

Abstract
Phosphodiesterase 4 (PDE4) is a promising target for the treatment of Parkinson's disease (PD). However, the underlying mechanism has not yet been well elucidated. Additionally, most of current PDE4 inhibitors produce severe nausea and vomiting response in patients, which limit their clinical application. FCPR16 is a novel PDE4 inhibitor with little emetic potential. In the present study, the neuroprotective effect and underlying mechanism of FCPR16 against cellular apoptosis induced by 1-methyl-4-phenylpyridinium (MPP+) were examined in SH-SY5Y cells. FCPR16 (12.5–50 μM) dose-dependently reduced MPP+-induced loss of cell viability, accompanied by reductions in nuclear condensation and lactate dehydrogenase release. The level of cleaved caspase 3 and the ratio of Bax/Bcl-2 were also decreased after treatment with FCPR16 in MPP+-treated cells. Furthermore, FCPR16 (25 μM) significantly suppressed the accumulation of reactive oxygen species (ROS), prevented the decline of mitochondrial membrane potential (Δψm) and attenuated the expression of malonaldehyde level. Further studies disclosed that FCPR16 enhanced the levels of cAMP and the exchange protein directly activated by cAMP (Epac) in SH-SY5Y cells. Western blotting analysis revealed that FCPR16 increased the phosphorylation of cAMP response element-binding protein (CREB) and protein kinase B (Akt) down-regulated by MPP+ in SH-SY5Y cells. Moreover, the inhibitory effects of FCPR16 on the production of ROS and Δψm loss could be blocked by PKA inhibitor H-89 and Akt inhibitor KRX-0401. Collectively, these results suggest that FCPR16 attenuates MPP+-induced dopaminergic degeneration via lowering ROS and preventing the loss of Δψm in SH-SY5Y cells. Mechanistically, cAMP/PKA/CREB and Epac/Akt signaling pathways are involved in these processes. Our findings indicate that FCPR16 is a promising pre-clinical candidate for the treatment of PD and possibly other oxidative stress-related neuronal diseases.
Keywords: Phosphodiesterase 4, FCPR16, Oxidative stress, Mitochondrial membrane potential, Parkinson's disease
Graphical abstract
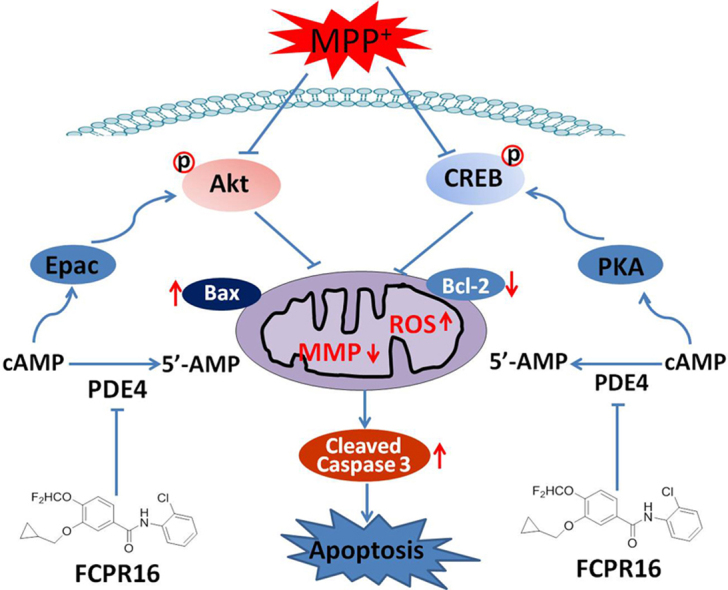
Highlights
-
•
FCPR16 protected SH-SY5Y cells against MPP+-induced apoptosis.
-
•
FCPR16 attenuated Δψm loss and ROS generation in SH-SY5Y cells treated with MPP+.
-
•
FCPR16 activated cAMP/PKA/CREB and Epac/Akt signaling pathways in SH-SY5Y cells.
-
•
Blocking cAMP/PKA/CREB or Epac/Akt pathways canceled the protective role of FCPR16.
1. Introduction
Parkinson's disease (PD) is a chronic neurodegenerative disorder caused by progressive dopaminergic neuronal death in the substantia nigra pars compacta within the midbrain [1]. The loss of dopaminergic neurons and dopamine storage in the striatum leads to movement disorder. Non-motor symptoms (such as progressive impairment of cognitive and sleep behavior disorder) are also frequently reported in PD patients [2], [3]. Currently, therapies for PD (such as rehabilitation, dopamine precursor, dopamine agonists and anti-cholinergic agents) can relieve the symptoms. However, there is no treatment available to halt or slow the dopaminergic cell death [2], [4]. On the other hand, although current medications provide symptom relief for a few years, many of these drugs produce unwanted side effects (such as levodopa-induced dyskinesias, on-off phenomenon, wearing off, hallucinations and delusions) that have not been well resolved [5]. The complicated pathology of PD and the lack of enduring therapies continue to be major limitations in the treatment of PD. This situation has motivated researchers to investigate novel targets and approaches [6]. In other words, studies identifying neuroprotective compounds for PD are still of high priority and urgently needed. Although the etiology of PD is poorly understood, dopaminergic neuronal apoptosis induced by enhanced oxidative stress in the brain is considered as one of the major contributors during the development of PD, especially in sporadic PD [7], [8]. Oxidative stress reflects an imbalance between excessive production of reactive free radical and deficits in antioxidant biosystem. The mitochondria are the main source of reactive oxygen species (ROS) and overproduction of intracellular ROS is usually elicited under the condition of mitochondrial dysfunction [9]. In the brain, overproduced ROS destroy the structure of neuronal cell membrane and impair the biological functions of lipids, proteins and DNA, which eventually trigger the apoptosis of neurons [9]. Specifically, in the development of PD, free radicals interact with several proteins involved in the pathology of PD (such as α-synuclein, and tau protein) and contribute to neuronal damage [10], [11], [12]. Multiple signaling pathways, including phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) and protein kinase A/cAMP response element-binding protein (PKA/CREB) pathways, are involved in the dopaminergic cell damage mediated by oxidative stress [8], [13], [14]. Oxidative stress can act as an initial trigger or is involved in the development of PD. Hence, neuroprotective agents which could block the oxidative stress-induced dopaminergic neuronal damage are supposed to be helpful to prevent the progress of PD.
Phosphodiesterase 4 (PDE4) inhibitors are potent and promising neuroprotectants against neurodegenrative diseases, mental disorders and acute brain injuries [15], [16], [17]. Our previous studies showed that inhibition of PDE4 by rolipram is effective to reverse Aβ-induced cognitive impairment and neuronal apoptosis in rats [18], and the neuroprotective effect of rolipram may be due to the antioxidative effects, as evidenced by the decreased level of ROS, and increased activity of antioxidant enzymes in mice treated with rolipram [19]. As for PD, PDE4 is highly expressed in the basal ganglia in the brain [20], and administration of PDE4 selective inhibitors has been shown to have protective effects against MPP+-induced neuronal loss in nigral neurons [21]. PDE4 inhibitor rolipram has also been shown to attenuate dopamine depletion in the striatum, and promote the survival of tyrosine hydroxylase-positive neurons in the substantia nigra in a PD animal model [22]. While the mechanism responsible for the protective effect of PDE4 inhibition against dopaminergic neuronal apoptosis is not well understood. Inhibition of PDE4 leads to the enhanced intracellular level of cAMP, which subsequently activates PKA and the exchange factor directly activated by cAMP (Epac) [23]. Activated PKA phosphorylates CREB at Ser133 and promotes its transcriptional activity [24]. Epac promotes the phosphorylation of Akt at Ser473 and increases its activity [25]. Dephosphorylation of CREB and Akt are involved in neuronal cell death and neurodegenerative disorders, including PD [26], [27]. Currently, PDE4 inhibitors have been viewed as potential agents for the treatment of neurodegenrative diseases [15], [23]. However, most of the available PDE4 inhibitors exert unpleasant and serious side effects, such as emesis and nausea, which hinder its clinical application [28]. Rolipram, a prototypic PDE4 inhibitor, was viewed as a potential neuroprotective agent, but rolipram therapy was poorly tolerated due to its side effects (nausea, vomiting and insomnia), which make it failure in the phase 2 clinical trials [29], [30]. So, further efforts are needed before PDE4 inhibitors with high therapeutic indices are available for treatment of neurodegenrative diseases, including PD.
FCPR16 is a novel selective PDE4 inhibitor synthesized in our lab [31], the chemical structure of FCPR16 is shown in Fig. 1A. The compound was designed based on following considerations: firstly, the catechol motif is responsible for binding to the glutamine residue at the PDE4 catalytic site, and this motif is widely found in a range of PDE4 inhibitors, including rolipram and roflumilast [31]. Hence, FCPR16 contains the catechol motif. Secondly, to get high selective PDE4 inhibitors, we referred to the design of FCPE07, another selective PDE4 inhibitor synthesized in our lab [32]. N-(2-chlorophenyl)formamide side chain in FCPR16 was designed and synthesized to improve the activity and selectivity. FCPR16 shows higher PDE4 inhibitory activity (IC50 = 90 nM) compared with canonical PDE4 inhibitor rolipram (IC50= 550 nM). We also found that FCPR16 had protective effects against cerebral ischemia-reperfusion injury through inhibiting inflammation and neuronal apoptosis in the brain in rats [33]. Most importantly, FCPR16 has low emetogenic potential, Our previous studies showed that oral administration of FCPR16 (3 mg/kg) did not induce vomiting in beagle dogs [33]. FCPR16 displays at least 600-fold selectivity for PDE4B and 1111-fold selectivity for PDE4D over other PDEs (PDE1-3 and PDE5-11) [31], while PDE4B is the most abundant isoform in the basal ganglia and PDE4D expression is also observed in the basal ganglia [34]. Based on these findings, we are interested to investigate the protective effect of FCPR16 in PD model.
Fig. 1.

FCPR16 suppressed MPP+-induced cell viability lose in SH-SY5Y cells. (A)The chemical structure of FCPR16. (B) SH-SY5Y cells were treated with various concentration (3.1–50 μM) of FCPR16 or 0.1% DMSO (vehicle control) for 1 h and then followed by incubation with MPP+ (500 μM) for 48 h and cell viability was measured using the CCK-8 assay. (C) Cells were pre-treated with FCPR16 (25 μM) or 0.1% DMSO (vehicle control) for 1 h and then incubated with or without 500 μM MPP+ for 48 h and the activity of lactate dehydrogenase (LDH) was detected by LDH cytotoxicity assay kit. (D) Cells were exposed to MPP+ (500 μM) in the absence and presence of FCPR16 for 48 h, apoptotic cells were determined by Hoechst assay, Bar = 200 µm. (E) Quantification of apoptotic cells, the number of apoptotic nuclei with condensed chromatin was counted from the photomicrographs and presented as a percentage of the total number of nuclei. Results are shown as the mean ± SD and represent three independent experiments. n = 3. ##P < 0.01, ###P < 0.001, compared with control group *P < 0.05, **P < 0.01, ***P<0.001, compared with MPP+ treated group.
MPP+ (1-methyl-4-phenylpyridinium) is a positively charged molecule. MPP+ induces massive oxidative stress through interfering with oxidative phosphorylation in mitochondria, leading to the damage of dopaminergic neurons [35]. SH-SY5Y cell line is widely used as an in vitro model of dopaminergic neurons for PD research, as this cell line exhibits multiple neuronal properties and a catecholaminergic phenotype of dopaminergic neurons. For example, SH-SY5Y cells express tyrosine hydroxylase, dopamine-beta-hydroxylase, dopamine receptors and dopamine transporter as well [36], [37]. Among several neurotoxins inducing PD pathology, the apoptosis of SH-SY5Y cells induced by MPP+ has been used as an in vitro experimental model for the study of PD [38], [39], [40]. Therefore, in the present study, we exposed SH-SY5Y cells to MPP+ to develop a cellular model of PD and investigated the protective effect of FCPR16 in this model, the possible mechanisms were investigated as well. We found that FCPR16 effectively enhanced cell viability in SH-SY5Y cells treated with MPP+. Mechanistically, our data showed that FCPR16 protected SH-SY5Y cells against MPP+-induced apoptosis, oxidative damage and the loss of mitochondrial membrane potential (Δψm). The protective effect of FCPR16 is abolished in the presence of inhibitors of PKA and Akt, indicating that FCPR16 elicits its protective effects via Epac/Akt and cAMP/PKA/CREB signaling pathways. These findings propose the possibility of animal pre-clinical studies for this compound.
2. Materials and methods
2.1. Materials
FCPR16, a selective PDE4 inhibitor, was kindly provided by Prof. Zhongzhen Zhou (Southern Medical University) with purity above 99%. The design, procedure for the synthesis and in vitro assay of FCPR16 for the inhibition of phosphodiesterases was reported by Zhou et al., [31]. H-89 and Perifosine (KRX-0401) were obtained from Selleck (Houston, TX, USA). Cell counting kit-8 (CCK-8) was purchased from Dojindo Corporation (Tokyo, Japan). Annexin V FITC apoptosis detection kit was from Merck-Calbiochem (St. Louis, MO, USA). Hoechst 33258, MPP+ and dimethylsulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolyl- carbocyanineiodide (JC-1), reactive oxygen species assay kit, lipid peroxidation MDA assay kit and LDH cytotoxicity assay kit were the products from Beyotime Institute of Biotechnology (Shanghai, China). BCA protein assay kit was obtained from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). anti-Bax and anti-Bcl-2 were purchased from Abcam (Cambridge, MA). anti-cleaved caspase-3, anti-Akt, anti-p-Akt (Ser473), anti-CREB, anti-GAPDH and anti-p-CREB (Ser133) were purchased from Cell Signaling Technology (MA, USA). Dulbecco's Modified Eagle's Medium (DMEM), Nutrient Mixture Ham's F-12 (DMEM/F12 1:1) medium, fetal bovine serum (FBS) and phosphate buffered saline (PBS) were bought from Gibco (Carlsbad, CA, USA).
2.2. Cell culture
SH-SY5Y cells were from American Type Culture Collection (ATCC, USA). Cells were cultured and maintained in Nutrient Mixture Ham's F-12 (DMEM/F12, 1:1) medium containing 10% (v/v) FBS and 1% penicillin/streptomycin in a humidified atmosphere of 5% CO2 and 95% air at 37 °C.
2.3. Cell viability
Cell viability was determined using the CCK-8 assay as previously reported. Briefly, SH-SY5Y cells were seeded in 96-well plate at 2 × 104 cells per well and allowed to adhere for 12 h at 37 °C with 5% CO2. Culture medium was removed and the cells were then treated with FCPR16 for 1 h before stimulation with MPP+. After 48 h incubation, the medium was removed and 10 μl of CCK-8 solution was added and incubated for 4 h at 37 °C. Absorbance was measured at 450 nm in a microplate reader (Synergy HT; BIOTEK, Broadview, IL).
2.4. Hoechst 33258 Staining
To study the morphological changes, SH-SY5Y cells were plated in 48-well plates pre-coated with poly-l-lysine. Cells were incubated with MPP+ (500 μM) in the absence or presence of FCPR16 (25 μM) for 48 h in 37 °C. After the treatment, cells were fixed with 4% paraformaldehyde for 30 min at room temperature. Cells were then washed with PBS and incubated with Hoechst 33258 (3 μg/ml) for 10 min at room temperature. Fluorescence was then observed by using a fluorescence microscope (Nikon, Japan).
2.5. Lactate dehydrogenase (LDH) release
Cytotoxicity was evaluated by detecting the release of LDH in the medium. Experiment was performed according to the supplier's instructions (LDH Cytotoxicity Assay Kit, Beyotime Institute of Biotechnology). In brief, FCPR16 was added into the medium 1 h before treatment with MPP+. Cells were incubated in CO2 incubator for 48 h. after treatment, 120 μl of culture medium from each well was transferred to another 96-well plate, 60 μl of reaction solution was then added into each well and the plate was placed on a shaker for 30 min at room temperature. The release of LDH into the medium was measured by detecting the optical density (OD) at 490 nm and 600 nm using a microplate reader (Synergy HT; BIOTEK, Broadview, IL).
2.6. Flow cytometric analysis of apoptosis
Flow cytometry was used to assess cellular apoptosis. An Annexin V FITC apoptosis detection kit (Merck-Calbiochem, St. Louis, MO, USA) was used to detect apoptotic and necrotic cells according to the supplier's instructions. In brief, SH-SY5Y cells were seeded in 6-well plate and treated as described above. After treatment, the cells were harvested and washed twice with ice-cold phosphate buffered saline (PBS), cells were then centrifuged and suspended in Annexin-binding buffer. The cell supernatant was stained with 100 μl Annexin V/7-ADD solution and incubated for 20 min at room temperature. The number of apoptotic or necrotic cells was analyzed using a flow cytometry. Early stage of apoptosis, late stage of apoptosis or necrosis, cellular debris and viable cells were identified as annexin V (+)/7-ADD (−), annexin V (+)/7-ADD (+), annexin V (−)/7-ADD (+) and annexin V (−)/7-ADD (−), respectively. Fluorescence intensities were analyzed using a flow cytometer (BD FACSVerse™ Flow Cytometer, BD Biosciences, San Jose, CA, USA). Each experiment was repeated 3 times.
2.7. Intracellular ROS measurement
The level of intracellular ROS was evaluated using DCFH-DA as a molecular probe. Cells were seeded in 6-well plate and pretreated with FCPR16 for 1 h, and then treated with MPP+ (500 µM) for 2 h. The cells were then incubated with DCFH-DA (10 µM) for 20 min at 37 °C in the dark, Cells were then washed with PBS and DCFH-DA fluorescence were visualized under a fluorescence microscope (Nikon, Tokyo, Japan). The excitation wavelength was set at 488 nm and the emission wavelength was 525 nm.
2.8. Mitochondrial membrane potential (Δψm) measurement
JC-1 is a cell-permeant dye which can enter and accumulate within mitochondria. In normal cells with normal Δψm, JC-1 aggregates and shows red fluorescence. While in apoptotic cells, the Δψm is decreased and JC-1 will show green fluorescence. Measurement of Δψm with JC-1 was used according to the manufacturer's instructions. Briefly, cells were seeded in 6-well plate and treated with FCPR16, followed by treatment with MPP+ (500 μM) for 24 h. Cells were then washed with PBS and suspended in 1 ml fresh medium. Cells were then incubated with 1 ml JC-1 staining solution for 20 min at 37 °C and 5% CO2. After washing with PBS twice, the fluorescence intensity was captured with an inverted fluorescence microscopy (Nikon, Japan), For red fluorescence, the fluorescence intensity was measured at Ex/Em: 525/590 nm. The green fluorescence intensity was measured at Ex/Em: 490/530 nm.
2.9. Malondialdehyde (MDA) levels
Lipid peroxidation in SH-SY5Y cells was determined by the level of malondialdehyde (MDA). SH-SY5Y cells were treated as described above. After treatment, cells were washed with PBS and lysed with lysis buffer. After centrifuge, the supernatant was collected and used for MDA detection. MDA level was measured following the manufacturer's protocol. A microplate reader (Synergy HT, BIOTEK, Broadview, IL) was used to measure the absorbance of cellular MDA at 532 nm. Protein concentrations were determined by BCA protein assay kit.
2.10. Western blotting
After treatment, cells were washed with ice-cold PBS and lysed in RIPA buffer (Beyotime) containing fresh protease and phosphatase inhibitors cocktail (Sigma-Aldrich). Lysed cells were centrifuged at 12,000 rpm for 10 min at 4 °C. The supernatant was collected and the concentration of total protein was measured using the BCA protein assay (Pierce). The protein extracts were then heat at 95 °C for 10 min and an equivalent of 30 μg protein was separated by electrophoresis in 8–12% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE; Beyotime). The proteins were then transferred to PVDF membranes (Millipore), and nonspecific bindings were blocked with 5% non-fat dry milk in TBST for 2 h at room temperature. After washing with TBST for four times, the membranes were incubated with primary antibodies at 4 °C overnight. After incubation with primary antibodies, membranes were washed four times with TBST and incubated with secondary antibodies for 1 h at room temperature. Protein signals of interest were detected using enhanced chemiluminescence reagent (ECL, Millipore) and analyzed by exposure to film. Densitometric analysis of the protein signals was carried out using Image J 6.0 software (NIH).
2.11. Statistical analysis
Data are presented as mean ± SD from three or more independent experiments. Data were assessed using one-way ANOVA followed by Bonferroni's post hoc test. (SPSS 19.0 software, Armonk, NY, USA). Graphs were plotted with the Graphpad Prism 5.0 (GraphPad Software, La Jolla, CA). A probability value of p ˂ .05 was considered statistically significant.
3. Results
3.1. FCPR16 protected against MPP+-induced cytotoxicity in SH-SY5Y cells
To evaluate the neuroprotective effect of FCPR16, we initially evaluated the cytotoxicity of MPP+ in SH-SY5Y cells in our experimental conditions. We found that MPP+ significantly reduced the cell viability in a dose-dependent manner (Supplementary Fig. 1). The cell viability was reduced by 48.34% at 500 μM MPP+. Thus, we used this concentration (500 μM MPP+) in following experiments. Subsequently, the SH-SY5Y cells were pretreated with FCPR16 (3.1–50 µM) for 1 h, followed by incubation with MPP+ (500 µM) for 48 h, and cell viability was determined by CCK-8 assay. As shown in Fig. 1B, compared with control group, the cell viability decreased to 51.74 ± 3.98% after being exposed to MPP+ (P < 0.01), pretreatment with 12.5–50 μM of FCPR16 increased cell viability in a dose-dependent manner in MPP+-treated SH-SY5Y cells. The pretreatment with 25 µM FCPR16 significantly reversed the MPP+-induced reduction in cell viability (70.40 ± 8.85%, P < 0.01). Based on this data, FCPR16 at the dose of 25 µM were evaluated further. LDH is cytosolic enzyme which is widely used as an indicator of cellular toxicity [41], [42]. We then investigated the level of LDH in the culture medium, and we found that treatment with MPP+ enhanced the leakage of LDH into the medium (159.22 ± 7.42% compared to control, P < 0.01), while FCPR16 significantly decreased the release of LDH (139.62 ± 17.57% compared to control, P < 0.05), suggesting that FCPR16 effectively blocked the cytotoxicity induced by MPP+. Hoechst 33258 Staining was then used to observe morphological changes after treatment with FCPR16 (25 µM) and MPP+ (500 µM). Cell shrinkage and nuclear condensation were observed in the MPP+-treated cells (Fig. 1E). However, FCPR16 pretreatment attenuated the morphological damage caused by MPP+ and resulted in a significant increase in the number of apoptotic cells (P < 0.05) (Fig. 1F). Taken together, these results suggest that FCPR16 protects SH-SY5Y cells against cellular cytotoxicity induced by MPP+.
3.2. FCPR16 suppressed MPP+-induced apoptosis in SH-SY5Y cells
To investigate the effect of FCPR16 and MPP+ on the apoptosis of SH-SY5Y cells, we then moved to detect apoptosis-related indicators. Flow cytometry was used to study the percentage of apoptotic cells. As shown in Fig. 2A, 25 µM FCPR16 did not exhibit toxicity in SH-SY5Y cells after 48 h incubation, which was consistent with the data that 3.1–50 µM had no effect on cell viability (Supplementary Fig. 2). The total percentage of early and late apoptotic cells in the control group, FCPR16 group, MPP+ group and MPP+ + FCPR16 group was 6.50%, 6.50%, 43.95% and 22.10% respectively. Our data showed that FCPR16 significantly reduced the ratio of apoptosis triggered by MPP+ (Fig. 2B). To further confirm the involvement of apoptosis, SH-SY5Y cells were harvested and apoptosis-related proteins were analyzed by Western blot. Fig. 2C showed that the levels of cleaved caspase 3 and Bax were robustly increased after treatment with MPP+, while FCPR16 attenuated the expression of both cleaved caspase 3 and Bax in SH-SY5Y cells (Fig. 2C and E). Bcl-2 functions as an anti-apoptotic protein in various cells. In the present study, we also found that FCPR16 enhanced the protein level of Bcl-2, and the ratio of Bcl-2/Bax were significantly increased by FCPR16 in MPP+-treated cells (Fig. 2C and D). These results indicate that treatment with FCPR16 attenuates apoptosis induced by MPP+ in SH-SY5Y cells.
Fig. 2.

FCPR16 attenuated MPP+-induced cell apoptosis in SH-SY5Y cells. Cells were pretreated with FCPR16 (25 μM) for 1 h, and then were exposed to 500 μM MPP+ for 24 h, the apoptosis of SH-SY5Y cells was determined by flow cytometry (A and B). (C) SH-SY5Y cells were pretreated with 25 μM FCPR16, and then were incubated with 500 μM MPP+ for 48 h. Expression of cleaved caspase 3, Bax and Bcl-2 was analyzed by Western blot. The relative levels of Bcl-2/Bax (D) and cleaved caspase 3/GAPDH (E) were determined by densitometry of the blots. Results are shown as the mean ± SD and represent three independent experiments. n = 3. #P < 0.05 ##P < 0.01, ###P<0.001 compared with control. **P < 0.01 compared with MPP+ treated group.
3.3. FCPR16 inhibited the reduction of Δψm and accumulation of intracellular ROS induced by MPP+
Mitochondrial oxidation-reduction (REDOX) activity and Δψm are involved in MPP+-induced dopaminergic neurotoxicity, and amelioration of mitochondrial dysfunction has been proposed as an effective way to slow progressive dopaminergic neurodegeneration [43]. Therefore, the Δψm in SH-SY5Y cells was evaluated by detecting the red/green fluorescent intensity ratio of JC-1 staining. As shown in Fig. 3A and B, treatment with MPP+ significantly increased green fluorescence intensity in SH-SY5Y cells (11.40 ± 2.99%, P < 0.01), indicating that MPP+ significantly reduced Δψm. Whereas pretreatment with FCPR16 (25 µM) remarkably reduced green fluorescence intensity and increased red fluorescence intensity in SH-SY5Y cells (49.01 ± 5.44%, P < 0.05). These results indicate that treatment with FCPR16 is effective to restore the reduced Δψm by MPP+ in SH-SY5Y cells.
Fig. 3.

FCPR16 attenuated MPP+-induced mitochondrial membrane potential (Δψm) loss and the increase of oxidative stress in SH-SY5Y cells. (A) After pre-treatment with 25 μM FCPR16 for 1 h, SH-SY5Y cells were incubated with or without 500 μM MPP+ for another 24 h, Δψm was determined by the JC-1 assay. Bar = 100 µm. (B) Histogram indicates the JC-1 polymermonomer fluorescence ratio after MPP+ insult in the presence or absence of FCPR16. (C) After pre-treatment with 25 μM FCPR16 for 1 h, SH-SY5Y cells were incubated with or without 500 μM MPP+ for another 2 h, The levels of reactive oxygen species (ROS) were determined by fluorescence intensity of DCFH-DA in SH-SY5Y cells. Bar = 100 µm. (D) Histogram indicates the ROS level after MPP+ insult in the presence or absence of FCPR16. (E) After pre-treatment with 25 μM FCPR16 for 1 h, SH-SY5Y cells were incubated with or without 500 μM MPP+ for another 48 h, lipid peroxidation in SH-SY5Y cells was determined by the level of malondialdehyde (MDA). Results are shown as the mean ± SD and represent three independent experiments. n = 3. ##P<0.01, ###P < 0.001 compared with control. *P < 0.05, **P < 0.01 compared with MPP+ treated group.
Mitochondria are the major source of ROS in various mammalian cells and excessive production of ROS in the mitochondria disrupts normal redox signaling [44]. Having known that FCPR16 is beneficial for the recovery of Δψm, we then moved to analyze the production of ROS in SH-SY5Y cells using DCFH-DA fluorescence assay. As shown in Fig. 3C and D, treatment with FCPR16 alone did not affect the production of ROS, while exposure to MPP+ increased intracellular fluorescence density dramatically in SH-SY5Y cells (126.54 ± 4.32%, P < 0.01). In contrast, pretreatment with FCPR16 significantly inhibited the accumulation of intracellular ROS induced by MPP+ (106.91 + 3.05%, P < 0.05). As free radicals cause membrane lipid peroxidation, we also analyzed the level of MDA in the present study. As shown in Fig. 3E, MPP+ stimulates the production of MDA significantly (12.07 ± 1.39 nM/mg protein, P < 0.001), while the increased MDA was prevented by FCPR16 treatment (8.91 ± 1.20 nM/mg protein, P < 0.05). These results suggest that FCPR16 preserves mitochondrial function, suppresses ROS production and inhibits excessive oxidation in SH-SY5Y cells.
3.4. FCPR16 enhanced cAMP accumulation and Epac2 expression in SH-SY5Y cells
FCPR16 is a potent PDE4 inhibitor [31], cAMP is a substrate of PDE4 and inhibition of PDE4 is supposed to increase the concentration of cAMP. We then examined the effect of FCPR16 on cAMP production in SH-SY5Y cells. As expected, treatment with FCPR16 for 160 min increased intracellular cAMP levels, and the concentration of cAMP increased 3.72-fold in cells treated with 25 μM FCPR16 (Fig. 4A). Epac is a direct target for cAMP and Epac2 is highly expressed in dopaminergic neurons [45], [46]. In the present study, we found that treatment of cells with 500 μM MPP+ for 4 h decreased the expression of Epac2 to 38% compared to the control group (P < 0.001). In contrast, pre-treatment with FCPR16 significantly increased the level of Epac2 to 58% (P < 0.05) (Fig. 4B). These results imply that inhibition of PDE4 by FCPR16 enhances the levels of cAMP and Epac2 in SH-SY5Y cells.
Fig. 4.

Effects of FCPR16 on intracellular levels of cAMP and Epac-2. (A) SH-SY5Y cells were incubated with media alone or indicated concentration of FCPR16 for 160 min. Lysates of the cells were assayed using cAMP ELISA kit. Results are shown as the mean ± SD and represent three independent experiments. n = 3. *P < 0.05, ***P < 0.001 compared with control group. (B) Cells were pre-incubated with by FCPR16 (25 µM) for 1 h, and then cells were treated with MPP+ (500 µM) for 4 h. Levels of Epac-2 were measured in cellular extracts using Western blot. Data shown are mean ± SD, ###P < 0.001 compared with control group, *P < 0.05 compared with MPP+-treated group.
3.5. FCPR16 promoted the phosphorylation of Akt and CREB in a time- and concentration-dependent manner in SH-SY5Y cells
We have proved that inhibition of PDE4 by FCPR16 increased the level of intracellular cAMP in SH-SY5Y cells. cAMP elevation activated two different signaling pathways, namely PKA-dependent CREB phosphorylation and Epac-dependent Akt phosphorylation [47], [48]. We therefore tested whether CREB and Akt pathways are involved in the protective effect of FCPR16 in SH-SY5Y cells. Cells were treated with FCPR16 (25 µM) for 0–160 min and the phosphorylation of Akt and CREB was evaluated by Western blot. As shown in Fig. 5A, C and D, FCPR16 markedly increased the phosphorylation of Akt and CREB in a time-dependent manner. Treatment with various concentration of FCPR16 (3.1–25 µM) for 80 min also increased the phosphorylation of Akt and CREB in a dose-dependent manner (Fig. 5B, E and F). These findings suggest that FCPR16 activates both Akt and CREB signaling pathways in SH-SY5Y cells.
Fig. 5.

FCPR16 stimulated the phosphorylation of Akt and CREB. (A) SH-SY5Y cells were treated with 25 μM FCPR16 for various times, (B) SH-SY5Y cells were treated with FCPR16 in different concentrations. Expression of phosphated Akt and CREB were analyzed by immunoblotting. (C-F) Relative levels of p-Akt versus total Akt and p-CREB versus total CREB in each sample determined by densitometry of the blots. Densitometric analysis of the immunoblot was expressed as a percentage of control. Results are shown as the mean ± SD and represent three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 compared with control.
3.6. FCPR16 stimulated the phosphorylation of Akt and CREB SH-SY5Y cells treated with MPP+
The phosphorylation and activation of Akt and CREB are essential for cell survival [47]. To further study the role of FCPR16 on the activation of Akt and CREB in SH-SY5Y cells treated with MPP+, we firstly investigated the role of MPP+ on the phosphorylation of both Akt and CREB. SH-SY5Y cells were incubated with MPP+ for 2 or 4 h, we found that MPP+ significantly inhibited the phosphorylation of Akt and CREB in SH-SY5Y cells (Supplementary Fig. 3), which was consistent with the finding that MPP+ decreased cell viability. We then asked whether FCPR16 could reverse the effect of MPP+ on the phosphorylation of Akt and CREB. SH-SY5Y cells were treated with FCPR16 (25 µM) for 1 h, followed by incubation with MPP+ for 4 h. We found that FCPR16 reversed the reduction of the phosphorylated Akt and CREB induced by MPP+ (Fig. 6A and B), indicating that the inhibitory effect of MPP+ was reversed by FCPR16. We also found that the Akt inhibitor KRX-0401 had no significant effect on the phosphorylation of Akt, while KRX-0401 blocked the stimulatory role of FCPR16 on the phosphorylation of Akt (Fig. 6A and B). Similarly, PKA inhibitor H-89 significantly blocked the role of FCPR16 on the phosphorylation of CREB (Fig. 6C and D). These data suggest that the FCPR16 promotes the activation of Akt and CREB under both basal and stressed conditions.
Fig. 6.

KRX-0401 and H-89 blocked the phosphorylation of Akt and CREB induced by FCPR16, respectively, in SH-SY5Y cells. (A) Following treatment with KRX-0401 for 1 h, SH-SY5Y cells were exposed to MPP+ in the presence of FCPR16 (25 μM) for 4 h, and the phosphorylation of Akt was determined by Western blot. (B) Relative levels of p-Akt versus total Akt in each sample determined by densitometry of the blots. (C) Following treatment with H-89 for 1 h, SH-SY5Y cells were exposed to MPP+ in the presence of FCPR16 (25 μM) for 4 h, and the phosphorylation of CREB was determined by Western blot. (D) The phosphorylation of CREB was analyzed by immunoblotting. Densitometric analysis of the immunoblot was expressed as a percentage of control. Results are shown as the mean ± SD and represent three independent experiments. ##P < 0.01, ###P<0.001, compared with control. *P < 0.05 compared with MPP+, $P < 0.05, compared with (FCPR16 + MPP+) treated group.
3.7. Inhibition of Akt and PKA/CREB signaling pathways abolished the neuroprotective effects of FCPR16 against MPP+
Having established that FCPR16 can induce the activation of Akt and CREB signaling pathways in both basal condition and under the condition of MPP+ treatment, we next investigated whether these two signaling pathways mediate the protective action of FCPR16. SH-SY5Y cells were pretreated with or without a PKA inhibitor, H-89 (10 μM) and a Akt inhibitor, KRX-0401 (25 μM), in the presence or absence of 25 μM FCPR16, and the Δψm and ROS were determined by JC-1 staining and DCFH-DA fluorescence assay, respectively. In accordance with Fig. 3, MPP+ stimulated a significant increase of ROS and decrease of Δψm, FCPR16 (25 μM) reversed the effect of MPP+ and restored both ROS and Δψm in SH-SY5Y cells. Interestingly, as shown in Fig. 7, Fig. 8, pretreatment of cells with H-89 or KRX-0401 significantly blocked the effects of FCPR16 on ROS and Δψm (Fig. 7A–D). To further confirm the role of the Akt and PKA/CREB signaling pathways in the protective effect of FCPR16, we measured the cell viability under these conditions. As expected, MPP+ triggered a significant decreased in cell viability (51.86 ± 4.16%), while FCPR16 increased the cell viability (59.98 ± 2.80%, P < 0.05). Notably, inhibition of PKA by H-89 eliminated the pro-survival effect of FCPR16 against MPP+ (51.52 ± 3.96%, P < 0.05, Fig. 7E). Inhibition of Akt by KRX-0401 produced a similar effect on cell viability (Fig. 8E). Thus, these experiments support that FCPR16 protects SH-SY5Y cells against MPP+-induced cell death, and PKA/CREB and Akt signaling pathways mediate the protection of FCPR16.
Fig. 7.

PKA signaling involved in the effects of FCPR16 towards MPP+-induced deficits in mitochondrial membrane potential (Δψm) and oxidative damage. (A) SH-SY5Y cells were pretreated with 10 μM H-89 for 1 h before FCPR16 treatment for 1 h, and then incubated with or without 500 μM MPP+ for another 24 h. Δψm was determined by JC-1 assay. Bar= 100 µm. (B) Quantitive data of A. (C) SH-SY5Y cells were pretreated with 10 μM H-89 for 1 h before FCPR16 treatment for 1 h, and then incubated with or without 500 μM MPP+ for another 2 h. ROS level was determined using DCFH-DA as a molecular probe. Bar = 100 µm. (D) Quantitive data of C. (E) SH-SY5Y cells were pretreated with 10 μM H-89 for 1 h before FCPR16 treatment for 1 h, and then incubated with or without 500 μM MPP+ for another 48 h. Cell viability were measured by CCK-8 assay. ###P < 0.001 compared with control; *P < 0.05, * *P < 0.01 compared with MPP+; $$P < 0.01, compared with (FCPR16 + MPP+) treated group.
Fig. 8.

Akt signaling involved in the effects of FCPR16 towards MPP+-induced deficits in mitochondrial membrane potential (Δψm) and oxidative damage. (A) SH-SY5Y cells were pretreated with 25 μM KRX-0401 for 1 h before FCPR16 treatment for 1 h, and then incubated with or without 500 μM MPP+ for another 24 h. Δψm was determined by JC-1 assay. (B) Quantitive data of A. (C) SH-SY5Y cells were pretreated with 25 μM KRX-0401 for 1 h before FCPR16 treatment for 1 h, and then incubated with or without 500 μM MPP+ for another 2 h. ROS level was determined using DCFH-DA as a molecular probe. (D) Quantitive data of C. (E) SH-SY5Y cells were pretreated with 25 μM KRX-0401 for 1 h before FCPR16 treatment for 1 h, and then incubated with or without 500 μM MPP+ for another 48 h. Cell viability were measured by CCK-8 assay. ###P < 0.001 compared with control; *P < 0.05, **P < 0.01 compared with MPP+; $P < 0.05, $$P<0.01 compared with (FCPR16 + MPP+) treated group.
4. Discussion
In the present study, we provided evidence that treatment of SH-SY5Y cells with MPP+ significantly reduced cell viability, increased the accumulation of intracellular ROS, mitochondrial dysfunction and induced typical cellular apoptosis. However, the above changes were markedly reversed by the treatment with a novel PDE4 inhibitor FCPR16. Moreover, the FCPR16-induced neuroprotective effect was mediated by cAMP/PKA/CREB and Epac/Akt signaling pathways. As far as we know, this is the first work that shows the reduction of oxidative stress and mitochondrial dysfunction through inhibition of PDE4 in PD cellular model, and activation of cAMP/PKA/CREB and Epac/Akt signaling pathways contributes to the antioxidant role of PDE4 inhibition. Importantly, compared with canonical PDE4 inhibitors (such as rolipram), FCPR16 has no effect in inducing emesis, hence, FCPR16 has obvious advantages concerning the side effects and drug safety. We would like to point out that even though FCPR16 does not cause vomiting/nausea in rodents, other side effects have not been investigated for this compound. Side effects, such as weight loss and insomnia have been discovered from the clinical use of the second-generation PDE4 inhibitor roflumilast for the treatment of chronic obstructive pulmonary disease [49]. Whether FCPR16 will produce similar side effects is largely unknown. Hence, animal experiments are needed in the future to systematically evaluate the side effects of FCPR16. We would also like to point out that PDE4 distribution has been most studied in brain, lung and inflammatory cells [50], [51]. Inhibition of PDE4 in these cells results in an elevation of cAMP, which mediates neuroprotection, airway smooth muscle relaxation and anti-inflammatory effects in a variety animal models [50], [51]. In addition to distribution in above-mentioned tissues, PDE4 is also expressed in heart, liver, spleen and other tissues [52]. Tissue distribution is an important determinant of PDE4 function. In the present study, we mainly focused on the role of FCPR16 in SH-SY5Y cells and neurons. It remains unclear whether treatment with FCPR16 will produce effects in other tissues, and this question should be extensively addressed in the future studies.
MPP+ is a toxic molecule which selectively enters the substantia nigra dopaminergic neurons through dopamine transporter in the presynaptic membrane. Inside dopaminergic neurons, MPP+ accumulates in the inner membrane of mitochondrial and inhibits the activity of the mitochondrial complex I and the α-ketoglutarate dehydrogenase complex, thereby interfering with the normal function of the mitochondrial respiratory chain, resulting in loss of mitochondrial membrane potential and formation of ROS, and eventually lead to oxidative stress and cell death [53]. Oxidative stress has been recognized as an important pathological process of PD [7], [8]. Since degeneration of dopaminergic neurons induced by mitochondrial dysfunction and oxidative stress is one of the prominent features of PD, hence, whether FCPR16 could protect dopaminergic neurons against MPP+ induced injury was investigated firstly in the present study. Pretreatment with FCPR16 dose dependently prevented MPP+-induced reduction of cell viability in SH-SY5Y cells, On the other hand, under the condition of stress, pro-apoptotic molecule Bax translocates to the mitochondria and forms Bax dimers. The dimerization of pro-apoptotic proteins then results in altered Δψm, overproduction of ROS and the release of cytochrome c. Caspase family proteins (such as caspase 3) located in the cytosol are activated by cytochrome c [54]. Cleaved or activated caspase 3 is the executor of apoptosis, while overexpression of Bcl-2 is supposed to counter these effects [54]. In the present study, MPP+ was found to enhance the levels of both cleaved caspase-3 and Bax in SH-SY5Y cells, while the anti-apoptosis protein Bcl-2 was decreased after MPP+ challenge. However, the effect of which could be counteracted by addition of FCPR16. Consistently, both Δψm and ROS were restored by FCPR16. As amelioration of PD-related mitochondrial abnormalities and ROS is an alternative approach to delay or slow the progress of PD [55], our results implicated that FCPR16 could effectively prevent MPP+-induced Δψm loss, intracellular accumulation of ROS and the activation of caspase 3, suggesting that FCPR16 may exert neuroprotective effect through the preservation of mitochondrial dysfunction.
PDE4 is an enzyme selectively hydrolyses cAMP and three PDE4 isoforms (PDE4A/B/D) are abundant in the basal ganglia [20]. Studies have shown that PDE4 inhibition by rolipram promotes the survival of cultured rat dopaminergic neurons [56]. What's more, administration with PDE4 inhibitors in C57BL/6 mice substantially reduced dopamine depletion in the striatum induced by MPTP [21]. Additionally, administration of rolipram improved the memory deficits in MPTP-induced animal model [57]. All these observations support that PDE4 inhibition is a potential strategy in the treatment of PD. Although the neuroprotective effects of PDE4 inhibitor rolipram have been studied both in vivo and in vitro, the anti-oxidative effect of PDE4 inhibitor in PD cellular model has never been reported. In the present study, we proved that inhibition of PDE4 by FCPR16 attenuated the overproduction of ROS and restored Δψm. Our study provides novel insights into the mechanisms underlying the protective effects of PDE4 inhibition in dopaminergic neurons. Additionally, astrocytes play direct and critical roles in mediating neuronal survival and function in PD [58], and astrocytes also mediates oxidative stress in the models of PD [59]. Interestingly, our previous study showed that FCPR16 suppressed the activation of astrocytes in the brain [33]. It is worth to study the role of FCPR16 or other PDE4 inhibitor in Redox regulation in astrocytes in both cellular and animal models of PD. As over-activated astrocytes and microglial cells and over-produced ROS are common contributors to the development of neurodegenerative diseases [60]. it is desirable in the future to test the protective role of FCPR16 in other neurodegenerative diseases. This hypothesis is also supported by the recent findings that cAMP-mediated signaling is reduced in aging and age-related diseases, while beneficial effects of PDE4 inhibitors were observed in aging and AD models [23].
In the present study, we also addressed the question of which intracellular signaling elements are involved in the FCPR16-mediated neuroprotective effect in SH-SY5Y cells exposed to MPP+. We revealed that PDE4 inhibition enhanced intracellular cAMP and the expression of Epac2. Enhancement of intracellular cAMP activates both PKA/CREB and Epac/Akt signal pathways [47], [48]. As expected, treatment with FCPR16 caused a time- and concentration-dependent activation of CREB and Akt. Our results suggest that FCPR16-triggered CREB and Akt activation is possibly a major step for the anti-oxidative role of FCPR16, as the roles of FCPR16 on ROS production and Δψm loss were dependent on the activation of CREB and Akt. Of note, our results do not exclude the involvement of any other neuroprotective mechanisms in the inhibition of MPP+-induced apoptosis by FCPR16. For example, besides Akt, Epac also promotes the phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) [61], whether ERK1/2 is involved in the anti-apoptotic effect of FCPR16 needs to be studied in future. Nevertheless, our results in vitro produce testable hypotheses for animal and clinical studies.
In this study, we provided evidence showing that FCPR16 protected SH-SY5Y cells against MPP+-induced cell death through limiting mitochondrial ROS production. It is predictable that FCPR16 may restore dopaminergic functions through alternative pathways. For example, the dopamine and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32) is highly expressed in neostriatum, and it has been identified as a pivotal target for dopamine signaling in striatal neurons [62]. Binding of dopamine with D1 receptors increases intracellular cAMP, leading to the activation of PKA and the subsequent phosphorylation of DARPP-32 at Thr34 [62]. Alternatively, inhibition of PDE4 by FCPR16 is supposed to enhance cAMP and thereby activates PKA. Hence, FCPR16 may act on DARPP-32 and thus manifest reduced parkinsonian phenotype. On the other hand, treatment with PDE4 inhibitor, such as FCPR16, could activate PKA-mediated CREB signaling and increase the release of brain-derived neurotrophic factors [24], which is a potent factor promoting the survival of existing neurons and thereby correct PD disabilities. All these potential pathways are deserved to be investigated in the next step. It is noteworthy that PD is a neurological disorder with evolving layers of complexity. This disease results from a complicated interplay of genetic and environmental factors affecting multiple fundamental cellular processes [63]. Mitochondrial oxidative stress is one of the cellular processes contributing to PD. Our findings support the role of FCPR16 in attenuating mitochondrial oxidative stress in PD cellular model. Other processes, such as genetic mutation, autophagy-lysosomal metabolism, ubiquitin–proteasome protein degradation, endoplasmic reticulum stress and unfolded protein response also contribute to the development of PD [63]. Our study could not support the effectiveness of FCPR16 when other processes, especially processes independent of oxidative stress, play a major role in the pathology of PD.
In our study, the data presented here are mainly obtained from SH-SY5Y cells. We also evaluated the protective effect of FCPR16 in cultured cortical neurons treated with MPP+. The data is shown in Supplementary Fig. 4, we found that MPP+ induced a dose-dependent apoptosis in cultured neurons, and 500 μM MPP+ caused an approximately 50% loss of cortical neurons (Supplementary Fig. 4A), while treatment with FCPR16 reversed the toxic effect of MPP+ and enhanced the cell viability in a dose-dependent manner (Supplementary Fig. 4A). However, it would be preferable to perform these experiments and get results from ventral midbrain dopaminergic neuron cultures. On the other hand, we mainly focused on the levels of ROS, mitochondrial membrane potential, apoptosis and the signaling pathways involved in the protective role of FCPR16, as dopaminergic neurons participates in the motor performance of patients with PD. behavioral tests, such as locomotor activity, rotarod, forepaw stride length, grid test, and pole test are deserved to be investigated in PD animal models treated with FCPR16 in the future.
5. Conclusions
In summary, our findings demonstrate that the novel PDE4 inhibitor FCPR16 could significantly protect against damaging pathways including oxidative stress, mitochondrial dysfunction and apoptosis in SH-SY5Y cells. we also found that FCPR16 prevented MPP+-induced neurotoxicity through activation of cAMP/PKA/CREB and Epac/Akt signaling pathways. As oxidative stress is likely to play an important role in the neurodegenerative pathogenesis of PD. Defects in mitochondrial functions in PD brain further provides connection to redox metabolism to PD. Our study may lead to development of mechanism-based therapeutics and improved pharmacotherapy for PD. It is reasonable to assume that FCPR16 is a potential candidate for further preclinical study aimed at the prevention and treatment of PD.
Acknowledgements
This work was supported by National Natural Science Fund of China (Grant numbers, 81301099 and 81373384).
Acknowledgments
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.02.008.
Contributor Information
Jiangping Xu, Email: jpx@smu.edu.cn.
Haitao Wang, Email: wht821@smu.edu.cn.
Appendix A. Supplementary material
Supplementary material
References
- 1.Zhai S., Tanimura A., Graves S.M., Shen W., Surmeier D.J. Striatal synapses, circuits, and Parkinson's disease. Curr. Opin. Neurobiol. 2017;48:9–16. doi: 10.1016/j.conb.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santiago J.A., Bottero V., Potashkin J.A. Biological and clinical implications of comorbidities in Parkinson's disease. Front Aging Neurosci. 2017;9:394. doi: 10.3389/fnagi.2017.00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaeffer E., Berg D. Dopaminergic therapies for non-motor symptoms in Parkinson's disease. CNS Drugs. 2017;31:551–570. doi: 10.1007/s40263-017-0450-z. [DOI] [PubMed] [Google Scholar]
- 4.Leggio L., Vivarelli S., L'Episcopo F., Tirolo C., Caniglia S., Testa N., Marchetti B., Iraci N. microRNAs in Parkinson's disease: from pathogenesis to novel diagnostic and therapeutic approaches. Int. J. Mol. Sci. 2017:18. doi: 10.3390/ijms18122698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agundez J.A., Garcia-Martin E., Alonso-Navarro H., Jimenez-Jimenez F.J. Anti-Parkinson's disease drugs and pharmacogenetic considerations. Expert Opin. Drug Metab. Toxicol. 2013;9:859–874. doi: 10.1517/17425255.2013.789018. [DOI] [PubMed] [Google Scholar]
- 6.Obeso J.A., Rodriguez-Oroz M.C., Goetz C.G., Marin C., Kordower J.H., Rodriguez M., Hirsch E.C., Farrer M., Schapira A.H., Halliday G. Missing pieces in the Parkinson's disease puzzle. Nat. Med. 2010;16:653–661. doi: 10.1038/nm.2165. [DOI] [PubMed] [Google Scholar]
- 7.Subramaniam S.R., Chesselet M.F. Mitochondrial dysfunction and oxidative stress in Parkinson's disease. Prog. Neurobiol. 2013;106–107:17–32. doi: 10.1016/j.pneurobio.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang T., Sun Q., Chen S. Oxidative stress: a major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson's disease and Alzheimer's disease. Prog. Neurobiol. 2016;147:1–19. doi: 10.1016/j.pneurobio.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Bhat A.H., Dar K.B., Anees S., Zargar M.A., Masood A., Sofi M.A., Ganie S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015;74:101–110. doi: 10.1016/j.biopha.2015.07.025. [DOI] [PubMed] [Google Scholar]
- 10.Zhou M., Xu S., Mi J., Ueda K., Chan P. Nuclear translocation of alpha-synuclein increases susceptibility of MES23.5 cells to oxidative stress. Brain Res. 2013;1500:19–27. doi: 10.1016/j.brainres.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 11.Terracciano C., Nogalska A., Engel W.K., Wojcik S., Askanas V. In inclusion-body myositis muscle fibers Parkinson-associated DJ-1 is increased and oxidized. Free Radic. Biol. Med. 2008;45:773–779. doi: 10.1016/j.freeradbiomed.2008.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schapira A.H. Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol. 2008;7:97–109. doi: 10.1016/S1474-4422(07)70327-7. [DOI] [PubMed] [Google Scholar]
- 13.Ramalingam M., Kim S.J. The neuroprotective role of insulin against MPP(+) -induced Parkinson's disease in differentiated SH-SY5Y cells. J. Cell Biochem. 2016;117:917–926. doi: 10.1002/jcb.25376. [DOI] [PubMed] [Google Scholar]
- 14.Baluchnejadmojarad T., Eftekhari S.M., Jamali-Raeufy N., Haghani S., Zeinali H., Roghani M. The anti-aging protein klotho alleviates injury of nigrostriatal dopaminergic pathway in 6-hydroxydopamine rat model of Parkinson's disease: involvement of PKA/CaMKII/CREB signaling. Exp. Gerontol. 2017;100:70–76. doi: 10.1016/j.exger.2017.10.023. [DOI] [PubMed] [Google Scholar]
- 15.Prickaerts J., Heckman P.R.A., Blokland A. Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer's disease. Expert Opin. Investig. Drugs. 2017;26:1033–1048. doi: 10.1080/13543784.2017.1364360. [DOI] [PubMed] [Google Scholar]
- 16.Jindal A., Mahesh R., Bhatt S. Etazolate, a phosphodiesterase-4 enzyme inhibitor produces antidepressant-like effects by blocking the behavioral, biochemical, neurobiological deficits and histological abnormalities in hippocampus region caused by olfactory bulbectomy. Psychopharmacology. 2015;232:623–637. doi: 10.1007/s00213-014-3705-0. [DOI] [PubMed] [Google Scholar]
- 17.Kraft P., Schwarz T., Gob E., Heydenreich N., Brede M., Meuth S.G., Kleinschnitz C. The phosphodiesterase-4 inhibitor rolipram protects from ischemic stroke in mice by reducing blood-brain-barrier damage, inflammation and thrombosis. Exp. Neurol. 2013;247:80–90. doi: 10.1016/j.expneurol.2013.03.026. [DOI] [PubMed] [Google Scholar]
- 18.Wang C., Yang X.M., Zhuo Y.Y., Zhou H., Lin H.B., Cheng Y.F., Xu J.P., Zhang H.T. The phosphodiesterase-4 inhibitor rolipram reverses Abeta-induced cognitive impairment and neuroinflammatory and apoptotic responses in rats. Int J. Neuropsychopharmacol. 2012;15:749–766. doi: 10.1017/S1461145711000836. [DOI] [PubMed] [Google Scholar]
- 19.Zhuo Y., Guo H., Cheng Y., Wang C., Wang C., Wu J., Zou Z., Gan D., Li Y., Xu J. Inhibition of phosphodiesterase-4 reverses the cognitive dysfunction and oxidative stress induced by Abeta25-35 in rats. Metab. Brain Dis. 2016;31:779–791. doi: 10.1007/s11011-016-9814-1. [DOI] [PubMed] [Google Scholar]
- 20.Perez-Torres S., Miro X., Palacios J.M., Cortes R., Puigdomenech P., Mengod G. Phosphodiesterase type 4 isozymes expression in human brain examined by in situ hybridization histochemistry and[3H]rolipram binding autoradiography. Comparison with monkey and rat brain. J. Chem. Neuroanat. 2000;20:349–374. doi: 10.1016/s0891-0618(00)00097-1. [DOI] [PubMed] [Google Scholar]
- 21.Hulley P., Hartikka J., Abdel'Al S., Engels P., Buerki H.R., Wiederhold K.H., Muller T., Kelly P., Lowe D., Lubbert H. Inhibitors of type IV phosphodiesterases reduce the toxicity of MPTP in substantia nigra neurons in vivo. Eur. J. Neurosci. 1995;7:2431–2440. doi: 10.1111/j.1460-9568.1995.tb01041.x. [DOI] [PubMed] [Google Scholar]
- 22.Yang L., Calingasan N.Y., Lorenzo B.J., Beal M.F. Attenuation of MPTP neurotoxicity by rolipram, a specific inhibitor of phosphodiesterase IV. Exp. Neurol. 2008;211:311–314. doi: 10.1016/j.expneurol.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelly M.P. Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell Signal. 2018;42:281–291. doi: 10.1016/j.cellsig.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo H., Cheng Y., Wang C., Wu J., Zou Z., Niu B., Yu H., Wang H., Xu J. FFPM, a PDE4 inhibitor, reverses learning and memory deficits in APP/PS1 transgenic mice via cAMP/PKA/CREB signaling and anti-inflammatory effects. Neuropharmacology. 2017 doi: 10.1016/j.neuropharm.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Morales V., Luaces-Regueira M., Campos-Toimil M. The cAMP effectors PKA and Epac activate endothelial NO synthase through PI3K/Akt pathway in human endothelial cells. Biochem. Pharmacol. 2017;145:94–101. doi: 10.1016/j.bcp.2017.09.004. [DOI] [PubMed] [Google Scholar]
- 26.Yoo J.M., Lee B.D., Sok D.E., Ma J.Y., Kim M.R. Neuroprotective action of N-acetyl serotonin in oxidative stress-induced apoptosis through the activation of both TrkB/CREB/BDNF pathway and Akt/Nrf2/Antioxidant enzyme in neuronal cells. Redox Biol. 2017;11:592–599. doi: 10.1016/j.redox.2016.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh A.K., Kashyap M.P., Tripathi V.K., Singh S., Garg G., Rizvi S.I. Neuroprotection through rapamycin-induced activation of autophagy and PI3K/Akt1/mTOR/CREB signaling against amyloid-beta-induced oxidative stress, synaptic/neurotransmission dysfunction, and neurodegeneration in adult rats. Mol. Neurobiol. 2017;54:5815–5828. doi: 10.1007/s12035-016-0129-3. [DOI] [PubMed] [Google Scholar]
- 28.De Savi C., Cox R.J., Warner D.J., Cook A.R., Dickinson M.R., McDonough A., Morrill L.C., Parker B., Andrews G., Young S.S. Efficacious inhaled PDE4 inhibitors with low emetic potential and long duration of action for the treatment of COPD. J. Med. Chem. 2014;57:4661–4676. doi: 10.1021/jm5001216. [DOI] [PubMed] [Google Scholar]
- 29.Zeller E., Stief H.J., Pflug B., Sastre-y-Hernandez M. Results of a phase II study of the antidepressant effect of rolipram. Pharmacopsychiatry. 1984;17:188–190. doi: 10.1055/s-2007-1017435. [DOI] [PubMed] [Google Scholar]
- 30.Bielekova B., Richert N., Howard T., Packer A.N., Blevins G., Ohayon J., McFarland H.F., Sturzebecher C.S., Martin R. Treatment with the phosphodiesterase type-4 inhibitor rolipram fails to inhibit blood--brain barrier disruption in multiple sclerosis. Mult. Scler. 2009;15:1206–1214. doi: 10.1177/1352458509345903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Z.Z., Ge B.C., Zhong Q.P., Huang C., Cheng Y.F., Yang X.M., Wang H.T., Xu J.P. Development of highly potent phosphodiesterase 4 inhibitors with anti-neuroinflammation potential: design, synthesis, and structure-activity relationship study of catecholamides bearing aromatic rings. Eur. J. Med. Chem. 2016;124:372–379. doi: 10.1016/j.ejmech.2016.08.052. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Z.Z., Ge B.C., Chen Y.F., Shi X.D., Yang X.M., Xu J.P. Catecholic amides as potential selective phosphodiesterase 4D inhibitors: design, synthesis, pharmacological evaluation and structure-activity relationships. Bioorg. Med. Chem. 2015;23:7332–7339. doi: 10.1016/j.bmc.2015.10.033. [DOI] [PubMed] [Google Scholar]
- 33.Chen J., Yu H., Zhong J., Feng H., Wang H., Cheng Y., Zou Z., Huang C., Zhou Z., Zheng W. The phosphodiesterase-4 inhibitor, FCPR16, attenuates ischemia-reperfusion injury in rats subjected to middle cerebral artery occlusion and reperfusion. Brain Res. Bull. 2017;137:98–106. doi: 10.1016/j.brainresbull.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 34.Ramirez A.D., Smith S.M. Regulation of dopamine signaling in the striatum by phosphodiesterase inhibitors: novel therapeutics to treat neurological and psychiatric disorders. Cent. Nerv. Syst. Agents Med. Chem. 2014;14:72–82. doi: 10.2174/1871524914666141226103421. [DOI] [PubMed] [Google Scholar]
- 35.Przedborski S., Tieu K., Perier C., Vila M. MPTP as a mitochondrial neurotoxic model of Parkinson's disease. J. Bioenerg. Biomembr. 2004;36:375–379. doi: 10.1023/B:JOBB.0000041771.66775.d5. [DOI] [PubMed] [Google Scholar]
- 36.Ross R.A., Spengler B.A., Biedler J.L. Coordinate morphological and biochemical interconversion of human neuroblastoma cells. J. Natl. Cancer Inst. 1983;71:741–747. [PubMed] [Google Scholar]
- 37.Presgraves S.P., Ahmed T., Borwege S., Joyce J.N. Terminally differentiated SH-SY5Y cells provide a model system for studying neuroprotective effects of dopamine agonists. Neurotox. Res. 2004;5:579–598. doi: 10.1007/BF03033178. [DOI] [PubMed] [Google Scholar]
- 38.Rosa A.I., Fonseca I., Nunes M.J., Moreira S., Rodrigues E., Carvalho A.N., Rodrigues C.M.P., Gama M.J., Castro-Caldas M. Novel insights into the antioxidant role of tauroursodeoxycholic acid in experimental models of Parkinson's disease. Biochim Biophys. Acta. 2017;1863:2171–2181. doi: 10.1016/j.bbadis.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 39.Benassi B., Filomeni G., Montagna C., Merla C., Lopresto V., Pinto R., Marino C., Consales C. Extremely low frequency magnetic field (ELF-MF) exposure sensitizes SH-SY5Y cells to the pro-Parkinson's disease toxin MPP(.) Mol. Neurobiol. 2016;53:4247–4260. doi: 10.1007/s12035-015-9354-4. [DOI] [PubMed] [Google Scholar]
- 40.Moreira S., Fonseca I., Nunes M.J., Rosa A., Lemos L., Rodrigues E., Carvalho A.N., Outeiro T.F., Rodrigues C.M.P., Gama M.J. Nrf2 activation by tauroursodeoxycholic acid in experimental models of Parkinson's disease. Exp. Neurol. 2017;295:77–87. doi: 10.1016/j.expneurol.2017.05.009. [DOI] [PubMed] [Google Scholar]
- 41.Song Q., Gou W.L., Zou Y.L. FAM3A protects against glutamate-Induced toxicity by preserving calcium homeostasis in differentiated PC12 cells. Cell Physiol. Biochem. 2017;44:2029–2041. doi: 10.1159/000485943. [DOI] [PubMed] [Google Scholar]
- 42.Jagadish S., Hemshekhar M., NaveenKumar S.K., Sharath Kumar K.S., Sundaram M.S., Basappa, Girish K.S., Rangappa K.S. Novel oxolane derivative DMTD mitigates high glucose-induced erythrocyte apoptosis by regulating oxidative stress. Toxicol. Appl. Pharmacol. 2017;334:167–179. doi: 10.1016/j.taap.2017.09.008. [DOI] [PubMed] [Google Scholar]
- 43.Nakai M., Mori A., Watanabe A., Mitsumoto Y. 1-methyl-4-phenylpyridinium (MPP+) decreases mitochondrial oxidation-reduction (REDOX) activity and membrane potential (Deltapsi(m)) in rat striatum. Exp. Neurol. 2003;179:103–110. doi: 10.1006/exnr.2002.8056. [DOI] [PubMed] [Google Scholar]
- 44.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seino S., Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol. Rev. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- 46.Tong J., Liu X., Vickstrom C., Li Y., Yu L., Lu Y., Smrcka A.V., Liu Q.S. The Epac-phospholipase Cepsilon pathway regulates endocannabinoid signaling and cocaine-induced disinhibition of ventral tegmental area dopamine neurons. J. Neurosci. 2017;37:3030–3044. doi: 10.1523/JNEUROSCI.2810-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kwak H.J., Park K.M., Choi H.E., Chung K.S., Lim H.J., Park H.Y. PDE4 inhibitor, roflumilast protects cardiomyocytes against NO-induced apoptosis via activation of PKA and Epac dual pathways. Cell Signal. 2008;20:803–814. doi: 10.1016/j.cellsig.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 48.Joshi R., Kadeer N., Sheriff S., Friend L.A., James J.H., Balasubramaniam A. Phosphodiesterase (PDE) inhibitor torbafylline (HWA 448) attenuates burn-induced rat skeletal muscle proteolysis through the PDE4/cAMP/EPAC/PI3K/Akt pathway. Mol. Cell Endocrinol. 2014;393:152–163. doi: 10.1016/j.mce.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 49.Yuan L., Dai X., Yang M., Cai Q., Shao N. Potential treatment benefits and safety of roflumilast in COPD: a systematic review and meta-analysis. Int. J. Chron. Obstruct. Pulmon. Dis. 2016;11:1477–1483. doi: 10.2147/COPD.S106370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang H.T. Cyclic AMP-specific phosphodiesterase-4 as a target for the development of antidepressant drugs. Curr. Pharm. Des. 2009;15:1688–1698. doi: 10.2174/138161209788168092. [DOI] [PubMed] [Google Scholar]
- 51.Napoletano M., Norcini G., Pellacini F., Marchini F., Morazzoni G., Ferlenga P., Pradella L. The synthesis and biological evaluation of a novel series of phthalazine PDE4 inhibitors I. Bioorg. Med. Chem. Lett. 2000;10:2235–2238. doi: 10.1016/s0960-894x(00)00449-2. [DOI] [PubMed] [Google Scholar]
- 52.Zhang K.Y., Ibrahim P.N., Gillette S., Bollag G. Phosphodiesterase-4 as a potential drug target. Expert Opin. Ther. Targets. 2005;9:1283–1305. doi: 10.1517/14728222.9.6.1283. [DOI] [PubMed] [Google Scholar]
- 53.Blum D., Torch S., Lambeng N., Nissou M., Benabid A.L., Sadoul R., Verna J.M. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson's disease. Prog. Neurobiol. 2001;65:135–172. doi: 10.1016/s0301-0082(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 54.Gross A., McDonnell J.M., Korsmeyer S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 55.Vos M., Verstreken P., Klein C. Stimulation of electron transport as potential novel therapy in Parkinson's disease with mitochondrial dysfunction. Biochem. Soc. Trans. 2015;43:275–279. doi: 10.1042/BST20140325. [DOI] [PubMed] [Google Scholar]
- 56.Yamashita N., Hayashi A., Baba J., Sawa A. Rolipram, a phosphodiesterase-4-selective inhibitor, promotes the survival of cultured rat dopaminergic neurons. Jpn. J. Pharmacol. 1997;75:155–159. doi: 10.1254/jjp.75.155. [DOI] [PubMed] [Google Scholar]
- 57.Kinoshita K.I., Muroi Y., Unno T., Ishii T. Rolipram improves facilitation of contextual fear extinction in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse model of Parkinson's disease. J. Pharmacol. Sci. 2017;134:55–58. doi: 10.1016/j.jphs.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 58.Rappold P.M., Tieu K. Astrocytes and therapeutics for Parkinson's disease. Neurotherapeutics. 2010;7:413–423. doi: 10.1016/j.nurt.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Niranjan R., Rajasekar N., Nath C., Shukla R. The effect of guggulipid and nimesulide on MPTP-induced mediators of neuroinflammation in rat astrocytoma cells, C6. Chem. Biol. Interact. 2012;200:73–83. doi: 10.1016/j.cbi.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 60.Agostinho P., Cunha R.A., Oliveira C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer's disease. Curr. Pharm. Des. 2010;16:2766–2778. doi: 10.2174/138161210793176572. [DOI] [PubMed] [Google Scholar]
- 61.Sun W., Jiao W., Huang Y., Li R., Zhang Z., Wang J., Lei T. Exchange proteins directly activated by cAMP induce the proliferation of rat anterior pituitary GH3 cells via the activation of extracellular signal-regulated kinase. Biochem Biophys. Res. Commun. 2017;485:355–359. doi: 10.1016/j.bbrc.2017.02.075. [DOI] [PubMed] [Google Scholar]
- 62.Nishi A., Shuto T. Potential for targeting dopamine/DARPP-32 signaling in neuropsychiatric and neurodegenerative disorders. Expert Opin. Ther. Targets. 2017;21:259–272. doi: 10.1080/14728222.2017.1279149. [DOI] [PubMed] [Google Scholar]
- 63.Jiang P., Dickson D.W. Parkinson's disease: experimental models and reality. Acta Neuropathol. 2018;135:13–32. doi: 10.1007/s00401-017-1788-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material