

Abstract
Background
Osteoarthritis (OA) is the most common chronic disorder of joints; however, the key genes and transcription factors (TFs) associated with OA are still unclear. Through bioinformatics tools, the study aimed to understand the mechanism of genes associated with the development of OA.
Methods
Four gene expression profiling datasets were used to identify differentially expressed genes (DEGs) between OA and healthy control samples by a meta-analysis. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses were performed with Multifaceted Analysis Tool for Human Transcriptome (MATHT). Subsequently, a protein–protein interaction (PPI) network was constructed for these DEGs. Significant network modules were identified using ReactomeFIViz, and the pathway of each module was enriched using MATHT. In addition, TFs in the DEGs were identified.
Results
In total, 690 DEGs were identified between OA and healthy control samples, including 449 upregulated and 241 downregulated DEGs. Additionally, 622 nodes and 2752 interactions constituted the PPI network, including 401 upregulated and 221 downregulated DEGs. Among them, FOS, TWIST1, POU2F1, SMARCA4, and CREBBP were also identified as TFs. RT-PCR results showed that the expression levels of Fos, Twist1, Pou2f1, Smarca4, and Crebbp decreased in mice with OA. In addition, FOS, TWIST1, SMARCA4, and CREBBP were involved in the positive regulation of transcription from the RNA polymerase II promoter.
Conclusions
TWIST1, POU2F1, SMARCA4, and CREBBP may play an important role in OA pathology.
Keywords: Osteoarthritis, Transcription factor, Meta-analysis, Gene expression profiling datasets
Background
Osteoarthritis (OA) is the most common chronic disorder of joints, such as knee joints, hip joints, and small finger joints [1]. In China, approximately 10% of the total population experiences OA [2]. The main symptoms of patients with OA are pain, swelling, and joint deformity because of cartilage breakdown [3]. There is no cure due to the long-term nature of the disease and multiple mechanisms associated with it, but physical activity, improving joint mobility and flexibility using assistive devices, and surgery can help improve symptoms [4, 5]. Thus, studies on etiological factors of OA are important.
Many studies have reported on factors that contribute to the development of OA, such as obesity, fracture, surgery or ligament tears, and genes [6, 7]. Various genes make individuals more susceptible to OA. Researchers have found that fatty acid amide hydrolase (FAAH) expression is related to increased pain sensitivity and is upregulated in patients with knee OA than in people without OA [8]. Subsequently, FAAH inhibitors, such as URB597, PF-04457845, and OL-135, have been focused on for OA treatment [9–11]. In addition, long and short proteins, which are encoded by DVWA and related to knee OA susceptibility, are mainly expressed in articular cartilage [12].
Transcription factors (TFs) are proteins that control the rate of transcription in molecular biology, which regulates gene expression [13]. The expression of SAF-1, an inflammation-responsive TF, was found to be overexpressed in moderate-to-severely damaged OA cartilage tissues [14]. Therefore, screening the key genes and TFs associated with OA is important for determining OA pathology.
Through bioinformatics tools, the present study aimed to understand the mechanism of genes associated with the development of OA. Differentially expressed genes (DEGs) were identified using four gene expression profiling datasets, GSE55235, GSE55457, GSE1919, and GSE12021, based on a meta-analysis. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed with MATHT (www.biocloudservice.com). Subsequently, a protein–protein interaction (PPI) network was constructed for these DEGs. Significant network modules were identified using ReactomeFIViz, and the pathway of each module was enriched using MATHT. In addition, TFs in the DEGs were identified, and the key DEGs were verified using quantitative real-time polymerase chain reaction (qRT-PCR). These findings may provide a novel understanding of the molecular mechanisms underlying OA.
Methods
Data acquisition
Four datasets, GSE55235, GSE55457, GSE1919, and GSE12021, including human OA and healthy control samples were downloaded from the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) database. The characteristics of each dataset are shown in Table 1.
Table 1.
The characteristic of four datasets
| Accession | OA count | Normal count | Platform |
|---|---|---|---|
| GSE55235 | 10 | 10 | Affymetrix Human Genome U133A Array |
| GSE55457 | 10 | 10 | Affymetrix Human Genome U133A Array |
| GSE1919 | 5 | 5 | Affymetrix Human Genome U95A Array |
| GSE12021 | 20 | 13 | Affymetrix Human Genome U133A Array, Affymetrix Human Genome U133B Array |
OA osteoarthritis
Data preprocessing
Raw CEL files were read using the Affy package in R software (version 1.28.0, http://www.bioconductor.org/packages/release/bioc/html/affy.html) [15]. Subsequently, data preprocessing was performed with RMA [16], such as background correction, normalization, and expression calculation. Each probe ID was transformed into a gene symbol. The probes corresponded to gene symbols according to the latest annotation file. If any probe corresponded to multiple genes, its expression value was removed. If more than one probe corresponded to the same gene symbol, the mean of the probe was used as the expression level of the gene.
Identification of DEGs using a meta-analysis
The MetaDE package in R software was used to integrate the data in the four datasets [17], and the DEGs in OA samples were identified from genes in the control samples. The expression value of each gene on different platforms was evaluated for heterogeneity and unbias, including τ2 (estimated amount of heterogeneity) and Qpval (P values for the heterogeneity test). If τ2 = 0 and Qpval > 0.05, the gene was homogeneous and unbiased. The differential expression of genes was then detected, and only genes with P < 0.05 were considered significant. Subsequently, the false discovery rate (FDR) of each gene was calculated using Benjamini–Hochberg correction. Genes with τ2 = 0, Qpval > 0.05, P < 0.05, and FDR < 0.01 were identified as DEGs. On the basis of these DEGs, the log2 fold change (log2FC) was calculated for each gene in the four datasets. If log2FC > 0, gene expression was upregulated in OA samples; otherwise, it was downregulated in OA samples.
GO enrichment function and pathway analysis
To determine the DEGs involved in biological processes (BPs), cellular components (CCs), molecular functions (MFs), and pathways, GO and KEGG pathway enrichment analyses were performed with MATHT based on Fisher’s test. A P value of < 0.05 was considered significant.
Construction of the PPI network
The interaction between proteins was analyzed using STRING (version 10.0) for the DEGs using default parameters. The threshold value was required confidence (combined score) > 0.4. Subsequently, the PPI network was visualized using Cytoscape (version 3.2.0, http://www.cytoscape.org/). In the network, the node represents a protein, the line represents the interaction, and the degree represents the number of interactions. Then, CytoNCA in Cytoscape was used to analyze the network topology under the “without weight” condition. The degree centrality, betweenness centrality, and closeness centrality of each node were obtained.
Module analysis in the PPI network
The ReactomeFIViz app-applied MCL graph clustering algorithm was used to generate a subnetwork for a list of significant network modules [18]. In addition, the average Pearson correlation coefficient among genes involved in the same module was calculated. On the basis of the subnetwork, the pathway in each module was enriched using MATHT.
Construction of the transcriptional regulatory network
On the basis of the TF–target gene data from the Transcriptional Regulatory Relationships Unraveled by Sentence-based Text Mining website, the TFs in the DEGs were identified. The network was visualized using Cytoscape.
Animal model of OA
To establish the animal model of OA, male rats were randomly divided into control and model groups (30 rats per group). After acclimatization for 3 days, rats in the control group were given normal food without any other treatment. The left knee joints of rats in the model group were subjected to anterior cruciate ligament transaction, and the right knee joints served as the control [19]. Subsequently, the rats were sacrificed and the joints were harvested at 8 weeks post surgery.
Verification of qRT-PCR results
To confirm the results, expression levels of FOS, TWIST1, POU2F1, SMARCA4, and CREBBP were detected using qRT-PCR. Total RNA was extracted from the synovial tissues of rats using TRIzol reagent following the manufacturer’s instructions (TAKARA, Dalian, China) under low temperature. Subsequently, first-strand cDNA was prepared from the RNA obtained from synovial tissues using PrimeScript™RT Master Mix according to the manufacturer’s instructions (RR036A, TAKARA). Rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the endogenous control. Primers used for FOS, TWIST1, POU2F1, SMARCA4, and CREBBP and GAPDH were based on the rat sequences (Table 2). Relative amounts of mRNAs were obtained using the Relative Expression Software Tool.
Table 2.
The rat sequences of primers used for RT-PCR
| Primer | Sequences (5′–3′) |
|---|---|
| GAPDH-F | AGACAGCCGCATCTTCTTGT |
| GAPDH-R | CTTGCCGTGGGTAGAGTCAT |
| FOS-F | GTGACAGCCATCTCCACCAG |
| FOS-R | TCCTTTCCCTTCGGATTCTC |
| TWIST1-F | TTTCACAAGAATCAGGGCGTG |
| TWIST1-R | CCGTTGCCTCTGGGAATCTC |
| POU2F1-F | GAGCAGCGAGTCAAGATGAGA |
| POU2F1-R | GGGCTGCTTCTCAAAGTCCA |
| SMARCA4-F | GGACGCTGTGATCAAGTACA |
| SMARCA4-R | GGTTTCGGATGCGTTCCTTG |
| CREBBP-F | CCAGGCAGGTGTTTCACAG |
| CREBBP-R | ACAGGAGTGGATGGCTGAGT |
F Forward primer, R Reverse primer
Statistical analysis
The OA and control groups were compared using unpaired Student’s t test by SPSS Statistics V22.0 (SPSS Inc., Chicago, IL, USA). P < 0.05 was considered significant.
Results
Results of function and pathway enrichment
In total, 690 DEGs were identified between the OA and healthy control samples after the meta-analysis, including 449 upregulated and 241 downregulated DEGs. In the whole upregulated DEGs, 93 BP terms (vesicle-mediated transport, oxidation-reduction process, etc.), 38 CC terms (AP-2 adaptor complex, etc.), 18 MF terms (protein binding, endopeptidase activity, NADH dehydrogenase [ubiquinone] activity, etc.), and 17 pathways (metabolic pathways, oxidative phosphorylation, peroxisome, etc.) were enriched. The top five terms are shown in Fig. 1. In the downregulated DEGs, 85 BP terms (positive regulation of transcription from the RNA polymerase II promoter, transforming growth factor beta receptor signaling pathway, etc.), 14 CC terms (nucleoplasm, nucleus, cytoplasm, etc.), 24 MF terms (poly(A) RNA binding, protein binding, nucleotide binding, etc.), and 20 pathways (TNF signaling pathway, neurotrophin signaling pathway, and Jak-STAT signaling pathway) were obtained. The top five terms are shown in Fig. 2.
Fig. 1.
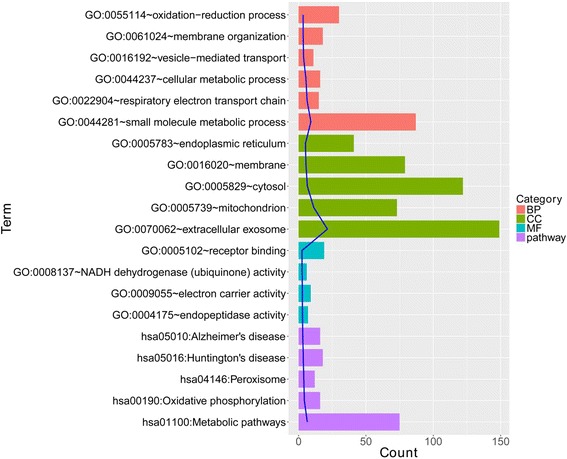
The top five biological process (BP), cellular component (CC), and molecular function (MF) terms and pathways of upregulated differentially expressed genes (DEGs)
Fig. 2.
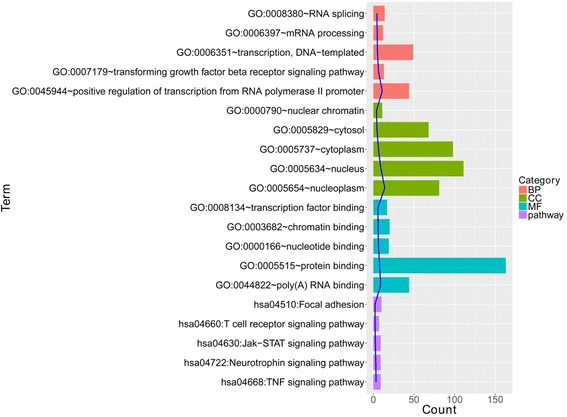
The top five BP, CC, and MF terms and pathways of downregulated DEGs
The PPI network
A total of 622 nodes and 2752 interactions constituted the PPI network, including 401 upregulated and 221 downregulated DEGs. The top 10 DEGs with a higher degree are shown in Table 3, such as FOS (degree = 55) and CREB-binding protein (CREBBP) (degree = 48), which are hub proteins. In addition, FOS interacted with CREBBP, TWIST1, and SMARCA4 and CREBBP interacted with SMARCA4 in the PPI network.
Table 3.
The top 10 of DEGs with higher degree
| Node | DC | Node | BC | Node | CC |
|---|---|---|---|---|---|
| UBC | 420 | UBC | 298,184.7 | UBC | 0.736655 |
| JUN | 77 | JUN | 10,611.95 | JUN | 0.494427 |
| HSP90AA1 | 75 | HSP90AA1 | 10,195 | HSP90AA1 | 0.491686 |
| MAPK1 | 60 | MAPK1 | 8585.318 | FOS | 0.476227 |
| FOS | 55 | KDM2A | 8230.978 | MAPK1 | 0.475498 |
| CDC42 | 52 | IL6 | 6267.055 | CDC42 | 0.473684 |
| CREBBP | 48 | CREBBP | 5555.968 | CREBBP | 0.470811 |
| IL6 | 45 | FOS | 5334.594 | IL6 | 0.469033 |
| YWHAZ | 44 | CDC42 | 5333.35 | BCL2L1 | 0.469033 |
| ACACB | 42 | CAT | 4848.764 | YWHAZ | 0.466567 |
DC degree centrality, BC betweenness centrality, CC closeness centrality
Pathways related to modules
The subnetwork was obtained after ReactomeFI analysis, including 157 nodes (108 upregulated and 49 downregulated DEGs) and 287 interactions, which belong to six different modules (Fig. 3). In addition, five modules were enriched by the following pathways: oxidative phosphorylation, MAPK signaling pathway, and metabolic pathways.
Fig. 3.
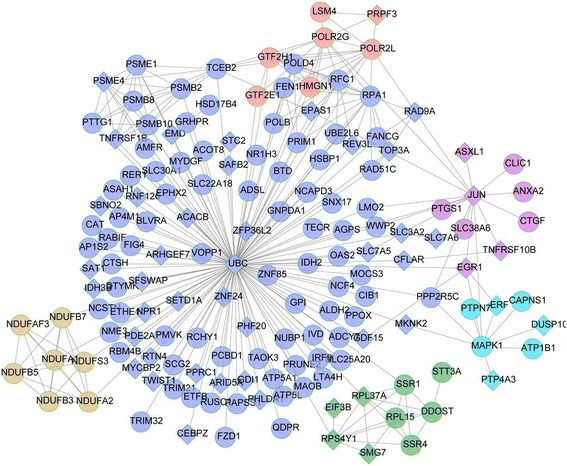
Significant network modules. The circle indicates upregulated DEGs, and the rhombus indicates downregulated DEGs. The different colors indicate different modules
The transcriptional regulatory network
There were 43 nodes and 47 link pairs in the transcriptional regulatory network, including 19 TFs (such as FOS, TWIST1, POU2F1, SMARCA4, and CREBBP), 16 upregulated DEGs, and 8 downregulated DEGs (Fig. 4).
Fig. 4.
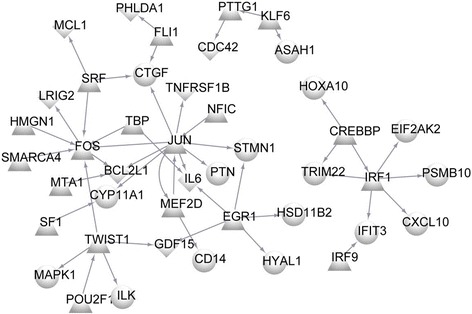
The transcriptional regulatory network. The circle indicates upregulated DEGs, the rhombus indicates downregulated DEGs, and the triangles indicate transcription factors
Expression levels of candidate genes connected with OA
As shown in Fig. 5a, b, the expression levels of TWIST1 and POU2F1 significantly decreased in rats with OA (P = 0.008), which confirmed the reliability of the bioinformatics method. In addition, the expression levels of SMARCA4 and CREBBP significantly decreased in rats with OA (P < 0.001) (Fig. 5c, d). Although FOS expression levels decreased in rats with OA, they were not significantly different (P = 0.307) (Fig. 5e).
Fig. 5.
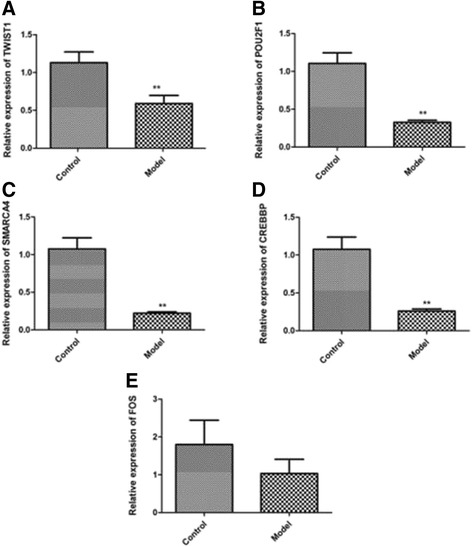
Relative expression levels of TWIST1 (a), POU2F1 (b), SMARCA4 (c), CREBBP (d), and FOS (e) in rats with osteoarthritis. *P < 0.05; **P < 0.01
Discussion
In this study, downregulated DEGs, such as FOS, TWIST1, POU2F1, SMARCA4, and CREBBP, were identified as TFs. RT-PCR showed that the expression levels of Fos, Twist1, Pou2f1, Smarca4, and Crebbp decreased in mice with OA. In addition, FOS, TWIST1, SMARCA4, and CREBBP were involved in the positive regulation of transcription from the RNA polymerase II promoter. In the PPI network, FOS interacted with CREBBP, TWIST1, and SMARCA4 and CREBBP interacted with SMARCA4.
TWIST1 encodes a basic helix–loop–helix TF that plays an important role in osteoblast metabolism and differentiation [20]. TWIST1, as a critical regulator of osteoblast differentiation in OA pathology, was also identified from the trabecular bone of patients with end-stage OA [21]. In addition, TWIST1 expression decreased in OA patients and was correlated with the inhibition of normal mineralization in OA patients [22]. Similarly, TWIST1 expression was downregulated in synovial tissues in OA. In a previous study, the downregulated gene TWIST1 was a target of the WNT signaling pathway [23], which is related to bone remodeling and pathologies such as OA [24]. Therefore, the downregulated gene TWIST1 is a key regulator in OA development. POU2F1 (also known as OCT1) downregulation facilitates osteosarcoma tumorigenesis [25]. Although POU2F1 has not been reported in OA pathology, it interacts with adenomatous polyposis coli, which negatively regulates the WNT pathway [26, 27]. In addition, POU2F1 can regulate TWIST1 expression in the transcriptional regulatory network. Therefore, POU2F1 might also be a candidate gene connected with OA pathology.
TWIST1 can regulate FOS expression in the transcriptional regulatory network, and TWIST1 and FOS interact with each other in the PPI network. Kinne et al. found that compared to patients with OA or normal joints, c-fos was highly expressed in the synovial membrane of patients with rheumatoid arthritis [28]. In addition, c-fos expression was detected in the superficial layer of cartilage only in 20% of OA patients [29]. A small molecule, harpagoside, as a therapeutic for preventing OA development, can inhibit IL-6 expression by blocking the expression of c-FOS in primary human OA chondrocytes [30]. However, FOS expression decreased in our study, probably because the sample sizes and patients from different countries in the present study were not the same as in previous studies. In the transcriptional regulatory network, SMARCA4 as a TF that modifies FOS was also downregulated in OA patients. Besides, FOS interacted with CREBBP (a hub gene) and SMARCA4 and SMARCA4 interacted with CREBBP in the PPI network. As reported, the upstream regulator SMARCA4 interacted with Nur77, which modulates inflammatory gene expression in the transcriptome of bone marrow-derived macrophages [31]. In samples from mice with OA, Smarca4 and Crebbp expression decreased compared to that in control samples. Therefore, SMARCA4 and CREBBP are candidate genes involved in OA pathology.
Conclusions
TWIST1, POU2F1, SMARCA4, and CREBBP may play an important role in OA pathology. Although the regulating interactions among them were obtained from bioinformatics analysis and require further validation, the results provided a guideline underlying the molecular mechanisms of OA and found a novel therapeutic target.
Acknowledgements
Not applicable.
Funding
None.
Availability of data and materials
The raw data were collected and analyzed by the author and are available upon reasonable request from the corresponding author.
Authors’ contributions
The author read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declares that there are no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Resnik CS, Bohndorf K. Arthritis. Musculoskeletal Diseases 2017-2020. Cham: Springer; 2017. p. 33-7.
- 2.Cheung RT, Ngai SP, Ho KK. Chinese translation and validation of the Oxford Knee Scale for patients with knee osteoarthritis. Hong Kong Physiotherapy Journal. 2017;37:46–49. doi: 10.1016/j.hkpj.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chehab E, Asay J, Favre J, Andriacchi T. Features of gait mechanics and biology that predict cartilage thickness change in a population at risk for age-related knee osteoarthritis. Osteoarthr Cartil. 2017;25:S20–S21. doi: 10.1016/j.joca.2017.02.048. [DOI] [Google Scholar]
- 4.Filardo G, Kon E, Longo UG, Madry H, Marchettini P, Marmotti A, et al. Non-surgical treatments for the management of early osteoarthritis. Knee Surg Sports Traumatol Arthrosc. 2016;24(6):1775–1785. doi: 10.1007/s00167-016-4089-y. [DOI] [PubMed] [Google Scholar]
- 5.Harris KP, Driban JB, Sitler MR, Cattano NM, Balasubramanian E, Hootman JM. Tibiofemoral osteoarthritis after surgical or nonsurgical treatment of anterior cruciate ligament rupture: a systematic review. J Athl Train. 2017;52(6):507–517. doi: 10.4085/1062-6050-49.3.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Polyakova J, Zavodovsky B, Seewordova L, et al. THU0476 Pathogenic Relationship Between Osteoarthritis, Overweight and Inflammation. Ann Rheum Dis. 2015;74(Suppl 2):372.2-372.
- 7.Thomas AC, Hubbard-Turner T, Wikstrom EA, Palmieri-Smith RM. Epidemiology of posttraumatic osteoarthritis. J Athl Train. 2017;52(6):491–496. doi: 10.4085/1062-6050-51.5.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richardson D, Pearson RG, Kurian N, Latif ML, Garle MJ, Barrett DA, et al. Characterisation of the cannabinoid receptor system in synovial tissue and fluid in patients with osteoarthritis and rheumatoid arthritis. Arthritis research & therapy. 2008;10(2):R43. doi: 10.1186/ar2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sagar DR, Staniaszek LE, Okine BN, Woodhams S, Norris LM, Pearson RG, et al. Tonic modulation of spinal hyperexcitability by the endocannabinoid receptor system in a rat model of osteoarthritis pain. Arthritis & Rheumatology. 2010;62(12):3666–3676. doi: 10.1002/art.27698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huggins JP, Smart TS, Langman S, Taylor L, Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153(9):1837–1846. doi: 10.1016/j.pain.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 11.Schlosburg JE, Kinsey SG, Lichtman AH. Targeting fatty acid amide hydrolase (FAAH) to treat pain and inflammation. AAPS J. 2009;11(1):39–44. doi: 10.1208/s12248-008-9075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakajima M, Miyamoto Y, Ikegawa S. Cloning and characterization of the osteoarthritis-associated gene DVWA. J Bone Miner Metab. 2011;29(3):300–308. doi: 10.1007/s00774-010-0230-z. [DOI] [PubMed] [Google Scholar]
- 13.Latchman DS. Transcription factors: an overview. International Journal of Biochemistry & Cell Biology. 1997;29(12):1305. doi: 10.1016/S1357-2725(97)00085-X. [DOI] [PubMed] [Google Scholar]
- 14.Ray A, Ray BK. An inflammation-responsive transcription factor in the pathophysiology of osteoarthritis. Biorheology. 2008;45(3-4):399. [PubMed] [Google Scholar]
- 15.Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- 16.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 17.Qi C, Hong L, Cheng Z, Yin Q. Identification of metastasis-associated genes in colorectal cancer using metaDE and survival analysis. Oncol Lett. 2016;11(1):568–574. doi: 10.3892/ol.2015.3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu G, Dawson E, Duong A, et al. ReactomeFIViz: a Cytoscape app for pathway and network-based data analysis. F1000Research. 2014;3:146. [DOI] [PMC free article] [PubMed]
- 19.Hayami T, Pickarski M, Zhuo Y, Wesolowski GA, Rodan GA, Duong LT. Characterization of articular cartilage and subchondral bone changes in the rat anterior cruciate ligament transection and meniscectomized models of osteoarthritis. Bone. 2006;38(2):234–243. doi: 10.1016/j.bone.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 20.Lee MS, Lowe GN, Strong DD, Wergedal JE, Glackin CA. TWIST, a basic helix-loop-helix transcription factor, can regulate the human osteogenic lineage. J Cell Biochem. 1999;75(4):566. doi: 10.1002/(SICI)1097-4644(19991215)75:4<566::AID-JCB3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 21.Hopwood B, Tsykin A, Findlay DM, Fazzalari NL. Microarray gene expression profiling of osteoarthritic bone suggests altered bone remodelling, WNT and transforming growth factor-β/bone morphogenic protein signalling. Osteoarthritis & Cartilage. 2007;9(5):R100. doi: 10.1186/ar2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumarasinghe D, Perilli E, Tsangari H, Truong L, Kuliwaba J, Hopwood B, et al. Critical molecular regulators, histomorphometric indices and their correlations in the trabecular bone in primary hip osteoarthritis. Osteoarthr Cartil. 2010;18(10):1337–1344. doi: 10.1016/j.joca.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 23.Hopwood B, Tsykin A, Findlay DM, Fazzalari NL. Microarray gene expression profiling of osteoarthritic bone suggests altered bone remodelling, WNT and transforming growth factor-β/bone morphogenic protein signalling. Arthritis research & therapy. 2007;9(5):R100. doi: 10.1186/ar2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Investig. 2006;116(5):1202. doi: 10.1172/JCI28551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie C-H, Cao Y-M, Huang Y, Shi Q-W, Guo J-H, Fan Z-W, et al. Long non-coding RNA TUG1 contributes to tumorigenesis of human osteosarcoma by sponging miR-9-5p and regulating POU2F1 expression. Tumor Biol. 2016;37(11):15031–15041. doi: 10.1007/s13277-016-5391-5. [DOI] [PubMed] [Google Scholar]
- 26.Wang P, Dai M, Xuan W, Mceachin RC, Jackson AU, Scott LJ, et al. SNP function portal: a web database for exploring the function implication of SNP alleles. Bioinformatics. 2006;22(14):523–529. doi: 10.1093/bioinformatics/btl241. [DOI] [PubMed] [Google Scholar]
- 27.Ting WC, Chen LM, Pao JB, Yang YP, You BJ, Chang TY, et al. Common genetic variants in Wnt signaling pathway genes as potential prognostic biomarkers for colorectal cancer. PLoS One. 2013;8(2):e56196. doi: 10.1371/journal.pone.0056196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kinne RW, Boehm S, Iftner T, Aigner T, Vornehm S, Weseloh G, et al. Synovial fibroblast-like cells strongly express jun-B and C-fos proto-oncogenes in rheumatoid- and osteoarthritis. Scand J Rheumatol Suppl. 1995;101(s101):121. doi: 10.3109/03009749509100913. [DOI] [PubMed] [Google Scholar]
- 29.Tsuji M, Hirakawa K, Kato A, Fujii K. The possible role of c-fos expression in rheumatoid cartilage destruction. J Rheumatol. 2000;27(7):1606–1621. [PubMed] [Google Scholar]
- 30.Haseeb A, Leigh D, Haqqi T. A small molecule harpagoside inhibits IL-1beta-induced expression of IL-6 by blocking the expression of C-FOS in primary human osteoarthritis chondrocytes. Osteoarthr Cartil. 2015;23:A155–A156. doi: 10.1016/j.joca.2015.02.910. [DOI] [Google Scholar]
- 31.Hamers AA, Argmann C, Moerland PD, Koenis DS, Marinković G, Sokolović M, et al. Nur77-deficiency in bone marrow-derived macrophages modulates inflammatory responses, extracellular matrix homeostasis, phagocytosis and tolerance. BMC Genomics. 2016;17(1):162. doi: 10.1186/s12864-016-2469-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data were collected and analyzed by the author and are available upon reasonable request from the corresponding author.