

Abstract
As a promising molecular process for selectively inhibiting cancer cells without inducing acquired drug resistance, enzyme-instructed self-assembly (EISA) usually requires relatively high dosages. Despite its discovery 30 years ago, the translation of the knowledge about NF-κB signaling into clinic remains complicated due to the broad roles of NF-κB in cellular regulation. Here we show that integrating EISA and NF-κB targeting boosts the efficacy of EISA over an order of magnitude without compromising selectivity against cancer cells. That is, in-situ enzymatic self-assembly of a tetrapeptide results in nanofibers, which hardly affect cell viability, but lead to inductive expression of tumor necrosis factor receptor 2 (TNFR2) and decreased expression of three key proteins at the up-stream of NF-κB pathway in the cancer cells. Adding the inhibitors targeting NF-κB further decreases the expressions of those up-stream proteins, which turns the otherwise innocuous nanofibers to being lethal to the cancer cells, likely causing necroptosis. As the first case of using supramolecular processes to enable synthetic lethality, this work illustrates a versatile approach to translate key regulatory circuits into promising therapeutic targets.
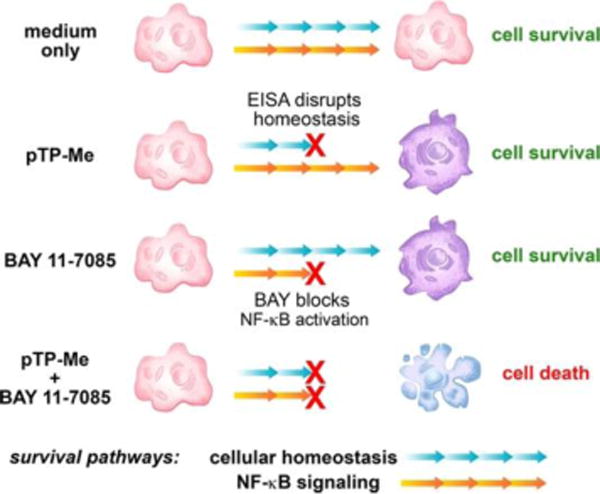
Graphical abstract
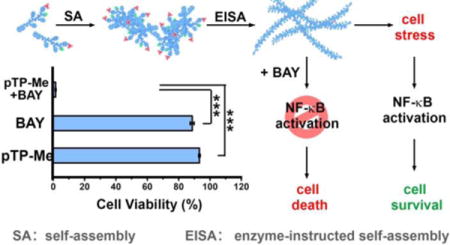
Introduction
Despite the significant advances in cancer biology and cancer therapy, cancer remains a challenge in drug development due to its great complexity.1 The elucidation of the mechanisms2 of drug resistance to current chemotherapy reveals the inherent drawbacks of molecular targeting therapy and the demands of new approaches for cancer therapy.3 Contrasting to targeting only a specific enzyme or protein, the integration of enzymatic reaction and molecular self-assembly4 leads to a multiple-step molecular process (termed as enzyme-instructed self-assembly (EISA))5 that is able to interact with multiple cellular targets and selectively inhibit cancer cells5c,6 without inducing acquired drug resistance.7 In fact, EISA is an inherent cellular process that affords spatiotemporal control of higher order structures from nanoscales to micron scales.8 Moreover, EISA is general and applicable for a wide range of substrates (e.g., peptides,9 carbohydrates,5c and nanoparticles10) and different enzymes (e.g., phosphatases,5b proteases,11 esterases,6c and glycosidases12), and is finding increased number of applications (e.g., efficient adjuvant for a vaccine,13 imaging of enzyme activity,14 monitoring autophagy,15 inhibiting bacteria,16 and removing unwanted iPS cells17).
However, EISA usually requires relatively high concentration for inhibiting cancer cells (e.g., about 1.0 mM5c) or killing iPS cells (e.g., 300 μM17). To address this limitation, we combine EISA with targeting the transcription factor nuclear factor-κB (NF-κB) and expect that their synergistic action would make NF-κB targeting selective and EISA effective against cancer cells. Being discovered 30 years ago,18 NF-κB, as a family of related protein hetero- or homodimers, exhibits remarkable capabilities for regulating the transcription of hundreds of target genes.19 Because activation of NF-κB is an essential feature of the survival of cancer cells during treatment, which contributes to cancer drug resistance, considerable efforts have focused on targeting NF-κB for cancer therapy. In fact, one of FDA-approved drugs for treating multiple myeloma, bortezomib (BTZ), works by blocking NF-κB activation.20 This success inspired the development of NF-κB inhibitors to sensitize cancer cells of solid tumors to anticancer drugs.21 A notable example of such development is BAY 11-7085 (BAY), which blocks proteasomal degradation of IκBα,22 allowing it to sequester NF-κB in an inactivated state.23 Therefore, we choose to integrate EISA with NF-κB inhibitors, such as BAY and BTZ for boosting the activity of EISA. Two fundamental biological facts support this choice: (i) Intracellular or pericellular peptide nanofibrils cause cell stress,10a,24 which results in up-regulation of transcription factors;25 (ii) NF-κB, as a rapidly inducible transcription factor,18–19 is critical for the survival response of cells to stress.26 Thus, blocking NF-κB signaling should sensitize cancer cells to the nanofibers formed by EISA (Scheme 1). In fact, our previous study has indicated the synergism of NF-κB inhibitors and pericellular nanofibers for apoptosis, however, the concentrations of the precursors required are still high10a and the relevant mechanism remains to be elucidated.
Scheme 1.
EISA in cellular milieu causes cell stress to activate NF-κB signaling for cell survival and the combination of EISA and NF-κB targeting effectively inhibit cancer cells.
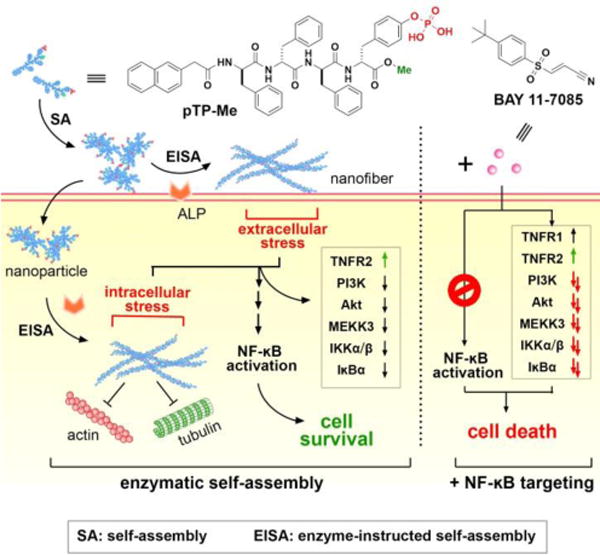
Here, this study shows exceptionally strong synergism between EISA and NF-κB targeting when EISA of a C-terminal methylated phosphotetrapeptide (pTP-Me) results in extra- and intracellular nano-assemblies (Scheme 1). Self-assembling to form nanoparticles to facilitate cell uptake27 and intracellular EISA and being dephosphorylated in cellular milieu of cancer cells (e.g., Saos-2), pTP-Me becomes TP-Me, which forms nanofibers and results in inductive expression of tumor necrosis factor receptor 2 (TNFR2)28 and decreased the expression of three key proteins (PI3K,29 Akt,30 and MEKK331) at the up-stream of NF-κB signaling in the cancer cells. Though exhibiting low cytotoxicity, the precursors drastically decrease cancer cell viability (about an order of magnitude) in the presence of the inhibitors targeting NF-κB signaling. Preliminary mechanistic study indicates that the combination of the EISA of pTP-Me and NF-κB inhibitors further decreases the expressions of those up-stream proteins, which eventually results in the death of Saos-2 via necroptosis. As the first report to focus on combining supramolecular chemistry with a chemical biology approach, this work illustrates the use of molecular processes to translate key regulatory circuits into promising therapeutic targets.
Results and Discussion
Molecular design and enzymatic self-assembly
Scheme 2 shows the structure of the precursor, pTP-Me, which consists of a tetrapeptide (D-Phe-D-Phe-D-Phe-D-Tyr (DFDFDFDY)) as the self-assembling motif, a phosphate on D-Tyr as the enzymatic trigger, a 2-acetylnaphthyl group at the N-terminal and a methyl group at the C-terminal of the peptide as the additional motifs for promoting self-assembly in water. To examine the roles of EISA and methylation, we synthesized three controls: pTP, without C-terminal methylation; TP-Me, lacking enzymatic trigger; TP, without both phosphate and methyl group (Scheme 2, Figs. S1-S11). Being generated after enzymatic dephosphorylation of pTP-Me by ALP, TP-Me forms a hydrogel with higher storage modulus (G’) than that of the soft hydrogel formed by pTP at the same condition (i.e., concentration (wt%), pH, and amount of ALP), indicating that methylation of the peptide indeed significantly increases self-assembling ability (Figs. S12, S13). In certain cases, a small portion of precursors being dephosphorylated would result in gelation. Since pTP-Me has much lower critical micelle concentration (CMC) than that of pTP (vide infra), it is reasonable for the mixture of TP-Me and pTP-Me to form a stronger gel than that of TP and pTP even though more pTP has been dephosphorylated than pTP-Me has. While it supports that formation of nanofibers contributes to cell death, this rheological observation also permits other factors to contribute to cell death.
Scheme 2.
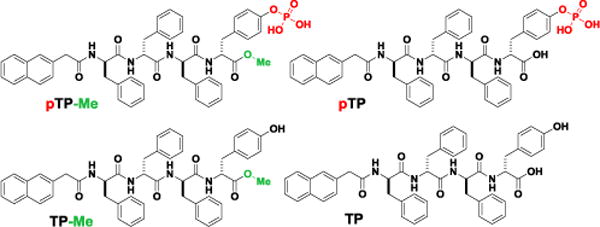
Molecular structures of pTP-Me, pTP, TP-Me, and TP.
Transmission electron microscopy (TEM) reveals that, at 100 μM (i.e., a working concentration in cellular environment), pTP-Me itself already self-assembles to form nanoparticle in water, and undergoes EISA to form nanofibers after the addition of ALP (Figure 1A). While pTP hardly aggregates itself at 100 μM, it forms nanofibers after EISA (Fig. S14). Being concentration-dependent, static light scattering (SLS) signals of the solutions of pTP-Me increase considerably after adding ALP (Figure 1B), suggesting significant aggregation. Notably, pTP-Me solutions exhibit considerable SLS signals around 7.5-15 μM before ALP treatment and around 1.9-3.7 μM after EISA, agreeing with the CMC of pTP-Me before (i.e., 14.8 μM) and after (i.e., 2.5 μM) the addition of ALP (Figure 1C, D). pTP exhibits similar SLS signal trend (Figure 1E), but with higher CMCs (128.66 μM and 8.69 μM before and after adding ALP, respectively) (Figure 1F, G). These results validate the enhanced self-assembly ability conferred by the methyl group at the C-terminal of DFDFDFDY. Because dephosphorylation plays an essential role in the multiple-step process of EISA, we monitored the dephosphorylation of pTP-Me and pTP treated by ALP at physiological conditions. As shown in Figure 1H, ALP dephosphorylates pTP-Me at a much slower relative rate than dephosphorylating pTP, likely due to the structural hinder of methyl group and the aggregation of pTP-Me at the concentration higher than its CMC.
Figure 1.
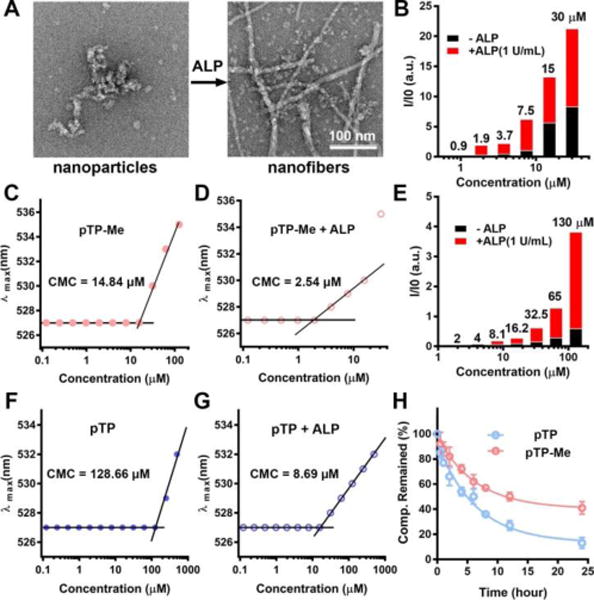
Before and after adding ALP (1 U/mL), (A) TEM images of the nanoparticles and nanofibers in the solutions of precursor pTP-Me (100 μM, pH 7.4), (B) SLS signals (at 30°) of the solution of pTP-Me and (C, D) CMCs of pTP-Me. (E) SLS signals (at 30°) of the solution of pTP and (F, G) CMCs of pTP before and after adding ALP (1 U/mL). (H) Time-dependent course of dephosphorylation of pTP-Me and pTP (500 μM) treated by ALP (0.01 U/mL).
Cytotoxicity of the designed molecules
Contrary to our observation of the cytotoxicity of a methylated phosphotripeptide,32 pTP-Me, though having higher self-assembling ability than pTP, exhibits less cytotoxicity than pTP does against several cancer cell lines (e.g., HeLa, Saos-2, SK-OV-3, and A2780cis) (Figure 2, Table 1). While pTP has an IC50 of 100, 120, 300, and 500 μM against HeLa, Saos-2, A2780cis, and SK-OV-3 cells, respectively, which largely agree with the ALP activity from these cells,33 the IC50 of pTP-Me against these two cells lines are higher than 500 μM (Table 1). Since the self-assembly ability and enzymatic dephosphorylation together dictate the biological activities of these precursors, the weaker cytotoxicity is likely due to the slower dephosphorylation rate of pTP-Me (Figure 1H). For example, after being incubated with ALP for 24 h, 87 % and 55 % of pTP and pTP-Me turn into TP and TP-Me, respectively. The unexpected low cytotoxicity pTP-Me makes it an ideal candidate to test the synergistic effect of pTP-Me and BAY for killing cancers.
Figure 2.
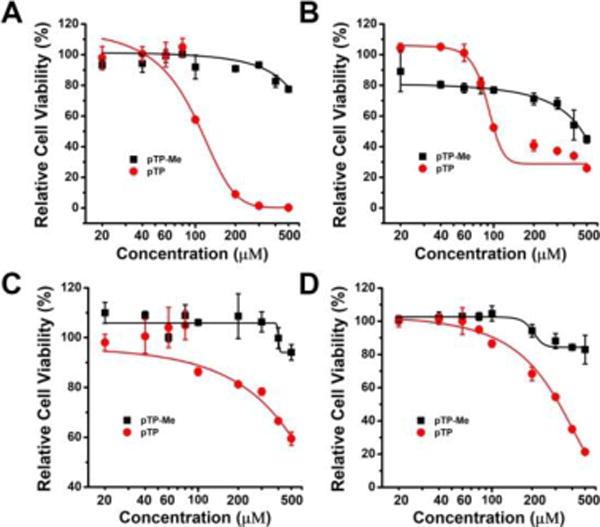
48-hour relative cell viability of (A) Saos-2, (B) HeLa, (C) SK-OV-3, and (D) A2780cis cells treated by pTP-Me and pTP at different concentrations (20, 40, 60, 80, 100, 200, 300, 400, and 500 μM).
Table 1.
IC50 summary of pTP-Me and pTP against different cells lines shown in Figure 2.
| Cell lines | IC50 (μM) | |
|---|---|---|
| pTP-Me | pTP | |
| Saos-2 | >500 | 120 |
| HeLa | >500 | 100 |
| SK-OV-3 | >500 | 500 |
| A2780cis | >500 | 300 |
Synergism between EISA and NF-κB targeting
The combination of pTP-Me (20-500 μM) and a NF-κB inhibitor (BAY, 8-20 μM) potently inhibit Saos-2 cells (Figure 3A) while neither of them alone significantly inhibits the Saos-2 cells (Figure 2A, Figure 3B) at the concentrations used for the combination. Notably, the combination of 20 μM pTP-Me (i.e., 17.7 μg/ml) and 16 μM BAY (i.e., 4.0 μg/ml) inhibits more than 95% Saos-2 cells, suggesting an exceptionally strong synergism. An ALP inhibitor (e.g., DQB35) is able to rescue 60% of Saos-2 cells treated by the combination of pTP-Me and BAY (Figure 3C), indicating the involvement of EISA for the synergistic inhibition of the cancer cells.
Figure 3.
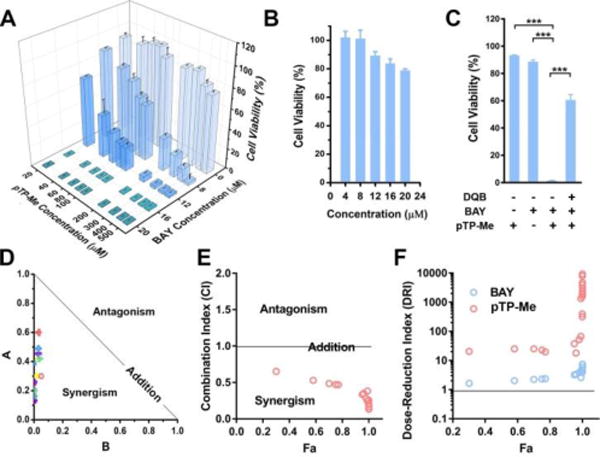
(A) 48-hour cell viability of Saos-2 cells incubated with pTP-Me (20, 40, 60, 80, 100, 200, 300, 400, and 500 μM) in combination with BAY (0, 8, 12, 16, and 20 μM). (B) 48-hour cell viability of Saos-2 cells incubated with BAY at different concentrations (4, 8, 12, 16, and 20 μM). (C) ALP inhibitor (DQB, 10 μM) significantly rescues Saos-2 cells treated by the combination of pTP-Me (100 μM) and BAY (10 μM). ***: p<0.001. (D) Normalized Isobologram,34 (E) Combination Index (CI)34 plot and (F) Dose-Reduction Index (DRI)34 plots for the combination of pTP-Me and BAY. All the analysis shown in (C-E) was based on the combination: pTP-Me 20-500 μM and BAY 12, 16, 20 μM, 27 data points in total.
To quantitatively evaluate the therapeutic effect of the combination of pTP-Me and BAY, we calculated the Combination Index (CI) (Table 2), a term quantitatively depicts synergism (CI < 1), additive effect (CI = 1), and antagonism (CI > 1), and plotted Dose-Reduction Index (DRI)-Fa diagram and normalized isobologram, according to Chou-Talalay method.34 As shown in the normalized isobologrm (Figure 3C), all these 27 combinations locate on the lower-left of the hypotenuse, confirming the synergistic effect (Table 3). Moreover, most of the data points in CI-Fa plot fall into the range of CI < 0.3 (Figure 3D), indicating strong synergism (Table 2). Since a major aim for achieving synergism in drug combination is dose reduction for reducing toxicity to host, DRI denotes how many fold of dose-reduction is allowed for each drug due to synergism when compared with each drug alone. When inhibition (Fa) is very close to 1 (100% inhibition), the calculated DRI value will become extremely large, which in most case is meaningless. Thus, according to the data points on the left side of the DRI diagram (Figure 3E), there is a 10-20 fold and 2-fold dose-reductions for pTP-Me and for BAY, respectively, further confirming the exceptionally strong synergism between EISA (of pTP-Me) and NF-ĸB targeting.
Table 2.
Determination of synergism and antagonism using CI analysis
| CI | Description |
|---|---|
| < 0.1 | Very strong synergism |
| 0.1 - 0.3 | Strong synergism |
| 0.3 – 0.7 | Synergism |
| 0.7 - 0.85 | Moderate synergism |
| 0.85 – 0.9 | Slight synergism |
| 0.9 – 1.1 | Nearly additive |
| 1.1 – 1.2 | Slight antagonism |
| 1.2 – 1.45 | Moderate antagonism |
| 1.45 – 3.3 | Antagonism |
| 3.3 – 10 | Strong antagonism |
| > 10 | Very strong antagonism |
Table 3.
Determination of synergism and antagonism using isobologram.
| Data Points | Illustrated Diagnosis |
|---|---|
| On the lower-left of the hypotenuse | Synergism |
| On the upper-right of the hypotenuse | Antagonism |
| On the hypotenuse | Addictive effect |
We also examined synergistic effect on the combination of pTP-Me and BAY in the case of HeLa cells, which is less significant than that on Saos-2 cells. As shown in Figure 4A, pTP-Me itself exhibits low cell inhibitory activity, but kills most of the HeLa cells when being combined with 20 μM BAY. However, the combination becomes almost ineffective when BAY concentration decreases to 16 μM. According to Chou-Talalay analysis (Figure 4B, C, D), most of the data points still locate in the area of synergism, but some are very close to or even on or above the hypotenuse representing additive effects in isobologram graph and have a calculated CI value equals to 1 or slightly below 1, indicating weaker synergism than that on Saos-2 cells. In addition, the DRI values are also lower than that on Saos-2 cell (Figure 3E). This result originates from the different ALP expressions between these two cell lines,36 implying the cell selectivity of this combination, which is reasonable since different cancer cells likely depend on different survival mechanisms.
Figure 4.
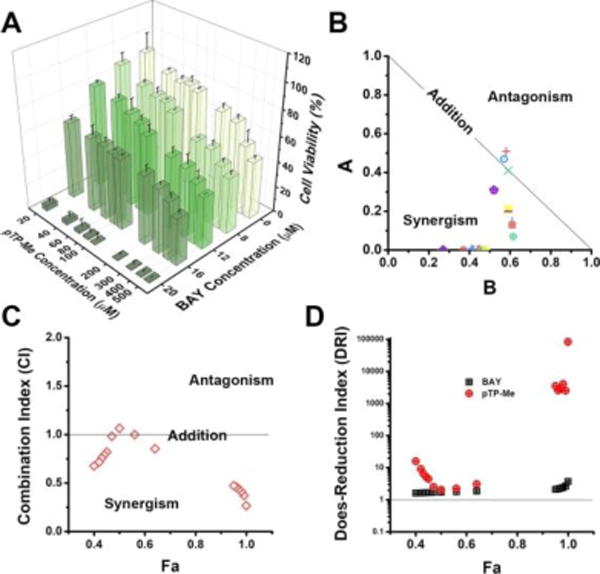
(A) 48-hour cell viability of HeLa cells incubated with pTP-Me (20, 40, 60, 80, 100, 200, 300, 400, and 500 μM) in combination with BAY (0, 8, 12, 16, and 20 μM). (B) Normalized Isobologram,34 (D) Combination Index (CI)34 plot and (D) Dose-Reduction Index (DRI)34 plots for the combination of pTP-Me and BAY. All the analysis shown in (C-D) was based on the combination: pTP-Me 20-500 μM and BAY 16, 20 μM, 18 data points in total.
The combination of pTP and BAY only exhibits weak synergism or additive effect on Saos-2 cells (Fig. S15), while only additive effect or even antagonism on HeLa cells (Fig. S16), further validating that the C-terminal methylation is critical for the observed high synergism. In addition, the synergism between pTP-Me and BAY occurs on other cancer cell line (e.g., A2780cis, a drug resistant ovarian cancer cell (Fig. S17)). Moreover, the combination of pTP-Me and BTZ, a different NF-κB inhibitor, also exhibits strong synergism although BTZ and BAY inhibit NF-κB signaling via different mechanisms (Figure 5).37 As shown in Figures 5A, pTP-Me, combined with only 100 nM BTZ, potently inhibits Saos-2 cells, while neither of them shows strong cytotoxicity alone (Fig. S18). CI-Fa plotting confirms very strong synergism between pTP-Me and BTZ (Figure 5B, Table 2). These observations suggest pTP-Me to be a promising therapeutic agent in the combination therapy that employs various NF-κB targeting molecules. There is little synergism observed between TP or TP-Me and BAY (Fig. S19), further indicating the importance of EISA and C-terminal methylation for the observed synergism.
Figure 5.
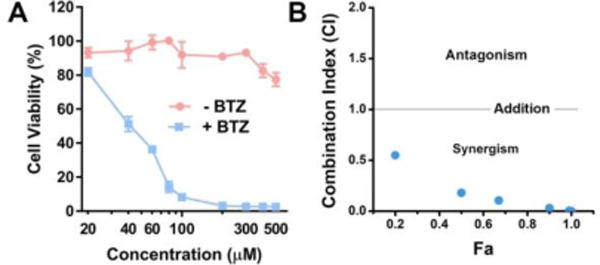
(A) 48-hour cell viability of Saos-2 cells incubated with different concentrations of pTP-Me (20, 40, 60, 80, 100, 200, 300, 400, and 500μM) in combination with BTZ (100 nM). (B) CI-Fa34 plotting for the combination of pTP-Me and BTZ. All the analysis shown in (B) was based on the combination: pTP-Me 20-500 μM and BTZ 100 nM, 9 data points in total.
Intracellular EISA
pTP-Me forms nanoparticles, which would be up-taken by the cells. To understand cellular distribution of pTP-Me, we synthesized f-pTP-Me as a structural analog of pTP-Me (Figure 6A, Fig. S6-S9). The imaging contrast conferred by the assemblies consisting of NBD-conjugated self-assembling peptides allows us to evaluate the location of EISA in cellular environment.38 As shown in Figure 6B, f-pTP-Me undergoes EISA on and inside Saos-2 cells to form extra- and intracellular nanofibers. This result implies that, besides acting as the substrate of extracellular EISA, pTP-Me allows intracellular EISA, likely due to that the assemblies/nanoparticles of pTP-Me, forming prior to EISA, favor cell uptake and undergo dephosphorylation inside cells. The precursor without C-terminal methylation, f-pTP, forms much more extra- and intracellular fluorescent nanofibers than f-pTP-Me does under the same condition (Fig. S20), which explains the higher cytotoxicity of pTP than that of pTP-Me.
Figure 6.
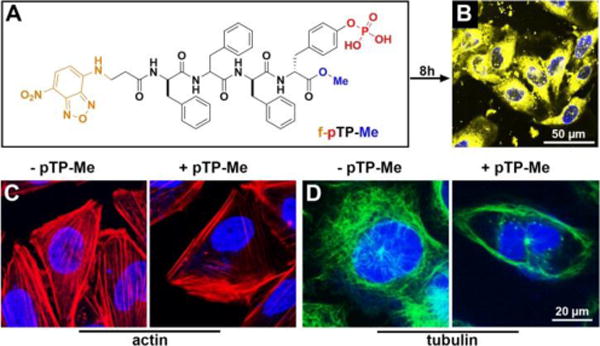
(A) Molecular structure of f-pTP-Me. (B) Fluorescent image of Saos-2 cells treated by f-pTP-Me (400 μM) for 8 h. (C) (D) The distribution of (C) actin and (D) tubulin in Saos-2 cells treated by pTP-Me (100 μM) for 4 hours.
Examining the changes of actin filaments and microtubules in Saos-2 cells treated by pTP-Me suggests that the formation of nanofibers dramatically disrupts cytoskeletons (Figures 6C, 6D, S21, and S22). That is, in the cells treated by pTP-Me, only few actin filaments and microtubules attach to the cell membrane while there are numerous well-defined actin filaments and microtubule stretching through the control cells. The addition of BAY barely influences the polymerization of either actin or tubulin and hardly aggravates the disruption of cytoskeletons in cells treated by pTP-Me (Figs. S21 and S22). These results suggest that the resulting extra- and intracellular nanofibers from EISA of pTP-Me cause considerable cell stress (Scheme 1).
Expression of signaling molecules
Using western blot (WB), we investigated the possible mechanisms underlying the anticancer activity of the combination of pTP-Me and BAY against Saos-2 cells by focusing on NF-ĸB signaling, an important factor and potential target in cancer therapy.39 The stresses conferred by EISA of pTP-Me alone lead to an inductive expression of TNFR2 and decreased expression of three key proteins—PI3K, Akt,3030 and MEKK3—at the up-stream of NF-κB pathway in Saos-2 cells as a response to the cellular stress. Moreover, the expressions of IKKα, IKKβ, and IĸBα, at down-stream of MEKK3, decline (Figure 7A, B (left panel)). These results indicate that the action of the nanofibers formed by EISA of pTP-Me results in the down regulation of Akt/PI3K/MEKK3 signaling, thus modulating constitutive and inducible NF-ĸB activation. The activation of NF-ĸB signaling, as the survival response to stress, keeps the cells alive (Scheme 1). Despite that BAY alone only increases the expression of TNFR1 and hardly influences the expressions of other proteins mentioned above (Figure 7A), the combination BAY with EISA, however, blocks NF-ĸB activation, thus further decreases the expressions of those up-stream proteins (i.e., PI3K, Akt,30 30 and MEKK3) and the down-stream proteins (i.e., IKKα, IKKβ, and IĸBα), which eventually results in cell death (Scheme 1 and Figure 7). The down regulation of Akt, MEKK3, IKKα, IKKβ, and IĸBα depends on the concentration of pTP-Me (Figure 8). Such a dose-dependent response supporting that the EISA of pTP-Me is able to modulate NF-ĸB signaling.
Figure 7.

(A) Western blot shows the protein variations in Saos-2 cells treated by pTP-Me, BAY, and the combination of pTP-Me and BAY 11-7085 for different time (e.g., 4, 8, 12, 24, 36 hours). (B) Quantification of protein variation over time in Saos-2 cells treated by pTP-Me or the combination of pTP-Me and BAY (Table S2, S3).
Figure 8.
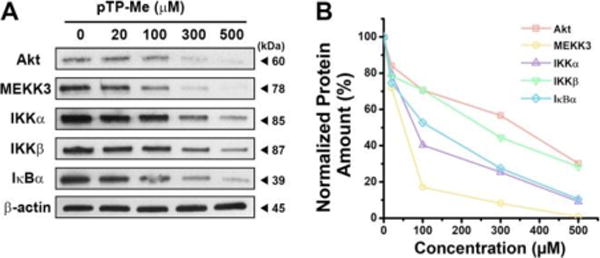
(A) Western blot shows the concentration dependent protein variations in Saos-2 cells treated by pTP-Me for 24 hours. (B) Quantification of protein variation in Saos-2 cells treated by pTP-Me at different concentrations for 24 hours (Table S4).
In a control experiment, treating Saos-2 cells by pTP also leads to an inductive expression of TNFR2, but barely influences the expression levels of Akt, PI3K, or MEKK3 (Fig. S23), as well as the expressions of those down-stream proteins (e.g., IKKα, IKKβ, and IĸBα). This observation indicates a different cell killing mechanism, which corresponds with different cytotoxicity of pTP-Me and pTP against Saos-2 cells and underscores the notion that self-assembly properties of small molecules is a critical factor to affect how their assemblies to modulate cell signaling. Meanwhile, since BAY increases the expression of TNFR1 (Figure 7A), the combination of pTP and BAY results an increase in both TNFR1 and TNFR2, but hardly causes any difference in the expression of other aforementioned signaling proteins over time. Although the lower CMC of pTP-Me likely is the key reason that pTP-Me has much better outcome compared with pTP in the combination with NF-κB inhibitors, other factors remain to be determined. We speculate that the aggregates of pTP-Me likely lead to more endoplasmic reticulum (ER) stress than that caused by pTP, which would decrease PI3K and Akt.40 In addition, except the increase in TNFR2, the variation of the protein expression (Fig. S24) of the HeLa cells treated by pTP-Me differ from those of Saos-2, confirming the selectivity of the combination of EISA and NF-kB targeting. Although the concept of the work is straightforward, the details of enzyme actions are rather sophisticated because both cell surface and intracellular phosphatases contribute to EISA, which may lead to the different activities of the combination against different cell lines. Moreover, the expression of esterases in HeLa cells may contribute to the moderate synergistic effect of pTP-Me and BAY on HeLa cells.
The modality of cell death
To examine the cell death mode induced by the combination of pTP-Me and BAY, we investigated the effects of zVAD-fmk, a pan-caspase inhibitor,41 and Nec-1, a necroptosis inhibitor,42 on cell death caused by pTP-Me. zVAD-fmk aggravates the Saos-2 cell death caused by pTP-Me, but hardly affects the cytotoxicity of the combination of pTP-Me and BAY; Nec-1 barely changes the cytotoxicity of pTP-Me, but rescues the cells treated by the combination of pTP-Me and BAY (Figure 9, Fig. S25). These results indicate that the combination of pTP-Me and BAY results in necroptosis of Saos-2 cells.
Figure 9.
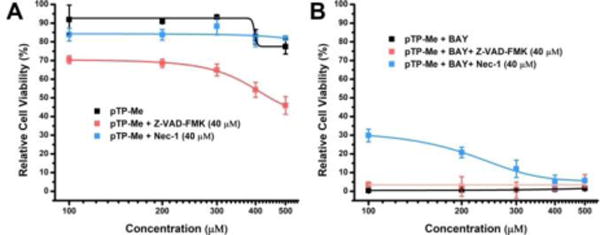
48-hour cell viability of Saos-2 cells incubated with (A) pTP-Me (100, 200, 300, 400, and 500 μM) or (B) the combination of pTP-Me and BAY (20 μM) with and without the presence of pan-caspase inhibitor (Z-VAD-FMK, 40 μM) or necroptosis inhibitor (Nec-1, 40 μM).
Drug resistance test
Combinational therapy is particularly attractive as it may result in a decrease in the onset of drug resistance. We designed an easy experiment to test whether the combination of EISA and NF-ĸB targeting will lead to acquired drug resistance. We maintained the Saos-2 cells in complete growth medium with BAY (4 μM), pTP-Me (100 μM), or the combination of BAY (4 μM) and pTP-Me (100 μM) in three culture dishes for 4 weeks. Four weeks later, we tested the cytotoxicity of BAY, pTP-Me and the combination of pTP-Me and BAY on both wild type cells and the cells pretreated by BAY and pTP-Me at suboptimal dosages. As shown in Figure 10A and 10B, constant treatment of BAY or pTP-Me alone both evokes resistance in Saos-2 cells. For example, the IC50 of BAY increases from 27 μM to 38 μM and 500 μM pTP-Me hardly show any cytotoxicity after 4 weeks. Meanwhile, the constant exposure of Saos-2 cells to the combination of BAY and pTP-Me hardly causes any drug resistance (Figure 10 C). This result indicates that the integration of EISA with NF-ĸB targeting promises a fundamentally new approach for developing anticancer therapeutics that may counter drug resistance.
Figure 10.
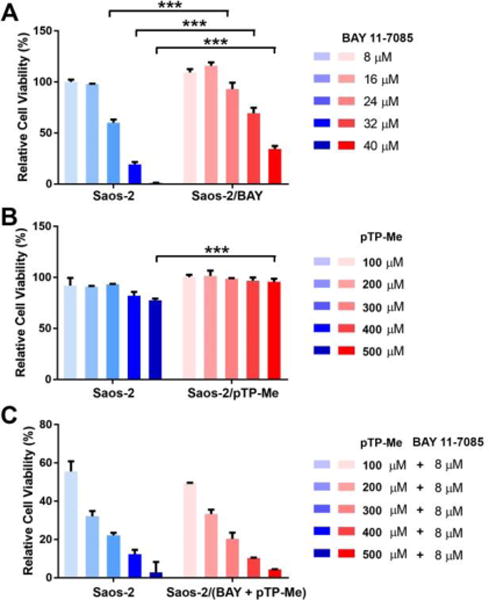
Relative cell viability (48-hour) of wide type Saos-2 cells (blue) or drugged Saos-2 cells (red) treated by (A) BAY 11-7085, (B) pTP-Me, and (C) the combination of pTP-Me and BAY 11-7085. Drugged Saos-2 cells were constantly maintained in complete growth medium with (A) 4 μM BAY 11-7085, (B) 100 μL pTP-Me and (C) the combination of 4 μM BAY 11-7085 and 100 μL pTP-Me for 4 weeks.
Conclusion
In conclusion, we demonstrate the exceptionally strong synergism between the nanofibers formed by EISA and NF-κB targeting, which turns the innocuous nanofibers into the cell death signals to cause necroptosis in cancer cells. The ability of EISA involved with pTP-Me for modulating the expression of TNFR2, PI3K, Akt, MEKK3, IKKs, and IĸB is significant because these proteins are the key proteins in cell signaling circuitry. On the other hand, NF-ĸB activation is essential for cell survival under stress. By synergistically modulating NF-ĸB activation (e.g., regulating the key proteins: Akt/PI3K/MEKK3 and IKKα/β), this combination possibly disrupts all the supporting pathways for cancer cell survival, thus achieves strong synergistic effects for dosage and toxicity reduction (Figure 3), as well as for decrease of the emergence of drug resistance (Figure 10).
In essence, EISA generated nanofibers inside cells disrupt the cell homeostasis, which likely upregulate the cellular processes that restore the cellular homeostasis. Such disruption in homeostasis by the nanofibers, which yields a non-lethal growth impairment, combines with other types of perturbations (e.g., NF-ĸB blocking) to result in a form of “synthetic lethality” (Scheme 3).43 Besides NF-ĸB inhibitors, the perturbation can also be the overexpression of genes, the action of a chemical compound, or environmental change. The concept demonstrated in this work promises a new class of anticancer therapies and future anticancer strategies that will increasingly consist of combination therapies based on personalized tumor vulnerabilities. Meanwhile, the use of alkaline phosphatase-instructed self-assembly is particular important because of (i) the selectivity conferred by the overexpression of certain ALPs on cancer cells,6b (ii) the easy control of local formation of nanofibers at targeting sites, and (iii) the reduced probability of resistance. As a general process to disrupt the homeostasis of cancer cells selectively, EISA is able to combine with the inhibitors targeting other essential node proteins in other pathways for anticancer therapy. In addition, this work provides useful insights for understanding the cytotoxicity of aberrant protein or peptide aggregates in the context of NF-ĸB modulation and the insights into gene function and drug action.
Scheme 3.
Cancer cells that have lost key functions (e.g., cytoskeleton protein dynamics) become reliant on backup pathways (e.g., NF-κB signaling) so that “synthetic lethality” can be exploited to kill cancers.
Supplementary Material
Acknowledgments
This work is partially supported by NIH (R01CA142746), the W.M. Keck Foundation, NSF (DMR-1420382). J.Z. is an HHMI International Research Fellow.
Footnotes
Supporting Information
See supporting information for synthetic procedures, NMR and LC-MS characterization, and other supplementary results. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Hanahan D, Weinberg RA. cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.(a) Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C. Science. 2007;318:287–290. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]; (b) Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JPJ. Biol Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]; (c) Hayes JD, Pulford DJ. Crit Rev Biochem Mol Biol. 1995;30:521–600. doi: 10.3109/10409239509083491. [DOI] [PubMed] [Google Scholar]; (d) Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML. Nature. 2010;467:1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Negrini S, Gorgoulis VG, Halazonetis TD. Nat Rev Mol Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]; (f) Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Campbell LL, Polyak K. Cell cycle. 2007;6:2332–2338. doi: 10.4161/cc.6.19.4914. [DOI] [PubMed] [Google Scholar]; (h) Mantovani A, Allavena P, Sica A, Balkwill F. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]; (i) Vaupel P, Kallinowski F, Okunieff P. Cancer Res. 1989;49:6449–6465. [PubMed] [Google Scholar]
- 3.Huang L, Zhao Y, Zhang H, Huang K, Yang J, Han G. Angew Chem Int Ed. 2017 Ahead of Print. [Google Scholar]
- 4.(a) Acar H, Ting JM, Srivastava S, LaBelle JL, Tirrell MV. Chem Soc Rev. 2017;46:6553–6569. doi: 10.1039/c7cs00536a. [DOI] [PubMed] [Google Scholar]; (b) van Genabeek B, de Waal BFM, Gosens MMJ, Pitet LM, Palmans ARA, Meijer EW. J Am Chem Soc. 2016;138:4210–4218. doi: 10.1021/jacs.6b00629. [DOI] [PubMed] [Google Scholar]; (c) Jin W, Fukushima T, Niki M, Kosaka A, Ishii N, Aida T. Proc Natl Acad Sci U S A. 2005;102:10801–10806. doi: 10.1073/pnas.0500852102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shao Y, Jia H, Cao T, Liu D. Acc Chem Res. 2017;50:659–668. doi: 10.1021/acs.accounts.6b00524. [DOI] [PubMed] [Google Scholar]; (e) Zhang Q, Jiang Q, Li N, Dai L, Liu Q, Song L, Wang J, Li Y, Tian J, Ding B, Du Y. ACS Nano. 2014;8:6633–6643. doi: 10.1021/nn502058j. [DOI] [PubMed] [Google Scholar]; (f) Yu G, Zhao R, Wu D, Zhang F, Shao L, Zhou J, Yang J, Tang G, Chen X, Huang F. Polym Chem. 2016;7:6178–6188. doi: 10.1039/C6PY01402J. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Kato T, Kihara H, Uryu T, Fujishima A, Frechet JMJ. Macromolecules. 1992;25:6836–6841. [Google Scholar]; (h) Uchida J, Yoshio M, Sato S, Yokoyama H, Fujita M, Kato T. Angew Chem Int Edit. 2017;56:14085–14089. doi: 10.1002/anie.201707740. [DOI] [PubMed] [Google Scholar]; (i) Chen J, McNeil AJ. J Am Chem Soc. 2008;130:16496–16497. doi: 10.1021/ja807651a. [DOI] [PubMed] [Google Scholar]; (j) Minodani S, Owaki M, Sano S, Tsuzuki S, Yamanaka M. Chem Commun. 2015;51:12920–12923. doi: 10.1039/c5cc05227k. [DOI] [PubMed] [Google Scholar]; (k) Ozawa H, Fujigaya T, Niidome Y, Hotta N, Fujiki M, Nakashima N. J Am Chem Soc. 2011;133:2651–2657. doi: 10.1021/ja109399f. [DOI] [PubMed] [Google Scholar]; (l) Mallia VA, Weiss RG. J Phys Org Chem. 2014;27:310–315. [Google Scholar]; (m) Kim BJ, Han S, Lee KB, Choi IS. Adv Mater (Weinheim, Ger) 2017;29 doi: 10.1002/adma.201700784. n/a. [DOI] [PubMed] [Google Scholar]; (n) Nagy-Smith K, Beltramo PJ, Moore E, Tycko R, Furst EM, Schneider JP. ACS Cent Sci. 2017;3:586–597. doi: 10.1021/acscentsci.7b00115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Nagy-Smith K, Moore E, Schneider J, Tycko R. Proc Natl Acad Sci U S A. 2015;112:9816–9821. doi: 10.1073/pnas.1509313112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Yang Z, Liang G, Xu B. Acc Chem Res. 2008;41:315–326. doi: 10.1021/ar7001914. [DOI] [PubMed] [Google Scholar]; (b) Yang Z, Gu H, Fu D, Gao P, Lam JK, Xu B. Adv Mater. 2004;16:1440–1444. [Google Scholar]; (c) Pires RA, Abul-Haija YM, Costa DS, Novoa-Carballal R, Reis RL, Ulijn RV, Pashkuleva I. J Am Chem Soc. 2015;137:576–579. doi: 10.1021/ja5111893. [DOI] [PubMed] [Google Scholar]; (d) Yang ZM, Xu KM, Guo ZF, Guo ZH, Xu B. Adv Mater. 2007;19:3152–3156. [Google Scholar]; (e) Zhou Z, Xie X, Yi Q, Yin W, Kadi AA, Li J, Zhang Y. Org Biomol Chem. 2017 doi: 10.1039/c7ob01548h. Ahead of Print. [DOI] [PubMed] [Google Scholar]
- 6.(a) Zhou J, Xu B. Bioconjugate Chem. 2015;26:987–999. doi: 10.1021/acs.bioconjchem.5b00196. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhou J, Du X, Xu B. Angew Chem, Int Ed. 2016;55:5770–5775. doi: 10.1002/anie.201600753. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li J, Kuang Y, Shi J, Zhou J, Medina JE, Zhou R, Yuan D, Yang C, Wang H, Yang Z, Liu J, Dinulescu DM, Xu B. Angew Chem, Int Ed. 2015;54:13307–13311. doi: 10.1002/anie.201507157. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tanaka A, Fukuoka Y, Morimoto Y, Honjo T, Koda D, Goto M, Maruyama T. J Am Chem Soc. 2015;137:770–775. doi: 10.1021/ja510156v. [DOI] [PubMed] [Google Scholar]
- 7.Wang H, Feng Z, Wang Y, Zhou R, Yang Z, Xu B. J Am Chem Soc. 2016;138:16046–16055. doi: 10.1021/jacs.6b09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlier MF. Phil Trans R Soc B. 1992;336:93–97. doi: 10.1098/rstb.1992.0048. [DOI] [PubMed] [Google Scholar]
- 9.Gao Y, Yang Z, Kuang Y, Ma ML, Li J, Zhao F, Xu B. Biopolymers. 2010;94:19–31. doi: 10.1002/bip.21321. [DOI] [PubMed] [Google Scholar]
- 10.(a) Du X, Zhou J, Wang J, Zhou R, Xu B. ChemNanoMat. 2017;3:17–21. doi: 10.1002/cnma.201600258. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) MacLaughlin CM, Wilson BC, Hedley DW, Ding L, Cao P, Siddiqui I, Chen J, Jin C, Zheng G, Hwang DM. J Biomed Opt. 2016;21:84002. doi: 10.1117/1.JBO.21.8.084002. [DOI] [PubMed] [Google Scholar]
- 11.(a) Kalafatovic D, Nobis M, Son J, Anderson KI, Ulijn RV. Biomaterials. 2016;98:192–202. doi: 10.1016/j.biomaterials.2016.04.039. [DOI] [PubMed] [Google Scholar]; (b) Du X, Zhou J, Wu L, Sun S, Xu B. Bioconjugate Chem. 2014;25:2129–2133. doi: 10.1021/bc500516g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shigemitsu H, Hamachi I. Acc Chem Res. 2017;50:740–750. doi: 10.1021/acs.accounts.7b00070. [DOI] [PubMed] [Google Scholar]
- 13.Wang HM, Luo Z, Wang YCZ, He T, Yang CB, Ren CH, Ma LS, Gong CY, Li XY, Yang ZM. Adv Funct Mater. 2016;26:1822–1829. [Google Scholar]
- 14.Conley NR, Dragulescu-Andrasi A, Rao JH, Moerner WE. Angew Chem, Int Ed. 2012;51:3350–3353. doi: 10.1002/anie.201105653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin YX, Qiao SL, Wang Y, Zhang RX, An HW, Ma Y, Rajapaksha R, Qiao ZY, Wang L, Wang H. Acs Nano. 2017;11:1826–1839. doi: 10.1021/acsnano.6b07843. [DOI] [PubMed] [Google Scholar]
- 16.Yang Z, Liang G, Guo Z, Guo Z, Xu B. Angew Chem, Int Ed. 2007;46:8216–8219. doi: 10.1002/anie.200701697. [DOI] [PubMed] [Google Scholar]
- 17.Kuang Y, Miki K, Parr CJC, Hayashi K, Takei I, Li J, Iwasaki M, Nakagawa M, Yoshida Y, Saito H. Cell Chem Biol. 2017;24:685–694.e684. doi: 10.1016/j.chembiol.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 18.Sen R, Baltimore D. Cell. 1986;46:705–716. [PubMed] [Google Scholar]
- 19.Zhang Q, Lenardo MJ, Baltimore D. Cell. 2017;168:37–57. doi: 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A, Palombella V, Adams J, Anderson KC. Journal of Biological Chemistry. 2002;277:16639–16647. doi: 10.1074/jbc.M200360200. [DOI] [PubMed] [Google Scholar]
- 21.Nakanishi C, Toi M. Nature Reviews Cancer. 2005;5:297–309. doi: 10.1038/nrc1588. [DOI] [PubMed] [Google Scholar]
- 22.Baeuerle P, Baltimore D. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 23.Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, Gerritsen ME. Journal of Biological Chemistry. 1997;272:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- 24.Kuang Y, Long MJC, Zhou J, Shi JF, Gao Y, Xu C, Hedstrom L, Xu B. J Biol Chem. 2014;289:29208–29218. doi: 10.1074/jbc.M114.600288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou R, Kuang Y, Zhou J, Du X, Li J, Shi J, Haburcak R, Xu B. PLoS One. 2016;11:e0154126/0154121–e0154126/0154118. doi: 10.1371/journal.pone.0154126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pahl HL. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 27.Lock LL, Reyes CD, Zhang P, Cui H. J Am Chem Soc. 2016;138:3533–3540. doi: 10.1021/jacs.6b00073. [DOI] [PubMed] [Google Scholar]
- 28.Locksley RM, Killeen N, Lenardo MJ. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 29.(a) Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JFM. Curr Cancer Drug Targets. 2008;8:187–198. doi: 10.2174/156800908784293659. [DOI] [PubMed] [Google Scholar]; (b) Kaileh M, Vazquez E, MacFarlane AWIV, Campbell K, Kurosaki T, Siebenlist U, Sen R. PLoS One. 2016;11:e0146955/0146951–e0146955/0146916. doi: 10.1371/journal.pone.0146955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hers I, Vincent EE, Tavare JM. Cell Signal. 2011;23:1515–1527. doi: 10.1016/j.cellsig.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 31.Blank JL, Gerwins P, Elliott EM, Sather S, Johnson GL. J Biol Chem. 1996;271:5361–5368. doi: 10.1074/jbc.271.10.5361. [DOI] [PubMed] [Google Scholar]
- 32.Feng Z, Wang H, Du X, Shi J, Li J, Xu B. Chem Commun. 2016;52:6332–6335. doi: 10.1039/c6cc02282k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou J, Du X, Yamagata N, Xu B. J Am Chem Soc. 2016;138:3813–3823. doi: 10.1021/jacs.5b13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chou TC. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 35.Dahl R, Sergienko EA, Su Y, Mostofi YS, Yang L, Simao AM, Narisawa S, Brown B, Mangravita-Novo A, Vicchiarelli M, Smith LH, O’Neill WC, Millan JL, Cosford NDP. J Med Chem. 2009;52:6919–6925. doi: 10.1021/jm900383s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou J, Du X, Berciu C, He H, Shi J, Nicastro D, Xu B. Chem. 2016;1:246–263. doi: 10.1016/j.chempr.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adams J, Kauffman M. Cancer Invest. 2004;22:304–311. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- 38.Gao Y, Shi JF, Yuan D, Xu B. Nat Commun. 2012;3 doi: 10.1038/ncomms2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoesel B, Schmid JA. Mol Cancer. 2013;12:86. doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang W, Neo SP, Gunaratne J, Poulsen A, Boping L, Ong EH, Sangthongpitag K, Pendharkar V, Hill J, Cohen SM. Cell Signal. 2015;27:436–442. doi: 10.1016/j.cellsig.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 41.Julien O, Kampmann M, Bassik MC, Zorn JA, Venditto VJ, Shimbo K, Agard NJ, Shimada K, Rheingold AL, Stockwell BR, Weissman JS, Wells JA. Nat Chem Biol. 2014;10:969–976. doi: 10.1038/nchembio.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, Declercq W, Callewaert N, Prendergast GC, Degterev A, Yuan J, Vandenabeele P. Cell Death Dis. 2012;3:e437, 410. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Neil NJ, Bailey ML, Hieter P. Nat Rev. 2017;18:613–623. doi: 10.1038/nrg.2017.47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.