chromosome independently (i.e., we assumed there is no dependence between the different chromosomes) and merged them together to form a single block-diagonal matrix, which contains the LD covariance structure for all the chromosomes. VanRaden (2008) used a genome-wide scaling factor that is averaged across all SNPs. When the markers are assumed to be independent, this scaling factor can be seen as, 2pI (1 − pi), here, I is an identity matrix with the order of number of markers, p is a vector of allele frequncies. Thus, the scaled version of the LD-corrected genomic relationship matrix (Gld) is calculated as follows:
| 5 |
Here, matrix S contains the covariance structure of linkage disequilibrium pattern between the SNPs. Hence, in model (1), the random genetic effects can be assumed to follow a normal distribution as: , where is the genomic variance. The mixed model equation (Henderson 1984) for the model (1) is as follows:
| 6 |
Here, G corresponds to different GRMs (LD-corrected, VanRaden G matrix or LDAK) used in this study and . Scaling of GRM does not influence on the prediction accuracy of genomic breeding values but still influences on the variance components and therefore SNP-heritability estimates. We used the function kin.blup in R-package’rrBLUP’ (Endelman 2011) to solve Eq. (6) to estimate the genomic breeding values (g) and variance components (the function uses restricted maximum likelihood (Patterson and Thompson 1971) approach) using different GRMs with the G-BLUP model. For understanding the relationship between the genomic breeding values estimated using mixed model and the SNP effects estimated using WGR model (ridge regression) see Piepho et al. (2012).
See appendix for details of the different approaches to calculate the LD covariance for each marker pair in matrix S of Eq. (4).
Example analyses
To demonstrate the superiority of our new approach, we used the following publicly available datasets in maize, rice (real and simulated phenotype), mice, human (simulated phenotypes) and cattle (simulated phenotypes). Before the analysis, we removed the duplicated markers (which showed more than 99% correlation) and only retained SNPs with minimum allele frequency greater than 5%. We also compared our results to those obtained using the LDAK (version 4.9, Speed et al. 2012) and GCTA (Genome-wide Complex Trait Analysis, Yang et al. 2011) packages. LDAK uses LD weighting to improve the SNP-heritability estimation and genomic prediction accuracy whereas GCTA uses a different scaling than VanRaden approach (see Uemoto et al. 2015 for the differences in the scaling factor used by different methods). “The goal of the [LDAK weighting] is that the signal from each SNP is down weighted so that replication of its signal by neighboring SNPs can be compensated for” (Speed et al. 2012). VanRaden approach implicitly assumes SNP effects and allele frequencies are independent, which would be true under evolutionarily neutral model. GCTA implicitly assumes rare variants tend to have larger effects, consistent with a model of purifying/stabilizing selection. LDAK is available at www.ldak.org/ and GCTA under http://cnsgenomics.com/software/gcta/.
Simulated datasets
In order to show the superiority of our new approach, we simulated two datasets one with high LD (rice data) and another with relatively low LD (human data).
Simulated human dataset—low LD case
Simulated human phenotypes were generated using the GCTA package conditionally on real genotype data and selected 10% of the total loci (from the LD pruned subset) serving as QTLs, where the marker effects were generated from a standard normal distribution. For the simulation we obtained the human HapMap3 (available under www.sanger.ac.uk/resources/downloads/human/hapmap3.html) dataset and we selected population the Maasai in Kinyawa, Kenya (MKK) with 171 individuals for our study. To imitate low LD situation, we selected a subset of 3024 SNPs from the chromosome 22 based on LD pruning using the PLINK software. Here, the criterion for LD pruning was set so that we had about 3000 markers. Due to LD pruning the amount of LD in the dataset ended up being very low. Using the subset of SNPs we generated 100 simulation replicates of the phenotype with a heritability of 0.7 using the GCTA package.
Simulated rice dataset—high LD case
The rice data (following section we provide more details about the data set) showed very high LD and we used the 3290 SNPs from the second chromosome in order to simulate the phenotypes. Based on the subset of SNPs we created 100 simulation replicates of the phenotypes conditionally on real genotype data using the GCTA package with a heritability of 0.7. We selected 10% of the total loci (from the LD pruned subset) serving as QTLs and the marker effects were generated from a standard normal distribution.
Maize dataset
We used the maize’IBM 302 population’ (Sharopova et al. 2002; Lee et al. 2002), which was developed as part of the Maize Mapping Project, for the analysis. The population consists of 302 lines which had genotypes at 1252 simple sequence repeats (SSR) markers and phenotypes for the trait ’leaf greenness’. The dataset is available at http://archive.maizegdb.org/qtl-data.php. We selected this dataset because (1) it is an out-breeding population, (2) it was developed as a part of a mapping project and (3) its genetic map positions are available at high accuracy. Additionally, plotting the magnitude of LD against the genetic distance in the dataset resulted in a picture with a clear LD decay pattern (see Fig. 1) as was expected based on points 1 and 2 above. Hence, we used the maize’IBM 302 population’ to illustrate our LD decay approach together with the observed LD approach.
Fig. 1.
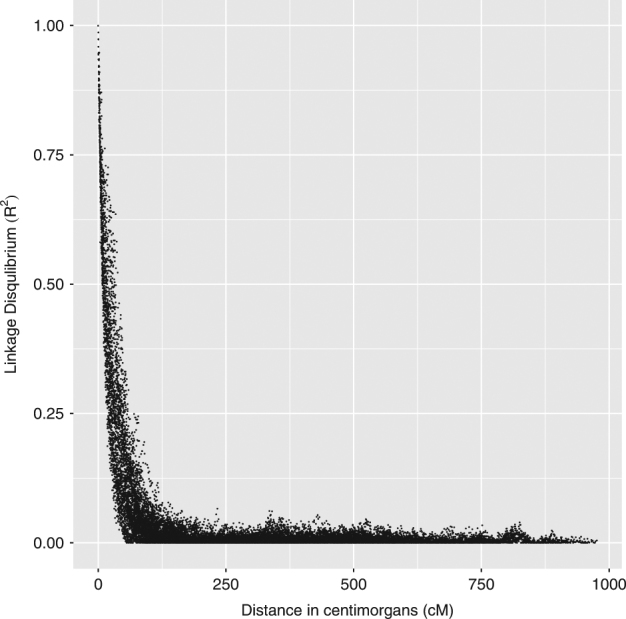
In maize, the linkage disequilibrium estimates (R2) between pairs of marker loci plotted against the genetic distance. To estimate the R2 values we used 128 polymorphic markers selected from the second chromosome
Rice dataset
This dataset consists of 413 diverse accessions of O. sativa (Zhao et al. 2011) collected from 82 different countries. The lines were genotyped using 36,901 SNP markers. Phenotypic information was available for 34 traits, and we analyzed the trait’amylose content’ in this study. The dataset is publicly available at http://www.ricediversity.org/data/. The phenotype information was missing for 20 lines, so the remaining 393 lines were used for our analysis. The computational complexity of estimating pairwise LD for all possible pairs of markers increases rapidly with increasing number of loci (even if it is being computed for each chromosome separately). Thus, after removing the monomorphic markers, we received 36,901 SNP markers in the rice dataset. Of these, we selected a subset of 3315 markers, which were as evenly distributed along the genome as possible, for the analysis. To ensure that we did not lose too much prediction accuracy in the smaller subset due to the marker selection, we calculated the correlation coefficient between the genome estimated breeding values (GEBVs) and the true phenotypes for the subset and the whole dataset using G-BLUP of VanRaden (VanRaden 2008). The obtained correlation coefficients were roughly comparable, at 0.86 and 0.88 for the subset (3315 SNPs) and the whole dataset (36,901 SNPs), respectively. We also examined possible exponential patterns of LD decay from the dataset but were unable to find any clear indications of this (see Fig. 2). Rice is a self-pollinating plant and moreover there is an admixture between populations in this dataset as reported by Zhao et al. (2011). So as shown in Fig. 2, it was apparent that there is no clear exponential decay of LD in the dataset. Therefore, we used this dataset to validate our observed LD approach only.
Fig. 2.
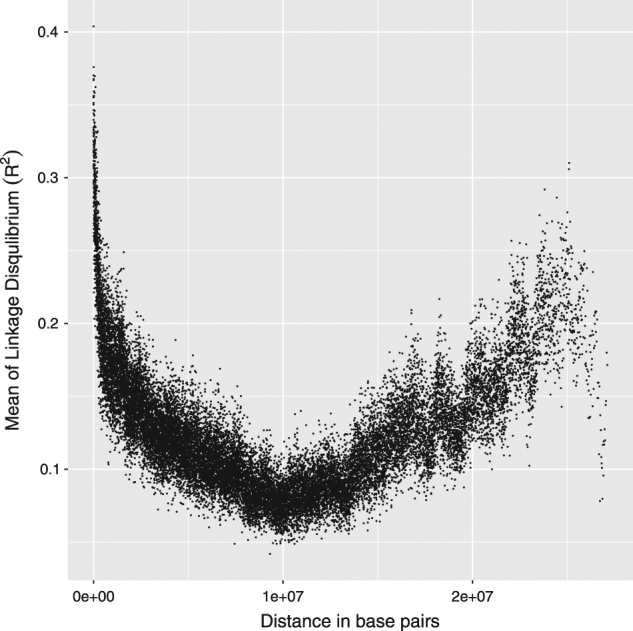
The mean LD decay plot of 2165 SNP markers selected from the 12th chromosome of the rice dataset. Here, the X-axis represents the distance in base pairs, and the Y-axis corresponds to the mean of pair wise linkage disequilibrium estimate (R2) values from a bin length of 100
Mice dataset
We selected datasets from animal studies to illustrate our new approach in the context of animal genetics. This dataset contains data from a heterogeneous stock mouse population (Valdar et al. 2006a) of 2527 individuals. In this population 1940 individuals are genotyped with 12,545 biallelic SNP markers. In this study, we concentrated on the trait ’body weight’, which was measured at the age of 6 weeks. Due to the computational complexity, for the mice dataset, we selected a subset of 2336 markers out of the total 12,545 markers. The selected markers were equally distributed along the genome. Then, we obtained the prediction accuracies for the subset (r = 0.77) and the whole dataset (r = 0.80) using G-BLUP of VanRaden (VanRaden 2008) approach, which indicated that the prediction accuracy was not significantly lower for the subset. The genotype and phenotype information of the individuals are available from http://mus.well.ox.ac.uk/GSCAN/index.shtml/.
Cattle dataset
This dataset is publicly available as a part of the R package’SynbreedData’ (Wimmer et al. 2015). In this dataset, 500 bulls were genotyped using 7250 SNP markers. The phenotype was generated using simulation and the pedigree relationships between the animals are known for the dataset. Thus, it is possible to compare the heritability estimates from the pedigree-based genetic relationships under the infinite locus model to that of marker-based genomic relationships under the finite locus model. For the cattle dataset, we took a subset of 3998 SNPs, which were equally distributed along the genome, for the analysis. Here, also we looked at the prediction accuracies for the subset and whole dataset. These were r = 0.83 and r = 0.80, respectively, according to the G-BLUP of VanRaden approach.
Results
Our primary interest was to illustrate the improved SNP-heritability estimation of our new Mahalanobis distance based LD-corrected genomic relationship matrix using the restricted maximum likelihood (REML) method. Additionally, we wanted to show the improved out-sample prediction accuracy of the LD-corrected genomic relationship matrix in the context of genome-enabled breeding value prediction using G-BLUP approach. To achieve these goals, we calculated the correlation coefficient (r) between the GEBVs and the true phenotypes and compared their values to those provided by the G-BLUP of VanRaden approach (VanRaden 2008). The results are presented in the following section.
SNP-heritability
In a recent study, Speed et al. (2012) pointed out the importance of accounting for LD while estimating the narrow-sense heritability using genome-wide SNP markers. Subsequently, they proposed a weighting approach for calculating an LD-corrected genomic relationship matrix for improved genomic heritability estimation. To this end, we compared our approaches to their approach to determine whether our Mahalanobis distance-based LD-corrected genomic relationship matrices could improve the heritability estimates using genome-wide markers. The narrow-sense SNP-heritabilities (h2) were estimated using our LD-corrected genomic relationship matrix in the G-BLUP model as: h2 = . Here, and are the genomic and residual variances, respectively. In following, GCTA and LDAK estimates were obtained using GCTA and LDAK packages respectively, whereas the other estimates were obtained using’rrBLUP’ package.
Estimation accuracy of SNP heritability in simulated data sets
Analysis of human simulated data
In order to compare the estimation accuracy of SNP heritability in human simulated data, first we estimated the narrow-sense heritability using 100 simulation replicates with different approaches. Figure 3 summarizes the box plots for the estimation errors (difference between the true and estimated heritability values) to visualize the estimation accuracy of different methods. Here, Y-axis corresponds to the differences between the true simulated and the estimated SNP heritability, whereas the X-axis corresponds to different GRM estimation methods. Based on results from Fig. 3, one can conclude that in the low LD situation, our observed LD approach provided same estimation accuracy like the competing methods (VanRaden and GCTA). Moreover, the estimation accuracy of LDAK was slightly better than that of our approach, VanRaden and GCTA. This is likely due to the fact that LDAK has been optimized for low LD situations.
Fig. 3.
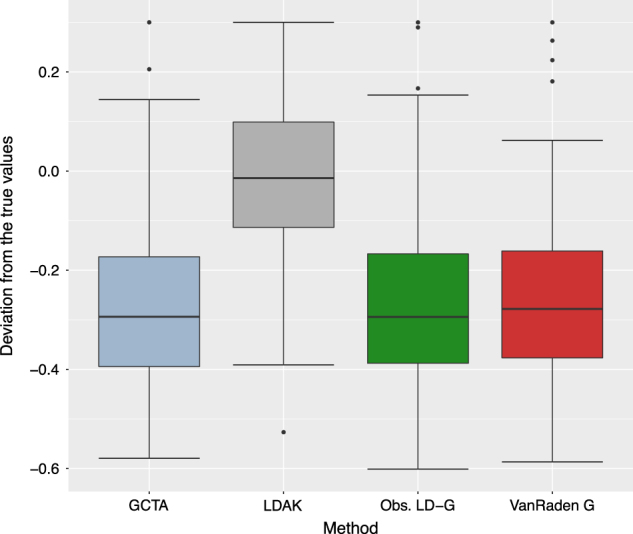
Low LD case: Box plots for the estimation error of heritability based on different approaches to calculate the genomic relationship matrix (GRM) using 100 simulation replicates with the human data. Here the Y-axis scale corresponds to the difference between the true simulated heritability and the estimated heritability values whereas X-axis corresponds to the different approaches to calculate the GRM
Analysis of rice simulated data
Figure 4 summarizes the box plots for the estimation errors (difference between the true and estimated heritability values) to visualize the estimation accuracy of different methods with the rice data, which corresponds to high LD situation. Here, Y-axis corresponds to the differences between the true simulated and the estimated SNP heritability, whereas the X-axis corresponds to different GRM estimation methods. From Fig. 4, one can conclude that in the presence of high LD, our observed LD approach provided better estimation accuracy than the competing methods (VanRaden, GCTA, and LDAK). Thus our approach seems to work better in presence of high LD.
Fig. 4.
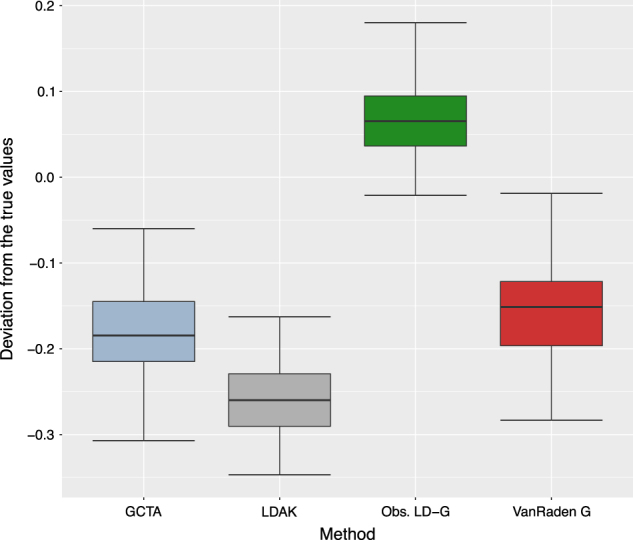
High LD case: Box plots for the estimation error of heritability based on different approaches to calculate the genomic relationship matrix (GRM) using 100 simulation replicates with the rice genotypes. Here the Y-axis scale corresponds to the difference between the true simulated heritability and the estimated heritability values whereas X-axis corresponds to the different approaches to calculate the GRM
SNP heritability in other datasets
Of the 302 lines in the maize dataset phenotype information for the trait ’leaf greenness’ was available for 270 lines. We also observed a clear exponential decay of LD (Fig. 1) in the dataset. We then used an expected LD decay-based approach (as a function of the genetic distance) to estimate the LD-corrected genomic relationship matrix. As shown in Fig. 1, the LD appeared to vanish rapidly after 20 cM. Therefore, in Eq. (7), we used different values of λ (between 20 and 30) to characterize the LD covariance structure. Following Eqs. (4) and (5), we calculated the scaled LD-corrected genomic relationship matrix (Gld) for each λ. We then estimated the correlation coefficient (r) between the GEBVs and the true phenotypes based on our new Gld matrices (for different λ values). We chose λ, which gave the highest correlation (for whole data set) and was at λ = 26.
The exponential decay of LD may be slightly different for different chromosomes, and finding the optimal λ is difficult and time-consuming. Therefore, we also used our observed LD-based approach to estimate the LD-corrected genomic relationship matrix. First, we calculated the pairwise LD estimates (D) for all marker pairs on each chromosome using the R-package called ‘genetics’ (Warnes and Leisch 2006) (note that some of the LD estimates were negative, and we kept those values unchanged). We then merged all chromosome-specific LD matrices together to form the LD covariance structure matrix S for the whole genome and estimated the unscaled LD-corrected genomic relationship matrix (Gld0) using Eq. (4), before scaling it using Eq. (7). By applying the REML estimation to our exponential LD decay based genomic relationship matrix, G-BLUP provided an estimated genomic heritability of 0.16, with = 3.67 and = 19.46. By contrast, the observed LD-corrected matrix led to an even higher SNP-heritability estimate of 0.22, with = 4.70 and = 16.51. The SNP-heritability estimate using a common genomic relationship matrix was approximately 0.14, with a genetic variance of 3.17 and a residual variance of 19.73. Similarly, the LDAK based SNP-heritability estimate was 0.28, with = 7.30 and = 18.66, whereas, the GCTA provided a heritability estimate of 0.14 with, = 3.14 and = 19.79. The high SNP-heritability estimate of LDAK for maize dataset, where degree of LD is low, may be due to the default settings of weighting parameters in LDAK which are optimized especially for low-LD datasets.
The rice population did not show any signs of an exponential LD decay pattern (Fig. 2), so we only used the observed LD-corrected genomic relationship matrix for the estimation of genomic heritabilities using the G-BLUP model. For the rice dataset where degree of LD is high, estimates from our approach were close to VanRaden approach. Whereas the SNP-heritability estimate of LDAK were lower than estimates of the other methods. This may be due to the fact that LDAK weighting parameters are tuned for the human datasets and these parameters may not be optimal for high-LD datasets. For the mice data, the pedigree-based heritability estimate (obtained using additive genetic relationship matrix calculated from the pedigree) provides natural comparison point for genomic heritability estimates. The genomic heritability 0.63 for mice subset of observed LD-G approach was more close to the pedigree-based estimate 0.74 as reported by Valdar et al. (2006b). The estimates of other approaches were lower (0.50–0.59). Also in cattle data, the genomic heritability 0.27 of observed LD-G approach was again the highest and most close to the pedigree-based estimate 0.41. The other approaches obtained lower values. Table 1 summarizes the genomic heritability estimates obtained using our LD-corrected and other (VanRaden, LDAK, and GCTA) genomic relationship matrices in G-BLUP model with different datasets.
Table 1.
The REML-estimated variance components and narrow-sense SNP-heritability estimates of the four G-BLUP mixed models (with differing genomic matrices) for the maize, rice, mice and cattle datasets
| Type of matrix | Maize | Rice | Mice | Cattle | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| h 2 | h 2 | h 2 | h 2 | |||||||||
| VanRaden G matrix | 3.17 | 19.73 | 0.14 | 0.39 | 0.46 | 0.46 | 8.12 | 5.68 | 0.59 | 48.68 | 193.90 | 0.20 |
| LDAK weighted G | 7.30 | 18.66 | 0.28 | 0.34 | 0.52 | 0.40 | 6.14 | 6.25 | 0.50 | 50.19 | 192.02 | 0.21 |
| GCTA | 3.14 | 19.79 | 0.14 | 0.38 | 0.49 | 0.43 | 7.01 | 6.04 | 0.54 | 51.25 | 190.67 | 0.21 |
| Expected LD-G | 3.50 | 19.46 | 0.15 | — | — | — | — | — | — | — | — | — |
| Observed LD-G | 4.7 | 16.51 | 0.22 | 0.36 | 0.42 | 0.46 | 9.81 | 5.68 | 0.63 | 60.27 | 186.20 | 0.27 |
| Pedigree based | — | — | — | — | — | — | — | — | 0.74 | 99.55 | 142.80 | 0.41 |
Additionally, similar estimates, based on the pedigree-derived additive genetic relationship matrices are given for the mice and cattle datasets
Genomic prediction
We also performed a genomic prediction (out-sample prediction) using five-fold cross-validation (Stone 1974) with four (rice, maize, mice and cattle) datasets. For the five-fold cross-validation (CV), we used 80% of the data points as the training set and the remaining 20% as the validation set. To remove the influence of random partitions on the accuracy, we repeated the cross-validation procedure 500 times and took the mean value; Table 2 summarizes these results. Our observed LD-based genomic relationship matrix provided better prediction accuracies for all datasets expect that of the maize population. It is already known that population structure has a profound effect on the genomic prediction accuracies. Our principal component analysis (PCA) (Jolliffe 2002) based plots (Fig. 5) indicated the presence of population structures for rice, mice and cattle datasets. So we also estimated the out-sample prediction based on 500 CV replicates by including the first three principal components in the model for the rice dataset, in order to assess the effect of population structure on prediction accuracy. However, this approach did not show any improvement to the prediction accuracy in rice dataset. We also applied stratified random sampling to assess the impact of the population structure on the genomic prediction accuracies with our datasets. To this end, we estimated the subpopulations using the k-means clustering with the R-package’adegenet’ (Jombart 2008). Then, from each subpopulation we selected 20% observations and formed the validation set. However, the subpopulation-based cross-validation with the different genomic relationship matrices (common G, LDAK weighted G and observed LD-G) did not show any improvement in the genomic prediction accuracies (results are not shown) compared to the random sampling cross-validation results (Table 2). Habier et al. (2007) showed that the presence of close relatives in the training dataset may inflate the genomic prediction accuracies. So we also checked for the proportion of close relatives in our real datasets by plotting the distribution of the upper off-diagonal elements of the genomic relationship matrix (Fig. 6). The plot for the rice population is bimodal so that many individuals have genomic relationship values close to −1, which may indicate the recent admixture in the population. From Fig. 6 it can be concluded that, close relatives are present in the rice dataset while maize, mice and cattle populations seem to be more unrelated.
Table 2.
Average out-sample prediction accuracy (measured as the correlation between the GEBVs and the true phenotypes) of four G-BLUP mixed models (with differing genomic matrices) determined by 5-fold cross-validation for the maize, rice, mice and cattle datasets calculated over 500 random partitions of data
| Type of matrix | Maize | Rice | Mice | Cattle |
|---|---|---|---|---|
| VanRaden G matrix | 0.22 | 0.44 | 0.50 | 0.18 |
| LDAK weighted G | 0.24 | 0.41 | 0.52 | 0.18 |
| Expected LD-G | 0.23 | — | — | — |
| Observed LD-G | 0.23 | 0.45 | 0.53 | 0.19 |
Fig. 5.

Population structure plot for the maize, rice, mice and cattle datasets. This scatter plot presents the first two principal components (PC1 and PC2) with each point representing a single individual. In the rice dataset, the individuals that do not clearly belong to any of the three original populations (corners) are considered to represent the out group—admixed individuals, and the corresponding points in the plot are indicated with a red color
Fig. 6.

Histograms of the upper off-diagonal elements of the genomic relationship matrix calculated using VanRaden (2008) approach in maize, rice, mice, and cattle datasets
The PCA plot based on the rice genotype information indicated the presence of an admixed population (marked in red in Fig. 5), which was already reported by Zhao et al. (2011). So we decided to compare the genomic prediction accuracies for the admixed population with different genomic relationship matrices. For that, we selected only the lines belonging to the admixed population as the validation set and obtained genomic prediction accuracies using 10-fold cross validation (note that there were insufficient amount of observations in the admixed population to perform 20% cross-validation, so we used 10% cross-validation) with 100 re-samples. In this case, our observed LD-based genomic relationship matrix showed improvement with a genomic prediction accuracy (r) of 0.38 compared to VanRaden G matrix (r = 0.32) and LDAK weighted G (r = 0.32).
Discussion
LD is the fundamental basis of association mapping studies as well as that of “Genomic selection” (Meuwissen et al. 2001). Moreover, LD plays a crucial role in evolutionary biology and provides information about the population history and the response to selection. Many studies have pointed out the importance of understanding the patterns and distributions of LD in humans (Flint-Garcia et al. 2003) as well as in other species, including animals (Qanbari et al. 2010) and plants (Ardlie et al. 2002). Meanwhile, it is evident that the pattens of LD in a population are strongly dependent on the population substructure (e.g, Gilad et al. 2002; Fricano et al. 2012; Chen et al. 2012). There are various methods proposed to correct for the population structure in association mapping studies (Yu et al. 2006; Kang et al. 2008; Sillanpää 2011b). However, less focus has been given to the importance of accounting for population structures in the estimation of genomic breeding values and heritability. Also, the relationship between epistasis and LD needs to be clarified in the estimation of genomic breeding values and heritability (e.g, Phillips 2008, Hemani et al. 2014). Arguably, LD can be considered a confounding factor along with the epistasis, population structure and genetic admixture.
In this study, a new approach to construct the genomic relationship matrix in the presence of strong dependence between collected SNPs is presented. This approach is based on, first estimating the pairwise observed LD from the marker data and then correcting the LD away from the SNPs using a Mahalanobis distance based formulation of the genomic relationship matrix. Based on our presented analyses, this method seems to be especially helpful for SNP-heritability estimation, but it also provides improvement for genomic prediction accuracy in the presence of population structure (because many of our tested datasets included strong population substructures). Our results also support substantial improvements for genomic prediction accuracy in populations that have experienced recent events of population admixture. This result is in contrast to the large number of other studies in which disappointingly low genomic prediction accuracies have been observed for admixed individuals (e.g., Vallee et al. 2014; Hidalgo et al. 2015). Why is genomic prediction accuracy usually much lower for admixed or multi-breed populations? In brief, if two populations in Hardy-Weinberg proportions and with divergent allele frequencies are analyzed together, as is performed in multi-breed genomic evaluations, the combined (or admixed) population may have a large amount of LD simply due to the process of combination (e.g., Ewens and Spielman 1995). This LD may then cause false positive signals for some loci, which do not have any connection to the studied trait in question. In the following section, we briefly discuss about population structure, which is mainly co-founded with the pattern of LD in the genome.
To study the impact and to correct for the population structure in genomic prediction as well as in the estimation of genomic heritability, de los Campos et al. (2010), Janss et al. (2012) and Guo et al. (2014), have considered the eigenvalue decomposition of G-matrix, which can be utilized to reparametrize the G-BLUP model to the form of principal component regression, in which the population structure can be separated and controlled. We argue that the population structure problem is essentially the LD problem in the data, and our observed LD-corrected G-BLUP model may be capable of doing something similar to principal component regression by transforming the estimated pairwise LD patterns away from the G-BLUP model. Our example with the rice data, in which a strong population admixture is present, suggests this type of behavior. However, sufficient validation of this claim will need to be completed in subsequent research.
In a conclusion, we hope that our method will open new avenues for SNP-heritability estimation as well as genomic prediction. Despite the good results, our method seems to generate more questions than answers. Moreover, unlike the competing methods our new approach is not able to include hundreds of thousands of markers in SNP heritability estimation due to the computational burden: (1) to estimate the pairwise LD for all marker pairs, and (2) to invert the LD structure matrix S of Eq. (4). In our example analysis, the pairwise LD calculation of the rice data with 393 lines and 3315 markers took around 90 min. For fast inversion of large S matrix, one could consider having non-zero LD values only at nearby markers or markers within a certain window/band, resulting in tridiagonal, five-diagonal, or banded matrix, where sparse matrix inversion techniques could be applied. Of course, it is very likely that other methods together with hundreds of thousands of SNPs may outperform our method with much smaller number of SNPs. Even though, there is a clear trade off between accuracy of the heritability estimation and the computation time, we still believe that our new proposed conceptual framework is able to provide the basis for GRM development towards improved SNP-heritability estimation.
Acknowledgements
We thank Doug Speed for the suggestions and comments which helped us to improve our manuscript and answering questions about LDAK package.
Appendix
Mahalanobis distance
We try to give insight here what is happening in Mahalanobis distance. It is important to note that multiplication with matrix inversion is kind of a similar operation than standardization (division with a square root of variance) for a single variable. First, let us consider normally distributed multivariate data with covariance matrix S, , where is a scaling factor. Then, consider Cholesky factorization for symmetric covariance matrix S = LL′ where L is a lower diagonal (or square root) matrix which is also called as a Cholesky factor of S. Squared Mahalanobis distance of Eq. (4) in the main text can then be represented using Cholesky factors as:
Here T = L−1(M − P)′ represent the transformed variables and T′T the squared Euclidean distance between transformed variables. In other words, the Mahalanobis distance accounts covariance between variables by transforming the data into uncorrelated form and then computing the ordinary Euclidean distance for it.
Different methods to calculate the LD between markers
Expected LD decay approach
In a random mating population, the magnitude of LD due to the physical linkage (with a close linkage of the two loci) depends on the exponential decay on the distance between the two loci. However, the LD may also be due to reasons other than the close linkage of the loci, such as selection, drift or population events. Therefore, exponential decay may not accurately describe all possible LD in the data. The genetic map distance between the loci is measured in Morgans and can be converted to recombination frequencies using map functions (e.g., the Haldane function). We therefore used the map position in an exponential function to estimate the covariance structure of the LD on each chromosome. Given, S, a matrix representing the covariance structure of the linkage disequilibrium, then the covariance (i, j) elements of S can be calculated as follows:
| 7 |
Here, dij is the map position or distances between (i, j) in Morgans and λ is the smoothing parameter controlling the rate of LD decay. The parameter λ is highly population specific, and the optimal λ can be chosen by cross-validation. Thus, we select the λ that gives the highest prediction accuracy.
Observed LD approach
The decay of the LD is affected by many factors, such as gene flow, genetic drift and selfing. In self-pollinating crops, LD decay is delayed due to selfing. The population history also has a tremendous impact on the patterns of LD (Pritchard and Przeworski 2001). Additionally, the presence of haplotype blocks (Daly et al. 2001; Patil et al. 2001; Wall and Pritchard 2003) makes the patterns of LD highly unpredictable and noisy. Thus, the expected decay of LD may not be true in all the populations under consideration. We therefore used the observed distribution of LD instead of the theoretical (exponential decay) distribution to obtain the covariance structure of LD, for populations that deviate from the exponential decay of LD. The amount of LD (D), between alleles (A and B) at two neighboring loci can be expressed as:
| 8 |
Here, PAB and PAPB are the observed and expected haplotype frequencies, respectively. The coefficient of LD (D) is a standard measure of LD and captures the extent of non-random allelic association between two loci. Different variants of D, including the standardized version (D′, Lewontin 1964) and a measure of the correlation coefficient (r2, Hill and Robertson 1968), have been proposed to quantify LD, to capture different attributes of nonrandom association. However, LD is a complex phenomenon and no single statistic can capture it (Hedrick 1987; Lewontin 1988; Slatkin 1994). All of these variants are directly related to D, so in our study, we used estimated D values to represent the covariance structure of LD between markers. For the calculation of the LD covariance structure (S) matrix, we first calculated the coefficient of disequilibrium for all marker pairs on each chromosome separately and then merged the values together in such a way that the LD is always zero between markers on different chromosomes.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no competing interests.
Footnotes
Conflict of interest
The authors declare that they have no competing interests.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Ardlie KG, Kruglyak L, Seielstad M. Patterns of linkage disequilibrium in the human genome. Nat Rev Genet. 2002;3:299–309. doi: 10.1038/nrg777. [DOI] [PubMed] [Google Scholar]
- Bhatia G. et al. Haplotypes of common SNPs can explain missing heritability of complex diseases. Preprint at bioRxiv 10.1101/022418 (2016)
- Browning SR, Browning BL. Population structure can inflate SNP-based heritability estimates. Am J Hum Genet. 2011;89:191–193. doi: 10.1016/j.ajhg.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Min D, Yasir TA, Hu YG. Genetic diversity, population structure and linkage disequilibrium in elite chinese winter wheat investigated with SSR markers. PLoS ONE. 2012;7:e44510. doi: 10.1371/journal.pone.0044510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti DV, Witte JS. Hierarchical modeling of linkage disequilibrum: genetic structure and spatial relations. Am J Hum Genet. 2003;72:351–363. doi: 10.1086/346117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES. High-resolution haplotype structure in the human genome. Nat Genet. 2001;29:229–232. doi: 10.1038/ng1001-229. [DOI] [PubMed] [Google Scholar]
- de los Campos G, Gianola D, Rosa GJ, Weigel KA, Crossa J. Semi-parametric genomic-enabled prediction of genetic values using reproducing kernel Hilbert spaces methods. Genet Res. 2010;92:295–308. doi: 10.1017/S0016672310000285. [DOI] [PubMed] [Google Scholar]
- de los Campos G, Hickey JM, Pong-Wong R, Daetwyler HD, Calus MP. Whole-genome regression and prediction methods applied to plant and animal breeding. Genetics. 2013;193:327–345. doi: 10.1534/genetics.112.143313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de los Campos G, Sorensen D, Gianola D. Genomic heritability: what is it? PLoS Genet. 2015;11:e1005048. doi: 10.1371/journal.pgen.1005048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de los Campos G, Vazquez AI, Fernando R, Klimentidis YC, Sorensen D. Prediction of complex human traits using the genomic best linear unbiased predictor. PLoS Genet. 2013;9:e1003608. doi: 10.1371/journal.pgen.1003608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maesschalck R, Jouan-Rimbaud D, Massart DL. The Mahalanobis distance. Chemom Intell Lab Syst. 2000;50:1–18. doi: 10.1016/S0169-7439(99)00047-7. [DOI] [Google Scholar]
- Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endelman JB. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome. 2011;4:250–255. doi: 10.3835/plantgenome2011.08.0024. [DOI] [Google Scholar]
- Ewens WJ, Spielman RS. The transmission/disequilibrium test: history, subdivision, and admixture. Am J Hum Genet. 1995;57:455. doi: 10.1002/ajmg.1320570319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber O, Kadmon R. Assessment of alternative approaches for bioclimatic modeling with special emphasis on the Mahalanobis distance. Ecol Modell. 2003;160:115–130. doi: 10.1016/S0304-3800(02)00327-7. [DOI] [Google Scholar]
- Fernando R, Cheng H, Sun X, Garrick D. A comparison of identity-by-descent and identity-by-state matrices that are used for genetic evaluation and estimation of variance components. J Animal Breed Genet. 2017;134:213–223. doi: 10.1111/jbg.12275. [DOI] [PubMed] [Google Scholar]
- Flint-Garcia SA, Thornsberry JM, IV B. Structure of linkage disequilibrium in plants. Ann Rev Plant Biol. 2003;54:357–374. doi: 10.1146/annurev.arplant.54.031902.134907. [DOI] [PubMed] [Google Scholar]
- Fricano A, Bakaher N, Del Corvo M, Piffanelli P, Donini P, Stella A, et al. Molecular diversity, population structure, and linkage disequilibrium in a worldwide collection of tobacco (Nicotiana tabacum L.) germplasm. BMC Genet. 2012;13:1. doi: 10.1186/1471-2156-13-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridley BL, Jenkins GD. Localizing putative markers in genetic association studies by incorporating linkage disequilibrium into Bayesian hierarchical models. Hum Hered. 2010;70:63–73. doi: 10.1159/000313852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G. Rare and common variants: twenty arguments. Nat Rev Genet. 2012;13:135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y, Rosenberg S, Przeworski M, Lancet D, Skorecki K. Evidence for positive selection and population structure at the human MAO-A gene. Proc Natl Acad Sci U S A. 2002;99:862–867. doi: 10.1073/pnas.022614799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DB. The importance of synthetic associations will only be resolved empirically. PLoS Biol. 2011;9:e1001008. doi: 10.1371/journal.pbio.1001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Tucker DM, Basten CJ, Gandhi H, Ersoz E, Guo B, et al. The impact of population structure on genomic prediction in stratified populations. Theor Appl Genet. 2014;127:749–762. doi: 10.1007/s00122-013-2255-x. [DOI] [PubMed] [Google Scholar]
- Gusev A, Bhatia G, Zaitlen N, Vilhjalmsson BJ, Diogo D, Stahl EA, et al. Quantifying missing heritability at known GWAS loci. PLoS Genet. 2013;9:e1003993. doi: 10.1371/journal.pgen.1003993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habier D, Fernando R, Dekkers J. The impact of genetic relationship information on genome-assisted breeding values. Genetics. 2007;177:2389–2397. doi: 10.1534/genetics.107.081190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick PW. Gametic disequilibrium measures: proceed with caution. Genetics. 1987;117:331–341. doi: 10.1093/genetics/117.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemani G, Knott S, Haley C. An evolutionary perspective on epistasis and the missing heritability. PLoS Genet. 2013;9:e1003295. doi: 10.1371/journal.pgen.1003295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemani G, Shakhbazov K, Westra HJ, Esko T, Henders AK, McRae AF, et al. Detection and replication of epistasis influencing transcription in humans. Nature. 2014;508:249–253. doi: 10.1038/nature13005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Henderson CR, 1984: Applications of Linear Models in Animal Breeding. University of Guelph, Guelph, ON, Canada.
- Hidalgo AM, Bastiaansen JW, Lopes MS, Harlizius B, Groenen MA, de Koning DJ. Accuracy of predicted genomic breeding values in purebred and crossbred pigs. G3. 2015;5:1575–1583. doi: 10.1534/g3.115.018119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill W, Robertson A. Linkage disequilibrium in finite populations. Theor Appl Genet. 1968;38:226–231. doi: 10.1007/BF01245622. [DOI] [PubMed] [Google Scholar]
- Hodge VJ, Austin J. A survey of outlier detection methodologies. Artif Intell Rev. 2004;22:85–126. doi: 10.1023/B:AIRE.0000045502.10941.a9. [DOI] [Google Scholar]
- Jakobsdottir J, Gorin MB, Conley YP, Ferrell RE, Weeks DE. Interpretation of genetic association studies: markers with replicated highly significant odds ratios may be poor classifiers. PLoS Genet. 2009;5:e1000337. doi: 10.1371/journal.pgen.1000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janss L, de Los Campos G, Sheehan N, Sorensen D. Inferences from genomic models in stratified populations. Genetics. 2012;192:693–704. doi: 10.1534/genetics.112.141143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolliffe IT (2002) Principal Component Analysis, second edn, Springer Series in Statistics, NewYork
- Jombart T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24:1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, et al. Efficient control of population structure in model organism association mapping. Genetics. 2008;178:1709–1723. doi: 10.1534/genetics.107.080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Grueneberg A, Vazquez AI, Hsu S, de los Campos G, et al. Will big data close the missing heritability gap? Genetics. Genetics. 2017;207(3):1135–1145. doi: 10.1534/genetics.117.300271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Sharopova N, Beavis WD, Grant D, Katt M, Blair D, et al. Expanding the genetic map of maize with the intermatedb73× mo17 (ibm) population. Plant Mol Biol. 2002;48:453–461. doi: 10.1023/A:1014893521186. [DOI] [PubMed] [Google Scholar]
- Legarra A. Comparing estimates of genetic variance across different relationship models. Theor Popul Biol. 2016;107:26–30. doi: 10.1016/j.tpb.2015.08.005. [DOI] [PubMed] [Google Scholar]
- Lewontin R. The interaction of selection and linkage. i. general considerations; heterotic models. Genetics. 1964;49:49–67. doi: 10.1093/genetics/49.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin R. On measures of gametic disequilibrium. Genetics. 1988;120:849–852. doi: 10.1093/genetics/120.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Altman RB. Finding haplotype tagging SNPs by use of principal components analysis. Am J Hum Genet. 2004;75:850–861. doi: 10.1086/425587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalanobis PC. On the generalized distance in statistics. Proc Natl Inst Sci. 1936;2:49–55. [Google Scholar]
- Malo N, Libiger O, Schork NJ. Accommodating linkage disequilibrium in genetic-association analyses via ridge regression. Am J Hum Genet. 2008;82:375–385. doi: 10.1016/j.ajhg.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Zaykin DV, Xu CF, Wagner M, Ehm MG. Selection of genetic markers for association analyses, using linkage disequilibrium and haplotypes. Am J Hum Genet. 2003;73:115–130. doi: 10.1086/376561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen THE, Hayes BJ, Goddard ME. Prediction of total genetic value using genome-wide dense marker maps. Genetics. 2001;157:1819–1829. doi: 10.1093/genetics/157.4.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AF, Krzanowski WJ. The Mahalanobis distance and elliptic distributions. Biometrika. 1985;72:464–467. doi: 10.1093/biomet/72.2.464. [DOI] [Google Scholar]
- Ober U, Ayroles JF, Stone EA, Richards S, Zhu D, Gibbs RA, et al. Using whole-genome sequence data to predict quantitative trait phenotypes in Drosophila melanogaster. PLoS Genet. 2012;8:e1002685. doi: 10.1371/journal.pgen.1002685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil N, Berno AJ, Hinds DA, Barrett WA, Doshi JM, Hacker CR, et al. Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science. 2001;294:1719–1723. doi: 10.1126/science.1065573. [DOI] [PubMed] [Google Scholar]
- Patterson D, Thompson R. Recovery of inter-block information when block sizes are unequal. Biometrika. 1971;58:545–554. doi: 10.1093/biomet/58.3.545. [DOI] [Google Scholar]
- Phillips PC. Epistasis the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet. 2008;9:855–867. doi: 10.1038/nrg2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piepho H, Ogutu J, Schulz-Streeck T, Estaghvirou B, Gordillo A, Technow F. Efficient computation of ridge-regression best linear unbiased prediction in genomic selection in plant breeding. Crop Sci. 2012;52:1093–1104. doi: 10.2135/cropsci2011.11.0592. [DOI] [Google Scholar]
- Pritchard JK, Przeworski M. Linkage disequilibrium in humans: models and data. Am J Hum Genet. 2001;69:1–14. doi: 10.1086/321275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qanbari S, Pimentel E, Tetens J, Thaller G, Lichtner P, Sharifi A, et al. The pattern of linkage disequilibrium in German Holstein cattle. Anim Genet. 2010;41:346–356. doi: 10.1111/j.1365-2052.2009.02011.x. [DOI] [PubMed] [Google Scholar]
- Resende MF, Muñoz P, Resende MD, Garrick DJ, Fernando RL, Davis JM, et al. Accuracy of genomic selection methods in a standard data set of loblolly pine (Pinus taeda L.) Genetics. 2012;190:1503–1510. doi: 10.1534/genetics.111.137026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharopova N, McMullen MD, Schultz L, Schroeder S, Sanchez-Villeda H, Gardiner J, et al. Development and mapping of SSR markers for maize. Plant Mol Biol. 2002;48:463–481. doi: 10.1023/A:1014868625533. [DOI] [PubMed] [Google Scholar]
- Shen X. The curse of the missing heritability. Front Genet. 2013;4:225. doi: 10.3389/fgene.2013.00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Alam M, Fikse F, Rönnegård L. A novel generalized ridge regression method for quantitative genetics. Genetics. 2013;193:1255–1268. doi: 10.1534/genetics.112.146720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillanpää MJ. On statistical methods for estimating heritability in wild populations. Mol Ecol. 2011;20:1324–1332. doi: 10.1111/j.1365-294X.2011.05021.x. [DOI] [PubMed] [Google Scholar]
- Sillanpää MJ. Overview of techniques to account for confounding due to population stratification and cryptic relatedness in genomic data association analyses. Heredity. 2011;106:511–519. doi: 10.1038/hdy.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillanpää MJ, Bhattacharjee M. Bayesian association-based fine mapping in small chromosomal segments. Genetics. 2005;169:427–439. doi: 10.1534/genetics.104.032680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M. Linkage disequilibrium in growing and stable populations. Genetics. 1994;137:331–336. doi: 10.1093/genetics/137.1.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speed D, Balding DJ. Relatedness in the post-genomic era: is it still useful? Nat Rev Genet. 2015;16:33–44. doi: 10.1038/nrg3821. [DOI] [PubMed] [Google Scholar]
- Speed D, Hemani G, Johnson MR, Balding DJ. Improved heritability estimation from genome-wide SNPs. Am J Hum Genet. 2012;91:1011–1021. doi: 10.1016/j.ajhg.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone M. Cross-validatory choice and assessment of statistical predictions. J Royal Stats Soc B. 1974;36:111–147. [Google Scholar]
- Strandén I, Garrick D. Derivation of equivalent computing algorithms for genomic predictions and reliabilities of animal merit. J Dairy Sci. 2009;92:2971–2975. doi: 10.3168/jds.2008-1929. [DOI] [PubMed] [Google Scholar]
- Sun X, Su H, Garrick DJ. Improved accuracy of across-breed genomic prediction using haplotypes in beef cattle populations. Animal Ind Rep. 2016;662:26. [Google Scholar]
- Tsai MY, Hsiao C, Wen SH. A Bayesian spatial multimarker genetic random-effect model for fine-scale mapping. Ann Hum Genet. 2008;72:658–669. doi: 10.1111/j.1469-1809.2008.00459.x. [DOI] [PubMed] [Google Scholar]
- Uemoto Y, Sasaki S, Kojima T, Sugimoto Y, Watanabe T. Impact of QTL minor allele frequency on genomic evaluation using real genotype data and simulated phenotypes in japanese black cattle. BMC Genet. 2015;16:134. doi: 10.1186/s12863-015-0287-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdar W, Solberg LC, Gauguier D, Burnett S, Klenerman P, Cookson WO, et al. Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat Genet. 2006;38:879–887. doi: 10.1038/ng1840. [DOI] [PubMed] [Google Scholar]
- Valdar W, Solberg LC, Gauguier D, Cookson WO, Rawlins JNP, Mott R, et al. Genetic and environmental effects on complex traits in mice. Genetics. 2006;174:959–984. doi: 10.1534/genetics.106.060004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee A, van Arendonk J, Bovenhuis H (2014) Accuracy of genomic prediction using two admixed crossbred populations. In: 10th World Congress on Genetics Applied to Livestock Production. Asas.
- VanRaden P. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–4423. doi: 10.3168/jds.2007-0980. [DOI] [PubMed] [Google Scholar]
- Visscher PM, Medland SE, Ferreira MA, Morley KI, Zhu G, Cornes BK, et al. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet. 2006;2:e41. doi: 10.1371/journal.pgen.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD, Pritchard JK. Haplotype blocks and linkage disequilibrium in the human genome. Nat Rev Genet. 2003;4:587–597. doi: 10.1038/nrg1123. [DOI] [PubMed] [Google Scholar]
- Warnes G, Leisch F (2006). genetics: population genetics. R Package, version 1.2. 1.
- Wimmer V, Albrecht T, Auinger HJ (2015) R Package synbreeddata.
- Wimmer V, Lehermeier C, Albrecht T, Auinger HJ, Wang Y, Schön CC. Genome-wide prediction of traits with different genetic architecture through efficient variable selection. Genetics. 2013;195:573–587. doi: 10.1534/genetics.113.150078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Bakshi A, Zhu Z, Hemani G, Vinkhuyzen AA, Lee SH, et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat Genet. 2015;47:1114–1120. doi: 10.1038/ng.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zeng J, Goddard ME, Wray NR, Visscher PM. Concepts, estimation and interpretation of SNP-based heritability. Nat Genet. 2017;49:1304–1310. doi: 10.1038/ng.3941. [DOI] [PubMed] [Google Scholar]
- Yang W, Tempelman RJ. A Bayesian antedependence model for whole genome prediction. Genetics. 2012;190:1491–1501. doi: 10.1534/genetics.111.131540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi H, Breheny P, Imam N, Liu Y, Hoeschele I. Penalized multimarker vs. single-marker regression methods for genome-wide association studies of quantitative traits. Genetics. 2015;199:205–222. doi: 10.1534/genetics.114.167817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Pressoir G, Briggs WH, Bi IV, Yamasaki M, Doebley JF, et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet. 2006;38:203–208. doi: 10.1038/ng1702. [DOI] [PubMed] [Google Scholar]
- Zaitlen N, Pasaniuc B, Sankararaman S, Bhatia G, Zhang J, Gusev A, et al. Leveraging population admixture to characterize the heritability of complex traits. Nat Genet. 2014;46:1356–1362. doi: 10.1038/ng.3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Liu J, Ding X, Bijma P, de Koning DJ, Zhang Q. Best linear unbiased prediction of genomic breeding values using a trait-specific marker-derived relationship matrix. PLoS ONE. 2010;5:e12648. doi: 10.1371/journal.pone.0012648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Tung CW, Eizenga GC, Wright MH, Ali ML, Price AH, et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun. 2011;2:467. doi: 10.1038/ncomms1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A. 2012;109:1193–1198. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk O, Schaffner SF, Samocha K, Do R, Hechter E, Kathiresan S, et al. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci U S A. 2014;111:E455–E464. doi: 10.1073/pnas.1322563111. [DOI] [PMC free article] [PubMed] [Google Scholar]