

Abstract
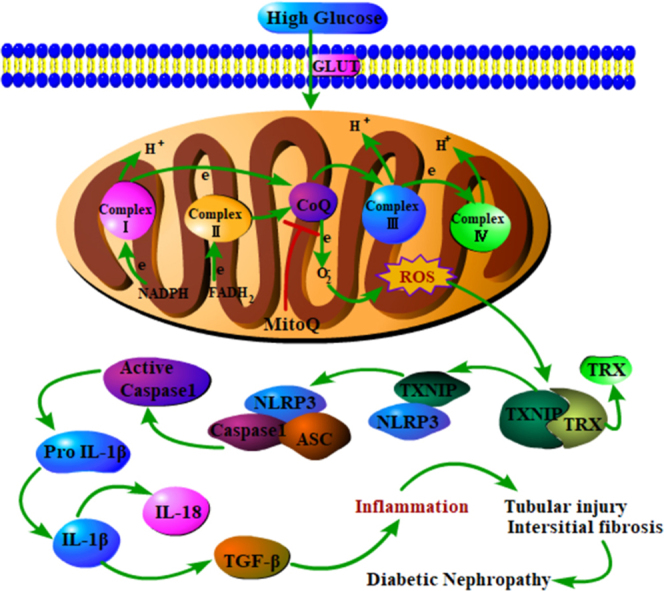
NLRP3/IL-1β activation via thioredoxin (TRX)/thioredoxin-interacting protein (TXNIP) following mitochondria ROS (mtROS) overproduction plays a key role in inflammation. However, the involvement of this process in tubular damage in the kidneys of patients with diabetic nephropathy (DN) is unclear. Here, we demonstrated that mtROS overproduction is accompanied by decreases in TRX expression and TXNIP up-regulation. In addition, we discovered that mtROS overproduction is also associated with increases in NLRP3/IL-1β and TGF-β expression in the kidneys of patients with DN and db/db mice. We reversed these changes in db/db mice by administering a peritoneal injection of MitoQ, an antioxidant targeting mtROS. Similar results were observed in human tubular HK-2 cells subjected to high-glucose (HG) conditions and treated with MitoQ. Treating HK-2 cells with MitoQ suppressed the dissociation of TRX from TXNIP and subsequently blocked the interaction between TXNIP and NLRP3, leading to the inhibition of NLRP3 inflammasome activation and IL-1β maturation. The effects of MitoQ were enhanced by pretreatment with TXNIP siRNA and abolished by pretreatment with monosodium urate (MSU) and TRX siRNA in vitro. These results suggest that mitochondrial ROS-TXNIP/NLRP3/IL-1β axis activation is responsible for tubular oxidative injury, which can be ameliorated by MitoQ via the inhibition of mtROS overproduction.
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen; BW, body weight; BG, blood glucose; DCFDA, dichlorodihydrofluorescein diacetate; DN, diabetic nephropathy; ECL, enhanced chemiluminescence; EM, electron microscopy; EMT, epithelial–mesenchymal transition; HbA1C, hemoglobin A1c; HE, hematoxylin-eosin; IF, immunofluorescence; IHC, immunohistochemistry; KW, kidney weight; LDL, low-density lipoprotein; MDA, malondialdehyde; MFI, median fluorescence intensity; MMP, mitochondrial transmembrane potential; MMT, methyl methane-thiosulphonate; MPTP, mitochondrial permeability transition pore; MSU, monosodium urate; MtROS, mitochondria reactive oxygen species; PAS, periodic acid-Schiff; Scr, serum creatinine; TC, total cholesterol; TG, triglyceride; TBARS, thiobarbituric acid reactive substances; TPP, triphenylphosphonium; TRX, thioredoxin; TXNIP, thioredoxin-interacting protein; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end-labeling; 8-OHdG, 8-oxo-deoxyguanosine; UA, uric acid
Keywords: Diabetic nephropathy, Mitochondria, Reactive oxygen species (ROS), TRX/TXNIP, NLRP3 inflammasome, MitoQ
Graphical abstract
Highlights
-
•
Reactive oxygen species promotes renal damage in diabetic nephropathy.
-
•
Mitochondrial ROS- TXNIP-NLRP3 biological axis involved in tubular injury of DN.
-
•
Inhibition of mitochondrial ROS by MitoQ ameliorated the renal tubular injury.
1. Introduction
Diabetic nephropathy (DN) is a major complication of diabetes and has become the leading cause of end-stage renal disease worldwide [1]. There is a clear evidence showing that tubular injury plays a pivotal role in the pathogenesis of DN [2], [3] and is closely related to its clinical manifestations [4]. Hyperglycemia-induced tubular injury has been attributed to many mechanisms, such as increases in extracellular matrix expression, advanced glycation product generation and Wnt/β-catenin activity [5], [6]. Danesh FR et al. recently reported that mitochondrial reactive oxygen species (mtROS) production is increased in the kidneys of db/db mice; these authors monitored a redox-sensitive green fluorescent protein biosensor (roGFP) in real time in vivo and found that the biosensor was expressed specifically in the mitochondrial matrix (db/dbmt-roGFP) [7]. Moreover, our previous studies, as well as other previous studies, have shown that mtROS overproduction induced apoptosis activation and tubular cell injury under high-glucose (HG) conditions [8], [9]. Therefore, mtROS overproduction plays a critical role in tubular damage in DN. However, Sharma K. et al. reported that superoxide production as assessed by in vivo real-time transcutaneous fluorescence, confocal microscopy and electron paramagnetic resonance analysis was reduced in the kidneys of mice with streptozotocin (STZ)-induced type 1 diabetes [10]. These controversial results might be attributable to differences between the animal models used in these studies. In addition, the precise mechanism responsible for the effects of ROS on tubular damage in DN has yet to be elucidated.
Several reports have shown that the NLRP3 inflammasome is closely associated with DN [11], [12]. However, the precise mechanism(s) by which HG conditions induce NLRP3 inflammasome activation still needs to be clarified. NLRP3 inflammasome activation by mtROS plays a central role in cellular responses [13], [14], [15]. Mitochondrial dysfunction has been regarded as a fundamental factor in the triggering of NLRP3-mediated inflammation [16], and mtROS overproduction is a critical contributor to NLRP3 inflammasome activation [15]. Thioredoxin-interacting protein (TXNIP) is a regulator of oxidative stress that is involved in cell proliferation, differentiation and apoptosis. TXNIP also inhibits the antioxidant activity of the endogenous antioxidant thioredoxin (TRX) by binding to the protein. Both TRX and TXNIP are expressed in the cytoplasm and mitochondria [17], where they regulate the NLRP3 inflammasome as well as cell apoptosis [13], [18], suggesting that the TXNIP/NLRP3 inflammasome play a key role in the pathogenesis of DN. However, the involvement of the TXNIP/NLRP3 inflammasome in the development of DN in tubulopathy remains largely unknown.
MitoQ is an orally bioavailable mitochondrial-targeted antioxidant that consists of a lipophilic triphenylphosphonium (TPP) cation covalently linked to the ubiquinone of the mitochondrial respiratory chain. MitoQ protects against a range of oxidation-related diseases, including cardiovascular disease [19], metabolic syndrome [20], Alzheimer's disease [21] and DN [22]. In this study, we applied MitoQ as a scavenger of mtROS to determine the role and regulatory effects of the TXNIP/NLRP3/IL-1β axis in tubular injury in DN. We demonstrated that TXNIP/NLRP3/IL-1β axis activation plays a key role in tubular injury in DN.
2. Materials & methods
2.1. Antibodies and other reagents
Antibodies targeting the following proteins were used in this study: NLRP3 (sc-66846, rabbit polyclonal, Santa Cruz Biotechnology, USA; ab4207, goat polyclonal, Abcam, USA), TRX (ab26320, rabbit polyclonal/ab16965, mouse monoclonal, Abcam, USA), TXNIP (sc-166234, mouse monoclonal, Santa Cruz Biotechnology, USA; ab215366, rabbit polyclonal, Abcam, USA), COX IV (ab14744, mouse monoclonal, Abcam, USA), Fibronectin (FN) (ab45688, rabbit monoclonal, Abcam, USA), Caspase-1 (ab1872, rabbit polyclonal, Abcam, USA), IL-1β (ab9722, rabbit polyclonal, Abcam, USA), IL-18 (#10663-1-AP, rabbit polyclonal, Proteintech, USA), Collagen I (M38, mouse monoclonal, DSHB Biosciences, USA; ab23446, mouse monoclonal, Abcam, USA), and Collagen IV (ab6586, rabbit polyclonal, Abcam, USA). MitoQ was obtained from FOCUS Biomolecules, and monosodium urate (MSU), TXNIP siRNA, TRX siRNA, DHE, MitoTracker Red, MitoSOX Red and DCFDA were purchased from Invitrogen.
2.2. Clinical data
Patients with DN (n = 15) and control patients with minor primary lesions who did not have diabetes (n = 15) were recruited for the study. Kidney biopsy tissues were stained with periodic acid-Schiff (PAS), and mitochondrial morphological changes were detected by electron microscopy (EM), as described in ref [23]. Tubulo-interstitial and glomerular injury were evaluated and scored based on the Tervaet semiquantitative scoring system, as described in Ref. [24]. All experiments described above were performed under the supervision and with the approval of the Institutional Human Experimentation Ethics Committee, Second Xiangya Hospital, Central South University.
2.3. Animal experimental design
Eight-week-old diabetic male db/db mice from the Aier Matt Experimental Animal Company (Suzhou, China) were randomly divided into a DN group that received MitoQ treatment (DN+MitoQ) (n = 12) and a DN group (n = 12). Eight-week-old db/m mice served as controls (n = 12). The mice in the DN+MitoQ group were intraperitoneally injected with MitoQ twice a week for 12 weeks (5 mg/kg), and the mice in the control and DN groups were injected with a 0.1 mol/L sodium citrate solution. The mice were sacrificed at 24 weeks of age, and the kidneys were harvested as discussed previously [22]. All procedures were conducted in accordance with the relevant institutional guidelines of the Animal Experimentation Ethics Committee of Second Xiangya Hospital of Central South University.
2.4. Proteomic analysis
Proteomic analysis of the kidney tissues was performed as described in ref [25]. Briefly, kidney proteins were extracted, alkylated by methyl methane-thiosulphonate (MMTS), digested by incubation with trypsin and then labeled with iTRAQ reagents. The proteins were also subjected to strong cation exchange (SCX) chromatography and high-performance liquid chromatography (HPLC) experiments. The proteins were identified and quantified by using Mascot v2.3 (Matrix Science, London, UK).
2.5. Quantitative real-time PCR
RNA was extracted from the kidney tissues and was reverse-transcribed into cDNA using a TaKaRa cDNA Synthesis Kit. Real-time PCR was performed using SYBR premix EXTaqTM reagents (TaKaRa). The sequences of the primers (from Sangon Biotech Corporation, Shanghai, China) for FN, Collagen I and Collagen IV were as follows: FN (Mouse), 5′-CTTT GGCAGTGGTCATTTCAG-3′ (sense) and 5′-TGGTAGGTCTTCCCATCGTCA-3′ (antisense) (product size: 142 bp); Collagen I (Mouse), 5′-CGDCATCAAGGTCTACTGC-3′ (sense) and 5′-GAATCCA TCGGTCATGCTCT-3′ (antisense); and Collagen IV (Mouse), 5′-ATGTCAATGGCACCCATCAC-3′ (sense) and 5′-CTTCAAGGTGGACGGCGTAG-3′ (antisense). The data are presented as fold changes (2-ΔΔCt).
2.6. Morphological analysis
Briefly, 4-μm-thick kidney tissue sections were prepared for hematoxylin-eosin (H&E), PAS and Masson trichrome staining, as described in ref [26].
2.7. Immunohistochemistry (IHC)
Kidney tissue sections from humans or mice were prepared for IHC, as described in ref [22]. The tissues were incubated with anti-NLRP3 (1:200), anti-TRX (1:100), anti-TXNIP (1:100), anti-Caspase-1 (1:200), anti-IL-1β (1:200) or anti-IL18 (1:100) antibodies.
2.8. Analysis of renal apoptosis
Apoptosis was detected by terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) staining in situ, according to the manufacturer's instructions [22].
2.9. Western blot (WB) analysis
Equal amounts of extracted kidney protein or cellular protein were subjected to SDS-PAGE, as described in ref [27]. The following primary antibodies were used in the experiment: anti-NLRP3 (1:500), anti-TRX (1:500), anti-IL-1β (1:500), anti-Caspase-1 (1:500), anti-TXNIP (1:500), anti-FN (1:2000), anti-Collagen I (1:2000) and anti-β-actin (1:6000). Antibody-antigen complexes were detected with enhanced chemiluminescence (ECL) (Amersham Pharmacia Biotech, Uppsala, Sweden).
2.10. Cell line
The HK-2 cell line, a human proximal tubular epithelial cell line, was purchased from ATCC and maintained in DMEM/F12, as described in ref [26]. The cells were treated with different concentrations of D-glucose and with or without MitoQ, siRNA, inhibitors or activators.
2.11. Cell transfection
HK-2 cells were pre-transfected with TXNIP or TRX siRNA using Lipofectamine 2000 (Life Technologies, USA), as previously described [22].
2.12. Assessment of MPTP expression and mitochondrial H2O2 production
Mitochondrial permeability transition pore (MPTP) expression was detected by a Mitochondrial Calcium Fluorescence Detection Kit (Genmed Scientifics Inc.), and mitochondrial H2O2 production was assessed with scopoletin fluorescence, as described in ref [28].
2.13. Analysis of Caspase-1, IL-1β, IL-18 and TGF-β activity
Caspase-1 (R&D Systems, USA), IL-1β, IL-18 and TGF-β (Ray Biotech, USA) expression and activity levels in the above cells were detected with the corresponding ELISA Kits, according to the manufacturers’ instructions [27], [29].
2.14. Measurement of mitochondrial ROS and mitochondrial membrane potential
Intracellular superoxide production was detected with dichlorodihydrofluorescein diacetate (DCF-DA), and mtROS levels were determined by MitoSOX Red (Molecular Probes). Mitochondrial membrane potential (MMP) was assessed with tetramethylrhodamine, ethyl ester (TMRE) dye (Molecular Probes), as previously described [9], [30].
2.15. Cell immunofluorescence (IF)
HK-2 cells were seeded onto round glass dishes and grown to near confluency, and then they were subjected to various treatments. The cells were first immersed in MitoTracker Red (1:1000, Molecular Probe, Invitrogen) solution diluted in serum-free DMEM/F12 buffer for 8 min in the dark. After being washed thrice with PBS, the cells were fixed with 4% paraformaldehyde for 5 min, permeabilized with ice-clod methanol for 10 min at − 20 ℃ and incubated in blocking buffer (1% BSA, 0.3% Triton X-100 in PBS, pH 7.4) for 1 h. The cells were subsequently incubated with primary antibody solution for 2 h at room temperature. The cells were then washed thrice with PBS and incubated with FITC- or Rhodamine-conjugated secondary antibody solution for 1 h in the dark. The cell nuclei were stained with Hoechst (1:25000) for 1 min, and then images were obtained by a confocal laser scanning microscope (Zeiss LSM 780), as described in ref [27].
2.16. Confocal microscopy
Confocal microscopy was carried out with an LSM 780 META laser scanning microscope (Zeiss, Thornwood, NY). Laser excitation of the green, red and blue fluorescence was performed sequentially using 488-, 568- and 405-nm lasers, respectively. The wavelengths of the emission filters used for the green, red and blue fluorescence were 490–578 nm, 565–712 nm, and 410–495 nm, respectively. The analysis was performed using LSM 510 software (Zeiss), as described in ref [31].
2.17. Immunoprecipitation (IP)/WB analysis
IP/WB analysis was carried out as described previously [32].
2.18. Flow cytometry analysis
Apoptosis was measured using an Annexin V-FITC Apoptosis Detection Kit (BD Bioscience). NLRP3 localization in the mitochondria was detected with COX IV and anti-NLRP3 antibody staining. Flow cytometry was used to test the interactions between NLRP3 and TXNIP and between TXNIP and TRX using anti-NLRP3, anti-TRX, and anti-TXNIP antibodies, as described in ref [33]
2.19. Luminex cytokine assays
IL-1β, IL-18, FN and Collagen I expression levels in HK-2 cells were measured with commercial Milliplex Human Cytokine Kits, according to the manufacturer's instructions (R&D Systems, Luminex Human Magnetic Assay, USA), and were detected with a Bio-Plex Suspension Array System (MAP™ Technology, Austin, Texas USA). The data were acquired and analyzed as the median fluorescence intensity (MFI), as described in ref [34].
2.20. Statistical analysis
Statistical analysis of the experimental data was performed using SPSS 13.0 software, and the results are presented as the mean ± SE. Measurement data were analyzed by one-way analysis of variance (one-way ANOVA). Multiple comparisons of the means of two samples were performed using independent-samples T-tests, and the correlations between two variables were assessed using Pearson correlation analysis. P < 0.05 indicated that the difference was statistically significant.
3. Results
3.1. Biological characteristics of and kidney morphological changes in patients with DN
Patients with DN exhibited noticeably higher blood glucose, hemoglobin A1c (HbA1C) and 24-h urine protein levels than control subjects, as shown in Table 1. There was no difference in age or sex between the two groups. PAS staining revealed that mesangial expansion and focal tubular atrophy had occurred in the kidneys of patients with DN (Fig. 1 Aa, b), while DHE staining showed that ROS levels in the kidneys of patients with DN were elevated compared with those in the kidneys of control subjects (Fig. 1Ae, f). Severe glomerulosclerosis and tubulo-interstitial injury were noted in the kidneys of patients with DN (Fig. 1B). Furthermore, IF analysis showed that NLRP3 expression in the kidneys of patients with DN was increased compared with that in the kidneys of control subjects (Fig. 1Ac, d and C). Additionally, NLRP3 expression was positively correlated with tubulo-interstitial damage (r = 0.752, Fig. 1D), glomerular damage (r = 0.644, Fig. 1E), urine protein (r = 0.674, Fig. 1F) and oxidative stress (r = 0.742, Fig. 1G). EM revealed that extensive foot process fusion, significant mitochondrial fragmentation and damage (Fig. 1H) and prominent increases in the numbers of tubular cells with fragmented mitochondria (Fig. 1I) had occurred in the kidneys of patients with DN. Moreover, oxidative stress levels as measured by fluorescence intensity were positively correlated with the numbers of tubular cells with mitochondrial fragmentation (r = 0.704, Fig. 1J).
Table 1.
Clinical characteristics of the patients with DN and the patients with minor lesions who did not have diabetes.
| Clinical data | Control | DN |
|---|---|---|
| N | 15 | 15 |
| BMI (kg/m2) | 23 ± 3.4 | 24.9 ± 4.0 |
| Blood glucose (mmol/L) | 4.50 ± 0.51 | 8.5 ± 2.3* |
| HBA1c (%) | 4.78 ± 0.49 | 8.1 ± 0.6* |
| Total cholesterol (mmol/L) | 6.32 ± 0.12 | 7.14 ± 0.23* |
| Triglyceride (mmol/L) | 2.67 ± 0.42 | 4.15 ± 1.8* |
| LDL (mmol/l) | 2.1 ± 0.61 | 2.8 ± 0.9* |
| ALT (U/L) | 18.3 ± 4.2 | 17.8 ± 5.6 |
| AST (IU/L) | 24.2 ± 9.2 | 23.9 ± 8.2 |
| Albumin (g/l) | 38.9 ± 3.9 | 32.2 ± 1.8* |
| Scr (μmol/L) | 92.30 ± 8.86 | 94.23 ± 7.87 |
| BUN (mmol/L) | 5.81 ± 0.34 | 6.01 ± 0.23 |
| UA (μmol/L) | 350 ± 68 | 402 ± 87 |
| Urine protein (g/24 h) | 3.15 ± 1.28 | 4.37 ± 1.12* |
| Red blood cells count in urine sediment | 14750 ± 1023 | 10233 ± 1124 |
| Systolic pressure (mmHg) | 118.24 ± 4.30 | 147.32 ± 2.3* |
| Diastolic pressure (mmHg) | 81.70 ± 1.72 | 94.46 ± 2.32* |
Note: HBA1c, glycosylated hemoglobin; LDL, low-density lipoprotein; ALT, alanine aminotransferase; AST, aspartate aminotransferase; Scr, serum creatinine; BUN, blood urea nitrogen; UA, uric acid. *p < 0.05, vs control. Values are the mean ± SE. n = 15.
Fig. 1.

Tubular injury, NLRP3 expression and ROS production are enhanced in the kidney tissues of patients with DN.A: PAS and DHE staining showing that tubular injury, interstitial fibrosis (top panels) and ROS production (bottom panels) are enhanced in the kidney tissues of patients with DN. IF revealed that NLRP3 expression is increased in patients with DN (middle panels) (magnification × 400). B: Tubulo-interstitial damage and glomerular damage scores. C: Quantification of DHE signal intensity and NLRP3 expression. D-G: Analysis of the correlations between NLRP3 expression and tubulo-interstitial damage (D), glomerular damage (E), urine protein (F) and oxidative stress (G) in the kidneys of patients with DN. H: Representative image of the electron microscopy analysis. The number of tubular cells with mitochondrial fragmentation was notably increased in the renal tissues of patients with DN (Hd, f) compared with those of control patients (Hc, e) (magnification × 10,000).I: Relative percentage of renal tubular cells with fragmented mitochondria. J: Correlation between mitochondrial fragmentation in tubular cells and oxidative stress. *P < 0.05, #P < 0.05 versus control. Values are the mean ± SE. n = 15.
3.2. TRX and TXNIP, NLRP3, IL-1β and IL-18 expression in the kidney
IHC and semi-quantification demonstrated that the kidneys of patients with DN had markedly higher TXNIP, IL-1β, IL-18 and NLRP3 expression but lower TRX expression than the kidneys of control subjects (Fig. 2A and B). Oxidative stress was positively correlated with TXNIP (r = 0.365, Fig. 2D), IL-1β (r = 0.531, Fig. 2E) and IL-18 expression (r = 0.412, Fig. 2F) but was negatively correlated with TRX expression (r = −0.545, Fig. 2C). Additionally, NLRP3 expression was positively correlated with TXNIP (r = 0.623, Fig. 2H), IL-1β (r = 0.590, Fig. 2I) and IL-18 expression (r = 0.560, Fig. 2J) and negatively correlated with TRX expression (r = −0.577, Fig. 2G).
Fig. 2.

TRX, TXNIP, NLRP3, IL-1β and IL-18 expression is altered in the kidney tissues of patients with DN.A: Immuno-histochemical analysis of TRX, TRX, TXNIP, NLRP3, IL-1β and IL-18 expression in the kidney tissues of patients with DN and controls (magnification × 200).B: Semi-quantification of IHC staining for TRX, TXNIP NLRP3, IL-1β and IL-18. C-F: Analysis of the correlations between oxidative stress and TRX (C), TXNIP (D), IL-1β (E) and IL-18 expression (F) in patients with DN. G-J: Analysis of the correlations between NLRP3 expression and TRX (G), TXNIP (H), IL-1β (I) and IL-18 expression (J) in patients with DN. Values are the mean ± SE.*P < 0.05 versus control. n = 15.
3.3. MitoQ reversed the biochemical parameter alterations and morphological changes noted in the kidneys of db/db mice
There was no significant difference in body weight or kidney weight between the db/db mice treated with MitoQ for 20 weeks and the untreated db/db mice (Table 2, Fig. 3A). Blood glucose (Table 2, Fig. 3B), urine protein (Table 2, Fig. 3C), blood urea nitrogen, serum creatinine, cholesterol and triglyceride levels (Table 2) were increased in untreated db/db mice compared with control mice; however, all of these increases were significantly attenuated following treatment with MitoQ. Enhancements of urinary H2O2, thiobarbituric acid reactive substances (TBARS), 8-oxo-deoxyguanosine (8-OHdG) and renal malondialdehyde (MDA) content were also attenuated by MitoQ treatment (Fig. 3D-G). H&E and PAS staining showed that glomerular mesangial matrix proliferation was enhanced in db/db mice compared with control mice; however, this enhancement was alleviated in db/db mice treated with MitoQ (Fig. 3H-c vs H-b and H-f vs H-e). Further analysis demonstrated that tubulo-interstitial fibrosis, ROS generation and cell apoptosis (apoptotic cells indicated by the arrow) were enhanced in the kidneys of db/db mice compared with those of control mice (Fig. 3Hh-n vs Hg-m); however, these enhancements were dramatically attenuated following MitoQ administration (Fig. 3Hi-o vs Hh-n). These results were confirmed by the quantification analysis (Fig. 3I). The alterations in renal oxidative stress levels and apoptotic cell numbers noted in db/db mice (Fig. 3J) were significantly reversed by MitoQ treatment. In addition, renal oxidative stress was found to be strongly positively correlated with tubulo-interstitial injury (r = 0.663, Fig. 3K). MitoQ dramatically ameliorated the basement membrane thickening, foot process fusion and mitochondrial fragmentation noted in the kidneys of the db/db mice (Fig. 3L and M).
Table 2.
Characteristics of the different groups mice (X ± S).
| Group | db/m | db/db | db/db+MitoQ |
|---|---|---|---|
| N | 12 | 12 | 12 |
| BW (g) | 23.3 ± 1.5 | 52.6 ± 3.0* | 49.0 ± 1.8* |
| KW (mg) | 118 ± 4.2 | 203 ± 22.1* | 189 ± 19.3*# |
| BS (mmol/l) | 5.35 ± 0.5 | 33.1 ± 2.2* | 29.8 ± 2.6*# |
| BUN (mmol/l) | 6.32 ± 0.64 | 9.15 ± 1.52* | 7.23 ± 1.03*# |
| Scr (mg/dl) | 0.12 ± 0.01 | 0.17 ± 0.04* | 0.14 ± 0.07*# |
| TC (mmol/l) | 1.22 ± 0.32 | 3.25 ± 0.84* | 3.08 ± 0.74*# |
| TG (mmol/l) | 0.63 ± 0.23 | 1.8 ± 0.13* | 1.62 ± 0.25*# |
Note: BW, body weight; KW, kidney weight, BG, blood glucose; TC, total cholesterol; TG, triglyceride. *p < 0.05, vs db/m group;#p < 0.05,vs db/db group. Values are the mean ± SE. n = 12.
Fig. 3.

Effects of MitoQ on morphological and functional characteristics, as well as oxidative stress and apoptosis, in the kidneys of db/db mice.A: Body weight changes in db/m, db/db and db/db mice treated with MitoQ for 8–20 weeks. B: Blood glucose levels. C: Proteinuria levels. D: Urinary H2O2 levels. E: Urinary TBARS levels. F: Urinary 8-OHdG levels. G: Renal MDA concentrations. H: Kidney sections stained with H&E, PAS, Masson trichrome, DHE and TUNEL (magnification × 400). I: Tubulo-interstitial damage and glomerular damage scores. (J) Oxidative stress densities and TUNEL-positive cell percentages in the kidneys of the different groups. K: Analysis of the correlation between oxidative stress and tubulo-interstitial damage (r = 0.663, P < 0.05). L: EM analysis shows that the kidney tissues of db/db mice displayed obvious basement membrane thickening and notable mitochondrial morphological changes in compared with those of db/m mice (Lb vs La and Le vs Ld). These changes were reversed by MitoQ treatment (Lc vs Lb and Lf vs Le) (magnification × 10,000). M: Relative percentages of fragmented mitochondria in the three groups. The data are presented as the mean ± SE; r: correlation coefficient; *P < 0.05 vs control group; #P < 0.05 vs db/db group. n = 12.
3.4. MitoQ attenuated TXNIP, NLRP3 and IL-1β up-regulation and inhibited FN and Collagen I and IV expression in the kidneys of db/db mice
The proteomics analyses showed that NLRP3, TXNIP, Caspase-1 and IL-1β levels in the protein extracts of db/db mouse kidneys were higher than those in the protein extracts of control mouse kidneys; however, TRX levels in the protein extracts of db/db mouse kidneys were lower than those in the protein extracts of control mouse kidneys (Fig. 4A). Similar results were observed in the IHC experiments. All of the above alterations were partially attenuated by MitoQ treatment (Fig. 4B and C). Correlation analysis showed that the NLRP3 expression was positively correlated with TXNIP (r = 0.695, Fig. 4E), Caspase-1 (r = 0.674, Fig. 4F), and IL-1β (r = 0.694, Fig. 4G) expression but was negatively correlated with TRX expression (r = −0.707, Fig. 4D). These results were confirmed by WB analysis (Fig. 4H and I). Moreover, MitoQ treatment significantly reduced FN, Collagen I, and Collagen IV mRNA and protein levels (Fig. 5A–G). Correlation analysis demonstrated that NLRP3 expression was strongly positively correlated with Collagen I (r = 0.652, Fig. 4H), Collagen IV (r = 0.761, Fig. 4I) and FN expression (r = 0.590, Fig. 4J). These results indicated that MitoQ suppresses the development of fibrosis by blocking the TXNIP/NLRP3/IL-1β axis.
Fig. 4.

Effects of MitoQ on renal NLRP3, TRX, TXNIP, Caspase-1 and IL-1β expression in db/db mice.A: Renal expression of the proteins detected by iTRAQ-based quantitative proteomics analysis in db/m and db/db mice. Green represents down-regulation, red represents up-regulation, and black represents no change. The bar code (− 1–1) at the top shows the color scale of the log2 values. B: Immunohistochemical analysis of NLRP3 (Ba-c), TRX (Bd-f), TXNIP (Bg-i), Caspase-1 (Bj-l) and IL-1β expression (Bm-o) in kidney tissues. (magnification × 200). C: Quantification of NLRP3, TRX, TXNIP, Caspase-1 and IL-1β expression in tissues from the different groups. D-G: Analysis of the correlations between NLRP3 expression and TRX, TXNIP, Caspase-1 and IL-1β expression: r (NLRP3/TRX) = −0.707 (D), r (NLRP3/TXNIP) = 0.695 (E), r (NLRP3/Caspase-1) = 0.674 (F) and r (NLRP3/IL-1β) = 0.694 (G). H: Western blotting analysis of NLRP3 (H, first line), TRX (H, second line), TXNIP (H, third line), Caspase-1 (H, fourth line) and IL-1β expression (H, last line) in the kidneys in the different groups of mice. I: Densitometric analysis of the WB analysis results. Values are the mean ± SE; r: correlation coefficient; *P < 0.05 vs control group; #P < 0.05 vs db/db group. n = 12.
Fig. 5.

Effects of MitoQ on FN, Collagen I and Collagen IV expression in the kidney tissues of db/db mice.A-C: Relative mRNA expression levels of FN (A), Collagen I (B) and Collagen IV (C) in the kidneys in the different groups of mice. D: Western blotting analysis of FN (upper panel), Collagen I (middle panel) and Collagen IV (bottom panel) expression in the kidneys in the different groups. E: Densitometric analysis of the WB analysis results. The FN, Collagen I and Collagen IV signals were normalized to the β-actin signals for the same samples to determine the fold-change relative to the control (Ctrl), whose expression level was set as 1. F: IF showing Collagen I (Fa-c), Collagen IV (Fd-f) and FN expression (Fg-i) in kidney sections from the different groups (magnification × 200). G: The bar graphs represent the semi-quantified fluorescence intensity of Collagen I, Collagen IV and FN. H-J: Analysis of correlations between NLRP3 expression and Collagen I, Collagen IV and FN expression: r (NLRP3/Collagen I) = 0.652 (D), r (NLRP3/Collagen IV) = 0.761 (E) and r (NLRP3/FN) = 0.590 (F). Values are the mean ± SE; *P < 0.05, vs control group; #P < 0.05 vs HG group. n = 12.
3.5. MitoQ mitigated ROS generation and mitochondrial dysfunction and blocked TXNIP/NLRP3/IL-1β signaling pathway activation in HK-2 cells exposed to HG conditions
As shown in the upper panel of Fig. 6A, exposure to HG conditions increased oxidative stress levels as measured by DCFDA staining in HK-2 cells; however, this effect was abolished by MitoQ administration. Exposure to HG conditions also reduced MMP and increased mtROS levels, as shown by TMRE and MitoSOX Red staining, respectively. These changes were reversed in cells pretreated with MitoQ (Fig. 6A, middle and bottom panels). The effects of MitoQ were dose dependent (Fig. 6B, C and D). Pretreatment with MitoQ also decreased the intracellular levels of H2O2 in a dose-dependent manner in cells exposed to an HG environment (Fig. 6E). Similar results were noted when MPTP expression was examined (Fig. 6F). WB analysis showed that exposure to HG conditions increased NLRP3, TXNIP, Caspase-1, IL-1β, Collagen I and FN expression levels and decreased TRX expression levels in a dose-dependent manner in HK-2 cells (Fig. 6G and H). However, these changes were significantly reversed by MitoQ (Fig. 6G line 5 vs line 3 and 6 H). Further investigation showed that MitoQ attenuated the alterations in the expression of these proteins in a dose-dependent manner (Fig. 6I and J). Additionally, MitoQ pretreatment alleviated the abovementioned changes in NLRP3, TRX and TXNIP expression in the mitochondria of HK-2 cells exposed to HG conditions and prevented the translocation of NLRP3 to the mitochondria (Fig. 7A–D).
Fig. 6.

Effects of MitoQ on NLRP3, TRX and TXNIP expression and intracellular ROS production in HK-2 cells exposed to different concentrations of D-glucose. A: Representations of intracellular ROS levels (A, upper panel), MMP (A, middle panel) and mitochondrial ROS levels (A, bottom panel) in HK-2 cells subjected to HG treatment with or without MitoQ pretreatment. B-F: Quantification of intracellular ROS production as measured with DCFDA staining (B), MMP as measured with TMRE staining (C), mitochondrial ROS production as measured with MitoSox Red staining (D), cellular H2O2 production (E) and Ca2+ loading for MPTP opening (F) in HK-2 cells treated with or without different concentrations of MitoQ. G-J: WB and densitometric analysis of NLRP3, TRX, TXNIP, Caspase-1, IL-1β, Collagen I and FN expression in HK-2 cells exposed to different concentrations of D-glucose and treated with or without MitoQ. Values are the mean ± SE, *P < 0.05, vs control group; #P < 0.05 vs HG group. n = 3.
Fig. 7.

Effects of MitoQ on the associations between NLRP3 expression and TRX and TXNIP expression in HK-2 cells subjected to an HG environment. A-D: IF analysis showing MMP and NLRP3 (A), TRX (B), and TXNIP expression (C) in HK-2 cells exposed to HG conditions and pretreated with or without MitoQ. D: Quantification of NLRP3, TRX and TXNIP expression in HK-2 cells exposed to HG and pretreated with or without MitoQ. E-H: Confocal immunofluorescence images and semi-quantification showing the co-localization of TRX and TXNIP (E, G) and TXNIP and NLRP3 (F, H) in HK-2 cells exposed to HG conditions and pretreated with or without MitoQ. I-K: Flow cytometry and quantification analysis of COX IV and NLRP3 expression (I), TRX-TXNIP interactions (J), and TXNIP-NLRP3 interactions (K) in HK-2 cells cultured under HG conditions and pretreated with or without MitoQ. L, M: Co-IP and WB analysis showing the interactions between TXNIP and NLRP3 and between TXNIP and TRX in HK-2 cells subjected to HG conditions and treated with MitoQ or MSU. N-P: Quantification analysis of the interactions between TXNIP and NLRP3 (N, P) and between TXNIP and TRX (O) in HK-2 cells cultured under HG conditions following MitoQ or MSU administration. Values are the mean ± SE; *P < 0.05 vs control group; #P < 0.05 vs HG group. n = 3.
3.6. MitoQ attenuated the co-localization of and interactions between TXNIP and TRX, as well as the co-localization of and interactions between TXNIP and NLRP3
Excessive mtROS production causes TRX to dissociate from its binding protein, TXNIP, which subsequently binds to NLRP3 and promotes NLRP3 inflammasome activation [15]. Thus, we assessed the alterations in the co-localization of TXNIP and TRX and TXNIP and NLRP3 using IF staining. The co-localization of TXNIP and NLRP3 was increased in HK-2 cells exposed to an HG environment; in contrast, the co-localization of TRX and TXNIP was reduced in HK-2 cells exposed to HG environment. These changes were reversed by treatment with MitoQ (Fig. 7E-H). These results were confirmed by flow cytometry analysis (Fig. 7I-K). IP/WB assays utilizing NLRP3, TRX and TXNIP antibodies showed that the interactions between TXNIP and TRX were significantly reduced in cells treated with HG; however, contrasting results were observed for the interactions between TXNIP and NLRP3. MitoQ administration dramatically reversed these changes in a time-dependent manner. No change was seen in TXNIP expression, which served as a loading control (Fig. 7L, N and O). Additionally, HG increased the interaction between TXNIP and NLRP3 (Fig. 7M, line 2 vs line 1); this effect was aggravated in cells treated with MSU, a selective NLRP3 activator (Fig. 7M, line 4 vs line 2), but was attenuated in cells treated with MitoQ (Fig. 7M, line 5 vs line 2). The inhibitory effect of MitoQ on the interaction between TXNIP and NLRP3 was partially blocked by pretreatment with MSU (Fig. 7M, line 7 vs line 5), suggesting that MitoQ regulated the binding of TXNIP to NLRP3 by suppressing mtROS production in HK-2 cells exposed to HG conditions.
3.7. MitoQ down-regulated FN, Collagen I and TGF-β expression by suppressing the biological TXNIP/NLRP3/IL-1β axis in HK-2 cells exposed to HG conditions
Cell fluorescence staining demonstrated that MitoQ down-regulated FN and Collagen I expression in HK-2 cells exposed to HG conditions (Fig. 8A). WB analysis revealed that MitoQ reduced enhancements of cleaved Caspase-1, IL-1β and IL-18 expression in a dose-dependent manner in cells exposed to HG conditions (Fig. 8B) and altered the activity of Caspase-1, IL-1β, IL-18, TGF-β and TGF-β content and cell apoptosis (Fig. 8C–H).
Fig. 8.

Effects of MitoQ, MSU, TRX siRNA and TXNIP siRNA on TXNIP/NLRP3/IL-1β axis and pro-fibrotic protein expression and apoptosis in HK-2 cells cultured under HG conditions. A: IF showing FN (Aa-c) and Collagen I (Ad-f) expression in HK-2 cells subjected to HG treatment following MitoQ treatment. B: WB analysis of Caspase-1, cleaved IL-1β and IL-18 expression in HK-2 cells cultured under HG conditions and treated with different concentrations of MitoQ (50,100, 150 nM). C-H: ELISA of Caspase-1 (C), IL-1β (D), IL-18 (E), TGF-β (F) activity; TGF-β expression (G); and relative apoptosis (H) in HK-2 cells subjected to HG conditions and treated with different concentrations of MitoQ (50, 100, 150 nM). I: Luminex analysis of IL-1β, IL-18, FN and Collagen I expression in HK-2 cells subjected to an HG environment and treated with MitoQ, MSU or TXNIP siRNA. J, K: WB and densitometric analysis of NLRP3, IL-1β, IL-18 and FN expression in HK-2 cells exposed to an HG environment and pretreated with MitoQ, TRX siRNA or TXNIP siRNA. Values are the mean ± SE; *P < 0.05 vs control group; #P < 0.05 vs HG group. n = 3.
To determine whether MitoQ regulated the expression of inflammatory cytokines and profibrotic signals via the mtROS/TXNIP/NLRP3 axis, we carried out siRNA experiments. Luminex assay demonstrated that IL-1β, IL-18, FN, and Collagen I protein expression levels were extremely high in HK-2 cells exposed to an HG environment (Fig. 8I bar 2 vs bar 1); however, these alterations were reversed by treatment with MitoQ or TXNIP siRNA (Fig. 8I bar 4 vs bar 2 and bar 8 vs bar 2) and enhanced by treatment with MSU (Fig. 8I bar 6 vs bar 2). The down-regulatory effects of MitoQ on the expression of these proteins were partially blocked when the agent was administered in combination with MSU in cells exposed to HG conditions (Fig. 8I bar 9 vs bar 4); however, they were remarkably enhanced by TXNIP siRNA (Fig. 8I bar 10 vs bar 4). Similar results were obtained by WB analysis. The expression profile of NLRP3 was similar to that of IL-1β. The changes in the expression of these proteins noted in HK-2 cells treated with TXNIP siRNA contrasted with those observed in cells transfected with TRX siRNA (Fig. 8J and K).
4. Discussion
Signals released by the mitochondria regulate and promote the inflammatory response during aging [35], and long-lived species display lower ROS production and/or higher antioxidant capacity [36]. Cellular ROS are also generated by the NADPH oxidase (NOX) system; however, many studies have demonstrated that NOX-derived ROS can activate NLRP3 inflammasomes [37] and that NLRP3 inflammasome activation triggered by TXNIP/NADPH oxidase signaling is also involved in podocyte injury under HG conditions [38]. In addition, the inhibition of Nox4-responsive oxidative stress and the ROS-sensitive NLRP3 signaling pathway under HG conditions protects against endothelial cell dysfunction both in vivo and in vitro [39]. These data clearly indicate that NOX plays a role in NLRP3 inflammasome activation. However, multiple lines of evidence indicate that the mitochondria are a major intracellular sources of ROS (90%) [40]. Moreover, previous studies by our laboratory and others have shown that mitochondrial ROS production plays a key role in the development of DN [22], [26], [41]. Thus, in this study we focused mainly on elucidating the mechanisms of NLRP3 inflammasome activation by mtROS and demonstrated that mitochondrial dysfunction activated the TXNIP/NLRP3/IL-1β biological axis in the kidney, a change associated with tubular damage and renal fibrosis in patients with DN and db/db mice. We also demonstrated that MitoQ treatment reduced mtROS levels and therefore inhibited the TXNIP/NLRP3/IL-1β signaling pathway, which led to the alleviation of kidney injury in db/db mice. We also demonstrated that MitoQ suppressed the dissociation of TXNIP from TRX and prevented the binding of TXNIP to NLRP3, thereby inhibiting NLRP3 inflammasome activation. These data suggested that the activation of the mtROS-TXNIP/NLRP3/IL-1β biological axis plays an essential role in kidney tubular injury in DN and that as an inhibitor of this pathway, MitoQ may serve as a therapeutic agent capable of facilitating DN regression.
Chronic inflammation and inflammasome activation have been reported to play an essential role in the development of DN [42], and previous studies have shown that both the mRNA and the protein expression of NLRP3, ASC and IL-1β were enhanced in PBMCs [43]; NLRP3 has been suggested to be strongly associated with renal tubular epithelial–mesenchymal transition (EMT), which is facilitated by enhancements of TGF-β1 signaling [44]. With regard to the regulation of the NLRP3 inflammasome, multiple studies have demonstrated that mtROS are major activators of the NLRP3 inflammasome [13], [45]. Additionally, TXNIP has been shown to play a role in NLRP3 inflammasome activation [46], [47]. Activated NLRP3-mediated inflammation cleaves the precursors of IL-1β and IL-18, which leads to an immuno-inflammatory response [48], [49]. Recent studies have revealed that TRX and TXNIP play a role in the pathogenesis of DN. HG conditions have been shown to reduce TRX activity by increasing the expression of TXNIP, which acts as an inhibitor of TRX [50]. Transgenic mice overexpressing TRX exhibited less mesangial matrix expansion and tubular injury in the kidney [51]. Conversely, increased TXNIP expression has been associated with Collagen IV accumulation in the kidneys of STZ-induced diabetic mice [52]. Collectively, these findings suggest that alterations in the TXNIP/NLRP3/IL-1β biological axis participate in the development of DN. However, whether NLRP3 is directly activated by mtROS via the modulation of the interactions between TRX and TXNIP remains unknown.
To verify the role of the mtROS-TXNIP-NLRP3-IL-1β biological axis in the development of DN, we examined mitochondrial morphology and function; mtROS production; and TRX, TXNIP, NLRP3 and IL-1β expression in the kidney tissues of patients with DN and db/db mice. The results showed that mitochondrial fragmentation and ROS production were increased in kidneys of patients with DN, as were the expression levels of the above proteins; furthermore, the above results showed that these changes were interrelated (Fig. 1, Fig. 2). Our findings are consistent with the notion that mitochondrial dysfunction under hyperglycemia plays a central role in the development of diabetic complications, including DN [53], [54], and they are also in agreement with the hypothesis that mtROS overproduction-induced oxidative tubular injury and cell apoptosis play a crucial role in the pathogenesis of DN [9], [32]. These observations also indicate that mtROS production is related to TXNIP/NLRP3/IL-1β biological axis activation in the kidneys of patients with DN.
To confirm the role of mtROS in the modulation of the activation of the TXNIP/NLRP3/IL-1β biological axis in DN, we applied the mitochondria-targeted antioxidant MitoQ to prevent ROS overproduction within the organelle. MitoQ has been reported to ameliorate ROS overproduction, suppress NLRP3 inflammasome activation and decrease IL-1β hypersecretion in a chronic ethanol-induced mouse macrophage model [55] and experimental mouse colitis [56]. Here, we showed that blood glucose and proteinuria levels were improved in db/db mice following 12 weeks of intraperitoneal injections of MitoQ (Fig. 3 and Table 2). Notably, treatment with MitoQ efficiently attenuated kidney injury and improved mitochondrial fragmentation, ROS generation, and cell apoptosis in the kidney in db/db mice (Fig. 3), observations consistent with those of previous studies on type 1 diabetes [57] and metabolic syndrome in mice [20]. Proteomics analysis showed that the expression levels of the components of the TXNIP/NLRP3/IL-1β biological axis were altered in the kidney tissues of db/db mice; these findings were confirmed by both IHC and WB analysis (Fig. 4). Notably, all the protein expression changes were partially attenuated by treatment with MitoQ; these effects were accompanied by decreases in FN and Collagen I expression (Fig. 4, Fig. 5). These results suggested that the decreases in mtROS levels in the kidneys of db/db mice induced by MitoQ attenuated TXNIP/NLRP3/IL-1β signaling pathway activation and thus alleviated renal cell apoptosis and fibrosis. Furthermore, the results indicated that mtROS-mediated TXNIP/NLRP3 pathway activation participated in the pathogenesis of DN.
TXNIP binds to NLRP3 via the leucine-rich repeat domain after it dissociates from TRX, and then it activates the NLRP3 inflammasome in response to oxidative stress [15]. The inhibition of TXNIP by TXNIP siRNA in podocytes was shown to prevent NLRP3-mediated inflammation activation under hyperhomocysteinemia conditions [37]. Moreover, emerging evidence suggests that TXNIP/NLRP3 inflammasome activation is responsible for endothelial cell inflammation and cell death [58], [59]. Based on these observations, we hypothesized that a similar event may also occur in tubular cells exposed to an HG environment. In support of this hypothesis, we showed that HG stimulation increased mtROS generation and TXNIP, NLRP3 and IL-1β protein expression and decreased TRX protein expression in HK-2 cells and that all of these changes were partially reversed by MitoQ treatment (Fig. 6). Furthermore, in agreement with a previous report showing that NLRP3 inflammasome activation promotes the redistribution of NLRP3 to the perinuclear space, where it co-localizes with mitochondria [13], using confocal microscopy, we observed that NLRP3 was localized mainly in cytoplasmic granular structures in cells not treated with HG but was translocated mainly to the mitochondria in HK-2 cells treated with HG (Fig. 7). Similarly, as shown in a previous study, uric acid crystals induced the dissociation of TXNIP from TRX, resulting in the binding of TXNIP to NLRP3 in an ROS-sensitive manner [15]. In this study, we showed that HG reduced the interactions between TRX and TXNIP but increased the interactions between NLRP3 and TXNIP. These alterations were reversed by treatment with MitoQ but were aggravated by treatment with MSU (Fig. 7). To confirm the effect of the mtROS-TXNIP-NLRP3-IL-1β biological axis on HG-induced immuno-inflammation, we knocked down TRX or TXNIP with siRNA. The results showed that the abovementioned alterations in NLRP3, IL-1β, IL-18, FN expression and relative activity in HK-2 cells exposed to HG conditions were reversed by treatment with MitoQ; however the effects of MitoQ were abolished by transfection with TRX siRNA or treatment with MSU but were enhanced by TXNIP siRNA (Fig. 8). These findings indicated that the modulation of TXNIP expression by mtROS plays a central role in NLRP3 inflammasome activation in tubular injury in DN.
Recent studies have shown that the binding of TXNIP to NLRP3 is a key signaling mechanism necessary for NOX-derived ROS-mediated NLRP3 inflammasome formation in glomerular injury under hyperhomocysteinemia [37]. In addition, Nox4 inhibition significantly attenuated ROS production and TXNIP, NLRP3, Caspase-1 and IL-1β protein expression in rats treated with an HG diet [39], indicating that NOX-induced ROS production may play a critical role in the TXNIP/NLRP3 biological axis. We propose that the inhibition of mtROS production and NOX expression in diabetic mice may result in a phenotype whose strength is sufficient to inhibit NRLP3 inflammasome activation; this hypothesis should be confirmed in a future study.
In summary, we provided evidence showing that mtROS-TXNIP-NLRP3-IL-1β pathway activation is a critical contributor to kidney tubular injury in DN. These results revealed that the pathogenesis of DN is driven by a novel mechanism in which high levels of mtROS or the suppression of TRX expression interrupt the interactions between TRX and TXNIP, thereby promoting the binding of TXNIP to NLRP3, resulting in NLRP3/IL-1β signaling pathway activation and leading to apoptosis and fibrosis. Additionally, this study confirmed that MitoQ is a possible therapeutic option in the management of DN and other related diseases. Finally, this study indicated that TXNIP is a very important molecule that modulates oxidative stress-induced injury in DN. We plan to generate TXNIP-knockout mice, which we will treat with a high-fat diet, to further demonstrate the role of the TXNIP/NLRP3 biological axis in development of DN.
Acknowledgments
Y.H. and X.X. generated the data for the manuscript and wrote part of the manuscript. C.T., P.G., X.C., X. X., M.Y. and S.K.Y. generated the data for the manuscript. X.Z., S.Y., F. L. and L.X. discussed the results of the study. Y.S.K. and L.S. edited the manuscript. Y.H. and X.X. performed statistical analyses of the data. L.S. is the guarantor of this work and, as such, had full access to all the study data and takes responsibility for its integrity and for the accuracy of the data analysis.
This work was supported by the General Program of the National Natural Science Foundation of China (NSFC) (81470960), the Key Program of the NSFC (81730018), the National Key R&D Program of China (2016YFC1305501) and the Key Research & Development Plan of Hunan Province (2016JC2061). The study was also supported by NIH grant DK60635.
Acknowledgments
Author disclosure statement
No competing financial interests exist.
References
- 1.Chan G., Tang S.C. Current practices in the management of diabetic nephropathy. J. R. Coll. Phys.Edinb. 2013;43:330–332. doi: 10.4997/JRCPE.2013.413. (quiz 333) [DOI] [PubMed] [Google Scholar]
- 2.Thomas M.C., Burns W.C., Cooper M.E. Tubular changes in early diabetic nephropathy. Adv. Chronic Kidney Dis. 2005;12:177–186. doi: 10.1053/j.ackd.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 3.Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011;300:R1009–R1022. doi: 10.1152/ajpregu.00809.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu X., Xiong X., Yuan S., Xiao L., Fu X., Yang Y., Tang C., He L., Liu F., Sun L. Validation of the interstitial fibrosis and tubular atrophy on the new pathological classification in patients with diabetic nephropathy: a single-center study in China. J. Diabetes Complicat. 2016;30:537–541. doi: 10.1016/j.jdiacomp.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Hu C., Sun L., Xiao L., Han Y., Fu X., Xiong X., Xu X., Liu Y., Yang S., Liu F., Kanwar Y.S. Insights into the mechanisms involved in the expression and regulation of extracellular matrix proteins in diabetic nephropathy. Curr. Med. Chem. 2015;22:2858–2870. doi: 10.2174/0929867322666150625095407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanwar Y.S., Sun L., Xie P., Liu F.Y., Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu. Rev. Pathol. 2011;6:395–423. doi: 10.1146/annurev.pathol.4.110807.092150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galvan D.L., Badal S.S., Long J., Chang B.H., Schumacker P.T., Overbeek P.A., Danesh F.R. Real-time in vivo mitochondrial redox assessment confirms enhanced mitochondrial reactive oxygen species in diabetic nephropathy. Kidney Int. 2017 doi: 10.1016/j.kint.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Awad A.S., You H., Gao T., Cooper T.K., Nedospasov S.A., Vacher J., Wilkinson P.F., Farrell F.X., Brian Reeves W. Macrophage-derived tumor necrosis factor-alpha mediates diabetic renal injury. Kidney Int. 2015;88:722–733. doi: 10.1038/ki.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun L., Xiao L., Nie J., Liu F.Y., Ling G.H., Zhu X.J., Tang W.B., Chen W.C., Xia Y.C., Zhan M., Ma M.M., Peng Y.M., Liu H., Liu Y.H., Kanwar Y.S. p66Shc mediates high-glucose and angiotensin II-induced oxidative stress renal tubular injury via mitochondrial-dependent apoptotic pathway. Am. J. Physiol. Ren. Physiol. 2010;299:F1014–F1025. doi: 10.1152/ajprenal.00414.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dugan L.L., You Y.H., Ali S.S., Diamond-Stanic M., Miyamoto S., DeCleves A.E., Andreyev A., Quach T., Ly S., Shekhtman G., Nguyen W., Chepetan A., Le T.P., Wang L., Xu M., Paik K.P., Fogo A., Viollet B., Murphy A., Brosius F., Naviaux R.K., Sharma K. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J. Clin. Investig. 2013;123:4888–4899. doi: 10.1172/JCI66218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schroder K., Zhou R., Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science. 2010;327:296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- 12.Wang C., Pan Y., Zhang Q.Y., Wang F.M., Kong L.D. Quercetin and allopurinol ameliorate kidney injury in STZ-treated rats with regulation of renal NLRP3 inflammasome activation and lipid accumulation. PloS One. 2012;7:e38285. doi: 10.1371/journal.pone.0038285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 14.Shimada K., Crother T.R., Karlin J., Dagvadorj J., Chiba N., Chen S., Ramanujan V.K., Wolf A.J., Vergnes L., Ojcius D.M., Rentsendorj A., Vargas M., Guerrero C., Wang Y., Fitzgerald K.A., Underhill D.M., Town T., Arditi M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou R., Tardivel A., Thorens B., Choi I., Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 16.Kepp O., Galluzzi L., Kroemer G. Mitochondrial control of the NLRP3 inflammasome. Nat. Immunol. 2011;12:199–200. doi: 10.1038/ni0311-199. [DOI] [PubMed] [Google Scholar]
- 17.Yoshihara E., Masaki S., Matsuo Y., Chen Z., Tian H., Yodoi J. Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 2014;4:514. doi: 10.3389/fimmu.2013.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y., Yang J., Chen M.H., Wang Q., Qin M.J., Zhang T., Chen X.Q., Liu B.L., Wen X.D. Ilexgenin A inhibits endoplasmic reticulum stress and ameliorates endothelial dysfunction via suppression of TXNIP/NLRP3 inflammasome activation in an AMPK dependent manner. Pharmacol. Res. 2015;99:101–115. doi: 10.1016/j.phrs.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 19.Graham D., Huynh N.N., Hamilton C.A., Beattie E., Smith R.A., Cocheme H.M., Murphy M.P., Dominiczak A.F. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54:322–328. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- 20.Mercer J.R., Yu E., Figg N., Cheng K.K., Prime T.A., Griffin J.L., Masoodi M., Vidal-Puig A., Murphy M.P., Bennett M.R. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/-/ApoE-/- mice. Free Radic. Biol. Med. 2012;52:841–849. doi: 10.1016/j.freeradbiomed.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 21.McManus M.J., Murphy M.P., Franklin J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer's disease. J. Neurosci.: Off. J. Soc. Neurosci. 2011;31:15703–15715. doi: 10.1523/JNEUROSCI.0552-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao L., Xu X., Zhang F., Wang M., Xu Y., Tang D., Wang J., Qin Y., Liu Y., Tang C., He L., Greka A., Zhou Z., Liu F., Dong Z., Sun L. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017;11:297–311. doi: 10.1016/j.redox.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao L., Zhu X., Yang S., Liu F., Zhou Z., Zhan M., Xie P., Zhang D., Li J., Song P., Kanwar Y.S., Sun L. Rap1 ameliorates renal tubular injury in diabetic nephropathy. Diabetes. 2014;63:1366–1380. doi: 10.2337/db13-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tervaert T.W., Mooyaart A.L., Amann K., Cohen A.H., Cook H.T., Drachenberg C.B., Ferrario F., Fogo A.B., Haas M., de Heer E., Joh K., Noel L.H., Radhakrishnan J., Seshan S.V., Bajema I.M., Bruijn J.A., Renal S. Pathology,Pathologic classification of diabetic nephropathy. J. Am. Soc. Nephrol. 2010;21:556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- 25.Sui W., Tang D., Zou G., Chen J., Ou M., Zhang Y., Dai Y. Differential proteomic analysis of renal tissue in lupus nephritis using iTRAQ reagent technology. Rheumatol. Int. 2012;32:3537–3543. doi: 10.1007/s00296-011-2207-1. [DOI] [PubMed] [Google Scholar]
- 26.Yang S., Zhao L., Han Y., Liu Y., Chen C., Zhan M., Xiong X., Zhu X., Xiao L., Hu C., Liu F., Zhou Z., Kanwar Y.S., Sun L. Probucol ameliorates renal injury in diabetic nephropathy by inhibiting the expression of the redox enzyme p66Shc. Redox Biol. 2017;13:482–497. doi: 10.1016/j.redox.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun L., Dutta R.K., Xie P., Kanwar Y.S. myo-Inositol oxygenase overexpression accentuates generation of reactive oxygen species and exacerbates cellular injury following high glucose ambience: a new mechanism relevant to the pathogenesis of diabetic nephropathy. J. Biol. Chem. 2016;291:5688–5707. doi: 10.1074/jbc.M115.669952. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Piotrkowski B., Fraga C.G., de Cavanagh E.M. Mitochondrial function and nitric oxide metabolism are modified by enalapril treatment in rat kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;292:R1494–R1501. doi: 10.1152/ajpregu.00540.2006. [DOI] [PubMed] [Google Scholar]
- 29.Hillegass J.M., Miller J.M., MacPherson M.B., Westbom C.M., Sayan M., Thompson J.K., Macura S.L., Perkins T.N., Beuschel S.L., Alexeeva V., Pass H.I., Steele C., Mossman B.T., Shukla A. Asbestos and erionite prime and activate the NLRP3 inflammasome that stimulates autocrine cytokine release in human mesothelial cells. Part. Fibre Toxicol. 2013;10:39. doi: 10.1186/1743-8977-10-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhan M., Usman I.M., Sun L., Kanwar Y.S. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J. Am. Soc. Nephrol. 2015;26:1304–1321. doi: 10.1681/ASN.2014050457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gordon L.I., Burke M.A., Singh A.T., Prachand S., Lieberman E.D., Sun L., Naik T.J., Prasad S.V., Ardehali H. Blockade of the erbB2 receptor induces cardiomyocyte death through mitochondrial and reactive oxygen species-dependent pathways. J. Biol. Chem. 2009;284:2080–2087. doi: 10.1074/jbc.M804570200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun L., Xie P., Wada J., Kashihara N., Liu F.Y., Zhao Y., Kumar D., Chugh S.S., Danesh F.R., Kanwar Y.S. Rap1b GTPase ameliorates glucose-induced mitochondrial dysfunction. J. Am. Soc. Nephrol. 2008;19:2293–2301. doi: 10.1681/ASN.2008030336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi H., Wang Y., Li X., Zhan X., Tang M., Fina M., Su L., Pratt D., Bu C.H., Hildebrand S., Lyon S., Scott L., Quan J., Sun Q., Russell J., Arnett S., Jurek P., Chen D., Kravchenko V.V., Mathison J.C., Moresco E.M., Monson N.L., Ulevitch R.J., Beutler B. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016;17:250–258. doi: 10.1038/ni.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Staples E., Ingram R.J., Atherton J.C., Robinson K. Optimising the quantification of cytokines present at low concentrations in small human mucosal tissue samples using Luminex assays. J. Immunol. Methods. 2013;394:1–9. doi: 10.1016/j.jim.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hill S., Van Remmen H. Mitochondrial stress signaling in longevity: a new role for mitochondrial function in aging. Redox Biol. 2014;2:936–944. doi: 10.1016/j.redox.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi Y., Pulliam D.A., Liu Y., Hamilton R.T., Jernigan A.L., Bhattacharya A., Sloane L.B., Qi W., Chaudhuri A., Buffenstein R., Ungvari Z., Austad S.N., Van Remmen H. Reduced mitochondrial ROS, enhanced antioxidant defense, and distinct age-related changes in oxidative damage in muscles of long-lived Peromyscus leucopus. Am. J. Physiol. Regul., Integr. Comp. Physiol. 2013;304:R343–R355. doi: 10.1152/ajpregu.00139.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abais J.M., Xia M., Li G., Chen Y., Conley S.M., Gehr T.W., Boini K.M., Li P.L. Nod-like receptor protein 3 (NLRP3) inflammasome activation and podocyte injury via thioredoxin-interacting protein (TXNIP) during hyperhomocysteinemia. J. Biol. Chem. 2014;289:27159–27168. doi: 10.1074/jbc.M114.567537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao P., He F.F., Tang H., Lei C.T., Chen S., Meng X.F., Su H., Zhang C. NADPH oxidase-induced NALP3 inflammasome activation is driven by thioredoxin-interacting protein which contributes to podocyte injury in hyperglycemia. J. Diabetes Res. 2015;2015:504761. doi: 10.1155/2015/504761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang W., Wu Q.H., Sui Y., Wang Y., Qiu X. Rutin protects endothelial dysfunction by disturbing Nox4 and ROS-sensitive NLRP3 inflammasome. Biomed. Pharmacother. = Biomed. Pharmacother. 2017;86:32–40. doi: 10.1016/j.biopha.2016.11.134. [DOI] [PubMed] [Google Scholar]
- 40.Kowaltowski A.J., de Souza-Pinto N.C., Castilho R.F., Vercesi A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009;47:333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 41.Tang S.C., Yiu W.H., Lin M., Lai K.N. Diabetic nephropathy and proximal tubular damage. J. Ren. Nutr.: Off. J. Counc. Ren. Nutr. Natl. Kidney Found. 2015;25:230–233. doi: 10.1053/j.jrn.2014.10.020. [DOI] [PubMed] [Google Scholar]
- 42.Shoelson S.E., Lee J., Goldfine A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee H.M., Kim J.J., Kim H.J., Shong M., Ku B.J., Jo E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62:194–204. doi: 10.2337/db12-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang W., Wang X., Chun J., Vilaysane A., Clark S., French G., Bracey N.A., Trpkov K., Bonni S., Duff H.J., Beck P.L., Muruve D.A. Inflammasome-independent NLRP3 augments TGF-beta signaling in kidney epithelium. J. Immunol. 2013;190:1239–1249. doi: 10.4049/jimmunol.1201959. [DOI] [PubMed] [Google Scholar]
- 45.Subramanian N., Natarajan K., Clatworthy M.R., Wang Z., Germain R.N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153:348–361. doi: 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oslowski C.M., Hara T., O'Sullivan-Murphy B., Kanekura K., Lu S., Hara M., Ishigaki S., Zhu L.J., Hayashi E., Hui S.T., Greiner D., Kaufman R.J., Bortell R., Urano F. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012;16:265–273. doi: 10.1016/j.cmet.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen G., Shaw M.H., Kim Y.G., Nunez G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu. Rev. Pathol. 2009;4:365–398. doi: 10.1146/annurev.pathol.4.110807.092239. [DOI] [PubMed] [Google Scholar]
- 48.Shahzad K., Bock F., Dong W., Wang H., Kopf S., Kohli S., Al-Dabet M.M., Ranjan S., Wolter J., Wacker C., Biemann R., Stoyanov S., Reymann K., Soderkvist P., Gross O., Schwenger V., Pahernik S., Nawroth P.P., Grone H.J., Madhusudhan T., Isermann B. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015;87:74–84. doi: 10.1038/ki.2014.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mirza R.E., Fang M.M., Weinheimer-Haus E.M., Ennis W.J., Koh T.J. Sustained inflammasome activity in macrophages impairs wound healing in type 2 diabetic humans and mice. Diabetes. 2014;63:1103–1114. doi: 10.2337/db13-0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Advani A., Gilbert R.E., Thai K., Gow R.M., Langham R.G., Cox A.J., Connelly K.A., Zhang Y., Herzenberg A.M., Christensen P.K., Pollock C.A., Qi W., Tan S.M., Parving H.H., Kelly D.J. Expression, localization, and function of the thioredoxin system in diabetic nephropathy. J. Am. Soc. Nephrol. 2009;20:730–741. doi: 10.1681/ASN.2008020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamada Y., Miyata S., Nii-Kono T., Kitazawa R., Kitazawa S., Higo S., Fukunaga M., Ueyama S., Nakamura H., Yodoi J., Fukagawa M., Kasuga M. Overexpression of thioredoxin1 in transgenic mice suppresses development of diabetic nephropathy. Nephrol., Dial., Transplant.: Off. Publ. Eur. Dial. Transplant. Assoc. - Eur. Ren. Assoc. 2007;22:1547–1557. doi: 10.1093/ndt/gfm099. [DOI] [PubMed] [Google Scholar]
- 52.Fang S., Jin Y., Zheng H., Yan J., Cui Y., Bi H., Jia H., Zhang H., Wang Y., Na L., Gao X., Zhou H. High glucose condition upregulated Txnip expression level in rat mesangial cells through ROS/MEK/MAPK pathway. Mol. Cell. Biochem. 2011;347:175–182. doi: 10.1007/s11010-010-0626-z. [DOI] [PubMed] [Google Scholar]
- 53.Chen W., Zhao M., Zhao S., Lu Q., Ni L., Zou C., Lu L., Xu X., Guan H., Zheng Z., Qiu Q. Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: a novel inhibitory effect of minocycline. Inflamm. Res.: Off. J. Eur. Histamine Res. Soc. 2017;66:157–166. doi: 10.1007/s00011-016-1002-6. [DOI] [PubMed] [Google Scholar]
- 54.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 55.Hoyt L.R., Randall M.J., Ather J.L., DePuccio D.P., Landry C.C., Qian X., Janssen-Heininger Y.M., van der Vliet A., Dixon A.E., Amiel E., Poynter M.E. Mitochondrial ROS induced by chronic ethanol exposure promote hyper-activation of the NLRP3 inflammasome. Redox Biol. 2017;12:883–896. doi: 10.1016/j.redox.2017.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dashdorj A., yothi K.R.J., Lim S., Jo A., Nguyen M.N., Ha J., Yoon K.S., Kim H.J., Park J.H., Murphy M.P., Kim S.S. Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med. 2013;11:178. doi: 10.1186/1741-7015-11-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chacko B.K., Reily C., Srivastava A., Johnson M.S., Ye Y., Ulasova E., Agarwal A., Zinn K.R., Murphy M.P., Kalyanaraman B., Darley-Usmar V. Prevention of diabetic nephropathy in Ins2(+/)(-)(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem. J. 2010;432:9–19. doi: 10.1042/BJ20100308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X.Q., Nigro P., World C., Fujiwara K., Yan C., Berk B.C. Thioredoxin interacting protein promotes endothelial cell inflammation in response to disturbed flow by increasing leukocyte adhesion and repressing Kruppel-like factor 2. Circ. Res. 2012;110:560–568. doi: 10.1161/CIRCRESAHA.111.256362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mohamed I.N., Hafez S.S., Fairaq A., Ergul A., Imig J.D., El-Remessy A.B. Thioredoxin-interacting protein is required for endothelial NLRP3 inflammasome activation and cell death in a rat model of high-fat diet. Diabetologia. 2014;57:413–423. doi: 10.1007/s00125-013-3101-z. [DOI] [PMC free article] [PubMed] [Google Scholar]