

Abstract
Purpose
Systemic increases in reactive oxygen species, and their association with inflammation, have been proposed as an underlying mechanism linking obesity and age-related macular degeneration (AMD). Studies have found increased levels of oxidative stress biomarkers and inflammatory cytokines in obese individuals; however, the correlation between obesity and retinal inflammation has yet to be assessed. We used the leptin-deficient (ob/ob) mouse to further our understanding of the contribution of obesity to retinal oxidative stress and inflammation.
Methods
Retinas from ob/ob mice were compared to age-matched wild-type controls for retinal function (electroretinography) and gene expression analysis of retinal stress (Gfap), oxidative stress (Gpx3 and Hmox1), and complement activation (C3, C2, Cfb, and Cfh). Oxidative stress was further quantified using a reactive oxygen species and reactive nitrogen species (ROS and RNS) assay. Retinal microglia and macrophage migration to the outer retina and complement activation were determined using immunohistochemistry for IBA1 and C3, respectively. Retinas and sera were used for metabolomic analysis using QTRAP mass spectrometry.
Results
Retinal function was reduced in ob/ob mice, which correlated to changes in markers of retinal stress, oxidative stress, and inflammation. An increase in C3-expressing microglia and macrophages was detected in the outer retinas of the ob/ob mice, while gene expression studies showed increases in the complement activators (C2 and Cfb) and a decrease in a complement regulator (Cfh). The expression of several metabolites were altered in the ob/ob mice compared to the controls, with changes in polyunsaturated fatty acids (PUFAs) and branched-chain amino acids (BCAAs) detected.
Conclusions
The results of this study indicate that oxidative stress, inflammation, complement activation, and lipid metabolites in the retinal environment are linked with obesity in ob/ob animals. Understanding the interplay between these components in the retina in obesity will help inform risk factor analysis for acquired retinal degenerations, including AMD.
Introduction
In 2014, the World Health Organization reported that more than 1.9 billion adults (18 years or older) were classified as overweight, with 600 million reported as obese. This level of obesity equates to almost 10% of the global population. Obesity is a serious nutritional problem known to increase the risk and morbidity of many diseases, including diabetes, cardiovascular disease, and cancer (reviewed in [1]). Obesity has also been linked to several eye diseases, including age-related macular degeneration (AMD) [2]. The retina undergoes several pathophysiological changes with age, including photoreceptor and RPE cell death, activation and migration of microglia and macrophages into the outer retina, accumulation of lipofuscin in the RPE, thickening of Bruch’s membrane, and the presence of drusen (reviewed in [3]). The onset and progression of AMD are strongly linked to complement system activation, a critical inflammatory component of the innate immune system (reviewed in [4]). Complement dysregulation, assessed with genetic polymorphisms and complement protein deposition in AMD retinas (reviewed in [4,5]), is a major risk factor for developing this disease.
Several population studies have reported an association between high body mass index (BMI) and AMD [6-10]. A higher BMI has also been associated with the progression to the more advanced forms of AMD, which can lead to severe vision loss [11-13]. In obese patients, increased levels of oxidative stress biomarkers and inflammatory cytokines have been detected in plasma and muscle [14,15]. Longitudinal studies and animals models also show that weight loss, dietary restriction, and exercise decrease these biomarkers [16]. Moreover, studies have shown that obesity induces oxidative stress, microglial activation, inflammation, and disruption of the blood–brain barrier in mouse brains [17-19]. A correlation between obesity and complement activation has been demonstrated in several tissues, including nerve biopsies from patients with diabetic polyneuropathy (reviewed in [20]), but little is known in ocular tissues. Although these data are tantalizing, the correlation between obesity and retinal inflammation is not fully understood.
In this study, we used an established genetic model of obesity, leptin-deficient (ob/ob) mice [21,22], to demonstrate a correlation between obesity, oxidative stress, and inflammation in retinal tissue. We showed that ob/ob animals exhibited lower retinal function and increased retinal stress, which was correlated with increased oxidative stress. No changes in the total number of microglia and macrophages in the ob/ob mice were detected; however, an increased distribution of these immune cells in the outer retina indicated possible low-grade chronic retinal inflammation. We also showed that key components and regulators of complement activation (C2, Cfb, and Cfh) were modulated in ob/ob animals. Last, we showed changes in several metabolites involved in oxidative stress and inflammation in the sera and retinas of ob/ob mice. This work lays a strong foundation for exploring the link between obesity and AMD and demonstrates a possible mechanistic role for complement activation in obesity, leading to the progression of inflammatory diseases, such as AMD.
Methods
Experimental animals
All experiments were conducted in accordance with the ARVO Statement for Use of Animals in Ophthalmic and Vision Research and were approved by the Australian National University (ANU) Animal Experimentation Ethics Committee (Application ID: 2014/56). Leptin (ob/ob) mutants and wild-type (wt) littermate control mice, on the C57BL/6J background, were used for the experiments at 70–90 postnatal days (animals obtained from Australian Phenomics Facility, ANU, Canberra, Australia; animal ID: 6490). Animals were born and raised in a 12 h:12 h light-dark cycle of 5 lux with free access to food and water.
Electroretinography
Full-field scotopic electroretinography (ERG) was performed to assess the retinal function of the leptin-deficient (ob/ob) mice and the littermate controls. Animals were dark adapted overnight and anesthetized with an intraperitoneal injection of ketamine (100 mg/kg; Troy Laboratories, Sydney, Australia) and xylazine (10 mg/kg; Troy Laboratories). The pupils were dilated with 1% atropine sulfate (Bausch + Lomb, Rochester, NY). ERG was performed using a single paradigm to elicit mixed responses (rod and cone) according to previously established methodology [23]. The a- and b-wave amplitudes were measured over a stimulus intensity range of −4.4 − 1.9 log cd·s·m-2. Data are expressed as the mean wave amplitude ± standard error of the mean (SEM; µV). Statistical significance (p<0.05) was determined using two-way analysis of variance (ANOVA) with Tukey’s multiple comparisons post-hoc test, using Prism 5 software (GraphPad, La Jolla, CA).
Measurements of blood glucose and glycated hemoglobin
The wt and ob/ob mice were weighed, and peripheral blood was collected for measurements of blood glucose and glycated hemoglobin (HbA1c). Blood was collected in sterile EDTA-treated tubes (#367838, BD Vacutainer; BD, Franklin Lakes, NJ) and stored at 4 °C. Blood glucose levels were measured using an Accu-Chek Performa blood glucose meter (Roche Diabetes Care, Sydney, Australia). HbA1c was measured using a Mouse Hemoglobin A1c Kit (#80310; Crystal Chem, Elk Grove Village, IL) according to the manufacturer’s instructions. HbA1c levels were read on a POLARstar Omega Spectrophotometer (BMG LABTECH, Ortenberg, Germany). Samples were averaged with n=8 per biological group, and statistical significance (p<0.05) was measured using a Student t test.
Tissue collection and preparation
Animals were euthanized via cervical dislocation. For preparation of samples for histology, the left eye from each animal was enucleated with the superior surface marked and fixed in 4% paraformaldehyde for 3 h. Eyes were cryopreserved in 15% sucrose overnight before embedding and cryosectioning at 12 µm in the parasagittal plane (superior–inferior). To enable comparison across animals, only histological sections containing the optic nerve (ON) head were used for analysis.
For preparation of molecular samples, the retina from the right eye of each animal was excised through a corneal incision and placed in RNAlater solution (Thermo Fisher Scientific, Waltham, MA) at 4 °C overnight and then stored at −80 °C. Total RNA was extracted from the retinal samples using a combination of an RNAqueous Micro Isolation Kit (Thermo Fisher Scientific) and TRIzol (Thermo Fisher Scientific) according to the manufacturer’s protocol. The concentration and purity of the RNA samples were determined using an ND-1000 spectrophotometer (Thermo Fisher Scientific).
Immunohistochemistry
Sections were incubated in 10% normal goat serum (Sigma-Aldrich, St. Louis, MO) for 1 h at room temperature (RT), followed by overnight incubation with the primary antibodies (Table 1) at 4 °C. Antigen retrieval (RevealIt-Ag; ImmunoSolutions, Brisbane, Australia) was performed only for the IBA1 antibody, before blocking with normal goat serum, for 1 h at 37 °C. All sections were incubated with appropriate fluorophore-conjugated secondary antibodies for 4 h at RT, before staining with bisbenzimide to identify the cellular layers and mounting with Aqua-Poly/Mount (Thermo Fisher Scientific). Visualization of immunofluorescence and image acquisition were performed using an A1+ confocal microscope (Nikon, Tokyo, Japan). The IBA1 antibody was conjugated to Alexa Fluor 647 (Thermo Fisher Scientific) for colocalization of the IBA1 and C3 antibodies (raised in rabbits).
Table 1. Primary and secondary antibodies used for immunohistochemistry.
| Antibody | Catalogue Number | Host | Dilution | Source |
|---|---|---|---|---|
| IBA1 |
019-19741 |
Rabbit |
1:500 |
Wako Pure Chemical Industries (Osaka, Japan) |
| GFAP |
Z0334 |
Rabbit |
1:500 |
Dako (Sydney, Australia) |
| C3 |
ab11887 |
Rabbit |
1:500 |
Abcam (Cambridge, United Kingdom) |
| Rhodopsin |
AB9279 |
Rabbit |
1:500 |
Merck-Millipore (Darmstadt, Germany) |
| Alexa Fluor 488 |
A31627 |
Goat |
1:1000 |
Thermo Fisher Scientific (Waltham, MA) |
| Alexa Fluor 594 | A31631 | Goat | 1:500 | Thermo Fisher Scientific |
IBA1-positive microglia and macrophages were counted across the retinal cryosections (n=6 per experimental group). For each animal, two retinal sections were counted, and all values averaged together and analyzed with a Student t test. The outer nuclear layer (ONL) thickness was calculated by counting the number of photoreceptor rows (500 µm from the ON on the superior side of the retina). Three counts for each section (n=5 animals per group) were used in the analysis, and the averages taken and compared using a Student t test.
Analysis of gene expression
Following purification of RNA, cDNA was synthesized using the Tetro cDNA Synthesis Kit (Bioline, London, UK) according to the manufacturer’s protocol. A 20 μl reaction mixture was prepared using 1 μg of purified RNA, 500 ng Oligo dT primer, and 200 U reverse transcriptase. Quantitative real-time PCR (qPCR) was performed using mouse-specific TaqMan hydrolysis probes (Table 2; Thermo Fisher Scientific) and TaqMan Gene Expression Master Mix (Thermo Fisher Scientific). Amplification of each sample was performed in technical duplicates, performed using a QuantStudio 12 K Flex RT–PCR machine (Applied Biosystems, Warrington, PA). Data were analyzed using Expression Suite v1.0.3 software (Thermo Fisher Scientific), and the analysis was performed using the comparative cycle threshold (Ct) method (ΔΔCt). Values are presented as a log twofold change. Target genes were normalized to two reference genes, glyceraldehyde-3-phosphate dehydrogenase (Gapdh) and actin beta (Actb). Statistical significance (p<0.05) was determined using one-way ANOVA with Tukey’s multiple comparison post-test (n=3 per experimental group).
Table 2. Taqman hydrolysis probes used for qPCR.
| Gene Symbol | Gene Name | Catalog Number | Entrez Gene ID |
|---|---|---|---|
| Gfap |
Glial fibrillary acidic protein |
Mm01205647_g1 |
14580 |
| Cntf |
Ciliary neurotrophic factor |
Mm00446376_m1 |
12803 |
| Fgf2 |
Fibroblast growth factor 2 |
Mm01285715_m1 |
14173 |
| C3 |
Complement component 3 |
Mm00437858_m1 |
12266 |
| C2 |
Complement component 2 |
Mm00442726_m1 |
12263 |
| Cfb |
Complement factor B |
Mm00433918_g1 |
14962 |
| Cfh |
Complement factor H |
Mm01299248_m1 |
12628 |
| Gpx3 |
Glutathione peroxidase 3 |
Mm00492427_m1 |
14778 |
| Hmox1 |
Heme oxygenase 1 |
Mm01536933_m1 |
15368 |
| Gapdh |
Glyceraldehyde-3-phosphate dehydrogenase |
Mm99999915_g1 |
14433 |
| Actb | Actin, Beta | Mm01205647_g1 | 11461 |
ROS and RNS assays
To measure reactive oxygen species (ROS) and reactive nitrogen species (RNS), both retinas from each animal were harvested and immediately homogenized in 500 µl of 0.1% Tween-20 in PBS (1X; 155 mM NaCl, 1 mM KH2PO4, 3 mM Na2HPO4-7H2O, pH 7.4), spun down at 10,000 ×g for 5 min, and the supernatant removed. Fifty microliters of each supernatant was used in the OxiSelect In Vitro ROS/RNS Assay Kit (Cell Biolabs, San Diego, CA) according to the manufacturer’s instructions to measure the ROS and RNS content of the isolates from freshly harvested retinas. The fluorescence was measured at 530 nm using a plate reader (Tecan Infinite Pro 200, Männedorf, Switzerland). Samples were averaged with n=6 per biological group and statistical significance (p<0.05) was measured.
FACS isolation of primary microglia
Isolation of rat retinal microglia was performed using fluorescence-activated cell sorting (FACS) following a modified protocol previously published [24,25]. Retinas were collected from dim-reared adult (P90) Wistar rats through a corneal incision and placed into chilled Hank’s Balanced Salt Solution (HBSS). Retinas were then mechanically dissociated with a scalpel blade followed by enzymatic digestion using a 0.2% papain digestion cocktail as previously described [24,25]. Samples were then neutralized and resuspended in staining buffer containing a Phycoerythrin/Cyanine7 (PE/Cy7)-conjugated anti-rat antibody to CD11b (Biolegend, San Diego, CA) and incubated for 40 min at 4 °C. Following staining, the cells were washed, resuspended, and filtered before sorting through a BD FACSAria II (BD Biosciences, Franklin Lakes, NJ). Approximately 4,000 cells were sorted per sample. Isolated microglia were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich), 1% antibiotic-antimycotic (Thermo Fisher Scientific), 3% L-glutamine (Thermo Fisher Scientific), and a granulocyte-macrophage colony stimulating factor (GM-CSF, 5 ng/ml; Stem Cell Technologies, Vancouver, Canada) with 5% CO2 at 37 °C. Media was replaced every 3–4 days until cells reached 80% confluency.
Validation of primary microglia using PCR
Standard PCR for validation of microglial cell markers was performed, testing for the absence of glial fibrillary acidic protein (Gfap) and rhodopsin (Rhod). The PCR reaction mixture was prepared using the MyTAQ kit (Bioline). To each reaction mixture, 10 μl of 5X MyTAQ Reaction Buffer, 0.25 μl of MyTAQ Polymerase, 0.5 μl of cDNA template (40 ng/μl), 1 μl of forward primer, 1 μl of reverse primer, and 37.25 μl of DNase-free water was added. The primers used for amplification of Gfap are as follows: forward primer 5′-AGT CGC TGG AGG AGG AGA T-3′ and reverse primer 5′-TGA GGT GGC CTT CTG ACA-3′. The final product length of Gfap was 642 bp. The primers used for amplification of Rhod are as follows: forward primer 5′-CTT CCC CAT CAA CTT CCT C-3′ and reverse primer 5′-CCC AGT GGR TTC TTG CC-3′. The final product length of Rhod was 834 bp. Samples were amplified in a thermocycler for 1 min at 95 °C followed by a further 15 s at 95 °C, 15 s at 60 °C, 10 s at 72 °C, and finally, a further 1 min at 72 °C. Samples were then run on a 1% agarose gel for 40 min.
Mass spectrometry analysis
The ob/ob and control mice (n=6) were randomly culled with cervical dislocation, and blood was collected after decapitation. Serum, obtained after blood clotting at 4 °C, was stored at −80 °C until analysis. Both retinas from each animal were collected in a 0.5 ml chilled Precellys tube (Bertin Corp, Rockville, MD) prefilled with ceramic beads. To each retina sample, 175 μl of a cold mixture of methanol:water (140:35, v:v) was added for tissue homogenization and metabolite extraction. Tissue was homogenized using a Precellys 24 homogenizer (Bertin Corp) with two cycles of grinding (40 s at 4440 ×g, followed by 30 s at 3780 ×g) at 4 °C. The resulting homogenate was centrifuged at 16,000 ×g for 5 min, and 140 µl of the supernatant was transferred to a second tube and spin-dried. Extracts were stored at −80 °C until needed for metabolomics analysis.
Targeted quantitative metabolomics analysis was performed using the AbsoluteIDQ p180 kit (Biocrates Life Sciences AG, Innsbruck, Austria) and the QTRAP 5500 (SCIEX, Framingham, MA) mass spectrometer (MS). This enables quantification of up to 188 endogenous metabolites, including lipids and polar molecules. Flow injection analysis (FIA-MS/MS) was used for quantifying carnitine, acylcarnitines, lipids, and sugar, while liquid chromatography (LC) was employed to separate amino acids and biogenic amines before MS quantification. All reagents used in this analysis were of LC-MS grade (VWR, Radnor, PA; Merck, Kenilworth, NJ). For retinal sample preparation, 30 µl of methanol was added to each sample and vortexed thoroughly for 5 min. Ten microliters of each retinal and serum sample was mixed with isotope-labeled internal standards and loaded on the 96-well plate. Metabolites were resuspended in ammonium acetate after filter spots had been dried under nitrogen flow and derivatized with phenylisothiocyanate (only for the quantification of amino acids and biogenic amines). Extracts were diluted with MS running solvent (MilliQ water for the high-performance liquid chromatography [HPLC] assay or a methanol solution for the FIA assay) before the FIA and LC-MS/MS analyses. Quality controls (QCs) at three concentrations (low, medium, and high) were included in the analysis. Analyst software (SCIEX) was used for MS data collection, and MetIDQ software (Biocrates Life Sciences AG) was used to control the entire assay workflow.
Before statistical analysis was performed, the raw data were examined to eliminate metabolites not accurately measured; for example, metabolites with a concentration outside the lower limit of quantitation (LLOQ) and the upper limit of quantitation (ULOQ). When more than 20% of concentration values were not accurately measured, the metabolite was not considered for statistical analysis. Multivariate analysis was performed using unit variance-scaled data. Principal component analysis (PCA) was used to detect sample groups and outliers. An orthogonal partial least-squares discriminant analysis (OPLS-DA), which is a supervised pattern recognition method, was then performed to maximize variation between groups and to determine variables contributing to this variation. The quality of models was validated by determining two parameters: R2 (goodness of fit) and cumulated Q2 (Q2cum, goodness of prediction). A threshold of 0.5 is widely accepted in classifying models as having good (Q2cum ≥0.5) or poor (Q2cum<0.5) predictive capabilities. The risk of overfitting and robustness was assessed with the intercept of the permutation plot. This intercept reflects the predictive capabilities (permQ2) of a model having the same number of components (or latent variables) and the same X matrix (metabolites) but a randomly permuted y vector (scrambled y) of response variables (that is, at the intercept, there is no correlation between the original y vector and the permuted y vector). Non-overfitted models have a negative permQ2 value [26]. If a predictive model (i.e., Q2cum ≥0.5) was obtained, the top variables were selected based on the variable importance for the projection (VIP) and loading values scaled as correlation coefficients (pcorr). VIP values summarize the importance of each variable for the OPLS-DA model, while the loadings are indicators of the relationship between the y variables (the leptin genotype) and the X variables (the matrix of the measured metabolites). Variables that have a VIP value higher than 1 are considered important for group discrimination in predictive models [27]. Plotting VIP versus pcorr values (“volcano” plot) enables selection of important variables in partial least-squares (PLS) models. Multivariate data analysis was conducted using SIMCA-P v.14.0 (Umetrics, Umeả, Sweden).
Results
Increased bodyweight of ob/ob is correlated to changes in retinal function and stress
Mice deficient in the leptin gene (ob/ob) were statistically significantly higher in bodyweight than littermate wt control mice (p<0.05, Figure 1A) and showed no change in blood glucose or glycated hemoglobin levels (Figure 1B,C). Analysis of the ONL thickness (Figure 1D,E) indicated that there were no changes in the number of photoreceptor in the ob/ob animals compared to the wt animals (p>0.05). Despite the lack of change in the photoreceptor layer size, there was a statistically significant change in retinal function (ERG); the ob/ob animals had a reduced a-wave (p<0.05, Figure 1F) and b-wave (p<0.05, Figure 1G) with the largest difference at the highest light intensity (1.9 Log cd.s/m2, Figure 1H). Assessment of cone function also indicated a statistically significant reduced response in the ob/ob animals at 1.9 Log cd.s/m2 (Figure 1I). Assessment of general retinal stress markers indicated altered expression of GFAP via qPCR and immunohistochemistry (Figure 1J–L). Gene expression analysis showed a statistically significant 1.5-fold change in GFAP in the ob/ob mice (p<0.05). There were no changes in the neuroprotective factors Cntf and Fgf2.
Figure 1.

Altered bodyweight was correlated with decreased retinal function and increased GFAP expression in leptin-deficient (ob/ob) mice. A: The ob/ob mice showed an approximate doubling in bodyweight (grams) at 90 postnatal days compared to the wild-type (wt) littermate controls (p<0.05, n=8). B, C: We observed no statistically significant changes in blood glucose (B) or glycated hemoglobin (HbA1c, C) levels in the ob/ob mice compared to the littermate wt control mice. D, E: No changes in outer nuclear layer (ONL) thickness were detected across the retina (B, n=5), with no difference between the superior and inferior retina observed (C, n=5). F–H: Functional studies showed a statistically significant decrease in a- (F) and b-wave (G) amplitudes in the ob/ob mice compared to the wt control mice. This difference was most noticeable at the highest stimulus intensity of 1.9 Log cd.s/m2 (H, n=3 wt, n=6 ob/ob). I: Photopic electroretinography (ERG; cones) showed a statistically significant difference in function between the wt control and ob/ob animals at 1.9 Log cd.s/m2 (F, n=6 wt, n=6 ob/ob). J: In the ob/ob mice compared to the wt control mice, a statistically significant increase was determined for the retinal stress marker Gfap (p<0.05, n=9), but no statistically significant change was detected for the retinal stress markers Cntf and Fgf2 (n=9). K, L: Immunohistochemistry for GFAP showed increased labeling (arrowheads) in the ganglion cell layer (GCL) and the inner plexiform layer (IPL) in the ob/ob retinas (K) compared to the wt control retinas (L). Statistical analysis was performed using a Student t test (A–D, I, J) and a two-way ANOVA with uncorrected Fisher’s least significant difference (LSD) post-test (F–H). * indicates a statistical significance and a p value of less than 0.05. INL, inner nuclear layer. Scale bars = 50 µm. Error bars are displayed as standard error of the mean (SEM).
Changes in inflammation and complement in the ob/ob mice
The overall number of retinal IBA1-positive microglia and macrophages did not change in the ob/ob mice (p>0.05, Figure 2A); however, there were noticeable changes in the distribution of IBA1-positive cells in the ob/ob retinas (Figure 2B–G). The wt animals showed IBA1-positive labeling primarily in the inner plexiform layer (IPL) with some cell processes extending into the ONL. No IBA1-positive nuclei were observed in the outer retinas of the wt control mice (Figure 2B). In contrast, the ob/ob mice had IBA1-positive cell nuclei in the ONL, with cell processes extending into the photoreceptor outer segments (OS) and the RPE (Figure 2C–G).
Figure 2.

Changes in morphology and location of IBA1-positive microglia and macrophages in ob/ob mice. A–C: There was no change in the overall numbers of IBA1-positive cells between the ob/ob retinas and the wild-type (wt) control retinas (A), with an occasional IBA1-positive cell process found in the outer nuclear layer (ONL) in the wt control mice (B) and ob/ob (C) mice. D–G: However, several IBA1-positive cells were found with nuclei in the outer retina in the ob/ob mice only, with processes extending into the outer segments (OS, D–F) and RPE (G). No IBA1-positive nuclei were observed in the outer retinas (ONL-RPE) of the wt control animals. Statistical analysis was performed using a Student t test. NS indicates no statistical significance (p>0.05). GCL, ganglion cell layer; INL, inner nuclear layer. Scale bars = 50 µm. Error bars are displayed as standard error of the mean (SEM).
No changes in the expression of C3 were observed in the ob/ob animals (Figure 3A), although statistically significant increases in expression, relative to the wt control mice, of C2 in the classical pathway of complement activation (eightfold increase, p<0.05) and Cfb in the alternative pathway (3.5-fold increase, p<0.05) were evident (Figure 3A). Cfh, a key negative regulator of the alternative pathway, was downregulated (twofold decrease, p<0.05). Immunohistochemistry for C3 (Figure 3B–E) showed no C3 expression in the wt retinas (Figure 3B) but showed C3 deposition in the outer retinas of the ob/ob mice (Figure 3C–E). Colocalization of C3 retinal expression with IBA1 (Figure 3F–N) showed a clear association between C3 expression and IBA1-positive cells indicating microglia and macrophage production of C3 in the outer retinas (photoreceptors and RPE) of ob/ob mice.
Figure 3.

Complement activation in ob/ob retinas. A: Statistically significantly altered gene expression of C2, Cfb, and Cfh (p<0.05, n=8–9) was detected in the ob/ob retinas compared to the wild-type (wt) control retinas, which are all involved in complement activation. No statistically significant difference was detected in C3 gene expression between the groups (n=12). B–E: Immunohistochemistry for C3 in the wt control retinas showed no C3 expression in any of the retinal sections analyzed (B, n=8). However, C3 expression (arrowheads) was detected in several cells in the ob/ob retinas, in the outer retina (C, E) and the outer plexiform layer (OPL, D). F–N: Colocalization of C3 (green) and IBA1 (purple) was observed in the ob/ob outer retinas (F–H), the OPL (I–K) and within the outer segments (OS, L and M). Statistical analysis was performed using a Student t test. * indicates a statistical significance of p<0.05. INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium. Scale bars = 50 µm. Error bars are displayed as standard error of the mean (SEM).
Increases in oxidative stress in ob/ob mice lead to complement activation
There was a clear and statistically significant increase in the number of ROS and RNS (Figure 4A) in the ob/ob mouse retinas compared to the wt control mouse retinas, with a 70% increase in activity detected with the ROS/RNS assay (p<0.05). Key oxidative stress genes were differentially regulated in the ob/ob retinas compared to the wt control retinas (p<0.05, Figure 4B), including the detoxifying hydrogen peroxide (H2O2) gene glutathione peroxidase 3 (Gpx3) and the antioxidant heme oxygenase 1 (Hmox1) gene. Primary microglia (isolated and cultured from whole rat retinas) were validated (Appendix 1) and treated with 1 mM H2O2 for 1 h to induce oxidative stress (Figure 4C,D), where cells exhibited no ramified processes. The expression of several complement activator and regulator genes was analyzed following H2O2 treatment (Figure 4E). Although we detected a decrease in C3 and Cfh expression, C2 and Cfb were increased (p<0.05).
Figure 4.
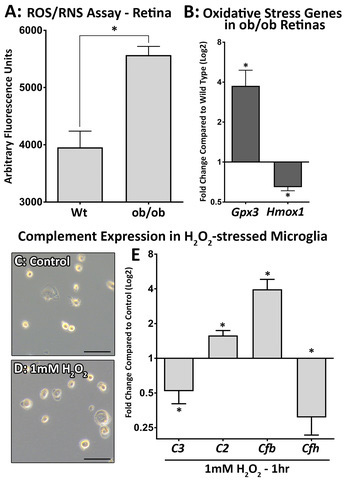
Increased oxidative stress correlates to changes in complement genes in ob/ob mice. A: A comparison of retinas isolated from the wild-type (wt) control mice to the retinas of the ob/ob mice indicated a statistically significant increase in reactive oxygen species (ROS) and reactive nitrogen species (RNS) production in the ob/ob retina (p<0.05, n=6). B: Gene expression analysis showed differential expression of oxidative stress markers in the ob/ob retinas compared to the wt control retinas, with increased Gpx3 and decreased Hmox1 expression detected (p<0.05, n=10). C–D: Primary microglial cells treated with 1 mM H2O2 exhibited no ramified processes, with slight morphological changes observed compared to controls. E: Gene expression analysis detected a statistically significant increase in C2 and Cfb expression (p<0.05, n=3), while the expression of C3 and Cfh was decreased following 1 mM H2O2 incubation for 1 h (p<0.05, n=3). Statistical analysis was performed using a Student t test. * indicates a p value of less than 0.05. Scale bars = 50 µm. Error bars are displayed as standard error of the mean (SEM).
Metabolomics of serum and retina in ob/ob mice
After validation of the quality controls, 128 metabolites in the serum and 136 metabolites in the retinal extract were retained for multivariate statistical analysis. PCA showed a clear grouping of samples according to the leptin genotype without outliers (Figure 5A). This separation was improved with the supervised OPLS-DA algorithm (Figure 5B), yielding a model explaining the variance in the X matrix (R2X=0.76) and in the y vector of the responses (R2Y=0.97) and with good predictive capabilities (Q2cum=0.82). The risk of overfitting was low as indicated by the negative permQ2 (permQ2=–0.62). The top discriminant metabolites for the wt and ob/ob serum were identified in the volcano plots as those having the greatest VIP (i.e., VIP>1) and absolute pcorr values (Figure 5C). In the ob/ob mouse serum, three metabolites were decreased, and 47 metabolites were increased compared to the wt control mouse serum.
Figure 5.
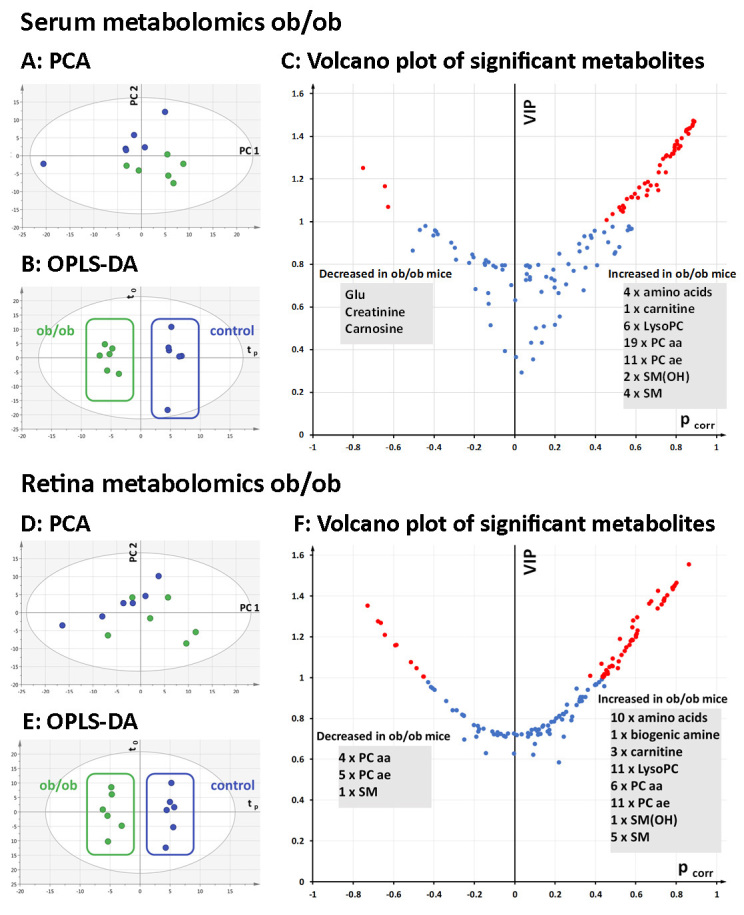
Multivariate analysis of metabolomics data from ob/ob and wt control mouse sera and retinas. A: Principal component analysis (PCA, PC1 versus PC2) of the serum samples showed a good separation between the ob/ob (green) and wild-type (wt) control (blue) mice along the second principal component (PC2). B: Orthogonal partial least-squares discriminant analysis (OPLS-DA) scatterplot displaying clear between-group separation along the predictive component (tp) in serum. C: Volcano plot showing top discriminant metabolites (highest variable importance for the projection (VIP) and pcorr values) between the ob/ob and wt control mice. Metabolites with a VIP value >1 are marked in red. Three metabolites decreased in the ob/ob mouse serum, and 47 increased compared to the wt control mice. D: Principal component analysis of the retinal samples (PC1 versus PC2) shows no outliers but a less clear separation between the ob/ob (green) and wt control retinal samples (blue). E: OPLS-DA scatter plot displaying clear between-group separation along the predictive component (tp). F: Volcano plot showing top discriminant metabolites (highest VIP and pcorr values) in between-group discrimination. Metabolites with a VIP value >1 are marked in red. Ten metabolites decreased in the ob/ob mouse retinas, and 48 increased compared to the wt control retinas. PCA, principal component analysis; PC1, first principal component; PC2, second principal component; tp, predictive component in the OPLS-DA model; to, orthogonal component in the OPLS-DA model; VIP, variable importance for the projection; pcorr, scaled loading values. AA, amino acids; BA, biogenic amine compounds; CC, carnitine compounds; Glu, glutamate; LysoPC, lyso-phosphatidylcholine; PC aa, diacyl phosphatidylcholine; PC ae, acyl-alkyl phosphatidylcholine; SM, sphingomyelin; SM(OH), hydroxy sphingomyelin.
PCA for retinal samples showed no outliers but a less clear separation between the leptin-deficient mice and the control mice (Figure 5D). However, the OPLS-DA method improved group discrimination (Figure 5E) in a model characterized by a correct approach of X and y variance (R2X=0.82 and R2Y=0.98, respectively), good predictive capabilities (Q2cum=0.70), and a low risk of overfitting (permQ2=–0.68). The top discriminant metabolites for the serum and retina models are identified in the volcano plots as those having the greatest VIP (i.e., VIP>1) and absolute pcorr values (Figure 5F). In the ob/ob retinas, ten metabolites were decreased, and 48 metabolites were increased compared to the wt control retinas.
In the ob/ob mice, the expression of several metabolites was altered compared to the wt control mice in the serum and retinal samples concurrently (Table 3), as well as the metabolites that changed in ob/ob serum only (Table 4) and the ob/ob retinas only (Table 5). This included several amino acids, such as glutamate, isoleucine, leucine, and tryptophan, as well as lipid molecules, such as lyso-phosphatidylcholine, diacyl phosphatidylcholine, acyl-alkyl phosphatidylcholine, hydroxy-sphingomyelin, and sphingomyelin.
Table 3. Metabolites that changed in ob/ob serum and retinas.
| Group | Metabolite | p corr | Serum VIP | p corr | Retina VIP |
|---|---|---|---|---|---|
| Amino Acids |
Glutamate |
-0.62848 |
1.07062 |
0.48521 |
1.09397 |
| |
Isoleucine |
0.789994 |
1.33623 |
0.585773 |
1.28116 |
| |
Leucine |
0.810948 |
1.34441 |
0.520356 |
1.19146 |
| |
Tryptophan |
0.878639 |
1.45113 |
0.676211 |
1.37675 |
| lyso-phosphatidylcholine |
lysoPC a C16:0 |
0.793288 |
1.36093 |
0.727204 |
1.35957 |
| |
lysoPC a C16:1 |
0.80441 |
1.36459 |
0.75586 |
1.40434 |
| |
lysoPC a C18:0 |
0.711927 |
1.23264 |
0.439303 |
1.00424 |
| |
lysoPC a C18:1 |
0.779201 |
1.32028 |
0.739892 |
1.37745 |
| |
lysoPC a C20:3 |
0.671639 |
1.17138 |
0.545529 |
1.13244 |
| |
lysoPC a C20:4 |
0.657354 |
1.14978 |
0.707013 |
1.3408 |
| diacyl-phosphatidylcholine |
PC aa C32:0 |
0.746435 |
1.23377 |
-0.58743 |
1.16226 |
| |
PC aa C36:1 |
0.864564 |
1.43952 |
-0.5154 |
1.07721 |
| |
PC aa C36:4 |
0.849357 |
1.43239 |
0.580902 |
1.18811 |
| |
PC aa C36:5 |
0.718598 |
1.26584 |
0.600343 |
1.20925 |
| |
PC aa C38:3 |
0.865695 |
1.43961 |
-0.67882 |
1.27696 |
| |
PC aa C38:5 |
0.882577 |
1.47481 |
0.552574 |
1.14977 |
| |
PC ae C34:1 |
0.525742 |
1.05543 |
-0.45293 |
1.00707 |
| acyl-alkyl phosphatidylcholine |
PC ae C36:4 |
0.614955 |
1.16171 |
0.584345 |
1.18598 |
| |
PC ae C36:5 |
0.534883 |
1.07635 |
0.800319 |
1.4662 |
| |
PC ae C38:0 |
0.746339 |
1.30686 |
0.574565 |
1.18106 |
| |
PC ae C40:6 |
0.60895 |
1.11477 |
-0.59195 |
1.15981 |
| |
SM (OH) C14:1 |
0.65887 |
1.18889 |
0.741898 |
1.38954 |
| hydroxy-sphingomyelin |
SM C16:0 |
0.79134 |
1.35024 |
0.781304 |
1.44328 |
| sphingomyelin |
SM C18:1 |
0.767981 |
1.30684 |
0.783581 |
1.43465 |
| SM C20:2 | 0.702222 | 1.17311 | 0.511015 | 1.04732 |
Table 4. Metabolites that changed in ob/ob serum only.
| Group | Metabolite | p corr | Serum VIP | p corr | Retina VIP |
|---|---|---|---|---|---|
| Amino Acids |
Valine |
0.7848 |
1.31971 |
0.204573 |
0.802158 |
| Biogenic Amines |
Carnosine |
-0.7523 |
1.25347 |
0.350184 |
0.905499 |
| |
Creatinine |
-0.6438 |
1.16845 |
0.091917 |
0.624234 |
| Carnitines |
Butyryl-L-carnitine |
0.70934 |
1.15011 |
0.118303 |
0.747896 |
| diacyl-phosphatidylcholine |
PC aa C32:1 |
0.86028 |
1.41343 |
0.336775 |
0.905788 |
| |
PC aa C32:2 |
0.53414 |
1.04884 |
0.253623 |
0.834408 |
| |
PC aa C34:1 |
0.85739 |
1.43615 |
0.321684 |
0.895455 |
| |
PC aa C34:2 |
0.58028 |
1.11623 |
-0.26064 |
0.820304 |
| |
PC aa C34:4 |
0.48431 |
1.03834 |
0.110975 |
0.739271 |
| |
PC aa C36:2 |
0.55357 |
1.10851 |
-0.06894 |
0.725665 |
| |
PC aa C36:3 |
0.80184 |
1.38013 |
-0.40703 |
0.949685 |
| |
PC aa C36:6 |
0.59324 |
1.13294 |
-0.25253 |
0.816078 |
| |
PC aa C38:4 |
0.8806 |
1.45839 |
0.009421 |
0.719502 |
| |
PC aa C38:6 |
0.84708 |
1.42543 |
0.136406 |
0.760069 |
| |
PC aa C40:4 |
0.54142 |
1.06771 |
-0.25239 |
0.81383 |
| |
PC aa C40:5 |
0.82655 |
1.39243 |
-0.15522 |
0.751357 |
| |
PC aa C40:6 |
0.88936 |
1.47078 |
-0.08527 |
0.720281 |
| acyl-alkyl phosphatidylcholine |
PC ae C38:3 |
0.45492 |
1.01024 |
0.181811 |
0.784762 |
| |
PC ae C38:4 |
0.64208 |
1.1811 |
-0.10392 |
0.72364 |
| |
PC ae C38:5 |
0.51624 |
1.06876 |
0.417416 |
0.97917 |
| |
PC ae C40:3 |
0.57574 |
1.1178 |
0.065519 |
0.729058 |
| |
PC ae C40:5 |
0.73397 |
1.29608 |
0.078598 |
0.72589 |
| |
PC ae C42:5 |
0.75062 |
1.31289 |
0.034568 |
0.72336 |
| hydroxy-sphingomyelin |
SM (OH) C22:1 |
0.65348 |
1.12435 |
-0.18174 |
0.737934 |
| sphingomyelin | SM C18:0 | 0.82008 | 1.35581 | 0.234547 | 0.795889 |
Table 5. Metabolites that changed in ob/ob retinas only.
| Group | Metabolite | p corr | Serum VIP | p corr | Retina VIP |
|---|---|---|---|---|---|
| Amino Acids |
Arginine |
0.517273 |
0.883953 |
0.37412 |
1.01052 |
| |
Phenylalanine |
0.577612 |
0.968697 |
0.60658 |
1.29648 |
| |
Proline |
0.572147 |
0.967029 |
0.71031 |
1.42675 |
| |
Serine |
0.089818 |
0.356669 |
0.58207 |
1.24897 |
| |
Threonine |
0.558031 |
0.961585 |
0.66561 |
1.36318 |
| |
Tyrosine |
0.497811 |
0.859313 |
0.42847 |
1.07025 |
| Biogenic Amines |
Histamine |
0.133188 |
0.435193 |
0.60836 |
1.2321 |
| Carnitines |
L-Carnitine |
-0.13254 |
0.667314 |
0.5999 |
1.20156 |
| |
Octadecenoyl-L-carnitine |
0.079876 |
0.435358 |
0.44321 |
1.00964 |
| |
Valeryl-L-carnitine |
- |
- |
0.52958 |
1.11339 |
| lyso-phosphatidylcholine |
lysoPC a C18:2 |
0.196199 |
0.726949 |
0.78953 |
1.44872 |
| |
lysoPC a C24:0 |
-0.09497 |
0.789297 |
0.48052 |
1.06013 |
| |
lysoPC a C26:1 |
0.407743 |
0.797227 |
0.43516 |
1.00356 |
| |
lysoPC a C28:0 |
0.179581 |
0.736869 |
0.48465 |
1.05785 |
| |
lysoPC a C28:1 |
0.254284 |
0.709321 |
0.44365 |
1.018 |
| diacyl-phosphatidylcholine |
PC aa C24:0 |
0.074606 |
0.731657 |
0.51499 |
1.08192 |
| |
PC aa C28:1 |
0.461476 |
0.905075 |
0.56906 |
1.16167 |
| |
PC aa C38:1 |
-0.39672 |
0.961062 |
-0.7301 |
1.35367 |
| |
PC aa C40:3 |
-0.22636 |
0.809281 |
0.44567 |
1.01657 |
| acyl-alkyl phosphatidylcholine |
PC ae C30:1 |
0.22234 |
0.558391 |
0.45905 |
1.0303 |
| |
PC ae C32:2 |
0.131107 |
0.673476 |
0.79237 |
1.45483 |
| |
PC ae C34:0 |
0.116072 |
0.799204 |
-0.6638 |
1.27003 |
| |
PC ae C34:2 |
-0.29146 |
0.823435 |
0.73692 |
1.38054 |
| |
PC ae C34:3 |
-0.38166 |
0.942243 |
0.43546 |
1.00916 |
| |
PC ae C36:1 |
0.073059 |
0.788656 |
-0.4865 |
1.047 |
| |
PC ae C40:1 |
0.374539 |
0.936429 |
-0.6445 |
1.21146 |
| |
PC ae C42:3 |
0.395941 |
0.958794 |
0.46826 |
1.05383 |
| |
PC ae C44:3 |
0.074266 |
0.792551 |
0.45358 |
1.03745 |
| |
PC ae C44:5 |
0.058851 |
0.756676 |
0.60351 |
1.21601 |
| |
PC ae C44:6 |
-0.14726 |
0.800369 |
0.86332 |
1.55599 |
| sphingomyelin |
SM C16:1 |
0.190844 |
0.776799 |
0.73965 |
1.37982 |
| |
SM C24:0 |
0.357199 |
0.879899 |
-0.4503 |
1.00622 |
| SM C24:1 | 0.216329 | 0.66833 | 0.46169 | 1.02116 |
Discussion
The association between obesity, inflammation, and retinal degenerative diseases, including AMD, has been shown in humans [2,8,28] and animal models [29-31]. However, the mechanisms underlying the increased risk of retinal degeneration in obesity, as well as the roles played by oxidative stress and complement activation, are unclear. In this study, we demonstrate a correlation between obesity, oxidative stress, and inflammation in retinal tissue. First, we showed that the ob/ob animals exhibited lower retinal function, which was correlated with an increase in general retinal stress and oxidative stress, compared with the wt control animals. Second, we demonstrated a change in the distribution of microglia and macrophages into the outer retina of the ob/ob animals, a hallmark of many retinal degenerations, including AMD. Third, the data showed that key regulators of the complement cascade (C2, Cfb, and Cfh) are regulated toward complement activation in ob/ob mice. Last, we showed that several lipid metabolites were differentially regulated in the serum and retinal samples taken from the ob/ob animals. This work lays strong foundations for exploring the link between obesity and AMD and demonstrates a possible mechanistic role for complement activation in obesity leading to the progression of AMD.
Modulation of oxidative stress and inflammation in ob/ob animals
In AMD, the balance between age-related stress or damage and the repair or remodeling functions of the local retinal immune system may be disturbed due to the altered regulation of immune processes (genetic predisposition) or excessive damage due to lifestyle factors (reviewed in [32]). Obesity may contribute to inflammation in the retina where symptoms progress with age, and promote the onset and progression of AMD. The present data suggest that obesity induces low-level oxidative stress in the retina, which can lead to chronic retinal inflammation over time [3].
Many facets of retinal aging, including a heightened inflammatory response, have been attributed to the increased production of ROS (reviewed in [33]), where it is hypothesized that regulation of ROS by antioxidant systems has the potential to prevent the progression of AMD [34]. The accumulation of ROS in fatty tissue is an important pathogenic mechanism in the development of obesity [15]. The present data suggest that an increase in ROS and general retinal stress in the ob/ob animals mediates retinal inflammation in obesity. We showed a decrease in retinal function in the ob/ob animals, although no difference in photoreceptor layer thickness was observed. However, we were able to detect an increase in general retinal stress (GFAP) and oxidative stress (Gpx3 and Hmox1), which was correlated with increased ROS and RNS production and microglia and macrophage migration to the outer retina in the ob/ob mice. It is likely that low-level changes in oxidative stress lead to an accumulation of ROS and RNS over time, prompting a change in the inflammatory status of the ob/ob retina. We have previously shown a correlation between oxidative stress and inflammation, and decreased retinal function in a model of retinal inflammation [23]. Despite the lack of photoreceptor cell death in the ob/ob retinas, lower retinal function indicates a low-level change in retinal homeostasis and a move toward a retinal environment conducive to degeneration.
Retinal complement activation in ob/ob animals
In this study, the increase in complement activators C2 and Cfb, and decrease in negative regulator Cfh in the ob/ob mouse retinas indicates that complement activation may be linked to obesity-induced oxidative stress and inflammation. Microglia and macrophages are a key source of complement synthesis in retinal aging and degeneration, contributing to the progression of retinal damage [35-38]. Numerous studies have shown that the recruitment of microglia, the resident immune cells of the retina, exacerbates retinal degeneration in animal models of AMD [25,39,40], diabetic retinopathy [41,42], and glaucoma [43]. In the ob/ob animals, we found migration of retinal microglia into the outer retina associated with C3 deposition and upregulation of complement activation. Further, induction of oxidative stress in primary isolated microglia resulted in a similar increase in complement activation, including an increase in C2 and Cfb, and a decrease in Cfh. Together, these results demonstrate obesity-related complement activation in the retina, mediated by microglia and macrophages.
Although C3 deposition by microglia and macrophages was detected with immunohistochemistry in the outer retinas of the ob/ob mice, we also found no change in the levels of retinal C3 gene expression in ob/ob mice. Similarly, in the H2O2-stimulated primary microglia, we observed upregulation of C2 and Cfb in response to H2O2 stimulation—indicative of complement system activation—but decreased C3 and Cfh gene expression. This finding is unexpected in light of data from other models of retinal damage we have investigated, in which C3 gene expression patterns mirrored the translation to protein [36-38]. It is possible that the addition of GM-CSF during the microglial culture process may have an effect on complement activation in vitro, or that changes in post-transcriptional regulation of C3 synthesis may explain the observed C3 deposition in the ob/ob mice. Further investigations are in progress to better understand these dynamics of C3 regulation.
Leptin-deficient ob/ob animals are hyper-responsive to infection and inflammation, suggesting an alternative role for leptin in regulating inflammation [44-47]. The effects of leptin in the retina have not been well studied; however, research has suggested that higher levels of serum leptin are inversely associated with AMD [48,49] suggesting a role for leptin that may impact retinal inflammation. Accordingly, further investigation into the role of leptin in the retina, as well as the use of alternative obesity models, including a high-fat diet, are required to fully substantiate these results.
Lipid metabolites in ob/ob mice
It is known that changes in cellular metabolic pathways are associated with obesity [50,51]. In this study, we analyzed the metabolites of serum and retinal tissue in wild-type control and ob/ob mice, and found numerous metabolites were differentially expressed in the obese mice. PUFAs and branched-chain amino acids (BCAAs) showed an altered expression pattern in the ob/ob mouse serum and retinas. In humans, several neurodegenerative diseases, including AMD, are linked to an increase in oxidative stress, which runs in parallel with lipid peroxidation of PUFAs [52]. The high lipid content of the photoreceptor outer segments, together with their high metabolic activity, makes photoreceptors highly susceptible to oxidative stress. It is possible that changes in the lipid composition of the retina may cause an increased level of oxidative damage, which may lead to the development of retinal degenerations.
The data suggest that leptin deficiency leads to an increased level of lipid metabolites, primarily PUFAs, in the serum and retinas of the ob/ob animals. These lipid metabolites include lyso-phosphatidylcholine (lysoPC), diacyl phosphatidylcholine (PC aa), acyl-alkyl phosphatidylcholine (PC ae), hydroxy sphingomyelin (SM-OH), and sphingomyelin (SM). Of major interest are the lysoPC, which can stimulate macrophage activation and phagocytosis [53], and are associated with neuroinflammation in diabetic retinopathy and AMD [54-56]. We have shown that ROS and RNS production is increased in ob/ob retinas. Taken together, the data suggest that changes in lipid metabolites in ob/ob mice may contribute to increased levels of oxidative stress in the retina.
In this study, we also observed an increase in BCAAs (leucine, isoleucine, and valine) in the ob/ob mouse serum or retinas. These findings may be due to a larger intake of food containing BCAAs in these hyperphagic mice; however, there is a strong correlation in the literature between increased BCAAs and obesity [57,58]. Due to the high incidence of insulin resistance associated with obesity, it remains unclear whether increased BCAAs are associated with obesity or insulin resistance [59]. Interestingly, increased BCAAs may have immunomodulatory effects on microglia, where cells cultured in high-BCAA-containing medium exhibit increased ROS production [60]. Additionally, these microglia produced decreased amounts of IGF-1, a neuroprotective and anti-inflammatory factor, suggesting a potential link between BCAAs and neuroinflammation [60].
Leptin-deficient (ob/ob) mice on all backgrounds are known to exhibit hyperphagia, hyperlipidemia, insulin resistance, reduced energy expenditure, and extreme obesity [61] and therefore, are considered a useful model for studying obesity and associated metabolic disturbances. It is known that the phenotype of ob/ob mice, including the levels of blood glucose and glycated hemoglobin (HbA1c), varies depending on the genetic background (reviewed in [61]). Ob/ob mice are considered obese at 4 weeks of age compared to wild-type littermate controls, and on the C57BL/6J background can have mild to severe hyperglycemia and hyperinsulinemia between 8 and 12 weeks of age [61]. However, mice on a C57BL/6J background do not develop severe type 2 diabetes to the extent of mice on the C57BL/KsJ [61] or the BTBR [62,63] background. Leptin-deficient mice on the C57BL/6J background can have no change to mild increases in blood glucose levels, depending on the animals’ age with the maximum glucose concentrations observed at about 5 months [21,22,64,65]. In addition, other studies have shown that ob/ob mice have statistically significantly increased blood glucose and HbA1c levels compared to wild-type or ob/+ mice on various genetic backgrounds, ages, and sexes [21,22,62,63,66]. In this set of experiments that used young adult animals (70-90 postnatal day), we did not observe any change in glucose or HbA1c levels in the colony of leptin-deficient animals. This result indicates the susceptibility to retinal degeneration in young leptin-deficient animals may not be directly related to blood glucose levels but attributed to a combination of other metabolic dysfunctions leading to obesity.
Conclusion
The economic and social burdens of AMD and obesity are steadily increasing, with growing evidence of a causational relationship of AMD and obesity. In this study, we showed that increased oxidative stress and inflammation in the retina are linked to obesity in leptin-deficient (ob/ob) animals. We demonstrated that oxidative stress is increased in ob/ob retinas, associated with increases in the expression of complement components upstream of C3 and changes in lipid metabolites in the retina. The findings provide insight into how obesity predisposes the retina to complement activation and points to oxidative damage-mediated mechanisms that may be addressed to some degree during aging with lifestyle approaches, including diet and exercise.
Acknowledgments
This study was supported by an Australian Government Research Training Program (RTP) Scholarship.
Appendix 1. Validation of primary isolated microglia as pure cultures.
A: Representative plots of the gating strategy used in fluorescence-activated cell sorting (FACS) to isolate CD11b+ cells (red population) from whole retinas. B-F: Microglial cells were immunolabelled with monocyte markers IBA1 (B, red) and CD45 (C, red), with nuclei counterstained with DAPI. Immunolabelling for other common retinal cell markers including Rhodopsin (Rhod, D), GFAP (E) and a negative control (-ve, F) showed no positive labeling for these markers. G-H: Primary microglial cells were also screened for other common retinal cell markers including Rhod (G) and Gfap (H) using PCR. There was no expression in any of the isolated microglial cell samples for Rhod or Gfap, however positive PCR banding was detected in a whole retinal sample indicating successful PCR amplification. Scale bars indicate 50 µm. To access the data, click or select the words “Appendix 1”
References
- 1.Pi-Sunyer X. The medical risks of obesity. Postgrad Med. 2009;121:21–33. doi: 10.3810/pgm.2009.11.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheung N, Wong TY. Obesity and eye diseases. Surv Ophthalmol. 2007;52:180–95. doi: 10.1016/j.survophthal.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Prog Retin Eye Res. 2009;28:348–68. doi: 10.1016/j.preteyeres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 4.Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, Hauser MA, Bowes Rickman C, Bok D, Hageman GS, Johnson LV. The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis re-visited. Prog Retin Eye Res. 2010;29:95–112. doi: 10.1016/j.preteyeres.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thakkinstian A, Han P, McEvoy M, Smith W, Hoh J, Magnusson K, Zhang K, Attia J. Systematic review and meta-analysis of the association between complementary factor H Y402H polymorphisms and age-related macular degeneration. Hum Mol Genet. 2006;15:2784–90. doi: 10.1093/hmg/ddl220. [DOI] [PubMed] [Google Scholar]
- 6.Hirvela H, Luukinen H, Laara E, Sc L, Laatikainen L. Risk factors of age-related maculopathy in a population 70 years of age or older. Ophthalmology. 1996;103:871–7. doi: 10.1016/s0161-6420(96)30593-9. [DOI] [PubMed] [Google Scholar]
- 7.Smith W, Mitchell P, Leeder SR, Wang JJ. Plasma fibrinogen levels, other cardiovascular risk factors, and age-related maculopathy: The Blue Mountains Eye Study. Arch Ophthalmol. 1998;116:583–7. doi: 10.1001/archopht.116.5.583. [DOI] [PubMed] [Google Scholar]
- 8.Klein BE, Klein R, Lee KE, Jensen SC. Measures of obesity and age-related eye diseases. Ophthalmic Epidemiol. 2001;8:251–62. doi: 10.1076/opep.8.4.251.1612. [DOI] [PubMed] [Google Scholar]
- 9.Zhang QY, Tie LJ, Wu SS, Lv PL, Huang HW, Wang WQ, Wang H, Ma L. Overweight, obesity, and risk of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2016;57:1276–83. doi: 10.1167/iovs.15-18637. [DOI] [PubMed] [Google Scholar]
- 10.Ersoy L, Ristau T, Lechanteur YT, Hahn M, Hoyng CB, Kirchhof B, den Hollander AI, Fauser S. Nutritional risk factors for age-related macular degeneration. BioMed Res Int. 2014;2014:413150. doi: 10.1155/2014/413150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schaumberg DA, Christen WG, Hankinson SE, Glynn RJ. Body mass index and the incidence of visually significant age-related maculopathy in men. Arch Ophthalmol. 2001;119:1259–65. doi: 10.1001/archopht.119.9.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seddon JM, Cote J, Davis N, Rosner B. Progression of age-related macular degeneration: association with body mass index, waist circumference, and waist-hip ratio. Arch Ophthalmol. 2003;121:785–92. doi: 10.1001/archopht.121.6.785. [DOI] [PubMed] [Google Scholar]
- 13.Clemons TE, Milton RC, Klein R, Seddon JM, Ferris FL., 3rd Age-Related Eye Disease Study Research, G. Risk factors for the incidence of Advanced Age-Related Macular Degeneration in the Age-Related Eye Disease Study (AREDS) AREDS report no. 19. Ophthalmology. 2005;112:533–9. doi: 10.1016/j.ophtha.2004.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vincent HK, Taylor AG. Biomarkers and potential mechanisms of obesity-induced oxidant stress in humans. Int J Obes. 2006;30:400–18. doi: 10.1038/sj.ijo.0803177. [DOI] [PubMed] [Google Scholar]
- 15.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–61. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dandona P, Mohanty P, Ghanim H, Aljada A, Browne R, Hamouda W, Prabhala A, Afzal A, Garg R. The suppressive effect of dietary restriction and weight loss in the obese on the generation of reactive oxygen species by leukocytes, lipid peroxidation, and protein carbonylation. J Clin Endocrinol Metab. 2001;86:355–62. doi: 10.1210/jcem.86.1.7150. [DOI] [PubMed] [Google Scholar]
- 17.Tucsek Z, Toth P, Sosnowska D, Gautam T, Mitschelen M, Koller A, Szalai G, Sonntag WE, Ungvari Z, Csiszar A. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: Effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J Gerontol A Biol Sci Med Sci. 2014;69:1212–26. doi: 10.1093/gerona/glt177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vieira AA, Michels M, Florentino D, Nascimento DZ, Rezin GT, Leffa DD, Fortunato JJ, Dal-Pizzol F, Barichello T, Quevedo J, Petronilho F. Obesity promotes oxidative stress and exacerbates sepsis-induced brain damage. Curr Neurovasc Res. 2015;12:147–54. doi: 10.2174/1567202612666150311111913. [DOI] [PubMed] [Google Scholar]
- 19.Bocarsly ME, Fasolino M, Kane GA, LaMarca EA, Kirschen GW, Karatsoreos IN, McEwen BS, Gould E. Obesity diminishes synaptic markers, alters microglial morphology, and impairs cognitive function. Proc Natl Acad Sci USA. 2015;112:15731–6. doi: 10.1073/pnas.1511593112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vlaicu SI, Tatomir A, Boodhoo D, Vesa S, Mircea PA, Rus H. The role of complement system in adipose tissue-related inflammation. Immunol Res. 2016;64:653–64. doi: 10.1007/s12026-015-8783-5. [DOI] [PubMed] [Google Scholar]
- 21.Ingalls AM, Dickie MM, Shell G. Obese, a new mutation in the house mouse. J Hered. 1950;41:317–8. doi: 10.1093/oxfordjournals.jhered.a106073. [DOI] [PubMed] [Google Scholar]
- 22.Lindström P. The physiology of obese-hyperglycemic mice. Sci World J. 2007;7:666–85. doi: 10.1100/tsw.2007.117. ob/ob mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Natoli R, Jiao H, Barnett NL, Fernando N, Valter K, Provis JM, Rutar M. A model of progressive photo-oxidative degeneration and inflammation in the pigmented C57BL/6J mouse retina. Exp Eye Res. 2016;147:114–27. doi: 10.1016/j.exer.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 24.Rutar M, Natoli R, Chia R, Valter K, Provis J. Chemokine-mediated inflammation in the degenerating retina is coordinated by Muller cells, activated microglia, and retinal pigment epithelium. J Neuroinflammation. 2015;12:8. doi: 10.1186/s12974-014-0224-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernando N, Natoli R, Valter K, Provis J, Rutar M. The broad-spectrum chemokine inhibitor NR58–3.14.3 modulates macrophage-mediated inflammation in the diseased retina. J Neuroinflammation. 2016;13:1–14. doi: 10.1186/s12974-016-0514-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Umetrics M. User guide to SIMCA. Malmö (Sweden): MKS Umetrics AB 2013 [Google Scholar]
- 27.Eriksson L, Kettaneh-Wold N, Trygg J, Wikström C, Wold S. Multi-and megavariate data analysis: Part I: Basic principles and applications. Umetrics Inc; 2006. [Google Scholar]
- 28.Habot-Wilner, Z, Belkin, M. Obesity is a risk factor for eye diseases. Harefuah 2005; 144:805–9, 21. [PubMed]
- 29.Toomey CB, Kelly U, Saban DR, Bowes Rickman C. Regulation of age-related macular degeneration-like pathology by complement factor H. Proc Natl Acad Sci USA. 2015;112:E3040–9. doi: 10.1073/pnas.1424391112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang RC, Shi L, Huang CC, Kim AJ, Ko ML, Zhou B, Ko GY. High-fat diet-induced retinal dysfunction. Invest Ophthalmol Vis Sci. 2015;56:2367–80. doi: 10.1167/iovs.14-16143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee JJ, Wang PW, Yang IH, Huang HM, Chang CS, Wu CL, Chuang JH. High-fat diet induces toll-like receptor 4-dependent macrophage/microglial cell activation and retinal impairment. Invest Ophthalmol Vis Sci. 2015;56:3041–50. doi: 10.1167/iovs.15-16504. [DOI] [PubMed] [Google Scholar]
- 32.Ambati J, Ambati BK, Yoo SH, Ianchulev S, Adamis AP. Age-related macular degeneration: Etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol. 2003;48:257–93. doi: 10.1016/s0039-6257(03)00030-4. [DOI] [PubMed] [Google Scholar]
- 33.Ding X, Patel M, Chan CC. Molecular pathology of age-related macular degeneration. Prog Retin Eye Res. 2009;28:1–18. doi: 10.1016/j.preteyeres.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Handa JT. How does the macula protect itself from oxidative stress? Mol Aspects Med. 2012;33:418–35. doi: 10.1016/j.mam.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karlstetter M, Scholz R, Rutar M, Wong WT, Provis JM, Langmann T. Retinal microglia: Just bystander or target for therapy? Prog Retin Eye Res. 2014;••• doi: 10.1016/j.preteyeres.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 36.Rutar M, Natoli R, Kozulin P, Valter K, Gatenby P, Provis JM. Analysis of complement expression in light-induced retinal degeneration: Synthesis and deposition of C3 by microglia/macrophages is associated with focal photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2011;52:5347–58. doi: 10.1167/iovs.10-7119. [DOI] [PubMed] [Google Scholar]
- 37.Rutar M, Valter K, Natoli R, Provis JM. Synthesis and propagation of complement C3 by microglia/monocytes in the aging retina. PLoS One. 2014;9:e93343. doi: 10.1371/journal.pone.0093343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Natoli R, Fernando N, Jiao H, Racic T, Madigan M, Barnett NL, Chu-Tan JA, Valter K, Provis J, Rutar M. Retinal macrophages synthesize C3 and activate complement in AMD and in models of focal retinal degeneration. Invest Ophthalmol Vis Sci. 2017;58:2977–90. doi: 10.1167/iovs.17-21672. [DOI] [PubMed] [Google Scholar]
- 39.Luhmann UF, Robbie S, Munro PM, Barker SE, Duran Y, Luong V, Fitzke FW, Bainbridge JW, Ali RR, MacLaren RE. The drusenlike phenotype in aging Ccl2-knockout mice is caused by an accelerated accumulation of swollen autofluorescent subretinal macrophages. Invest Ophthalmol Vis Sci. 2009;50:5934–43. doi: 10.1167/iovs.09-3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sennlaub F, Auvynet C, Calippe B, Lavalette S, Poupel L, Hu SJ, Dominguez E, Camelo S, Levy O, Guyon E, Saederup N, Charo IF, Rooijen NV, Nandrot E, Bourges JL, Behar-Cohen F, Sahel JA, Guillonneau X, Raoul W, Combadiere C. CCR2(+) monocytes infiltrate atrophic lesions in age-related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mol Med. 2013;5:1775–93. doi: 10.1002/emmm.201302692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ibrahim AS, El-Shishtawy MM, Pena A, Jr, Liou GI. Genistein attenuates retinal inflammation associated with diabetes by targeting of microglial activation. Mol Vis. 2010;16:2033–42. [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang L, Li Y, Payne J, Srivastava S, Fan X, Fung J, Li X, Kern TS, Lin F. Presence of retinal pericyte-reactive autoantibodies in diabetic retinopathy patients. Sci Rep. 2016;6:20341. doi: 10.1038/srep20341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bosco A, Inman DM, Steele MR, Wu G, Soto I, Marsh-Armstrong N, Hubbard WC, Calkins DJ, Horner PJ, Vetter ML. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2008;49:1437–46. doi: 10.1167/iovs.07-1337. [DOI] [PubMed] [Google Scholar]
- 44.Grunfeld C, Zhao C, Fuller J, Pollack A, Moser A, Friedman J, Feingold KR. Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. J Clin Invest. 1996;97:2152–7. doi: 10.1172/JCI118653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faggioni R, Fantuzzi G, Gabay C, Moser A, Dinarello CA, Feingold KR, Grunfeld C. Leptin deficiency enhances sensitivity to endotoxin-induced lethality. Am J Physiol. 1999;276:R136–42. doi: 10.1152/ajpregu.1999.276.1.R136. [DOI] [PubMed] [Google Scholar]
- 46.Faggioni R, Fantuzzi G, Fuller J, Dinarello CA, Feingold KR, Grunfeld C. IL-1 beta mediates leptin induction during inflammation. Am J Physiol. 1998;274:R204–8. doi: 10.1152/ajpregu.1998.274.1.R204. [DOI] [PubMed] [Google Scholar]
- 47.Faggioni R, Fuller J, Moser A, Feingold KR, Grunfeld C. LPS-induced anorexia in leptin-deficient (ob/ob) and leptin receptor-deficient (db/db) mice. Am J Physiol. 1997;273:R181–6. doi: 10.1152/ajpregu.1997.273.1.R181. [DOI] [PubMed] [Google Scholar]
- 48.Seshasai S, Liao J, Toh QC, Cheng CY, Cheung GC, Sethi S, Wong TY, Sabanayagam C. Serum leptin and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2015;56:1880–6. doi: 10.1167/iovs.14-15933. [DOI] [PubMed] [Google Scholar]
- 49.Evereklioglu C, Doganay S, Er H, Cekmen M, Ozerol E, Otlu B. Serum leptin concentrations are decreased and correlated with disease severity in age-related macular degeneration: A preliminary study. Eye (Lond) 2003;17:350–5. doi: 10.1038/sj.eye.6700345. [DOI] [PubMed] [Google Scholar]
- 50.Herman MA, She P, Peroni OD, Lynch CJ, Kahn BB. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem. 2010;285:11348–56. doi: 10.1074/jbc.M109.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Luis DA, Almansa R, Aller R, Izaola O, Romero E. Gene expression analysis identify a metabolic and cell function alterations as a hallmark of obesity without metabolic syndrome in peripheral blood, a pilot study. Clin Nutr. 2017 doi: 10.1016/j.clnu.2017.06.006. In press. [DOI] [PubMed] [Google Scholar]
- 52.Nowak JZ. Oxidative stress, polyunsaturated fatty acids-derived oxidation products and bisretinoids as potential inducers of CNS diseases: Focus on age-related macular degeneration. Pharmacol Rep. 2013;65:288–304. doi: 10.1016/s1734-1140(13)71005-3. [DOI] [PubMed] [Google Scholar]
- 53.Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113:717–30. doi: 10.1016/s0092-8674(03)00422-7. [DOI] [PubMed] [Google Scholar]
- 54.Hollyfield JG, Perez VL, Salomon RG. A hapten generated from an oxidation fragment of docosahexaenoic acid is sufficient to initiate age-related macular degeneration. Mol Neurobiol. 2010;41:290–8. doi: 10.1007/s12035-010-8110-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.West XZ, Malinin NL, Merkulova AA, Tischenko M, Kerr BA, Borden EC, Podrez EA, Salomon RG, Byzova TV. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature. 2010;467:972–6. doi: 10.1038/nature09421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng L, Han X, Shi Y. A regulatory role of LPCAT1 in the synthesis of inflammatory lipids, PAF and LPC, in the retina of diabetic mice. Am J Physiol Endocrinol Metab. 2009;297:E1276–82. doi: 10.1152/ajpendo.00475.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takashina C, Tsujino I, Watanabe T, Sakaue S, Ikeda D, Yamada A, Sato T, Ohira H, Otsuka Y, Oyama-Manabe N, Ito YM, Nishimura M. Associations among the plasma amino acid profile, obesity, and glucose metabolism in Japanese adults with normal glucose tolerance. Nutr Metab (Lond) 2016;13:5. doi: 10.1186/s12986-015-0059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wiklund P, Zhang X, Pekkala S, Autio R, Kong L, Yang Y, Keinänen-Kiukaanniemi S, Alen M, Cheng S. Insulin resistance is associated with altered amino acid metabolism and adipose tissue dysfunction in normoglycemic women. Sci Rep. 2016;6:24540. doi: 10.1038/srep24540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adams MK, Simpson JA, Aung KZ, Makeyeva GA, Giles GG, English DR, Hopper J, Guymer RH, Baird PN, Robman LD. Abdominal obesity and age-related macular degeneration. Am J Epidemiol. 2011;173:1246–55. doi: 10.1093/aje/kwr005. [DOI] [PubMed] [Google Scholar]
- 60.De Simone R, Vissicchio F, Mingarelli C, De Nuccio C, Visentin S, Ajmone-Cat MA, Minghetti L. Branched-chain amino acids influence the immune properties of microglial cells and their responsiveness to pro-inflammatory signals. Biochim Biophys Acta (BBA) - Molecular Basis of Disease. 2013;1832:650–9. doi: 10.1016/j.bbadis.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 61.Kennedy AJ, Ellacott KLJ, King VL, Hasty AH. Mouse models of the metabolic syndrome. Dis Model Mech. 2010;3:156–66. doi: 10.1242/dmm.003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Brien PD, Hur J, Hayes JM, Backus C, Sakowski SA, Feldman EL. BTBR ob/ob mice as a novel diabetic neuropathy model: Neurological characterization and gene expression analyses. Neurobiol Dis. 2015;73:348–55. doi: 10.1016/j.nbd.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O’Brien PD, Hur J, Robell NJ, Hayes JM, Sakowski SA, Feldman EL. Gender-specific differences in diabetic neuropathy in BTBR ob/ob mice. J Diabetes Complications. 2016;30:30–7. doi: 10.1016/j.jdiacomp.2015.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Drel VR, Mashtalir N, Ilnytska O, Shin J, Li F, Lyzogubov VV, Obrosova IG. The leptin-deficient (ob/ob) mouse: A new animal model of peripheral neuropathy of Type 2 diabetes and obesity. Diabetes. 2006;55:3335–43. doi: 10.2337/db06-0885. [DOI] [PubMed] [Google Scholar]
- 65.Makimura H, Mizuno TM, Roberts J, Silverstein J, Beasley J, Mobbs CV. Adrenalectomy reverses obese phenotype and restores hypothalamic melanocortin tone in leptin-deficient ob/ob mice. Diabetes. 2000;49:1917–23. doi: 10.2337/diabetes.49.11.1917. [DOI] [PubMed] [Google Scholar]
- 66.Dubuc PU, Keyes Scott B, Peterson CM. Sex differences in glycated hemoglobin in diabetic and non-diabetic C57BL/6 mice. Diabetes Res Clin Pract. 1993;21:95–101. doi: 10.1016/0168-8227(93)90055-a. [DOI] [PubMed] [Google Scholar]