

Abstract
Background
Angiogenesis is a process of formation of new blood vessels and is an important criteria demonstrated by cancer cells. Over a period of time, these cancer cells infect the other parts of the healthy body by a process called progression. The objective of the present article is to identify a drug molecule that inhibits angiogenesis and progression.
Methods
In this pursuit, ligand based pharmacophore virtual screening was employed, generating a pharmacophore model, Hypo1 consisting of four features. Furthermore, this Hypo1 was validated recruiting, Fischer’s randomization, test set method and decoy set method. Later, Hypo1 was allowed to screen databases such as Maybridge, Chembridge, Asinex and NCI and were further filtered by ADMET filters and Lipinski’s Rule of Five. A total of 699 molecules that passed the above criteria, were challenged against 4AG8, an angiogenic drug target employing GOLD v5.2.2.
Results
The results rendered by molecular docking, DFT and the MD simulations showed only one molecule (Hit) obeyed the back-to-front approach. This molecule displayed a dock score of 89.77, involving the amino acids, Glu885 and Cys919, Asp1046, respectively and additionally formed several important hydrophobic interactions. Furthermore, the identified lead molecule showed interactions with key residues when challenged with CDK2 protein, 1URW.
Conclusion
The lead candidate showed several interactions with the crucial residues of both the targets. Furthermore, we speculate that the residues Cys919 and Leu83 are important in the development of dual inhibitor. Therefore, the identified lead molecule can act as a potential inhibitor for angiogenesis and progression.
Electronic supplementary material
The online version of this article (10.1186/s12885-018-4050-1) contains supplementary material, which is available to authorized users.
Keywords: Angiogenesis, Progression, Back-to-front approach, VEGFR-2, MD simulations
Background
Initiation of tumour cell and its progression is a process which is performed by certain factors known as angiogenic factors [1]. Angiogenesis is a complex process during which the endothelial cells are involved in the generation of metallo-proteases, initiate cell migration, cell division, and further proliferation. Additionally, they are also responsible for the formation of the new cells [2]. More specifically, cancer has an ability of rapid cell growth and hence, it is evident that angiogenesis supports the cancer metastasis [1].It is therefore essential to identify the novel drug molecules, which could hinder angiogenesis.
Vascular Endothelial Growth Factors (VEGFs) demonstrate an essential role in angiogenesis and vasculogenesis [3] and therefore, portray to be an ideal drug targets for designing novel inhibitors. Typically, VEGFRs are the transmembrane proteins that are known to trigger angiogenesis through VEGF receptor signalling [4]. There are three different types of receptor tyrosine kinases (RTK) which display a high affinity towards VEGF, namely, VEGFR-1, VEGFR-2 and VEGFR-3, respectively. However, VEGFR-2 remains as the only protein kinase domain transmitting the angiogenic signals [5], while the VEGFR-1 revealed a reduced activity than VEGFR-2 [6, 7] and VEGFR-3 exhibits its role in governing the embryonic angiogenesis [8]. Therefore, VEGFR-2 emerges as an ideal protein target to identify new drugs. Moreover, VEGFR-2 has a crucial role in rheumatoid arthritis [9] inflammation [10] porosis [11], metastasis [12] and ocular neovascularization [13]. Accordingly, identification of novel drugs against VEGFR-2 might also have a curative effect on the aforementioned diseases.
Depending upon the binding patterns, the tyrosine kinase inhibitors can be grouped into type I and type II [14]. Type I inhibitors interact with the Adenosine Triphosphate (ATP) binding site of the kinase in its active form [15] and thereby, displaying reduced selectivity, while in type II, the inhibitors bind to the ATP site along with the allosteric, hydrophobic site [16] and exhibits high selectivity. This interaction happens during the inactive state of the kinases [17]. The conserved triad Asp-Phe-Gly (DGF) governs the active and the inactive states of the kinase enzymes. Generally, DGF-in conformation was noticed in the active condition, whereas in the DFG-out is projected during the inactive state [17]. The simple architecture of VEGFR-2 active site comprises of the front and the back pocket. ATP- binding front pocket has two key residues associated with it, Glu917 and Cys919. The back hydrophobic pocket has Glu885 and Asp1046. Glu885 is seated on the αC helix and the Asp1046 forms an important part of the triad [18, 19].
One of the highly significant characters of the cancer cells is being able to divide rapidly [20]. Cyclic-dependent kinases (CDKs), are crucial enzymes and contribute at large towards this process and belong to the serine-threonine kinases subfamily [21]. These kinases have gained popularity for their role in cell division, differentiation, neuronal functions, transcription and apoptosis [22]. Of all the known CDKs, CDK1-CDK13, CDK2 is widely studied protein as it has a crucial role to be played during the cell cycle progression more specifically during the G1 to S phase transition [22, 23]. However, this requires being associated with regulatory subunits such as cyclin A and E [23]. Cyclin A is complexed with CDK2 for the S-phase (Synthesis phase) progression while cyclin E is required during the retinoblastoma protein phosphorylation that provides the transition of G1/S (Gap1) [23, 24]. Additionally, reports exists on the inhibition of CDK2 that can eventually lead to the killing of the cancerous cells by deregulation of E2F-1 activity [25]. Therefore, it will be beneficial to identify a small molecule that can act both on the angiogenesis and on progression. Such a type of inhibition was earlier reported by Antony et al. [26].
The objective of the present study is to identify a novel inhibitor that has a potential to interact both with the VEGFR protein and with the CDK2 protein. In the present investigation, an attempt was made to screen inhibitors that bind more precisely in the back-to-front fashion of the target protein. This approach has been proven extremely successful with respect to certain kinases [27–29].
Although, both the proteins share a very little homology, they exhibit structural similarity within the ATP- binding region. Accordingly, the current study tries to exploit these important features in identifying the novel dual drug/inhibitor. The investigation proceeds with the initial identification of VEGFR-2 inhibitors and then challenging them with the CDK2 protein.
Methods
Preparation of the dataset
A systematic literature survey was conducted to extract the dataset for VEGFR-2 inhibitors [30]. From the obtained information of 63 diverse compounds [31–39], 24 structurally diverse compounds were referred to as training set and the remaining 39 were grouped into test set compounds. The training set compounds were employed to build the pharmacophore model while the test set was used to validate the same. The important criteria in choosing the training set compounds is the inclusion of the most active compounds into it such that they impart the most reliable information pertaining to the generated pharmacophore model. Additionally, the training set compounds exhibited a wide range of, half maximal inhibitory concentration, IC50, spanning between 0.2 to 45,000 nmol/L. Further, the compounds in the training set, (Fig. 1), were designated as most active (IC50 < 250 nmol/L, +++), moderately active (250 nmol/L ≤ IC50 < 5000 nmol/L, ++) and inactive (IC50 ≥ 5000 nmol/L, +), that was adapted based on the IC50values. The same criteria were followed for the test set compounds. Their corresponding two dimensional (2D) structures were sketched using the ChemSketch (http://www.acdlabs.com/ resources/ freeware/ chemsketch/ Advanced Chemistry Development (ACD) Inc., Toronto, Canada) and were translated to their three dimensional (3D) structures adapting the Discovery Studio v4.5 (DS).
Fig. 1.
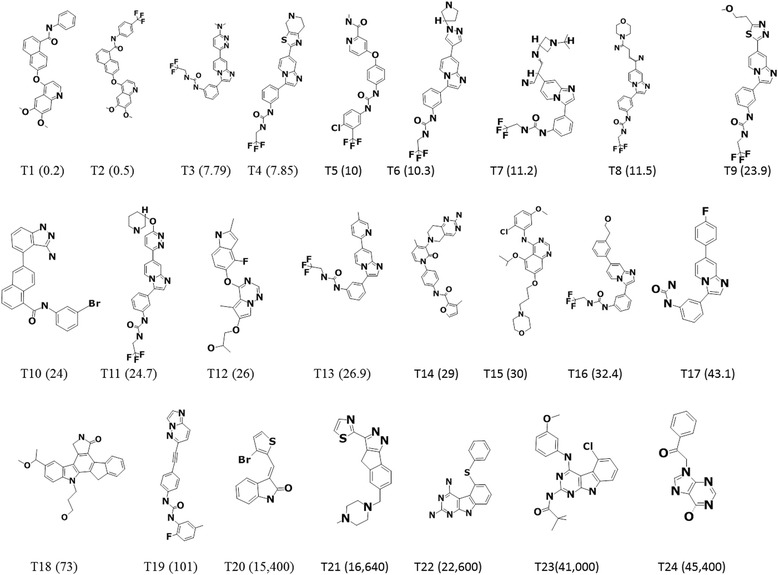
2D structures of 24 training set compounds and their IC50 values represented in parenthesis
Generation of the pharmacophore model
In the recent times, pharmacophore modelling takes advantage of being one of the most reliable methods in identifying novel leads for different targets. For the present investigation, the three dimensional quantitative structure-activity relationship (3D–QSAR) based pharmacophore model was generated using the Catalyst HypoGen algorithm provided with the DS v4.5. This exploits the chemical features of the training set compounds and the conformation with the least energy were developed employing the BEST algorithm. In order to generate the best pharmacophore model, the energy and the uncertainty value were fixed at 20 kcal/mol and 3, respectively [40]. Further, Feature Mapping protocol was employed for investigating into the chemical features and to recognize the common features present in the training set that could be essential in the pharmacophore generation. This protocol has an ability to construct pharmacophore features available with the training set compounds and further these identified features play a critical role in the generation of the model. Amongst the generated models, the best hypothesis was chosen based upon the Debnath’s method [41].
Validation of the generated pharmacophore model
With an aim to determine the predictive ability and its capability to identify the active compounds from that of the inactives, the selected pharmacophore was subjected to validation recruiting three different approaches such as, Fischer’s randomization, test set method, and the decoy set method. Fischer’s randomization was carried out alongside the pharmacophore generation, which prompts random spreadsheets based upon the selected level of confidence. For the present investigation, the confidence level was chosen to be 95%. The test and the decoy method of validations were conducted in order to understand if the generated pharmacophore was able to select the compounds in a similar manner as for the experimental activities. Ligand Pharmacophore Mapping protocol available on the DS was employed with Best Flexible algorithm. Test set was assembled with 39 structurally different compounds. The decoy set was instituted with a database of 710 compounds consisting of 15 active compounds. Following this, the enrichment factor (EF) and the goodness of fit score (GF) were computed using the formulae,
Here, Ha represents the total number of active compounds, Ht refers to total hits redeemed from the database, A refers to the total number of active compounds in the database, and D denotes the total number of molecules in the database.
Database screening for extracting the candidate compounds
Validated pharmacophore was employed as the 3D query to screen the databases such as Maybridge, Asinex, Chembridge, and NCI to retrieve novel scaffolds against angiogenesis, if the chemical compounds mapped with all the chemical features present in the pharmacophore. In this pursuit, the Ligand Pharmacophore Mapping protocol was used with Best–Flexible options.
Drug-likeness assessment
Drug-likeness assessment was performed to the retrieved compounds from the databases so as to assess their biological activities. Accordingly, to judge the compound for strong pharmacokinetic properties, ADMET [42] and Lipinski’s rule were applied. ADMET specifically evaluates if the compound can cross the Blood Brain Barrier (BBB), allowable solubility, good intestinal absorption with less toxicity. Therefore, the values 3, 3 and 0 were fixed for BBB, solubility and absorption, correspondingly and were computed adapting ADMET Descriptors module on the DS. Additionally, the Lipinski’s Rule of 5 [43] was applied to the above filtered compounds to quantify if the prospective drug molecules could be well absorbed. This rule recommends a compound should have less than 10 hydrogen bond acceptors, less than 5 hydrogen bond donor groups having a molecular weight of less than 500 Da with log p value of less than 5 with 10 rotatable bonds. All the compounds that satisfied the aforementioned criteria were forwarded for the docking studies.
Molecular docking studies
Challenging the potential screened lead molecules with the reliable drug target and to assess the degree of their binding affinities rendered in terms of the dock scores happens to be one of the most significant methodologies in drug discovery. Typically, this approach was deduced to assess the nature of the lead molecules in the active site and thereby its conformation. For the current study, Genetic Optimization for Ligand Docking v5.2.2 (GOLD) has been recruited [44, 45]. Target protein with the protein data bank (PDB) code, 4AG8, with high resolution of 1.95 Å, co-crystalled with axitinib was selected. Missing residues of the protein were rectified and all the hydrogen atoms were added, removing the water molecules [46]. Histidine tautomer orientations were placed in agreement with the crystal structure. The binding site of the protein was calculated for all the atoms that lie within the bound ligand around 15 Å. Furthermore, GOLD score was specified to understand the binding affinities between the ligand and the drug target, while the Chemscore was adapted for enumerating the rescoring function. Moreover, the GOLD score was initiated to generate 50 docking poses for each ligand and the reliable pose was selected based upon the highest dock score, molecular interactions and the hydrogen bonds that resulted between the ligand and the amino acid residues present at the active site of the protein molecule. Hereinafter, the most active compound in the training set was labeled as the reference compound.
The lead molecules identified after challenging against the 4AG8 would be further challenged with the CDK2 protein, 1URW, a potential target for cancer progression.
Density functional theory
DFT is one of the most dynamic methods adapted to calculate the electronic structure of matter and thus provides the most valuable information with respect to the selected inhibitors. DFT for the resultant docking molecules was performed using Becke, 3-parameter, Lee-Yang Parr (B3LYP) [47], available on the DS in order to evaluate their orbital energies such as highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO). HOMO refers to the electron donor and LUMO denotes the electron acceptor. DFT was executed together with the Hit compounds and the compounds from the training set.
Molecular dynamics simulations
To gain further insight into the protein-ligand interactions, the procured Hit compounds from the docking studies and the DFT were subjected to the Molecular Dynamics (MD) simulations along with the reference compound. The ligand topologies were generated utilizing the SwissParam [48–50], while the topologies of the protein were generated employing Chemistry at HARvard macromolecular Mechanics force-field (CHARMm ff) [51–54] implemented in Groningen Machine for Chemical Simulations (GROMACS) 5.0.6 [55]. Dodecahedron box was obtained and was solvated with three-site transferable intermolecular potential (TIP3P) water model followed by neutralizing the system with the counter ions. All the bad contacts were further removed by subjecting the system to pass through steepest descent algorithm at 10,000 steps with an upper limit of the force being lower than 1000 kJ/mol [56]. Following this, the equilibration was conducted by Number of particles, Volume and Temperature (NVT) [57] and Number of particles, Pressure and Temperature (NPT) [58] at 100 ps at 300 k and 100 ps at a pressure of 1 bar maintained by Parrinello-Rahman barostat and allowing the movement of the counter ions and the water molecules, constraining the protein backbone. Linear Constraint Solver for Molecular Simulations (LINCS) [59] algorithm was used to restrain heavy atom bonds and their respective hydrogen atoms. Particle Mesh Ewald (PME) [60] was utilized to compute the long rage electrostatic interaction and a cut-off distance of 12 Å was attributed for Coulombic and van der Waals interactions. MD simulations were performed for 30 ns storing the coordinate data for every 2 fs. Corresponding results were evaluated employing the Visula Molecular Dynamics (VMD) [61] and DS, respectively.
Results
HypoGen based pharmacophore model generation
Using the HypoGen algorithm provided with the DS, 24 training set compounds were employed to develop the pharmacophore model, (Figs. 1 and 2), which resulted in the generation of 10 hypotheses, Table 1, upon the utilization of 3D QSAR Pharmacophore Generation protocol available with the DS. The preferred features for the pharmacophore generation were hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), hydrophobic (HyP), hydrophobic aliphatic (Hy-Ali) and ring aromatic (RA).
Fig. 2.
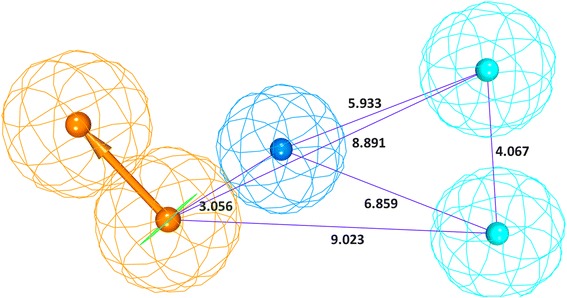
Hypo1 pharmacophore model with its corresponding features and geometry, Aromatic Rings (brown), Hydrophobic Aliphatic (blue), 2 Hydrophobic (cyan)
Table 1.
Statistical data of the generated hypothesis employing HypoGen
| Hypo no | Total costa | Cost difference | RMSDb | Correlation | Featuresc | Max fit |
|---|---|---|---|---|---|---|
| Hypo1 | 111.95 | 71.22 | 0.7 | 0.97 | HyAli, 2HyB,RA | 11.4 |
| Hypo 2 | 113.31 | 69.86 | 0.7 | 0.96 | HyAli, 2HyB,RA | 11.5 |
| Hypo 3 | 116.45 | 66.71 | 0.8 | 0.95 | HyAli,HyB,RA,HBA | 11.9 |
| Hypo 4 | 116.47 | 66.69 | 1.0 | 0.94 | HBA, HBD 2HyB | 10.7 |
| Hypo 5 | 117.11 | 66.05 | 0.9 | 0.94 | HyAli,HyB,RA,HBA | 11.5 |
| Hypo 6 | 119.51 | 63.65 | 1.0 | 0.93 | HBA,HBD,2HyB | 11.26 |
| Hypo 7 | 119.52 | 63.65 | 0.9 | 0.95 | HBA,2HyB,RA | 12.65 |
| Hypo 8 | 119.82 | 63.35 | 0.9 | 0.94 | HBA,Hy-Ali,HD,RA | 12.33 |
| Hypo 9 | 119.94 | 63.23 | 1.2 | 0.91 | HBA,Hy-Ali,2HyB, RA | 11.98 |
| Hypo10 | 120.52 | 62.65 | 1.1 | 0.91 | HBA,HBD,HY-Ali | 7.8 |
aCost difference between the null and the total cost. The null cost, the fixed cost and the configuration cost were found to be 183.177, 101.77 and 19.91, respectively
bRMSD-Root Mean Square Deviation
cHyP- Hydrophobic, RA- Ring Aromatic, Hy-Ali-Hydrophobic Aliphatic, HBA-Hydrogen Bond Acceptor, HBD-Hydrogen Bond Donor
From the generated models, Hypo1 was chosen as the best pharmacophore model as it satisfied the Debnath’s rules, which states that a good pharmacophore model should consists of high cost difference, good correlation, least RMSD and low cost values. The generated pharmacophore model composed of aromatic feature (RA), one hydrophobic aliphatic feature (Hy-Ali) and two hydrophobic (HyP) features.
Further, to evaluate the predictive ability of the Hypo1, it was allowed to examine the inhibitory activities of 24 training set compounds. The training set compounds were grouped into most active, moderately active and the least active compounds based upon their IC50 values as, (IC50 < 250 nmol/L, +++), (250 nmol/L ≤ IC50 < 5000 nmol/L, ++) (IC50 ≥ 5000 nmol/L, +), correspondingly. Hypo1 calculated the inhibitory activity values of the training set in accordance with the experimental values, Table 2. Furthermore, Hypo1 has successfully mapped to the most active compound and the most inactive compound, (Figs. 3 and 4).
Table 2.
Assessing the training set compound values for estimated and the experimental activities by Hypo1
| Name | Fit value | Experimental IC50 (nmol/L) | Predicted IC 50 (nmol/L) | Errora | Experimental scale | Predicted scale |
|---|---|---|---|---|---|---|
| Molecule1 | 10.47 | 0.2 | 0.191 | −1.04 | +++ | +++ |
| Molecule2 | 8.58 | 5.2 | 14.9 | 2.86 | +++ | +++ |
| Molecule3 | 8.40351 | 7.79 | 22.7 | 2.91 | +++ | +++ |
| Molecule4 | 8.4847 | 7.85 | 18.83 | 2.39 | +++ | +++ |
| Molecule5 | 7.6927 | 10 | 116.68 | 11.66 | +++ | +++ |
| Molecule6 | 8.4082 | 10.3 | 22.46 | 2.18 | +++ | +++ |
| Molecule7 | 8.95463 | 11.2 | 6.38 | −1.75 | +++ | +++ |
| Molecule8 | 8.44562 | 11.5 | 20.61 | 1.79 | +++ | +++ |
| Molecule9 | 8.42254 | 23.9 | 21.73 | −1.09 | +++ | +++ |
| Molecule10 | 8.004 | 24 | 56.97 | 2.37 | +++ | +++ |
| Molecule11 | 8.36497 | 24.7 | 24.816 | 1 | +++ | +++ |
| Molecule12 | 8.50932 | 26 | 17.79 | −1.46 | +++ | +++ |
| Molecule13 | 8.20893 | 26.9 | 35.54 | 1.32 | +++ | +++ |
| Molecule14 | 7.99567 | 29 | 58.08 | 2 | +++ | +++ |
| Molecule15 | 8.48967 | 29.51 | 18.62 | −1.58 | +++ | +++ |
| Molecule16 | 8.36485 | 32.4 | 24.82 | −1.3 | +++ | +++ |
| Molecule17 | 8.19005 | 43.1 | 37.12 | −1.16 | +++ | +++ |
| Molecule18 | 7.79069 | 73 | 93.11 | 1.27 | +++ | +++ |
| Molecule19 | 8.47375 | 101 | 19.31 | −5.22 | +++ | +++ |
| Molecule20 | 5.74562 | 15,400 | 10,329.50 | −1.49 | + | + |
| Molecule21 | 5.23885 | 16,600 | 33,178.10 | 1.99 | + | + |
| Molecule22 | 5.71617 | 22,600 | 11,054.20 | −2.04 | + | + |
| Molecule23 | 5.7456 | 41,200 | 10,330.10 | −3.98 | + | + |
| Molecule24 | 5.74648 | 45,000 | 10,309.20 | −4.36 | + | + |
aError, ratio of the predicted activity (Pred IC50) to the experimental activity (Exp IC50) or its negative inverse if the ratio is < 1
Fig. 3.
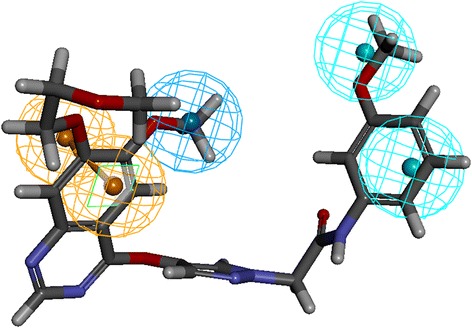
Most active compound (IC50 = 0.2) mapped to all the features
Fig. 4.
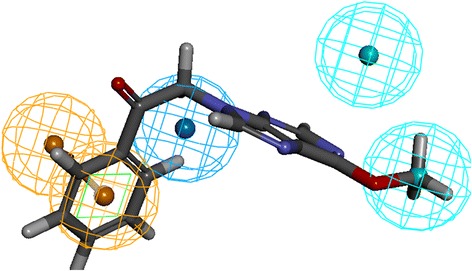
Most inactive compound (IC50 = 45,000) aligned to only three features
Validation of the pharmacophore model, Hypo1
In order to assess the quality of the generated pharmacophore, it was subjected to a series of validations such as, Fischer’s randomization method, test set method and the decoy test method.
Fischer’s randomization method
To ascertain the statistical robustness of Hypo1, Fischer’s randomization was performed that was run alongside the pharmacophore generation. A confidence level of 95% was selected which resulted in the formation of 19 spreadsheets. Thereafter, hypothesis significance was calculated employing the formula, [1-(1 + X)/Y] X100. Herein, X denotes sum of hypothesis, while Y indicates the total HypoGen runs, both the initial and the random runs. This method takes the advantage of assessing the correlation between the chemical structures and their corresponding biological activities, thereby quashing the chance correlation, and thus establishing that Hypo1 was not generated by chance. These results evidently state that the generated pharmacophore was of superior quality and thus, had least cost value as depicted in, Fig. 5.
Fig. 5.
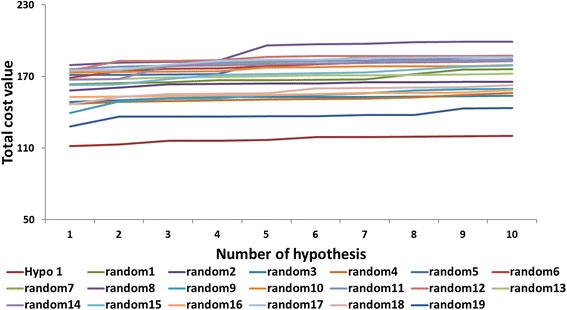
Validation by Fischer’s randomization method. Comparison of total cost of Hypo1 with the randomly generated 19 scrambles when 95% confidence level was selected
Test set validation
Test set method of validation was conducted to examine the ability of the Hypo1 in recognizing the compounds other than the training set and thus to classify them in the same order of the activity range. 39 structurally diverse compounds were collected and were grouped in accordance with the training set. Subsequently, the correlation coefficient (r) for the test set was computed as 0.95, while that of the training set compounds was 0.96, (Fig. 6). Additionally, Hypo1 ably estimated the activities of the external compounds, however, overestimated three moderately active compounds as active compounds, (Additional file 1). The test set validation results are a clear indicative of the fact that Hypo1 can be recruited to classify the external compounds as well.
Fig. 6.
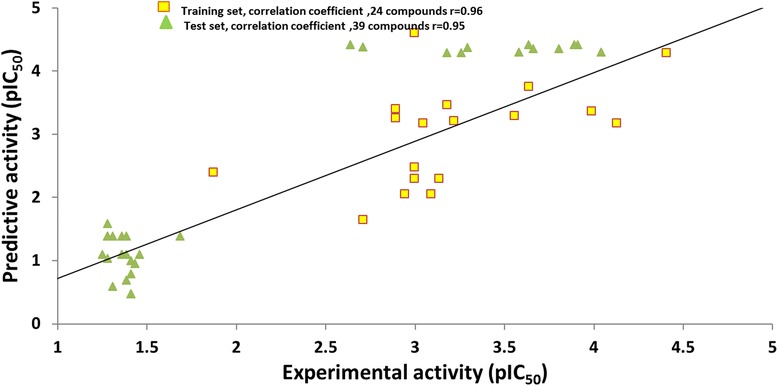
Correlation prediction of Hypo1 between the test set and the training set compounds
Decoy set validation
To further quantify the usefulness of the Hypo1, it was subjected to yet another validation process, called the decoy set method. This method was executed recruiting an external database. Accordingly, a database (D) of 710 molecules was instituted with an addition of 15 actives (A). Database screening was performed and as a result 17 Hit compounds were retrieved (Ht) with 14 actives (Ha) in it. Further, the EF and the GF were computed to be 44.17 and 0.79 respectively, authenticating the goodness of the Hypo1, Table 3. Moreover, the percentage of ratio of the actives was found to be 93, pronouncing the high ability of Hypo1 in screening.
Table 3.
Validation of Hypo1 by employing decoy set method
| S. no | Parameters | Values |
|---|---|---|
| 1 | Total number of molecules in database (D) | 710 |
| 2 | Total number of actives in database (A) | 15 |
| 3 | Total number of hit molecules from the database (Ht) | 17 |
| 4 | Total number of active molecules in hit list (Ha) | 14 |
| 5 | % Yield of active [(Ha/Ht) | 82 |
| 6 | % Ratio of actives [(Ha/A) X 100] | 93 |
| 7 | Enrichment Factor (EF) | 44.17 |
| 8 | False negatives (A-Ha) | 1 |
| 9 | False Positives (Ht–Ha) | 3 |
| 10 | Goodness of fit score (GH) | 0.79 |
*GH score between 0.6–0.8 is regarded as a good score
Identifying the novel lead molecules through virtual screening
Validated Hypo1 was employed to screen and retrieve the novel compounds against the VEGFR-2. The chemical features imbibed within the Hypo1 display an important role in identifying the new leads. Four databases, such as Chembridge, Maybridge, Asinex and NCI were selected for screening the dual inhibitors. Accordingly, Hypo1 has successfully mapped with 12,080, 14,521, 29,836 and 29,660 compounds from the aforementioned databases. Following which, compounds with a greater fit value than 8 were proceeded to the Lipinski’s Rule of Five and the ADMET studies. These two studies quantify the pharmacokinetics of a drug molecule and thus, is an essentiality for a drug molecule to qualify them. ADMET particularly computes BBB penetration, solubility, human intestinal absorption (HIA) solubility, plasma protein binding (PPB) and CytochromesP450 (CYPs) inhibition. The results were evaluated by setting the standard as 3, 3, and 0 for solubility, BBB and absorption, correspondingly. From the four databases, a total of 699 molecules were identified possessing the drug-like properties. The screened compounds along with the training set compounds were subjected to molecular docking analysis. The overall process of screening is represented pictorial form (Fig. 7).
Fig. 7.
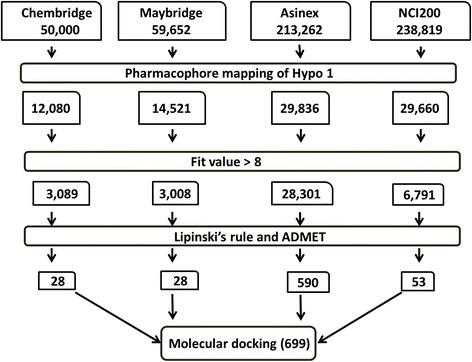
Schematic representation of methodology involved in virtual screening
Molecular docking studies
Genetic Optimization for Ligand Docking, (GOLD) v5.2.2 was recruited for performing the molecular docking assay. GOLD has an ability to conduct lead optimization and to determine the accurate binding pose analysis. Technically, GOLD operates by two scoring functions, Goldscore fitness, and the Chemscore. Goldscore is a fitness parameter that is governed by four components, such as; protein-ligand hydrogen bond energy (external H-bond), protein-ligand van der Waals (vdw) energy (external vdw), ligand internal vdw energy (internal vdw), ligand torsional strain energy (internal torsion), respectively. This score was optimized for understanding and predicting the position of the bound ligand. On the other hand, Chemscore was used for rescoring and further computes the total free energy change on the ligand binding. The aptness of the docking parameters were assessed by subjecting the co-crystal to dock into the selected active site. This generated a reasonable RMSD of 0.9 Å between the docked pose and the co-crystal thus ensuring the accuracy of the GOLD parameters. Therefore, these parameters were considered for docking the screened compounds into the active site of the target, (Additional file 2).
Docking of the screened ligands and the training set compounds was initiated after specifying the radius of the active site around the co-crystal to 15 Angstrom (Å) followed by allowing the generation of 50 conformations for each ligand. All those compounds which fall in the selected radius were considered for the succeeding steps. The docking studies were initially executed with 4AG8, a potential drug target for angiogenesis. The final selected lead compounds after the DFT and the MD simulations were challenged with the CDK2 protein, 1URW. This strategy was conceived to identify a common drug molecule for both angiogenesis and progression.
The angiogenesis target for the present study was 4AG8, retrieved for the Protein Data Bank (PDB, http://www.rcsb.org/pdb/home/home.do). The reference molecule has displayed a dock score of 89.77 and therefore, only those lead molecules whose scores were above the reference molecules were chosen for further investigation and thus, qualifying 18 screened molecules. These compounds were examined manually for the hydrogen bond interactions with the amino acids residues, Cys919, Glu917, Asp1046, and Glu885, respectively. Out of 18 screened lead molecules, only 11 exhibited the hydrogen bond interactions with the key residues. These 11 compounds, (Fig. 8), obeyed all the filters employed to identify an efficient lead candidate. These 11 molecules were subjected to the Density Functional Theory (DFT) analysis for further understanding their molecular orbital energies.
Fig. 8.
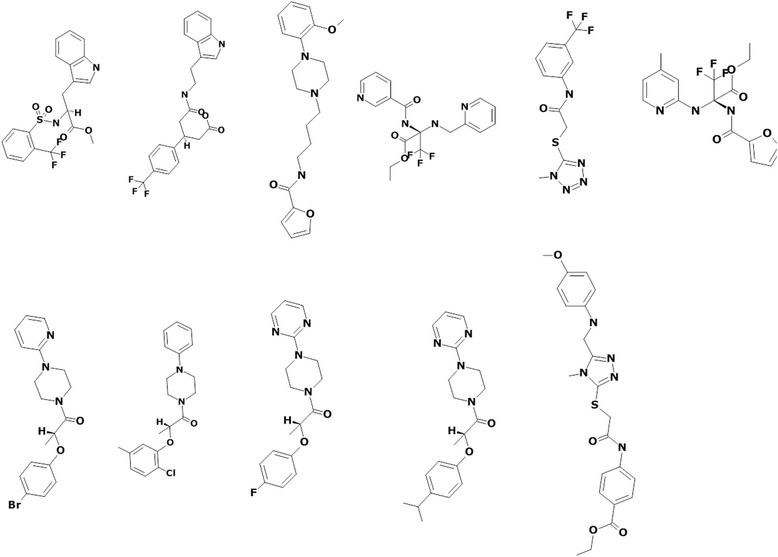
2D structures of final 11 compounds that comply to all the filters
Density functional theory studies
Highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) that ascribe the DFT were computed with six training set compounds (2 active, 2 moderately active and 2 inactive compounds) and 11 screened compounds. Small gap obtained, reports a compound to be highly reactive, while the larger gap signifies the presence of low reactivity of the compound to that of the target protein [62]. Accordingly, compound 4 has been selected as it demonstrated the least gap, Table 4, obeys the back to front approach and maps to all the pharmacophore features, (Fig. 9). Hereafter, is compound was named Hit.
Table 4.
Calculation of the orbital energy values of Hit compounds and training set compounds utilizing DFT. Only top four candidates are tabulated
| Name | HOMO (eV) | LUMO (eV) | ∆E (eV) | IC50 |
|---|---|---|---|---|
| Compound 1 | −0.17 | −0.02 | 0.14 | |
| Compound 2 | −0.18 | −0.06 | 0.11 | |
| Compound 3 | −0.19 | −0.07 | 0.12 | |
| Compound 4(Hit) | −0.15 | −0.08 | 0.07 | |
| T1 | −0.18 | − 0.10 | 0.08 | 0.2 |
| T2 | −0.20 | −0.11 | 0.09 | 5.2 |
| T12 | −0.18 | − 0.07 | 0.10 | 26 |
| T14 | −0.18 | −0.07 | 0.10 | 29 |
| T23 | −0.18 | −0.09 | 0.08 | 41,000 |
| T24 | −0.20 | −0.11 | 0.09 | 45,000 |
Fig. 9.
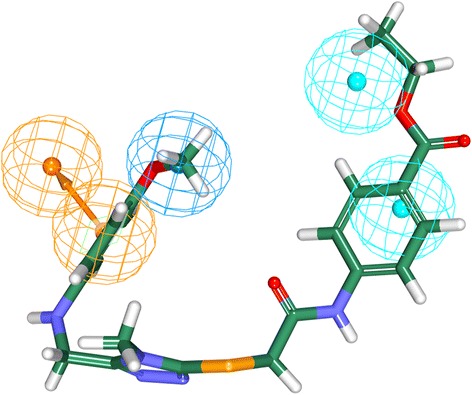
Mapping of the Hit compound to all the features of the pharmacophore. The compound is found to be well aligned with all the pharmacophore features
Molecular dynamics simulations
With an objective of assessing the binding stability of the final systems, the best conformations obtained from the docking were proceeded to MD simulations. Furthermore, MD simulation evaluates and delineates on their dynamic behaviour and with each other. The MD results were examined for the RMSD values, potential energy and the radius of gyration to assess the stability of the protein backbone. The RMSD values were found to be 1.0 nm, and 0.8 nm, respectively for the reference molecule and the Hit. Additionally, it can be observed that the RMSD profiles of the candidate compound was more stable than the reference, (Fig. 10). It was also noticed that the stability was attained after 20 ps for reference compound and while Hit 1 was stable after 25 ps, (Fig. 10). Furthermore, their stability was assessed by plotting the potential energy, (Fig. 11) and the radius of gyration, (Fig. 12). Both profiles have rendered results that showed no abnormal behaviour throughout the simulation. Furthermore, the protein-Hit complex was found to be more compact as compared with the protein-reference complex, (Fig. 12). Additionally, from the Fig. 12 it can be understood that there were a few aberrations exhibited by the protein- reference complex and was stable after 25 ns. On the contrary, the protein-Hit complex was found to be stable after 16 ns, showing no aberrations thereafter.
Fig. 10.
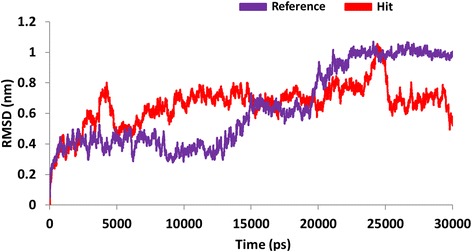
Quantifying the overall stability of the backbone during 30 ps. Purlpe denotes the protein-reference complex and red denotes the protein-Hit complex
Fig. 11.
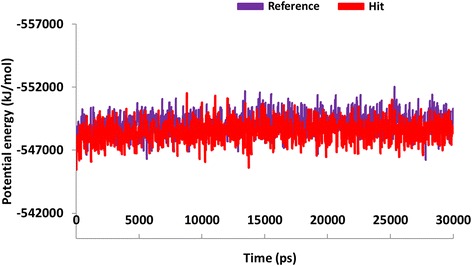
Potential energy of protein-reference (purple) complex and protein-Hit complex (red). The plots show that both the complexes were well converged between − 547,000 kJ/mol and −551,000 kJ/mol
Fig. 12.
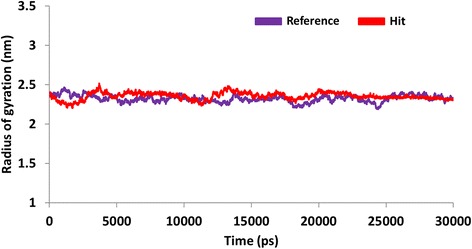
Radius of gyration profiles protein-reference (purple) complex and protein-Hit complex (red). The protein-Hit complex was observed to be stably compact than the reference
The binding mode analysis was performed retrieving the representative structures from the last 3 ns trajectories. Upon superimposition, it was observed that the binding pattern of the Hit was similar with the reference compound, (Fig. 13). Further inspecting the molecular interactions of the reference, it was revealed that the reference has formed three hydrogen bonds demonstrated by two key residues, Cys919 and Asp1046, respectively and displaying an acceptable bond length, Table 5. The HN atom of the Cys919 interacted with N7 atom of the ligand and NH and O atoms of Asp1046 have joined with O26 and H62 of the ligand, (Fig. 14a). Leu840 has participated in the π-sigma interaction, while the residues, Ala866, Ala866, Val898, Leu889, and Leu1035 were found to interact through the π-alkyl bond, (Additional file 3). The Hit has interacted with the protein target with three hydrogen bonds represented one each by Glu885, Cys919 and Asp1046, respectively, (Fig. 14b). HN atom of Cys919 has involved with the ligand’s O14 atom with a hydrogen bond length of 2.16 Å and the OE2 atom of Glu885 joined to the OE2 atom of the ligand displaying a bond length of 2.09 Å, respectively. Additionally, Leu840, Val848, Lys868, Leu1035 have interacted involving with the π-π bonds. Leu889 and Phe918 have interacted with the π-alkyl bond and thus make the ligand to be seated firmly within the active site groove, (Additional file 4). Moreover, from the large set of screened databases only one Hit was identified has the only retrieved compound that obeyed back-to-front pattern of binding and thus projecting itself as a unique lead compound, (Fig. 15). Correspondingly, it was observed that the Hit molecule has been positioned within the DFG motif comprising of Asp1046-Phe1047-Gly1048 residues sandwiched between Asp1046 and Glu885 on either side and hence crucial in conferring the reduction of angiogenesis. The Cys1045 has formed a π- sulfur bond that makes the ligand to be placed firmly with the active site. A hydrophobic interaction have additionally concentrated towards the DFG motif of the activation loop and thereby, distorts the allosteric change of the motif and inhibits the kinase activity [5] and therefore the Hit may act as type II inhibitor. Further details of the interactions are tabulated in Table 5. Furthermore, to gain insight into and attain a proper reasoning on the nature of the hydrogen bonds in the active site, the hydrogen bonds were recorded throughout the simulation time. Consequently, it was noticed that the reference has produced an average of 0.7 hydrogen bonds, while the Hit has displayed a higher average of 1.7, (Fig. 16).
Fig. 13.
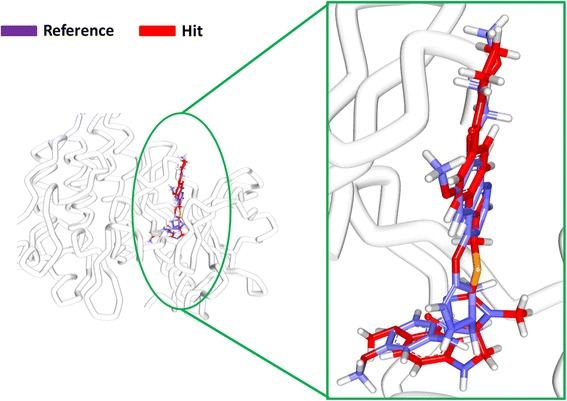
Assessment of the binding mode patterns of the reference (purple) and the Hit (red). The Hit obeys the same binding pattern of the reference compound
Table 5.
Intermolecular interactions between the VEGFR-2 inhibitors
| Name of the compound | Hydrogen bonds < 3 Å | van der Waals interactions | π-π | ||
|---|---|---|---|---|---|
| Residue atom | Ligand atom | Bond length | |||
| Reference | Cys919:HN Asp1046:NH Asp1046: O |
N7 O26 H62 |
2.2 2.0 2.0 |
Val848, Lys868, Glu885, Ile892, asn900, Leu901, Lys920, Phe921, Gly922, Asn923, Leu1019, leu1044, His1026 Phe1047 | Leu840, Ala866, Ala866, Val898, Leu889, Leu1035 |
| Hit | Glu885:OE2 Cys919: O Asp1046:HN |
H36 H35 N5 |
1.88 2.9 2.3 |
Ala866, Glu850, Ile888, Ile892, Val898, Val914, Lys920, Asn923, Gly922, Leu1019, His1026, Ile1044, Phe1047 | Leu840, Val848, Leu889, Lys868, Phe918, Leu1035 |
Fig. 14.
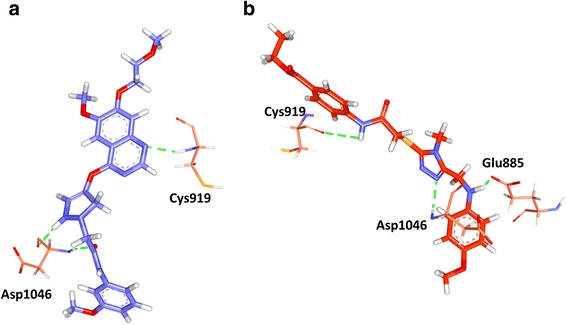
Molecular interaction between the reference- protein (purple) and Hit-protein (red). Green dotted lines indicate the hydrogen bonds. The residues are represented in orange stick model
Fig. 15.
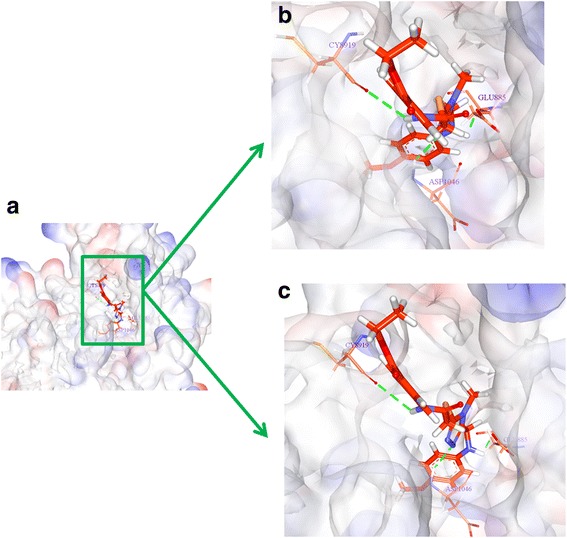
Back-to-front pattern of binding. a) Depiction of the presence of Hit in the active site pocket in back-to-front fashion. A clear enlarged cavity is represented in b and c. The ligand was found to be seated firmly with Cys919 from the top and is sandwiched with Glu885 and Asp1046
Fig. 16.
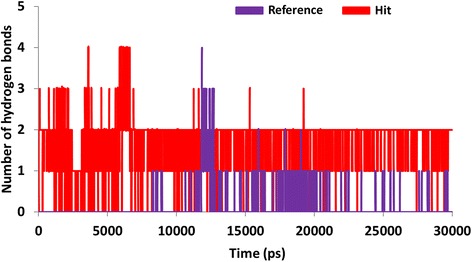
Estimation of the hydrogen bond interactions during the whole simulations. The refrence is represented in purple and red denotes the Hit. The reference has produced lower number of hydrogen bonds and predominantly seen during 11,000 ps ~ 20,000 ps. The Hit has shown regular bonds throughout the simulations
Challenging the identified lead molecule against CDK2 protein
After achieving the first objective, now the investigation proceeds to understand if the ligand is potential enough to act against cancer prognosis. Cyclin-Dependent Kinase 2(CDK2) has been attributed with certain cancer progressions, especially the oral cancer [63, 64]. Progression refers to the advancement of the disease during the course of the time. For the current study the protein 1URW, was retrieved from the protein data bank. This protein is in complex with imidazole [1,2-B] pyridazine inhibitor of 1.6 Å. The identified lead molecule form the above steps is now challenged with 1URW, such that a common drug could be identified.
All the steps, such as, protein preparation and ligand preparation were followed as per the aforementioned methods. The active site of the protein was plotted at 12 Å around the inbuilt co-crystal. To ensure the suitability of the docking parameters, the co-crystal was initially docked and an acceptable RMSD of 1.75 Å was generated, (Additional file 5) and the same parameters were considered for docking employing GOLD v5.2.2. The results were examined taking into consideration that the inhibitor lies within the selected active site sphere and forms a hydrogen bond with Leu83, Asp86 and Lys89. Amongst them, Leu83 is the core residue as reported earlier [26, 65]. To authenticate the binding of the ligand with the active site residues, the results generated by the co-crystal docking were taken into consideration. The best pose generated was escalated to MD simulation studies to assess the protein backbone stability and were read as root mean square deviation (RMSD). Subsequent results demonstrated that the cocrystal, reference and the Hit have displayed stable RMSD values that exist below 0.25 nm throughout 30 ns run, (Fig. 17).
Fig. 17.
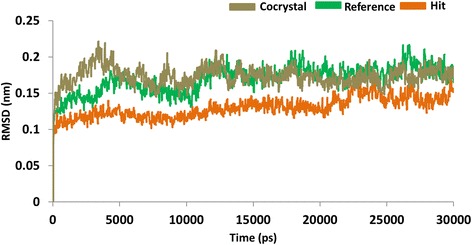
The RMSD backbone stability of the three systems during 30 ns run
The representative structures from the last 3 ns were extracted and superimposed for further analysis. Upon superimposition of the co-crystal, reference and the Hit have demonstrated a similar type of binding mode, (Fig. 18). The dock results revealed that the co-crystal has formed four hydrogen bonds with residues namely Leu83, Asp86 and Lys89 demonstrating a dock score of 53.05, while the reference and Hit has displayed a score of 52.56 and 64.34. Additionally, a number of hydrophobic bonds have been generated in the form of van der Waals interactions and π-π bonds as represented in Fig. 19a and Additional file 6. The reference compound on the other hand has rendered five hydrogen bonds one each by Glu12 and Gly16 and three bonds with Lys89. It was observed that only the Lys89 residue was noticed to participate as was seen in the crystal structure. Additionally, Glu12 formed a weak hydrogen bond of 3.0 Å and displayed no interaction with Leu83. This could be because the compound is a specific VEGFR-2 inhibitor. However, it exerts its effect through the involvement of Lys89 residue, (Fig. 19b: Additional file 7). The retrieved Hit has displayed three hydrogen bonds involving two crucial residues, Leu83 and Lys89 and further has generated an acceptable bond length of 2.1 Å and 2.4 Å, respectively, Fig. 19C. Additionally, it formed more number of hydrophobic interactions with the key resides, (Additional file 8). The details of the molecular interactions are tabulated in Table 6.
Fig. 18.
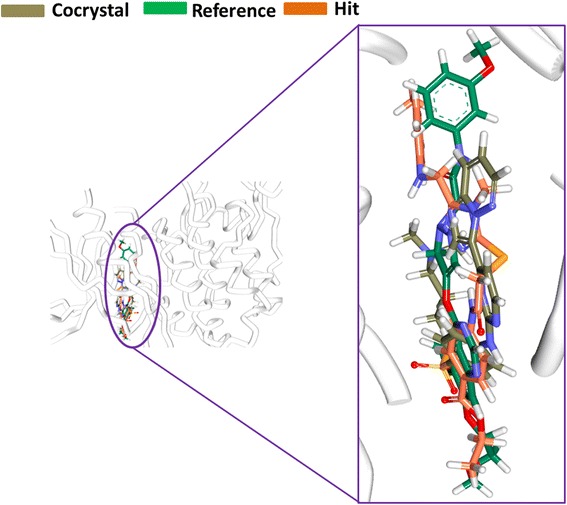
Binding mode assessment of compounds. The co-crystal is represented in gray, reference is denoted in green and the Hit in orange. All the three follow the same pattern
Fig. 19.
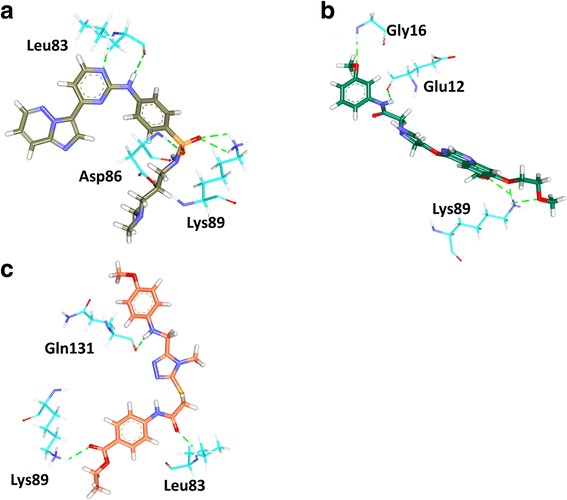
Intermolecular interactions between the ligand and the protein. The co-crystal is represented in gray, reference is denoted in green and the Hit in orange. Green dotted lines represent the hydrogen bonds. The protein residues are indicated in cyan
Table 6.
Intermolecular interaction between the protein and the ligands
| Name of the compound | Hydrogen bonds < 3 Å | van der Waals interactions | π-π | ||
|---|---|---|---|---|---|
| Residue atom | Ligand atom | Bond length | |||
| Co-crystal | Leu83: O Leu83:HN Asp86:HN Lys89:HZ2 Lys89:HZ3 |
H42 N20 O31 O32 O32 |
2.2 2.0 2.5 2.4 2.4 |
Gly11, Glu12, Lys20, Lys33, Val64, Glu81, Gln85, Lys88, Gln131, Asn132, Leu148, | Val18, Ala31, Phe80, Phe82, Leu134, Ala144 |
| Reference | Glu12: O Gly16:HN Lys89:HZ2 Lys89:HZ2 Lys89:HZ3 |
H43 O27 O34 O31 O29 |
3.0 2.2 2.3 1.9 2.5 |
Gly11, Tyr15, Val18, Gln85, Asp86, Asp127, Lys129, Gln131, Asn132, Leu134, Leu298, | Phe82 |
| Hit | Leu83:HN Lys89:HZ2 Gln131: O |
O25 O24 H36 |
2.1 2.4 2.2 |
Gly11, Glu12, Ala31, Lys33, Val64, Phe82, His84, Gln85, Asp86, Lys129, Asn132, Asp145, Ala144, Val164 | Ile10, Val18, Leu134, Leu298 |
Furthermore, the binding pockets of both the targets were evaluated to understand their similarity and was assessed using PocketMatch (PM) [66] that was executed through three aspects (a) representation of each site as sorted lists of distances between chosen points, (b) alignment of two sets of distance lists and (c) choosing a scoring scheme for arriving at a final score. The similarity is read as PM score which ranges from 0 to 1, where zero implies no similarity and 1 refers to complete similarity. The obtained results revealed the PM score to be 0.82 (0.6 or greater refers to almost similar). To further affirm the similarity of the active sites, the align structures protocol available on the DS was initiated and subsequently, the arrangement of the sequences, (Fig. 20) and the active site residues, (Fig. 21: Additional file 9) were evaluated. It was interesting to note that the majority of the key residues were conserved besides; the key residues Cys919 and Leu83 were complementary with each other and were instrumental in rendering the inhibitory activity in the corresponding drug targets. These findings further reinforce that they share similar binding pocket facilitating the use of common drug.
Fig. 20.

Sequence alignment of the protein targets representing angiogenesis (4AG8) and progression (1URW). The sequence identity and the sequence similarity were found to be 20.1 and 38.1, respectively. The active site shows higher degree of identity as demonstrated by the residues highlighted in yellow. The orange amino acids refer to the key inhibitory residues found to be complementary with each other
Fig. 21.
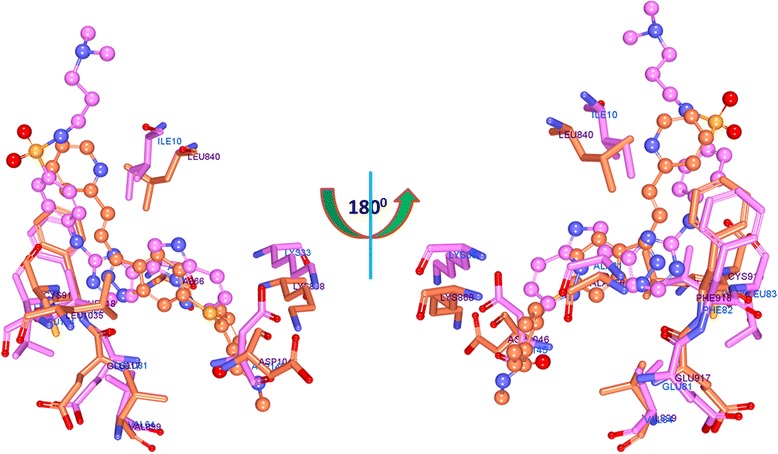
Comparison of the active sites of 4AG8 (orange) and 1URW (pink). The corresponding ligand molecule is represented in ball and stick model
Discussion
Receptor tyrosine kinases (RTKs) are a class of proteins that play a crucial role in cell proliferation, differentiation, metabolism and cell growth and survival. Among them, vascular endothelial growth factor receptor-2 (VEGFR-2) is important in angiogenic regulation and hence is a major target for repressing the cancer growth and metastasis [67–70] . Additionally, the uncontrolled development of these cancerous cells is the main reason for metastasis. Therefore, in the current study we aimed at identifying a small molecule from the large datasets that could selectively inhibit both the process, thus leading to suppression of cancer. Accordingly, only one lead candidate has been identified that binds in back-to-front approach with the VEGFR-2 and also inhibits the CDK2. The identified inhibitor successfully binds to the ATP site (front pocket) and also extends towards the hydrophobic site (back pocket) and hence categorizes itself has type -II inhibitor. Cys919 located at the front pocket holds firmly the ligand molecule by HN atom and facilitates the bulky benzene ring to be accommodated within the active site with a slight tilt formed at the tip of the ligand comprised of CH3 group, (Fig. 15b). The back pocket on the other end was observed to be held tightly with two hydrogen bonds and several hydrophobic bonds, (Fig. 15c: Additional file 4). The pentane ring of the ligand that is located at the ligands center was seen to be held with two hydrophobic bonds produced by Val848 and Lys868 besides displaying a hydrogen bond with Asp1046. Therefore, the presence pentene ring in the ligand apparently seems to be important in making this inhibitor of a type II class. Additionally, the sulphur group of Cys1045 has involved with the pentane ring. Additionally, the first benzene ring was held by Leu840 and Leu1035 and the second benzene ring was held by Leu889. Furthermore, it can be observed that the bulky groups present within the ligand have been held by either hydrogen bonds or the hydrophobic bonds, thus providing proper positioning for the ligand.
Furthermore, we challenged the identified Hit against CDK2 target whose inhibition can abrupt the progression of the cancer cells. The CDK2 displays four binding sites such as ATP binding site (competitive) and two non- competitive sites. Additionally, upon subjecting to the cyclin binding process gives rise to a new site caused due to the conformational changes in the ATP site and is labeled as the allosteric site (site IV). Amongst them the ATP binding site is highly conserved across all the human kinases [23] . Therefore, we focused on the ATP binding site to evaluate the potency of the identified ligand. This ATP binding site is a cleft located between the N-terminal domain consisting of beta-sheets (small lobe) and a C-terminal domain rich in helices (large lobe) [71], which can be further divided into three. The first one is the hydrophobic region comprising of Ile10, Ala31, Val64, Phe80, Glu81, Phe82, Leu83, Leu134 and Ala144. The second region consists of three amino acids, Val18, Asp86 and Gln131 and generally interacts with the ribose moiety of the ATP. The third region is made up of Lys33, Asp145 and Asn132. Additionally, two domains are joined by the hinge region constituting Glu81 and Leu83, respectively. The identified ligand has formed an hydrogen bond interactions with the key residues involving the Leu83 and Lys89 [65, 71]. An additional hydrogen bond was formed with the amino acid Gln131 that is located at the polar interaction site through its HN group. This pattern of binding is in agreement as described previously [71]. The residues belonging to the hydrophobic region consisting of Ile10, Val18 and Leu134 have joined with the ligand by the hydrophobic bonds. The residues, Val64, Ala144 of the hydrophobic interaction site and Lys33, Asp86 and Asp145 that belong to the polar interaction site have interacted with the ligand through the van der Waal’s interactions and thus facilitating the lead molecule to be seated firmly with in the active site. Furthermore, since the ATP binding sites are conserved across the kinases [71], this could be one reason for the complementarity that exists between Cyc919 of VEGFR2 and Leu83 of CDK2, (Fig. 20).
Conclusions
The present study aims at identifying a dual inhibitor that can repress the angiogenesis and progression. In order to achieve this, a systematic ligand-based pharmacophore modelling and subsequent large database screening was conducted. From the obtained lead molecules, further knowledge based screening was conducted to find the compounds that obeyed the back-to-front type of inhibitory mechanism employing the docking protocol and the interactions with the key residues. Consequently, results showed that only one compound has qualified this criterion. Upon further studies and applying the MD simulations, it was observed that the ligand-protein complex was relatively stable. Therefore this lead compound was challenged against 1URW, where the Hit was found to interact with the important residues seated at its active site. Our results also demonstrate that binding with Cys919 and Lue83 are important in designing a dual inhibitor for angiogenesis and progression. Taken together, we suggest that the Hit compound might be a potential lead candidate that can repress cancer angiogenesis and growth.
Additional files
Test set assessment. Assessing the test set values for estimated and the experimental activities by Hypo1. (DOCX 18 kb)
Cocrystal re-dock results of 4AG8. Overlapping of the co-crystal (cyan) onto the docked pose (orange). The binding pattern was found to be similar. (DOCX 181 kb)
2D interaction representation of the the reference compound and 4AG8. Detailed molecular interactions of the reference compound. (DOCX 420 kb)
2D interaction representation of the Hit compound and 4AG8. Detailed molecular interactions of the Hit compound. (DOCX 424 kb)
Docking of the co-crystal within the binding pocket of 1URW. Docking of the co-crystal within the binding pocket. Pink is the docked pose and green represents the co-crystal position. (DOCX 142 kb)
2D interaction representation of the co-crystal and 1URW. Detailed molecular interactions of the co-crystal compound. (DOCX 229 kb)
2D interaction representation of the reference compound and 1URW. Molecular interaction details of the reference compound. (DOCX 204 kb)
2D interaction representation of the Hit compound and 1URW. Molecular interaction details of the Hit compound. (DOCX 264 kb)
Active sites comparison. Comparison of the active site residues of 4AG8 and 1UMR. (DOCX 13 kb)
Acknowledgements
Not applicable
Funding
This research was supported by Next-Generation BioGreen 21 Program from Rural Development administration, Republic of Korea (Grant no. PJ01106202), and also supported by the Pioneer research Center Program through the National Research Foundation (NRF) funded by the Ministry o of Science, ICT and Future Planning (NRF-2015M3C1A3023028).
Availability of data and materials
All the data and the material are provided with the manuscript and the Additional files 1, 2, 3, 4, 5 and 6.
Abbreviations
- 2D
Two Dimensiional
- 3D QSAR
Three-dimensional quantitative structure-activity relationships
- 3D
Three Dimensional
- Å
Angstrom
- A
Number of actives
- ACD
Advanced chemistry development
- ADMET
Absorption, distribution, metabolism, excretion, toxicity
- ATP
Adenosine Triphosphate
- B3LYP
Becke, 3-parameter, Lee-Yang-Parr
- BBB
Blood Brain Barrier
- CDK
Cyclin-dependent kinases
- CHARMm
Chemistry at HARvard Macromolecular Mechanics
- CYPs
Cytochromes P450
- D
Total number of molecules in a database
- DFT
Density functional theory
- DGF
Asp-Phe-Gly
- DS
Discovery Studio
- EF
Enrichment Factor
- Ff
force field
- G1
Gap 1
- GF
Goodness of Fit
- GOLD
Genetic optimisation for ligand docking
- GROMACS
GROningen machine for chemical simulations
- Ha
Total Actives
- HBA
Hydrogen bond acceptor
- HBD
Hydrogen bond donor
- HIA
Human intestinal absorption
- HOMO
Highest occupied Molecular Orbitals
- Ht
Total Hits
- Hy Ali
Hydrophobic aliphatic
- HyP
HydroPhobic
- IC50
Half maximal inhibitory concentration
- LINCS
Linear constraint solver for molecular simulations
- LUMO
Lowest unoccupied molecular orbitals
- Max
Maximum
- MD
Molecular dynamics
- NCI
National cancer institute
- NPT
Number of particles, pressure, and temperature
- NVT
Number of particles, volume, and temperature
- PDB
Protein data bank
- PM
Pocket match
- PME
Patricle mesh ewald
- PPB
Plasma protein binding
- RA
Ring aromatic
- RKT
Receptor Tyrosine Kinases
- RMSD
Root mean square deviation
- S phase
Synthesis phase
- TIP3P
Three-site transferrable intermolecular potential
- VEGFRs
Vascular endothelial growth factor receptors
- VEGFs
Vascular endothelial growth factors
- VMD
Visual molecular dynamics
Authors’ contributions
KWL, SR and AB designed the project. SR and AZ wrote the manuscript. SR and KWL assessed the results. All the authors read and approve the manuscript.
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12885-018-4050-1) contains supplementary material, which is available to authorized users.
Contributor Information
Shailima Rampogu, Email: shailima.rampogu@gmail.com.
Ayoung Baek, Email: ayoung@gnu.ac.kr.
Amir Zeb, Email: zebamir85@gmail.com.
Keun Woo Lee, Email: kwlee@gnu.ac.kr.
References
- 1.Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2(3):213–219. doi: 10.2147/vhrm.2006.2.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ucuzian AA, Gassman AA, East AT, Greisler HP. Molecular mediators of angiogenesis. J Burn Care Res. 2010;31(1):158–175. doi: 10.1097/BCR.0b013e3181c7ed82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-Angiogenic therapies. Genes Cancer. 2011;2(12):1097–1105. doi: 10.1177/1947601911423031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koch S, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med. 2012;2(7):a006502. doi: 10.1101/cshperspect.a006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J, Zhou N, Luo K, Zhang W, Li X, Wu C, Bao J. In silico discovery of potential VEGFR-2 inhibitors from natural derivatives for anti-angiogenesis therapy. Int J Mol Sci. 2014;15(9):15994–16011. doi: 10.3390/ijms150915994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han KY, Dugas-Ford J, Lee H, Chang JH, Azar DT. MMP14 cleavage of VEGFR1 in the cornea leads to a VEGF-trap Antiangiogenic effect. Invest Ophthalmol Vis Sci. 2015;56(9):5450–5456. doi: 10.1167/iovs.14-16248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rahimi N. VEGFR-1 and VEGFR-2: two non-identical twins with a unique physiognomy. Front Biosci. 2006;11:818–829. doi: 10.2741/1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Witmer AN, van Blijswijk BC, Dai J, Hofman P, Partanen TA, Vrensen GF, Schlingemann RO. VEGFR-3 in adult angiogenesis. J Pathol. 2001;195(4):490–497. doi: 10.1002/path.969. [DOI] [PubMed] [Google Scholar]
- 9.Taylor PC. VEGF and imaging of vessels in rheumatoid arthritis. Arthritis Res. 2002;4(Suppl 3):S99–107. doi: 10.1186/ar582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fava GA. Affective disorders and endocrine disease. New insights from psychosomatic studies. Psychosomatics. 1994;35(4):341–353. doi: 10.1016/S0033-3182(94)71755-2. [DOI] [PubMed] [Google Scholar]
- 11.Detmar M. The role of VEGF and thrombospondins in skin angiogenesis. J Dermatol Sci. 2000;24(Suppl 1):S78–S84. doi: 10.1016/S0923-1811(00)00145-6. [DOI] [PubMed] [Google Scholar]
- 12.Lee YJ, Karl DL, Maduekwe UN, Rothrock C, Ryeom S, D'Amore PA, Yoon SS. Differential effects of VEGFR-1 and VEGFR-2 inhibition on tumor metastases based on host organ environment. Cancer Res. 2010;70(21):8357–8367. doi: 10.1158/0008-5472.CAN-10-1138. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Homayouni M. Vascular endothelial growth factors and their inhibitors in ocular Neovascular disorders. J Ophthalmic Vis Res. 2009;4(2):105–114. [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu KC, Sung TY, Lin CT, Chiu YY, Hsu JT, Hung HC, Sun CM, Barve I, Chen WL, Huang WC, et al. Anchor-based classification and type-C inhibitors for tyrosine kinases. Sci Rep. 2015;5:10938. doi: 10.1038/srep10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dar AC, Shokat KM. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu Rev Biochem. 2011;80:769–795. doi: 10.1146/annurev-biochem-090308-173656. [DOI] [PubMed] [Google Scholar]
- 16.Fedorov O, Muller S, Knapp S. The (un) targeted cancer kinome. Nat Chem Biol. 2010;6(3):166–169. doi: 10.1038/nchembio.297. [DOI] [PubMed] [Google Scholar]
- 17.Sanphanya K, Wattanapitayakul SK, Phowichit S, Fokin VV, Vajragupta O. Novel VEGFR-2 kinase inhibitors identified by the back-to-front approach. Bioorg Med Chem Lett. 2013;23(10):2962–2967. doi: 10.1016/j.bmcl.2013.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Honda T, Nagahara H, Mogi H, Ban M, Aono H. KDR inhibitor with the intramolecular non-bonded interaction: conformation-activity relationships of novel indole-3-carboxamide derivatives. Bioorg Med Chem Lett. 2011;21(6):1782–1785. doi: 10.1016/j.bmcl.2011.01.063. [DOI] [PubMed] [Google Scholar]
- 19.Bauer D, Whittington DA, Coxon A, Bready J, Harriman SP, Patel VF, Polverino A, Harmange JC. Evaluation of indazole-based compounds as a new class of potent KDR/VEGFR-2 inhibitors. Bioorg Med Chem Lett. 2008;18(17):4844–4848. doi: 10.1016/j.bmcl.2008.07.080. [DOI] [PubMed] [Google Scholar]
- 20.Jiang P, Du W, Wu M. Regulation of the pentose phosphate pathway in cancer. Protein Cell. 2014;5(8):592–602. doi: 10.1007/s13238-014-0082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15(6):122. doi: 10.1186/gb4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neganova I, Lako M. G1 to S phase cell cycle transition in somatic and embryonic stem cells. J Anat. 2008;213(1):30–44. doi: 10.1111/j.1469-7580.2008.00931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Zhang J, Gao W, Zhang L, Pan Y, Zhang S, Wang Y. Insights on structural characteristics and Ligand binding mechanisms of CDK2. Int J Mol Sci. 2015;16(5):9314–9340. doi: 10.3390/ijms16059314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79(4):551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 25.Chen Y-NP, Sharma SK, Ramsey TM, Jiang L, Martin MS, Baker K, Adams PD, Bair KW, Kaelin WG. Selective killing of transformed cells by cyclin/cyclin-dependent kinase 2 antagonists. Proc Natl Acad Sci U S A. 1999;96(8):4325–4329. doi: 10.1073/pnas.96.8.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Latham AM, Kankanala J, Fearnley GW, Gage MC, Kearney MT, Homer-Vanniasinkam S, Wheatcroft SB, Fishwick CW, Ponnambalam S. In silico design and biological evaluation of a dual specificity kinase inhibitor targeting cell cycle progression and angiogenesis. PLoS One. 2014;9(11):e110997. doi: 10.1371/journal.pone.0110997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baldwin I, Bamborough P, Haslam CG, Hunjan SS, Longstaff T, Mooney CJ, Patel S, Quinn J, Somers DO. Kinase array design, back to front: biaryl amides. Bioorg Med Chem Lett. 2008;18(19):5285–5289. doi: 10.1016/j.bmcl.2008.08.051. [DOI] [PubMed] [Google Scholar]
- 28.Iwata H, Oki H, Okada K, Takagi T, Tawada M, Miyazaki Y, Imamura S, Hori A, Lawson JD, Hixon MS, et al. A back-to-front fragment-based drug design search strategy targeting the DFG-out pocket of protein tyrosine Kinases. ACS Medl Chem Lett. 2012;3(4):342–346. doi: 10.1021/ml3000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Regan J, Breitfelder S, Cirillo P, Gilmore T, Graham AG, Hickey E, Klaus B, Madwed J, Moriak M, Moss N, et al. Pyrazole urea-based inhibitors of p38 MAP kinase: from lead compound to clinical candidate. J Med Chem. 2002;45(14):2994–3008. doi: 10.1021/jm020057r. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Yang S, Jiao Y, Liu H, Yuan H, Lu S, Ran T, Yao S, Ke Z, Xu J, et al. An integrated virtual screening approach for VEGFR-2 inhibitors. J Chem Inf Mod. 2013;53(12):3163–3177. doi: 10.1021/ci400429g. [DOI] [PubMed] [Google Scholar]
- 31.Kawakami JK, Martinez Y, Sasaki B, Harris M, Kurata WE, Lau AF. Investigation of a novel molecular descriptor for the lead optimization of 4-aminoquinazolines as vascular endothelial growth factor receptor-2 inhibitors: application for quantitative structure-activity relationship analysis in lead optimization. Bioorg Med Chem Lett. 2011;21(5):1371–1375. doi: 10.1016/j.bmcl.2011.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Potashman MH, Bready J, Coxon A, DeMelfi TM, Jr, DiPietro L, Doerr N, Elbaum D, Estrada J, Gallant P, Germain J, et al. Design, synthesis, and evaluation of orally active benzimidazoles and benzoxazoles as vascular endothelial growth factor-2 receptor tyrosine kinase inhibitors. J Med Chem. 2007;50(18):4351–4373. doi: 10.1021/jm070034i. [DOI] [PubMed] [Google Scholar]
- 33.Zhang L, Zheng Q, Yang Y, Zhou H, Gong X, Zhao S, Fan C. Synthesis and in vivo SAR study of indolin-2-one-based multi-targeted inhibitors as potential anticancer agents. Eur J Med Chem. 2014;82:139–151. doi: 10.1016/j.ejmech.2014.05.051. [DOI] [PubMed] [Google Scholar]
- 34.Caballero J, Munoz C, Alzate-Morales JH, Cunha S, Gano L, Bergmann R, Steinbach J, Kniess T. Synthesis, in silico, in vitro, and in vivo investigation of 5-[(1) (1)C] methoxy-substituted sunitinib, a tyrosine kinase inhibitor of VEGFR-2. Eur J Med Chem. 2012;58:272–280. doi: 10.1016/j.ejmech.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 35.Kuchar M, Oliveira MC, Gano L, Santos I, Kniess T. Radioiodinated sunitinib as a potential radiotracer for imaging angiogenesis-radiosynthesis and first radiopharmacological evaluation of 5-[125I] Iodo-sunitinib. Bioorg Med Chem Lett. 2012;22(8):2850–2855. doi: 10.1016/j.bmcl.2012.02.068. [DOI] [PubMed] [Google Scholar]
- 36.Bhide RS, Cai ZW, Zhang YZ, Qian L, Wei D, Barbosa S, Lombardo LJ, Borzilleri RM, Zheng X, Wu LI, et al. Discovery and preclinical studies of (R)-1-(4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-5- methylpyrrolo [2,1-f] [1,2,4] triazin-6-yloxy) propan- 2-ol (BMS-540215), an in vivo active potent VEGFR-2 inhibitor. J Med Chem. 2006;49(7):2143–2146. doi: 10.1021/jm051106d. [DOI] [PubMed] [Google Scholar]
- 37.Wang S, Midgley CA, Scaerou F, Grabarek JB, Griffiths G, Jackson W, Kontopidis G, McClue SJ, McInnes C, Meades C, et al. Discovery of N-phenyl-4-(thiazol-5-yl) pyrimidin-2-amine aurora kinase inhibitors. J Med Chem. 2010;53(11):4367–4378. doi: 10.1021/jm901913s. [DOI] [PubMed] [Google Scholar]
- 38.Kiselyov AS, Semenova M, Semenov VV. 3,4-Disubstituted isothiazoles: novel potent inhibitors of VEGF receptors 1 and 2. Bioorg Med Chem Lett. 2009;19(4):1195–1198. doi: 10.1016/j.bmcl.2008.12.078. [DOI] [PubMed] [Google Scholar]
- 39.Iwata H, Imamura S, Hori A, Hixon MS, Kimura H, Miki H. Biochemical characterization of a novel type-II VEGFR2 kinase inhibitor: comparison of binding to non-phosphorylated and phosphorylated VEGFR2. Bioorg Med Chem. 2011;19(18):5342–5351. doi: 10.1016/j.bmc.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 40.Sakkiah S, Thangapandian S, John S, Kwon YJ, Lee KW. 3D QSAR pharmacophore based virtual screening and molecular docking for identification of potential HSP90 inhibitors. Eur J Med Chem. 2010;45(6):2132–2140. doi: 10.1016/j.ejmech.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 41.Debnath AK. Pharmacophore mapping of a series of 2,4-diamino-5-deazapteridine inhibitors of Mycobacterium Avium Complex dihydrofolate reductase. J Med Chem. 2002;45(1):41–53. doi: 10.1021/jm010360c. [DOI] [PubMed] [Google Scholar]
- 42.Leeson PD, Davis AM, Steele J. Drug-like properties: guiding principles for design - or chemical prejudice? Drug Discov Today Techs. 2004;1(3):189–195. doi: 10.1016/j.ddtec.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 43.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Del Rev. 2001;46(1–3):3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 44.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267(3):727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 45.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein–ligand docking using GOLD. Proteins Struct Funct Bioinform. 2003;52(4):609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 46.Vardhini SRD. In silico evaluation for the potential naturally available drugs for breast cancer. J Recept Signal Transduct. 2014;34(3):174–179. doi: 10.3109/10799893.2013.868904. [DOI] [PubMed] [Google Scholar]
- 47.Kavitha R, Karunagaran S, Chandrabose SS, Lee KW, Meganathan C. Pharmacophore modeling, virtual screening, molecular docking studies and density functional theory approaches to identify novel ketohexokinase (KHK) inhibitors. Biosystems. 2015;138:39–52. doi: 10.1016/j.biosystems.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 48.Rampogu S, Baek A, Son M, Zeb A, Park C, Kumar R, Lee G, Kim D, Choi Y, Cho Y, et al. Computational exploration for lead compounds that can reverse the nuclear morphology in Progeria. Biomed Res Int. 2017;2017:15. doi: 10.1155/2017/5270940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zoete V, Cuendet MA, Grosdidier A, Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem. 2011;32(11):2359–2368. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- 50.Rampogu S, Son M, Park C, Kim H-H, Suh J-K, Lee KW. Sulfonanilide derivatives in identifying novel Aromatase inhibitors by applying docking, virtual screening, and MD simulations studies. Biomed Res Int. 2017;2017:17. doi: 10.1155/2017/2105610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mackerell AD., Jr Empirical force fields for biological macromolecules: overview and issues. J Comput Chem. 2004;25(13):1584–1604. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- 52.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 53.Zhu X, Lopes PEM, MacKerell AD. Recent developments and applications of the CHARMM force fields. Wiley Interdiscip Rev Comput Mol Sci. 2012;2(1):167–185. doi: 10.1002/wcms.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mallajosyula SS, Jo S, Im W, MacKerell AD., Jr Molecular dynamics simulations of glycoproteins using CHARMM. Methods Mol Biol (Clifton, NJ) 2015;1273:407–429. doi: 10.1007/978-1-4939-2343-4_25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 56.Bavi R, Kumar R, Rampogu S, Son M, Park C, Baek A, Kim HH, Suh JK, Park SJ, Lee KW. Molecular interactions of UvrB protein and DNA from helicobacter pylori: insight into a molecular modeling approach. Comput Biol Med. 2016;75:181–189. doi: 10.1016/j.compbiomed.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 57.Berendsen HJC, Postma JPM, WFv G, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 58.Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys. 1981;52(12):7182–7190. doi: 10.1063/1.328693. [DOI] [Google Scholar]
- 59.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18(12):1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 60.Darden T, York D, Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 61.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 62.Banavath HN, Sharma OP, Kumar MS, Baskaran R. Identification of novel tyrosine kinase inhibitors for drug resistant T315I mutant BCR-ABL: a virtual screening and molecular dynamics simulations study. Sci Rep. 2014;4:6948. doi: 10.1038/srep06948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mihara M, Shintani S, Nakahara Y, Kiyota A, Ueyama Y, Matsumura T, Wong DT. Overexpression of CDK2 is a prognostic indicator of oral cancer progression. Jpn J Cancer Res Gann. 2001;92(3):352–360. doi: 10.1111/j.1349-7006.2001.tb01102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: a perspective. Oncogene. 2005;24(17):2909–2915. doi: 10.1038/sj.onc.1208618. [DOI] [PubMed] [Google Scholar]
- 65.Byth KF, Cooper N, Culshaw JD, Heaton DW, Oakes SE, Minshull CA, Norman RA, Pauptit RA, Tucker JA, Breed J, et al. Imidazo [1,2-b] pyridazines: a potent and selective class of cyclin-dependent kinase inhibitors. Bioorg Med Chem Lett. 2004;14(9):2249–2252. doi: 10.1016/j.bmcl.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 66.Yeturu K, Chandra N. PocketMatch: a new algorithm to compare binding sites in protein structures. BMC Bioinform. 2008;9(1):543. doi: 10.1186/1471-2105-9-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zwick E, Bange J, Ullrich A. Receptor tyrosine kinases as targets for anticancer drugs. Trends Mol Med. 2002;8(1):17–23. doi: 10.1016/S1471-4914(01)02217-1. [DOI] [PubMed] [Google Scholar]
- 68.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29(6 Suppl 16):15–18. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 69.Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular endothelial growth factor and angiogenesis. Pharmacoll Rev. 2004;56(4):549–580. doi: 10.1124/pr.56.4.3. [DOI] [PubMed] [Google Scholar]
- 70.Schenone S, Bondavalli F, Botta M. Antiangiogenic agents: an update on small molecule VEGFR inhibitors. Curr Med Chem. 2007;14(23):2495–2516. doi: 10.2174/092986707782023622. [DOI] [PubMed] [Google Scholar]
- 71.Tripathi SK, Muttineni R, Singh SK. Extra precision docking, free energy calculation and molecular dynamics simulation studies of CDK2 inhibitors. J Theor Biol. 2013;334:87–100. doi: 10.1016/j.jtbi.2013.05.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Test set assessment. Assessing the test set values for estimated and the experimental activities by Hypo1. (DOCX 18 kb)
Cocrystal re-dock results of 4AG8. Overlapping of the co-crystal (cyan) onto the docked pose (orange). The binding pattern was found to be similar. (DOCX 181 kb)
2D interaction representation of the the reference compound and 4AG8. Detailed molecular interactions of the reference compound. (DOCX 420 kb)
2D interaction representation of the Hit compound and 4AG8. Detailed molecular interactions of the Hit compound. (DOCX 424 kb)
Docking of the co-crystal within the binding pocket of 1URW. Docking of the co-crystal within the binding pocket. Pink is the docked pose and green represents the co-crystal position. (DOCX 142 kb)
2D interaction representation of the co-crystal and 1URW. Detailed molecular interactions of the co-crystal compound. (DOCX 229 kb)
2D interaction representation of the reference compound and 1URW. Molecular interaction details of the reference compound. (DOCX 204 kb)
2D interaction representation of the Hit compound and 1URW. Molecular interaction details of the Hit compound. (DOCX 264 kb)
Active sites comparison. Comparison of the active site residues of 4AG8 and 1UMR. (DOCX 13 kb)
Data Availability Statement
All the data and the material are provided with the manuscript and the Additional files 1, 2, 3, 4, 5 and 6.