

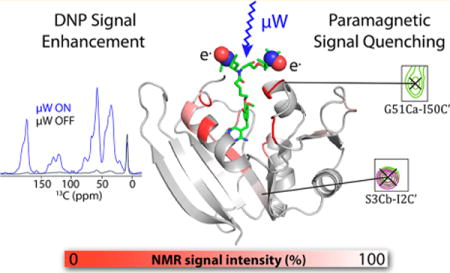
Abstract
We characterize the effect of specifically bound biradicals on the NMR spectra of dihydrofolate reductase from E. coli. Dynamic nuclear polarization methods enhance the signal-to-noise of solid state NMR experiments by transferring polarization from unpaired electrons of biradicals to nuclei. There has been recent interest in colocalizing the paramagnetic polarizing agents with the analyte of interest through covalent or noncovalent specific interactions. This experimental approach broadens the scope of dynamic nuclear polarization methods by offering the possibility of selective signal enhancements and the potential to work in a broad range of environments. Paramagnetic compounds can have other effects on the NMR spectroscopy of nearby nuclei, including broadening of nuclear resonances due to the proximity of the paramagnetic agent. Understanding the distance dependence of these interactions is important for the success of the technique. Here we explore paramagnetic signal quenching due to a bound biradical, specifically a biradical-derivatized trimethoprim ligand of E. coli dihydrofolate reductase. Biradical-derivatized trimethoprim has nanomolar affinity for its target, and affords strong and selective signal enhancements in dynamic nuclear polarization experiments. In this work, we show that, although the trimethoprim fragment is well ordered, the biradical (TOTAPOL) moiety is disordered when bound to the protein. The distance dependence in bleaching of NMR signal intensity allows us to detect numerous NMR signals in the protein. We present the possibility that static disorder and electron spin diffusion play roles in this observation, among other contributions. The fact that the majority of signals are observed strengthens the case for the use of high affinity or covalent radicals in dynamic nuclear polarization solid state NMR enhancement.
Graphical abstract
INTRODUCTION
Dynamic nuclear polarization (DNP) is an emerging method for sensitizing solid-state NMR experiments by transferring polarization from unpaired electrons to nearby nuclei.1–4 Due to the increased signal strength provided, DNP is becoming an important tool in surface chemistry,3 materials science,4 and structural biology.5–7 DNP is typically conducted at cryogenic temperatures in samples containing a paramagnetic compound. The ESR spectroscopic properties of the radical dictate, in part, how polarization is transferred to the nuclei, with different DNP mechanisms operative for different radicals. The cross effect is a three-spin mechanism involving two coupled electrons hyperfine coupled to at least one nucleus.8,9 This mechanism is efficient in favorable cases at high magnetic fields up to 800 MHz.10 The use of biradicals such as TOTAPOL11 and AMUPol,12 in which two nitroxide radicals are tethered to increase cross effect efficiency, has enabled excellent enhancements on many systems.13–15
While endogenous radicals such as organic16 or metal17 cofactors can provide DNP enhancement, most often a radical such as TOTAPOL is doped into a glycerol/water solvent matrix at millimolar concentrations with the analyte of interest. Recently, there has been interest in DNP experiments in which radicals are colocalized with the analyte of interest through covalent or noncovalent interactions (see review18 and references therein). The specificity of the interaction implies that sample perturbation can be controlled and reduced, since the radicals do not need to be codissolved at high concentrations with the biomolecule of interest. This approach enables DNP to be used in a broader range of sample conditions, such as membrane proteins in lipid bilayers,19–21 where the analyte of interest is not soluble in the traditional glycerol/water matrix and aggregation of the radical can be problematic. Moreover, selective enhancement of proteins using TOTAPOL affinity compounds22,23 demonstrates that DNP based on affinity compounds can be used to highlight dilute biomolecules in complex mixtures such as cellular lysates. Such experiments will enable structural and functional studies of proteins in their native environments.
The introduction of paramagnetic compounds can have other effects on nearby nuclei besides the desired polarization enhancement. These effects, which include shifts in the resonant frequency of the nuclei and increased nuclear relaxation, depend on the distance from the paramagnetic center. Contact shifts resulting from unpaired spin density centered at the nucleus are confined to a small radius around the unpaired electrons.24 Through-space pseudocontact shifts require an anisotropic electron g-tensor and can be ignored for nitroxide polarizing agents, for which the g-tensor anisotropy is too small to yield substantial effects.25
Paramagnetic relaxation enhancements (PREs) can, however, be considerable for the polarizing agents used in DNP studies. For nuclei within range, electron–nuclear couplings lead to increased effective transverse and longitudinal relaxation rates that broaden the nuclear resonances and lead to their decay during spin locks or other pulse sequence elements, sometimes beyond the limit of detection.26 This relaxation or “bleaching” due to nitroxides has been used extensively in NMR studies as a structural probe.27,28 In room temperature studies, these effects are typically analyzed within the framework outlined by the Solomon–Bloembergen equations,29,30 which predicts the paramagnet-induced T1 and T2 relaxation based on the correlation time τc of the electron–nuclear interaction. τc contains contributions from chemical exchange, electron T1e and T2e relaxation (≫T1n), and rotational correlation of the molecular system. In solid samples, the electron T1e is thought to drive relaxation in the absence of molecular rotation and chemical exchange. Under the conditions typically used for DNP—aqueous solvents at approximately 100 K—electronic relaxation is expected to change. High field measurements of both biradical and monoradical nitroxide compounds indicate that T1e increases considerably from the room temperature value of approximately 100 ns26 to the high microsecond/millisecond range at cryogenic temperatures,31–35 depending on nitroxide concentration and magnetic field.36,37 Moreover, in systems suitable for cross-effect DNP, an additional polarization quenching mechanism termed MAS-induced depolarization arises without the application of microwave (MW) irradiation.36,38,39 This effect is absent when microwaves are applied.
The paramagnetic effects described above manifest experimentally as a decrease in the NMR signal. The signal quenching due to nitroxides and other DNP polarizing agents at 100 K has been studied in a variety of systems ranging from small molecules to proteins with bound biradicals. Reduced MW-off signal intensity relative to a sample without paramagnetic cosolutes was initially observed in DNP samples of silica material impregnated with biradical.40 In samples of neurotoxin bound to a membrane transporter codissolved with TOTAPOL, solvent-exposed threonine peaks disappeared from 2D 13C–13C spectra.41 Oschkinat and co-workers measured the decrease in signal intensity for samples of proline codissolved with varying concentrations of TOTAPOL in a DNP matrix of water and glycerol and estimated that all nuclei closer than 10 Å to a radical center are bleached.42 Similarly, Corzilius et al.31 measured signal quenching for solutions of 13C-urea containing varying concentrations of polarizing agents and estimated a quenching radius of 21 Å for the monoradical TEMPO and 29 Å for TOTAPOL.
When the radical is bound to the protein of interest, paramagnetic bleaching and its distance dependence become very important, as the radical has a fixed distance from specific nuclei rather than stochastically bleaching through an assumedly random distribution of radicals. Ladizhansky and co-workers used methanethiosulfonate (MTS) chemistry to covalently attach TOTAPOL to cysteines in the membrane protein Anabaena sensory rhodopsin (ASR).20 In samples where every ASR monomer has an attached TOTAPOL, 70% attenuation of the overall 1H signal intensity, as measured via 1H–15N CP spectra, was observed. When TOTAPOL-derivatized ASR was diluted with unreacted monomers, signal quenching decreased. Bovine serum albumin has been observed to interact with TOTAPOL, as manifested in the dependence of DNP enhancements on the TOTAPOL:BSA ratio.43 At the TOTAPOL concentration associated with optimal enhancements, an NMR signal loss of ∼40% is observed. Neither of these studies correlated the observed bleaching with specific residues near the location of the biradical. By contrast, when Baldus and co-workers derivatized the membrane protein KcsA with AMUPol using MTS chemistry, specific peaks associated with residues spatially close to the derivatization site disappeared from 2D 15N–13C spectra.21
Here, we employ an affinity reagent for dihydrofolate reductase to study paramagnetic bleaching as a function of distance from a nitroxide biradical. Trimethoprim (TMP) is a high affinity ligand of E. coli dihydrofolate reductase (DHFR) that we previously derivatized with TOTAPOL for targeted DNP applications.22 While we previously reported reduced bulk signal bleaching with the bound biradical relative to concentrated TOTAPOL samples, localizing the biradical to the protein undoubtedly exerts paramagnetic relaxation effects on nearby residues. By mapping the pattern of signal attenuation for DNP samples of DHFR bound to the TOTAPOL-derivatized TMP, we are able to probe the spatial dependence of these effects. The distance dependence of the bleaching effect is of broad relevance to those interested in targeted DNP studies of biomolecules. Moreover, since TOTAPOL has been shown to bind to various biomolecules, including amyloid proteins,44 these results are of general interest for situations where binding of radicals is suspected.
MATERIALS AND METHODS
Protein Preparation and Purification
E. coli dihydrofolate reductase was overexpressed and purified as previously reported.22 After elution from the Ni-NTA affinity column, protein was concentrated and loaded onto a Superdex-75 HiLoad 16/60 FPLC column. Protein eluted at a sample volume of 80 mL, consistent with a molecular weight of 18 kDa. SDS-PAGE analysis indicated >95% purity. Protein was concentrated and flash-frozen for crystallography and NMR applications.
Specifically isotopically enriched DHFR was prepared as previously reported,22,45 with the modification that cells were grown to an OD600 of 0.6 in LB media and then transferred to amino-acid-defined media for overexpression of DHFR. Amino-acid-defined media was prepared with glycerol as the carbon source and 1.0 g/L of both isotopically enriched amino acids and all remaining essential amino acids at natural abundance. The purified protein was assessed to be >95% pure by SDS-PAGE. U–13C,15N,2H-DHFR was grown according to the protocol of Tugarinov and Kay.46
Crystallography
For experiments with TMP bound to DHFR, a 170 mM stock solution in DMSO was added to DHFR at approximately 10-fold molar excess and incubated for half an hour at 4 °C. For experiments with TMP-V-T, a 170 mM stock solution of TMP-V-T was added to the protein solution, whereupon a fine precipitate formed. EPR measurements of the supernatant from a solution prepared to be 2.5 mM gave an actual concentration of 0.82 mM. Therefore, solutions were prepared by adding the TMP-V-T stock to buffer, vortexing well to suspend the precipitate and saturate the solution, adding a concentrated protein stock, mixing gently, and incubating on ice for 30 min. Solutions were centrifuged at 18,000g for 10 min before setting up the crystallization trays; the supernatant was used for crystallization.
Diffraction quality crystals for TMP complex were grown using the microbatch method by mixing 3 μL drops of a 10 mg/mL DHFR/TMP solution with 3 μL of a solution containing 10% (w/v) PEG-8000 and 0.2 M (NH4)2SO3 and incubating under 12 μL of paraffin oil. Crystals for TMP-V-T were grown by microseeding from hanging drops prepared by mixing 1 μL of a 8 mg/mL DHFR/TMP-V-T solution with 1 μL of a reservoir solution containing 4% (w/v) PEG-8000, (0.2 M NH4)2SO3, and 4% (v/v) 1,1,1,3,3,3-hexafluoro-2-propanol.
X-ray diffraction data for the TMP complex were collected to 2.5 Å resolution at the Advanced Light Source (ALS). Diffraction images were processed with HKL2000.47 The structure was solved by molecular replacement with the program Phaser48 using the structure of DHFR in complex with NADPH as the search model. X-ray diffraction data for the TMP-V-T complex were collected to 1.95 Å resolution at the NE-CAT beamline of the Advanced Photon Source (APS). The structures were refined with PHENIX,49 and manual model rebuilding was carried out with Coot.50 Supporting Information Table 1 contains crystallographic parameters for both TMP and TMP-V-T crystals.
Solution NMR Experiments
Solution state NMR measurements were conducted on Bruker spectrometers at 500, 800, and 900 MHz 1H frequency at the New York Structural Biology Center. The sample temperature was 298 K. The DHFR sample was prepared in PBS at pH 7.3 with 5% D2O.
Using U–13C,15N,2H-DHFR, we assigned the solution NMR spectra for the ternary complex of DHFR with TMP/NADPH using triple resonance approaches, including TROSY-filtered HNCO, HNCA, HNCACB, and HN(CO)Ca experiments.51 Assignments were made using the algorithm PINE52 and validated manually by confirming inter-residue correlations for each residue using the HNCA, HNCACB, and HN(CO)CA experiments. To confirm the isotopic enrichment of selectively isotopically enriched samples, 2D 1H–15N and 1H–13C planes of HNCO and HNCA experiments were collected and compared with the assigned 3D spectra (Figure S8).
For paramagnetic relaxation and broadening studies, a sample of 300 μM U–13C/15N-DHFR was mixed with 300 μM TMP-V-T and 5 mM H2NADPH (to ensure cofactor saturation). 1H–15N HSQC experiments were collected in blocks to ensure that TMP-V-T was not reduced over time. Ascorbic acid was added (to a final concentration of 1 mM) to reduce TMP-V-T. The pH after addition of ascorbic acid was 7.2 (0.1 pH unit change). The sample was incubated at room temperature for 1 h, and then returned to the spectrometer. HSQC, HNCACB, and HNCO spectra were collected on the reduced sample to confirm assignments.
The HSQC spectra of the oxidized and reduced spectra were assigned using HNCO and HNCACb spectra collected on the reduced sample. The RMSD for 1H and 15N chemical shifts between the oxidized and reduced spectra is 0.001 and 0.008, respectively. The spectra were processed with 10 Hz exponential line broadening, and the center, amplitude, and line width were fit to a Lorentzian function in each dimension using the nonlinear fitting method in CCPN 2.4.53 Peak heights of the oxidized and reduced peaks were extracted, and the intensity ratio Iox/Ired was calculated by dividing the peak height of the oxidized sample by the peak height of the reduced sample. Intensity ratios were normalized (to account for dilution effects) by averaging the intensity ratio for residues 2, 128–133, and 155–158, which we estimate to be over 30 Å away from the paramagnetic center. As previously reported,54,55 intrinsic relaxation rates of the amide protons were estimated from the intrinsic line width of the reduced spectra and calculated using , where T2 is the relaxation time constant and R2 is 1/T2. Intensity ratios were corrected for the observed presence of a dilute diamagnetic component. R2PRE, the paramagnetic relaxation rate enhancement, was estimated from the intensity ratio and line width of the reduced spectra using the following equation: as in ref 54.
CW EPR Measurements
TMP-V-T stock solutions were diluted to 0.1 mM final concentration, and 10-fold molar excess of K3[Fe(SCN)6] was added to reoxidize any reduced radical. CW-EPR data were acquired at room temperature using a Bruker EMX X-band EPR spectrometer at a microwave frequency of 9.756 GHz, with a center field of 3480 G, a sweep width of 100 G, a modulation frequency of 100 kHz, a modulation amplitude of 1.00 G, a conversion time of 164 s, and a microwave power of 20.1 mW. The integrated intensity of the reoxidized sample was compared to that of a control without added K3[Fe(SCN)6] to determine the reduced population.
Molecular Modeling Calculations
Using the Maestro molecular modeling program, the crystal structure of the TMP-DHFR complex was derivatized manually with the linker and TOTAPOL moiety. Monte Carlo Multiple Minimum search methods were implemented in MacroModel56,57 to search the conformational space of the linker and TOTAPOL moiety. OPLS3 was used as the force field, and the GB/SA continuum solvation model was used with water as the solvent. Flexible atoms included the linker, TOTAPOL, and side chains within 10 Å of the TOTAPOL moiety. Atoms more than 6 Å away from flexible atoms were ignored. Output conformations were clustered, and the lowest energy conformations were reminimized using a Prime58 single point molecular mechanics energy calculation in the OPLS3 force field to get relative energies.
DNP Experiments
Table S2 contains details of sample composition for all samples characterized by DNP spectroscopy. DHFR samples containing TMP-V-T at 1:1 stoichiometry were prepared as previously reported22 by incubating 0.1 mM DHFR in 2H-PBS with approximately 8-fold molar excess of TMP-V-T. Unbound TMP-V-T was removed by coconcentrating and washing with 2H-PBS, followed by addition of 30 v/v d8-glycerol.22 For DNP measurements with the codissolved biradical AMUPol and bound cofactors, DHFR in 2H PBS was mixed with trimethoprim (170 mM in d6-DMSO) and, where noted, H2NADPH in 2H PBS, and incubated at 4 °C for 30 min. Then, 30% v/v d8-glycerol and AMUPol from a concentrated stock solution were added. All samples were centrifuged into 3.2 mm sapphire rotors for DNP measurement.
To assist in spectral assignments at low temperature, specifically isotopically enriched DHFR samples with 5 mM AMUPol, saturating TMP, and, where noted, H2NADPH, were prepared (see Table S2). Assignments were made using CaNCo spectra59 in combination with 2D 13C–13C correlation spectra at a range of mixing times (25–250 ms). The CaNCo spectra enable the Ca and N of Gly, Ala, and Ser to be correlated to the Co of the preceding amino acid (Leu, Pro, or Ile). Assignments were made on the basis of similarity to solution chemical shifts, as well as the available Cb chemical shifts (for serine) and Ca–Ca crosspeaks for I94/G95 (local sequence is IGG) and I5/A6 (local sequence is IAA). In total, 14 13Co–13Ca pairs can be confidently assigned to positions at a range of distances from the biradical, and two 13Ca–13Cb pairs (for L62 and S3) are available as well.
To calculate the intensity ratio ITMPVT/IAMUPol, DNP enhanced 13C–13C DARR spectra of LAPG and IAG isotopically enriched samples with 25 and 250 ms (IAG) and 50 ms (LAPG) mixing times were processed with 50 Hz of Gaussian line broadening; peak height, position, and line width were fit using nonlinear fitting methods in the CCPN60 software package. Peak heights were extracted and intensity ratios between AMUPol and TMP-V-T samples calculated. Intensity ratios for both the IAG and LAPG samples were normalized to the Cb–Ca crosspeak corresponding to S3, which was isotopically enriched in both samples, and well over 25 Å away from the nearest nitroxide in the MacroModel simulations. The chemical shifts for this peak are 56.6 and 66.3 for Ca and Cb, respectively, consistent with those expected for a β-sheet serine.61 Although DHFR has eight serines, the other two β sheet serines (S135 and S138) are distant from the biradical moiety as well, justifying the use of this peak as a normalization constant that permits direct comparison between the samples. Moreover, the normalization constant calculated from this peak agrees well with that derived from the average of the Co–Ca crosspeaks of I2/S3 and I82/A83 in the IAG sample—two peaks that are both similarly over 25 Å from the nearest nitroxide in the MacroModel simulations. Due to the paucity of normalization peaks, however, it must be noted that these intensity ratios are not quantitative and only serve as a relative measure of signal attenuation with the bound biradical. The variation in intensity ratio for normalization peaks between different spectra was approximately 15%; therefore, intensity ratios carry errors of ±0.15.
DNP-SSNMR experiments were performed on a 600 MHz (14.1 T) Bruker AVANCE III-DNP system at the New York Structural Biology Center (NYSBC), equipped with a 395 GHz gyrotron, a 3.2 mm HCN E-free probe, and a 1.9 mm HCN probe. MAS spinning frequencies of 11 kHz were used for all samples except for the spectrum shown in Figure 5b, which was collected at 22 kHz using the 1.9 mm probe. Our best estimate of sample temperature during DNP SSNMR experiments is 111 ± 2.5 K based on temperature measurements at the stator (not including the effects of decoupling and sample rotation). Typical temperatures for nitrogen gas flows were 98 ± 0.5 K for variable-temperature (VT) gas, 106 ± 1.2 K for bearing gas, and 104 ± 1 K for drive gas. Prior to experiments, gyrotron power was adjusted for maximum enhancement. All spectra were acquired with a recycle delay of 3 s. CP experiments were performed with a 10% tangential ramp, a 1H π/2 pulse time of 2.5 μs (100 kHz), and a proton decoupling field strength of 110 kHz.
Figure 5.
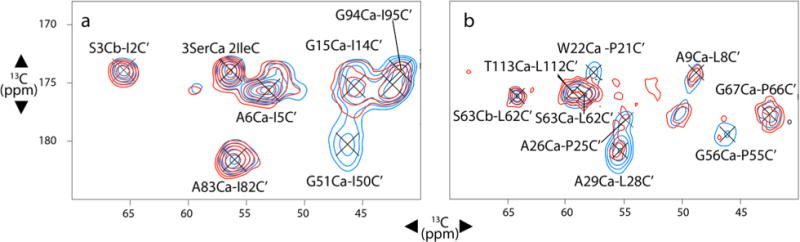
2D 13C–13C DARR correlation spectra for IAG isotopically enriched (see the Materials and Methods section) samples (a) and LAPG samples (b). The contour lines compare TMP-V-T (red) and AMUPol/TMP (blue). These data indicate site-dependent signal quenching due to the bound TMP-V-T. DARR mixing times are (a) 250 ms and (b) 100 ms. See Table S3 for a quantitative comparison of signal intensity. All spectra were collected at 600 MHz 1H frequency with a MAS frequency of 11 kHz, except for the blue AMUPol/TMP spectrum in part b, which was collected at 22 kHz MAS. Sample temperatures were 111 ± 2.5 K, with continuous microwave irradiation. 110 kHz SPINAL-6486 1H decoupling was applied during acquisition.
RESULTS AND DISCUSSION
TMP-V-T Ligand
In our previous study, TMP was conjugated to TOTAPOL using a one-carbon linker to form the nanomolar biradical affinity ligand trimethoprim-TOTAPOL (TMP-T).22 Here we use a similar compound we term TMP-V-T, in which a four-carbon methylene linker connects TMP at the 4′-methoxy position via an ether bond and TOTAPOL at the 4-amino position via an amide bond (see Figure 1 and Figure S1). EPR saturation binding experiments with TMP-V-T indicate that its affinity for DHFR is comparable to that of TMP-T (Figure S2), and that binding is stoichiometric when the protein and ligand concentrations are in the micromolar range. Moreover, enhancements on stoichiometric samples prepared with TMP-V-T are similar to those seen with TMP-T (see Table S2). TMP-V-T was always added to samples below its solubility of 0.8 mM, and samples with concentrations in the midmillimolar range were prepared by coconcentration as elaborated in the Materials and Methods section.
Figure 1.
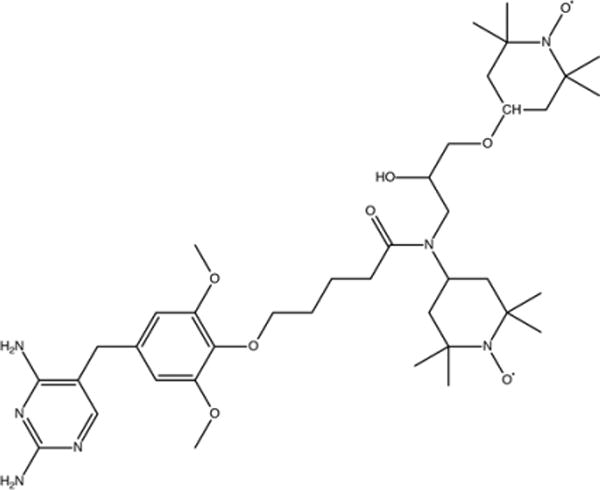
Chemical structure of the compound TMP-V-T, with trimethoprim linked covalently to TOTAPOL, which was used here as a biradical affinity reagent for DHFR.
TOTAPOL Moiety of TMP-V-T Is Disordered When Bound to DHFR
A structural model of TMP-V-T bound to DHFR is crucial for correlating the effects of the biradical on NMR spectra with the distance from the nucleus to the biradical. We therefore obtained crystal structures of DHFR bound to both TMP-V-T and the parent TMP. The two complexes crystallized in the same space group with nearly identical cell dimensions (Table S1). The crystal form, in space group P6122, is distinct from all previously reported crystal forms of DHFR (as tabulated in ref 62). The crystals with TMP and TMP-V-T diffracted to 2.5 and 1.95 Å, respectively. The overall structures are very similar to each other, with an RMSD of 0.3 Å for backbone atoms 10–13 and 26–159 (see Figure 2). Residues 10–24 were excluded because they have a much larger RMSD of 1.5 Å. These residues comprise the M20 binding loop, which sits like a lid on the folate-binding pocket where trimethoprim binds. In the TMP-V-T structure, density for this loop is poor, and residues 16–17 are disordered and could not be included in the final model.
Figure 2.
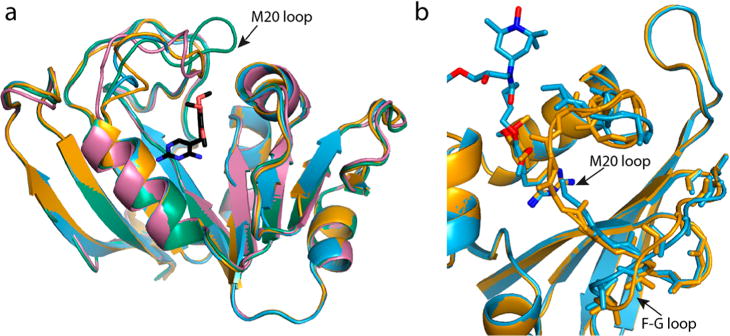
(a) Crystal structure of DHFR with TMP-V-T bound (blue) and with TMP bound (orange) from this work compared by superimposition with the previously published structure in which the M20 loop is closed (PDB accession code 1RX2, green) and with one where the loop is occluded (PDB accession code 1RX4, pink). TMP is shown in black. The loop in the TMP structure is more similar to the occluded loop conformation than to the closed loop conformation. (b) An expansion of the M20 and F-G loops of the new structures with TMP and TMP-V-T bound highlights the relative displacement of these residues in the otherwise similar structures. The location of the linker and TOTAPOL atoms is modeled and not based on crystal density because the density is quite low, as discussed below. The image was produced using the PyMOL Molecular Graphics System, version 1.8.4.2, Schrödinger, LLC.
The M20 loop is known to adopt several crystallographic conformations, termed the occluded, closed, and open conformations.62 This loop is also disordered in the apo state63 and when complexed with the inhibitor methotrexate. The structure of DHFR with the parent TMP is most similar to the occluded conformation (represented by PDB file 1RX2), with an overall backbone RMS of 1.0 Å (Figure 2). The structural differences between the TMP and TMP-V-T complexes occur in loops close to the expected location of the linker and TOTAPOL moiety. Importantly, there is very poor density for the linker and the TOTAPOL moiety in the TMP-V-T structure (Figure 3 and Figure S3). This is despite the fact that both structures have excellent density for the trimethoprim moiety. The benzene ring of trimethoprim is rotated only slightly between the two structures and the pyrimidine ring of trimethoprim is in the same location (Figure 2b), confirming that the binding mode for TMP-V-T is the same as that for the parent TMP. We conclude that the linker and TOTAPOL region experience disorder that prevents their observation in the crystal structure. The lack of crystal density observed here for the TOTAPOL and linker region is similar to previous crystallographic studies of nitroxides covalently attached to T4 lysozyme using the MTSL cysteine-reactive reagent.64 Similar to our findings, there is minimal density for the nitroxide rings in flexible, solvent exposed locations, indicating that the nitroxide is disordered.
Figure 3.
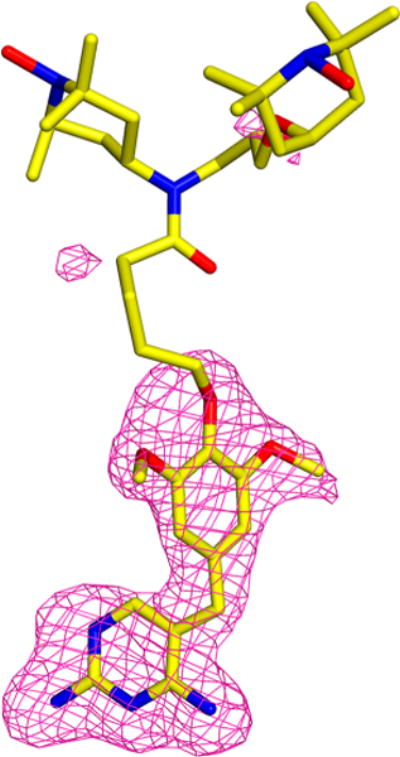
Fo–Fc omit map for the TMP-V-T region of the TMP-V-T crystal structure indicating that, while there is excellent density for the trimethoprim moiety, there is essentially no density for the linker and TOTAPOL. TMP-V-T is in yellow, and the positive contour of the omit map is shown in pink. The structure was refined without ligand present, and contour levels are set at 3 sigma. The image was produced using the PyMOL Molecular Graphics System, version 1.8, Schrödinger, LLC.
Solution NMR Paramagnetic Broadening Reveals a Limited Bleaching Radius
Since TMP-V-T is a paramagnetic ligand, in the absence of crystallographic coordinates for the ligand position, solution state paramagnetic relaxation enhancements (PRE) of the bound DHFR can be used to orient TMP-V-T vis a vis the nearby protein residues.55,65 The nitroxides of TMP-V-T will broaden the 1H amide resonances in a distance-dependent fashion, as dictated by the Solomon–Bloembergen equation for the modified paramagnetic relaxation enhancement R2PRE:29,37,66
| (1) |
τc includes contributions from electron relaxation, molecular tumbling, and chemical exchange. In solution state PRE experiments with nitroxides, τc is approximated as the rotational correlation time of the protein in solution (9.05 ns for DHFR67), as T1e of the nitroxide is considerably longer at 10−7–10−6 s.37
However, applying this equation to PREs observed with TMP-V-T will have numerous sources of error. Since TMP-V-T is a biradical, for any given site, the second nitroxide will provide an additional contribution to the PRE. The r−6 dependence of the PRE implies that the second nitroxide may generally be a small perturbation but will depend on the detailed conformation. Additionally, crystallography indicates that TMP-V-T is disordered and likely flexible. This will have two effects. First, the observed PRE is an ensemble average over all TMP-V-T conformations. Due to its r−6 dependence, the PRE will be biased toward the closest radical in the ensemble;65 thus, calculating high-quality structures from flexible paramagnetic tags often necessitates structure calculation from a conformational ensemble.65,68 This is especially relevant for TMP-V-T, where the TOTAPOL moiety can explore a large binding pocket (see Figure 2). Second, the internal motions of the electron–nuclear vector, including local protein motions and/or reorientation of the biradical moiety, will affect the τc and therefore the observed PRE. Clore and others65,69 have extended the Solomon–Bloembergen equations using the model-free formalism to incorporate an order parameter for the electron–nuclear vector. Clore and Iwahara show that flexibility leads to underestimation of R2PRE. However, since the distance between the nitroxide and the nucleus of interest is typically long, order parameters are generally large. Moreover, the distances calculated from R2PRE are fairly insensitive to the motional effects, permitting an approach that only considers the ensemble effect of the motions to be acceptable for some applications.65,68,70
When bound to TMP-V-T, the 1H–15N HSQC peaks of DHFR are reduced in intensity in a site-specific manner (Figure 4). This paramagnetic broadening is correlated with distance from the binding pocket of TMP-V-T, with the strongest intensity reductions for residues located in the two loops and helices that line the mouth of the binding pocket (Figure 4 and Figure S4). The pattern of paramagnetic relaxation enhancement is compatible with the heterogeneity observed in the crystallography and EPR, since no single conformation is likely to account for the R2PRE experienced by residues encompassing a 20 × 30 Å2 surface. For reference, the internitroxide distance in soluble TOTAPOL is 13.1 Å.71
Figure 4.
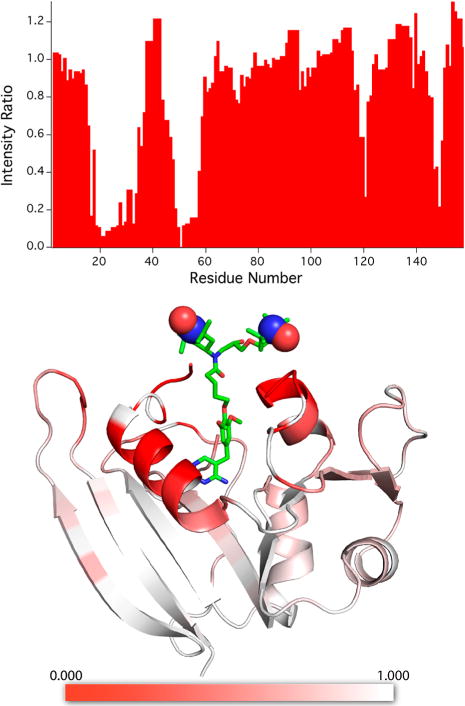
Bleaching of signals in solution state 1H–15N HSQC studies of U–15N,13C-DHFR:H2NADPH:TMP-V-T complex due to the affinity radical. At the top, the ratio of signal intensity Iox/Ired is displayed as a function of residue number for the oxidized paramagnetic complex relative to the reduced diamagnetic sample (see the Materials and Methods section for details). At the bottom, a heat map is used to encode intensity ratios Iox/Ired onto the structure of DHFR with TMP-V-T, demonstrating that the largest reductions in signal intensity occur for residues close to the expected location of the TOTAPOL moiety. Unassigned residues are shown in white. The structure of the TMP-V-T linker and TOTAPOL was built into the crystal structure by hand using the Maestro software (Schrödinger Release 2017-3: Maestro, Schrödinger, LLC, New York, 2017). The paramagnetic N–O groups are shown as spheres. The image was produced using the PyMOL Molecular Graphics System, version 1.8.4.2, Schrödinger, LLC.
We estimated the paramagnetic relaxation enhancement from the intensity ratio Iox/Ired and the line width of the reduced sample, as in a prior report54 (see the Materials and Methods section). For each site, we also estimated an effective distance to the nearest nitroxide using eq 1. As discussed above, these distances are subject to significant error due to the simplified model but are useful as a relative measure of nitroxide proximity.
We also compared the chemical shifts for DHFR samples where the only substitution was TMP-V-T for TMP (analogous to our isomorphous crystal structures). For the detected peaks, the chemical shifts are similar but not identical comparing the samples with H2NADPH/TMP and H2NADPH/TMP-V-T (Figure S5). The RMSD for assigned residues is 0.03 for 1H, 0.14 for 15N, and 0.15 for 13Co, larger than the experimental error in these measurements (0.01 for 1H, 0.1 for 15N, and 0.05 for 13Co). The largest chemical shift perturbations (CSPs) occur for the residues lining the opening of the binding pocket (namely, G51, N18, S49, I50, W22, M16, and M20), but CSPs also occur in other regions of the protein (Figure S6). Interestingly, significant CSPs are observed for H124 and T123, residues at the base of the displaced F–G loop shown in Figure 2. The largest CSP is 0.1 for 1H and 0.75 for 15N. DHFR chemical shifts are very sensitive to structure and ligation state, and structural changes typically induce large CSPs of ≥1.5 ppm (see ref 72). The small magnitude of the CSPs confirms that the overall conformation of DHFR is not significantly perturbed by the derivatization of TMP, consistent with our crystallographic results.
Specifically Isotopically Enriched DHFR with Bound TMP-V-T Enables Detailed Studies of Relaxation at Low Temperature
For samples at cryogenic temperatures (such as those commonly used for DNP experiments), additional line broadening associated with the lower temperature is expected. While PRE effects are expected to contribute in principle to the homogeneous line width, prior analyses indicate that the line width at cryogenic temperatures is generally inhomogeneous in nature73,74 and correlated with solvent exposure75,76 and conformational heterogeneity.77,78 Typical line widths for frozen solution samples of DHFR are 1.2–1.6 ppm for 13C and 3–4 ppm for 15N. Selective isotope enrichment79,80 is therefore an efficient tool15 for confident assignment of peaks in DNP samples.
To site-specifically study the bleaching due to TMP-V-T in DNP samples, we prepared two different selectively isotopically enriched samples of DHFR. One sample was prepared from bacteria grown in minimal media supplemented with 13Co-Ile + 15N/13Ca-Gly + 13Ca-Ala (herein referred to as IAG) and the second with 13Co-Leu + 13Co-Pro + 15N/13Ca-Ala + 15N/13Ca-Gly (herein referred to as LAPG). Serine is effectively enriched as well in both cases due to glycine metabolism.81 These combinations of amino acids were chosen so that, in 13C–13C experiments, we would observe pairs originating from 13Cai (of Gly, Ala, or Ser) to the 13Coi–1 (of Ile, Leu, or Pro). Similarly, in 13C–15N correlation experiments, we observe pairs originating from 15Ni (of Gly, Ala, or Ser) to the 13Coi–1 (of Ile, Leu, or Pro). These two isotope enrichment schemes select amino acid pairs that are at a range of distances from the expected location of the TMP-V-T binding site (Figure S7). Solution NMR experiments confirmed the expected isotopic enrichment (Figure S8), indicating that, for the IAG sample, scrambling was minimal. However, for the LAPG sample, scrambling of 13Co to glutamic acid and scrambling of 15N to threonine and tryptophan was observed. The resulting spectral overlap still gave resolved peaks in 2D 13C–13C DNP spectra (see Figure 5, Figure S9, and the Materials and Methods section for discussion of spectral assignments at low temperature).
Signal Bleaching with Bound TMP-V-T
To measure and quantify the signal reduction due to the bound nitroxides, 2D spectra of selectively enriched DNP samples with bound TMP-V-T were compared to reference spectra collected with 5 mM AMUPol and saturating TMP. Reference spectra were DNP enhanced to achieve sufficient signal-to-noise. Previous reports suggest that, at 5 mM radical concentration, generalized signal bleaching will be minimal.42 Moreover, similar absolute concentrations of AMUPol and TMP-V-T were used. Assuming that the biradical distributes evenly within the glycerol/water matrix, the two samples experience similar intermolecular quenching; in the TMP-V-T sample, each protein “sees” the radicals on neighboring protein molecules. The random distribution should be similar, given that each was equilibrated at room temperature. Therefore, the site-specific bleaching in the TMP-V-T complex should be the additional bleaching over the nearest-neighbor bleaching from nearby paramagnetic compounds. Moreover, in our initial report,22 we did not observe more bleaching (adjusted for concentration) in a 1D 1H–13C CP spectrum of superstoichiometric 3 mM DHFR:7 mM TMP-T samples relative to a 4.5 mM stoichiometric (1:1) sample, implying that, at low millimolar concentrations, stochastic bleaching from the 4 mM excess unbound TMP-T is not significant.
For each sample, 2D 13C–13C DARR82 experiments at a range of mixing times as well as 2D 15N–13C correlation spectra using both TEDOR83 and SPECIFIC-CP84 transfer sequences were collected. Figure 5 and Figure S9 compare the 13C–13C and 15N–13C correlation spectra, respectively, for samples with bound TMP-V-T to samples with soluble AMUPol and bound TMP. Notably, few peaks disappear when the radical is localized to the protein. Although solution NMR measurements indicated the presence of a small diamagnetic population, enhancements on the two specifically labeled samples were both 23, consistent with enhancements on other stoichiometric TMP-T samples22 (see Table S2). We assume that enhancement on the apo protein is lower than that on the bound protein, thereby reducing its contribution to the observed signal to <5%. CW EPR experiments in which we reoxidized the TMP-V-T stock solution used for DNP measurements indicated only a 3% signal increase upon reoxidation with K3[Fe(SCN)6], a mild reoxidizing agent that specifically reoxidizes the reduced compound,85 indicating that the radical was primarily paramagnetic.
To quantify the relative intensities between samples with TMP-V-T and samples with AMUPol and bound TMP, peak heights for all assigned peaks in the 13C–13C DARR correlation spectra were extracted for pairs of spectra collected and processed identically. Spectra with AMUPol/TMP and TMP-V-T were analogous except for enhancement factors and number of scans. We calculated an intensity ratio, ITMPVT/IAMUPol, by dividing the intensity in the TMP-V-T sample by the intensity in the AMUPol/TMP sample (see the Materials and Methods section), normalized by the intensity ratio for the S3 Ca–Cb peak, which experienced minimal signal attenuation and was enriched in both the IAG and LAPG samples (see the Materials and Methods section). Because we were limited in our normalization peaks, the intensity ratios are only useful for comparison between peaks in the TMP-V-T samples. However, using these comparisons of intensity, we grouped residues into three groups: those that experience the least (or no) signal attenuation, those that experience substantial but incomplete intensity attenuation, and those that experience complete signal attenuation. Figure 6 shows the plot of signal attenuation with TMP-V-T mapped onto a wire diagram of DHFR, while Figure S10 shows the correlation of the intensity ratios with distance from the TMP binding site. For these three groups, it is informative to compare the intensity ratios observed in the solid-state samples with those from solution NMR. Table S3 presents intensity ratios for IAG and LAPG samples, respectively, along with the intensity ratio for the amide position from the solution paramagnetic bleaching analysis and the calculated distance using eq 1. In Figure S11, the intensity ratios for solution and solid state are plotted versus residue number for facile comparison.
Figure 6.
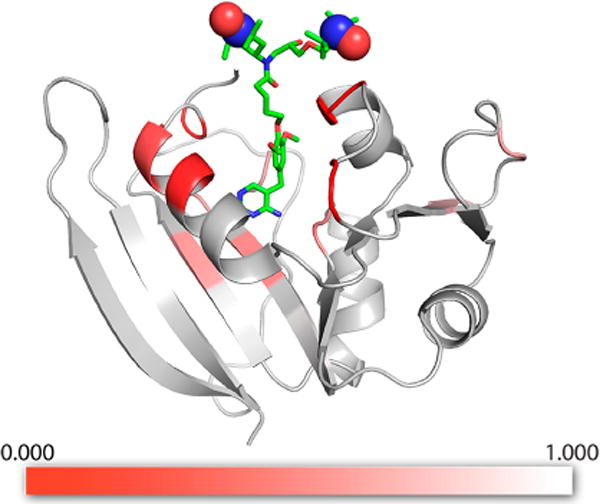
DHFR model with low temperature SSNMR bleaching information shown as a heat map. The ratio of signal intensity for specifically isotopically enriched DHFR samples with bound TMP-V-T relative to samples with exogenous AMUPol/TMP, observed using DNP-enhanced 13C–13C DARR spectra (ITMPVT/IAMUPol), is indicated by the color scale, red to white. The strongest reduction in signal intensity, or bleaching, is observed for residues proximal to the TMP-V-T binding pocket. The structure of the TMP-V-T linker and TOTAPOL was built into the crystal structure by hand using the Maestro software (Schrödinger Release 2017-3: Maestro, Schrödinger, LLC, New York, 2017), with the paramagnetic N–O groups represented as spheres. The image was produced using the PyMOL Molecular Graphics System, version 1.8.4.2, Schrödinger, LLC.
The residues that experience the least signal attenuation, with intensity ratios >0.98, are residues I2, S3, I82, and A83 (see Table S3). These residues similarly have Iox/Ired > 0.97 in solution NMR measurements, with calculated effective distances greater than 23 Å for the nearest nitroxide. Residues I50, G51, L21, and W22 are completely bleached in the DNP experiments and are also fully attenuated in the solution state measurement. Residues P55 and G56 are bleached in the DNP samples but are not assigned in the solution spectra. The remaining residues, namely, I5, A6, L8, A9, I14, G15, P25, A26, L28, A29, L62, S63, P66, G67, L112, and T113, are still observable but experience some level of signal attenuation relative to the samples prepared with AMUPOL. Residues A5, A6, L8, A9, L112, and T113 experience signal attenuation of 40–50% in DNP measurements, with only 10% signal attenuation in the solution measurements. Residue L62 experiences 10% more signal attenuation in the DNP experiment, while residues I14 and G15 experience similar signal attenuation in both experiments. Of special interest are residues P25, A26, L28, and A29: these are mostly attenuated in the solution measurements but exhibit intensity in the DNP solid-state measurements.
Taken in aggregate, the patterns of bleaching intensity suggest that the “blind zone” in DNP samples is similar to that in the solution NMR analysis. The only residues fully bleached are those proximal to the TOTAPOL moiety and similarly bleached at room temperature, indicating a bleaching radius of approximately 10.5 Å. Moreover, residues that have a nitroxide center within 23 Å, as indicated by signal attenuation in the solution NMR experiments, similarly experience signal attenuation in the DNP samples. In some favorable cases, residues are visible in the DNP samples that are mostly attenuated in the solution NMR experiments. A bleaching zone of 23 Å is consistent with estimates of the DNP bleaching radius from signal attenuation of proline samples with TOTAPOL42 but is surprisingly small, given long electron relaxation times at cryogenic temperatures.31–34,87 In solid samples, the electron T1e is thought to drive relaxation in the absence of molecular rotation. While there are no measures of T1e at this temperature and field (100 K and 14.1 T), T1e is expected to be in the mid-microsecond time scale based on measurements at similar fields and temperatures,31–34,87 which is several orders of magnitude slower than the estimated nitroxide T1e at room temperature (100 ns).88 Paramagnetic relaxation enhancement increases with longer correlation time.
On this basis of the available data, we cannot pinpoint the reason for the short bleaching radii observed in the SSNMR DNP experiments but would like to discuss several hypotheses. In concentrated DNP samples of the monoradicals trityl and 4-amino-TEMPO at very low temperatures (<20 K), the electron T2e was simulated to be much shorter than the T1e (microsecond as opposed to millisecond).89,90 Additionally, electron spin diffusion has been shown to be operative in concentrated nitroxide solutions suitable for DNP.32,89,90 Although the extent of electron spin diffusion is unclear at the temperatures and concentrations used here (110 K, 2–4 mM protein bound 1:1 to TOTAPOL and ≤8 mM overall nitroxide concentration), electron spin diffusion is likely to be of the same order or faster than the T1e.90 Relatively fast T2e and electron spin diffusion time constants could serve to reduce the paramagnetic relaxation enhancements, and significantly flatten their temperature dependence, and so might be relevant for understanding the results presented here. Testing or developing these arguments will require further computational and experimental investigations outside of the scope of this work. The overall sensitivity of detection and the DNP enhancement ratios likely reflect a number of rate constants in addition to the relaxation rate of the nucleus in question. Quantitative measurements of nuclear and electron relaxation times in samples with bound biradicals are needed to evaluate these aspects.
The conformational heterogeneity observed in the crystallographic DHFR/TMP-V-T samples is expected to contribute to the observed intensities; if a given resonance is bleached in one conformation, it may be unaffected in another. At 100 K, the various conformations are expected to mainly contribute to static disorder, rather than the dynamic disorder observed at room temperature. For each NMR line, conformers that place the nitroxide relatively farther away would then dominate the observed signal, while closer conformers have their signal quenched. Many sites are expected to be “unmasked” in this way. To evaluate this hypothesis, we considered the conformers accessible to TMP-V-T using Monte Carlo Multiple Minimum sampling (implemented in MacroModel56 software, see the Materials and Methods section). We identified two representative, distinct conformers at extreme positions of a major axis of the available open space (Figure S12). These conformers qualitatively agree with the observed solution NMR paramagnetic relaxation enhancements (Figure S13). Table S4 compares residue-specific distances for each representative conformation. For example, residues P25 and A26, which are partly bleached, are expected to be far from both nitroxides in at least some populated conformers (>15 Å). By contrast, I50 and G51, which are fully bleached, have a nearby nitroxide (<15 Å) in both predicted conformations. I5 and A6 (IAMUPol/ITMPVT = 0.5) experience considerable bleaching because in no conformer are they farther than 21 Å from a nitroxide. I82 and A83, by contrast, have strong signals (ITMPVT/IAMUPol = 0.99), presumably because in all expected conformations the nitroxides are distant (>27 Å away). While the complexity of the dynamics does not allow us to establish a quantitative relationship between distance and bleaching, we observe that significant signal attenuation occurs primarily at nitroxide distances under 15 Å and in some cases even closer, similar to the distances reported in room temperature studies.
Our observation of a relatively small blind zone in DNP samples with an attached biradical may not be universal for targeted DNP samples at 100 K. Specifically, Ladizhansky and co-workers20 observed a strong paramagnetic quenching effect of 70% for the Anabaena sensory rhodopsin (ASR) trimer derivatized with TOTAPOL in a 1:1 ratio. However, in that case, signal quenching was measured with MW off. As mentioned above, in biradical-containing samples without MW irradiation, there may be an additional depolarization effect that further reduces the observed signal.39,91 Additionally, ASR forms a trimer, thereby generating both intermonomer and intramonomer PREs, with many short intermonomer distances. First-shell contact shifts may also be operative in this study, as the biradical had a short linker based on the methanethiosulfonate attachment chemistry. Lastly, the protein was measured in lipid bilayer liposomes, and at a much higher overall nitroxide concentration for 1:1 protein:tag than that used here for TMP-V-T samples. PRE effects between different lipid bilayers cannot be discounted, and the polarization transfer to the lipid protons may not be as favorable as transfer to the solvent, which occurs preferentially in the TMP-V-T samples.
CONCLUSIONS
We demonstrate that the TOTAPOL moiety in the biradical affinity reagent TMP-V-T is disordered when bound to DHFR through crystallographic measurements. This disorder is expected to have static and dynamic forms depending on temperature and the time scale of the measurement. We measure the paramagnetic bleaching in DNP samples with bound TMP-V-T, and show that signals under 10 Å from the nitroxide center are fully bleached, signals between approximately 10 and 23 Å experience signal attenuation, and signals over 25 Å away appear unaffected. The radius of complete bleaching in DNP samples appears to be similar to that observed in solution PRE measurements, and we argue that static disorder plays a large role in this observation. We anticipate that the conformational heterogeneity observed with TMP-V-T will be a general property of nitroxide biradical tags, unless they are designed with rigidity in mind or immobilized upon binding to their target.
Our results with TMP-V-T could dictate design principles for targeted DNP. Namely, the nitroxide should be close enough to provide selective enhancement, yet solvent exposure and the resultant conformational heterogeneity may positively contribute to reduce PRE effects and enable site-specific assignment of proteins with attached biradicals. If a specific region of the protein is of interest, then the nitroxide affinity reagent should be placed at least 10–15 Å away to minimize signal quenching. Moreover, in such samples, the distance guidelines outlined here can serve as an assignment tool. The signal quenching can even be used as a qualitative structural restraint in targeted DNP samples, as argued previously by Baldus and co-workers.21 The utility of these restraints in studying the tertiary structure of large proteins, as well as protein–protein interactions, is clear from the abundant examples in room temperature solution- and solid-state NMR.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Science Foundation (NSF) (MCB1412253 to A.E.M.) and by the National Institute of Health Grant P41GM118302 for the Center on Macromolecular Dynamics by NMR Spectroscopy located at the New York Structural Biology Center (NYSBC). The data collected at NYSBC were enabled by a grant from NYSTAR and ORIP/NIH facility improvement grant CO6RR015495. The 600 MHz DNP/NMR spectrometer was purchased with funds from NIH grant S10RR029249. R.R. was supported by the National Institutes of Health Training Program in Molecular Biophysics T32GM008281. The authors thank Dr. Mike Goger, Dr. Boris Itin, and Dr. Shibani Battacharya of the New York Structural Biology Center for support with NMR instrumentation and many helpful discussions. The authors thank Makeda Tekle-Smith for help with synthesis of H2NADPH.
Footnotes
Published as part of The Journal of Physical Chemistry virtual special issue “Recent Advances in Connecting Structure, Dynamics, and Function of Biomolecules by NMR”.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.7b08274.
Details of crystallography parameters, EPR characterization, and supplementary figures (PDF)
ORCID
Rivkah Rogawski: 0000-0002-6438-2269
Ann E. McDermott: 0000-0002-9249-1649
Notes
The authors declare no competing financial interest.
References
- 1.Smith AN, Long JR. Dynamic Nuclear Polarization as an Enabling Technology for Solid State Nuclear Magnetic Resonance Spectroscopy. Anal Chem. 2016;88:122–132. doi: 10.1021/acs.analchem.5b04376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lilly Thankamony AS, Wittmann JJ, Kaushik M, Corzilius B. Dynamic Nuclear Polarization for Sensitivity Enhancement in Modern Solid-State NMR. Prog Nucl Magn Reson Spectrosc. 2017;102–103:120–195. doi: 10.1016/j.pnmrs.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Zagdoun A, Rossini AJ, Conley MP, Grüning WR, Schwarzwälder M, Lelli M, Franks WT, Oschkinat H, Copéret C, Emsley L, et al. Improved Dynamic Nuclear Polarization Surface-Enhanced NMR Spectroscopy through Controlled Incorporation of Deuterated Functional Groups. Angew Chem, Int Ed. 2013;52:1222–1225. doi: 10.1002/anie.201208699. [DOI] [PubMed] [Google Scholar]
- 4.Leskes M, Kim G, Liu T, Michan AL, Aussenac F, Dorffer P, Paul S, Grey CP. Surface Sensitive NMR Detection of the SEI Layer on Reduced Graphene Oxide. J Phys Chem Lett. 2017;8:1078–1085. doi: 10.1021/acs.jpclett.6b02590. [DOI] [PubMed] [Google Scholar]
- 5.McDermott A. Structure and Dynamics of Membrane Proteins by Magic Angle Spinning Solid-State NMR. Annu Rev Biophys. 2009;38:385–403. doi: 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- 6.Ward ME, Brown LS, Ladizhansky V. Advanced Solid-State NMR Techniques for Characterization of Membrane Protein Structure and Dynamics: Application to Anabaena Sensory Rhodopsin. J Magn Reson. 2015;253:119–128. doi: 10.1016/j.jmr.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 7.Comellas G, Rienstra CM. Protein Structure Determination by Magic-Angle Spinning Solid-State NMR, and Insights into the Formation, Structure, and Stability of Amyloid Fibrils. Annu Rev Biophys. 2013;42:515–536. doi: 10.1146/annurev-biophys-083012-130356. [DOI] [PubMed] [Google Scholar]
- 8.Wollan DS. Dynamic Nuclear Polarization with an Inhomogeneously Broadened ESR Line. I. Theory. Phys Rev B. 1976;13:3671–3685. [Google Scholar]
- 9.Wollan DS. Dynamic Nuclear Polarization with an Inhomogenously Broadened ESR Line. II. Experiment. Phys Rev B. 1976;13:3686–3696. [Google Scholar]
- 10.Mathies G, Caporini Ma, Michaelis VK, Liu Y, Hu K-N, Mance D, Zweier JL, Rosay M, Baldus M, Griffin RG. Efficient Dynamic Nuclear Polarization at 800 MHz/527 GHz with Trityl-Nitroxide Biradicals. Angew Chem, Int Ed. 2015;54:11770–11774. doi: 10.1002/anie.201504292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song C, Hu K-N, Joo C-G, Swager TM, Griffin RG. TOTAPOL: A Biradical Polarizing Agent for Dynamic Nuclear Polarization Experiments in Aqueous Media. J Am Chem Soc. 2006;128:11385–11390. doi: 10.1021/ja061284b. [DOI] [PubMed] [Google Scholar]
- 12.Sauvée C, Rosay M, Casano G, Aussenac F, Weber RT, Ouari O, Tordo P. Highly Efficient, Water-Soluble Polarizing Agents for Dynamic Nuclear Polarization at High Frequency. Angew Chem, Int Ed. 2013;52:10858–10861. doi: 10.1002/anie.201304657. [DOI] [PubMed] [Google Scholar]
- 13.van der Wel PCa, Hu K-N, Lewandowski J, Griffin RG. Dynamic Nuclear Polarization of Amyloidogenic Peptide Nanocrystals: GNNQQNY, a Core Segment of the Yeast Prion Protein Sup35p. J Am Chem Soc. 2006;128:10840–10846. doi: 10.1021/ja0626685. [DOI] [PubMed] [Google Scholar]
- 14.Sergeyev IV, Day LA, Goldbourt A, McDermott AE. Chemical Shifts for the Unusual DNA Structure in Pf1 Bacteriophage from Dynamic-Nuclear-Polarization-Enhanced Solid-State NMR Spectroscopy. J Am Chem Soc. 2011;133:20208–20217. doi: 10.1021/ja2043062. [DOI] [PubMed] [Google Scholar]
- 15.Lehnert E, Mao J, Mehdipour AR, Hummer G, Abele R, Glaubitz C, Tampe R. Antigenic Peptide Recognition on the Human ABC Transporter TAP Resolved by DNP-Enhanced Solid-State NMR Spectroscopy. J Am Chem Soc. 2016;138:13967–13974. doi: 10.1021/jacs.6b07426. [DOI] [PubMed] [Google Scholar]
- 16.Maly T, Cui D, Griffin RG, Miller AH. Dynamic Nuclear Polarization Based on an Endogenous Radical. J Phys Chem B. 2012;116:7055–7065. doi: 10.1021/jp300539j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wenk P, Kaushik M, Richter D, Vogel M, Suess B, Corzilius B. Dynamic Nuclear Polarization of Nucleic Acid with Endogenously Bound Manganese. J Biomol NMR. 2015;63:97–109. doi: 10.1007/s10858-015-9972-1. [DOI] [PubMed] [Google Scholar]
- 18.Rogawski R, McDermott A. New NMR Tools for Protein Structure and Function: Spin Tags for Dynamic Nuclear Polarization Solid State NMR. Arch Biochem Biophys. 2017;628:102–113. doi: 10.1016/j.abb.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith AN, Caporini Ma, Fanucci GE, Long JR. A Method for Dynamic Nuclear Polarization Enhancement of Membrane Proteins. Angew Chem, Int Ed. 2015;54:1542–1546. doi: 10.1002/anie.201410249. [DOI] [PubMed] [Google Scholar]
- 20.Voinov Ma, Good DB, Ward ME, Milikisiyants S, Marek A, Caporini Ma, Rosay M, Munro Ra, Ljumovic M, Brown LS, et al. Cysteine-Specific Labeling of Proteins with a Nitroxide Biradical for Dynamic Nuclear Polarization NMR. J Phys Chem B. 2015;119:10180–10190. doi: 10.1021/acs.jpcb.5b05230. [DOI] [PubMed] [Google Scholar]
- 21.Van Der Cruijsen EAW, Koers EJ, Hulse RE, Weingarth M, Ouari O, Perozo E, Tordo P, Baldus M. Biomolecular DNP-Supported NMR Spectroscopy Using Site-Directed Spin Labeling. Chem - Eur J. 2015;21:12971–12977. doi: 10.1002/chem.201501376. [DOI] [PubMed] [Google Scholar]
- 22.Rogawski R, Sergeyev IV, Li Y, Ottaviani MF, Cornish V, McDermott AE. Dynamic Nuclear Polarization Signal Enhancement with High-Affinity Biradical Tags. J Phys Chem B. 2017;121:1169–1175. doi: 10.1021/acs.jpcb.6b09021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Viennet T, Viegas A, Kuepper A, Arens S, Gelev V, Petrov O, Grossmann TN, Heise H, Etzkorn M. Selective Protein Hyperpolarization in Cell Lysates Using Targeted Dynamic Nuclear Polarization. Angew Chem, Int Ed. 2016;55:10746–10750. doi: 10.1002/anie.201603205. [DOI] [PubMed] [Google Scholar]
- 24.Bertini I, Luchinat C, Parigi G, Pierattelli R. NMR Spectroscopy of Paramagnetic Metalloproteins. ChemBioChem. 2005;6:1536–1549. doi: 10.1002/cbic.200500124. [DOI] [PubMed] [Google Scholar]
- 25.Griffith OH, Cornell DW, McConnell HM. Nitrogen Hyperfine Tensor and G Tensor of Nitroxide Radicals. J Chem Phys. 1965;43:2909–2910. [Google Scholar]
- 26.Nadaud PS, Helmus JJ, Höfer N, Jaroniec CP. Long-Range Structural Restraints in Spin-Labeled Proteins Probed by Solid-State Nuclear Magnetic Resonance Spectroscopy. J Am Chem Soc. 2007;129:7502–7503. doi: 10.1021/ja072349t. [DOI] [PubMed] [Google Scholar]
- 27.Knight MJ, Felli IC, Pierattelli R, Emsley L, Pintacuda G. Magic Angle Spinning NMR of Paramagnetic Proteins. Acc Chem Res. 2013;46:2108–2116. doi: 10.1021/ar300349y. [DOI] [PubMed] [Google Scholar]
- 28.Jaroniec CP. Structural Studies of Proteins by Paramagnetic Solid-State NMR Spectroscopy. J Magn Reson. 2015;253:50–59. doi: 10.1016/j.jmr.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Solomon I, Bloembergen N. Nuclear Magnetic Interactions in the HF Molecule. J Chem Phys. 1956;25:261–266. [Google Scholar]
- 30.Solomon I. Relaxation Processes in a System of Two Spins. Phys Rev. 1955;99:559–565. [Google Scholar]
- 31.Corzilius B, Andreas LB, Smith AA, Ni QZ, Griffin RG. Paramagnet Induced Signal Quenching in MAS-DNP Experiments in Frozen Homogeneous Solutions. J Magn Reson. 2014;240:113–123. doi: 10.1016/j.jmr.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siaw TA, Fehr M, Lund A, Latimer A, Walker Sa, Edwards DT, Han S-I. Effect of Electron Spin Dynamics on Solid-State Dynamic Nuclear Polarization Performance. Phys Chem Chem Phys. 2014;16:18694–18706. doi: 10.1039/c4cp02013h. [DOI] [PubMed] [Google Scholar]
- 33.Zagdoun A, Casano G, Ouari O, Der MS, Rossini AJ, Aussenac F, Yulikov M, Jeschke G, Cope C, Lesage A, et al. Large Molecular Weight Nitroxide Biradicals Providing Efficient Dynamic Nuclear Polarization at Temperatures up to 200 K. J Am Chem Soc. 2013;135:12790–12797. doi: 10.1021/ja405813t. [DOI] [PubMed] [Google Scholar]
- 34.Kubicki D, Casano G, Schwarzwalder M, Abel S, Sauvee C, Genevan K, Yulikov M, Rossini AJ, Jeschke G, Coperet C, et al. Rational Design of Dinitroxide Biradicals for Efficient Cross-Effect Dynamic Nuclear Polarization. Chem Sci. 2016;7:550–558. doi: 10.1039/c5sc02921j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimon D, Hovav Y, Feintuch A, Goldfarb D, Vega S. Dynamic Nuclear Polarization in the Solid State: A Transition between the Cross Effect and the Solid Effect. Phys Chem Chem Phys. 2012;14:5729–5743. doi: 10.1039/c2cp23915a. [DOI] [PubMed] [Google Scholar]
- 36.Mentink-Vigier F, Akbey Ü, Hovav Y, Vega S, Oschkinat H, Feintuch A. Fast Passage Dynamic Nuclear Polarization on Rotating Solids. J Magn Reson. 2012;224:13–21. doi: 10.1016/j.jmr.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 37.Bertini I, Luchinat C, Parigi G, Ravera E. NMR of Parmagnetic Molecules (Second ed), Applications to Metallobiomolecules and Models. Elsevier; Amsterdam: 2017. Relaxation; pp. 77–126. [Google Scholar]
- 38.Thurber KR, Tycko R. Theory for Cross Effect Dynamic Nuclear Polarization under Magic-Angle Spinning in Solid State Nuclear Magnetic Resonance; the Importance of Level Crossings. J Chem Phys. 2012;137:084508. doi: 10.1063/1.4747449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thurber KR, Tycko R. Perturbation of Nuclear Spin Polarizations in Solid State NMR of Nitroxide-Doped Samples by Magic-Angle Spinning without Microwaves. J Chem Phys. 2014;140:184201. doi: 10.1063/1.4874341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rossini AJ, Zagdoun A, Lelli M, Gajan D, Rascón F, Rosay M, Maas WE, Copéret C, Lesage A, Emsley L. One Hundred Fold Overall Sensitivity Enhancements for Silicon-29 NMR Spectroscopy of Surfaces by Dynamic Nuclear Polarization with CPMG Acquisition. Chem Sci. 2012;3:108–115. [Google Scholar]
- 41.Linden AH, Lange S, Franks WT, Akbey U, Specker E, Rossum B-J, van Oschkinat H. Neurotoxin II Bound to Acetylcholine Receptors in Native Membranes Studied by Dynamic Nuclear Polarization NMR. J Am Chem Soc. 2011;133:19266–19269. doi: 10.1021/ja206999c. [DOI] [PubMed] [Google Scholar]
- 42.Lange S, Linden AH, Akbey U, Franks WT, Loening NM, van Rossum B-J, Oschkinat H. The Effect of Biradical Concentration on the Performance of DNP-MAS-NMR. J Magn Reson. 2012;216:209–212. doi: 10.1016/j.jmr.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 43.Ravera E, Corzilius B, Michaelis VK, Luchinat C, Griffin RG, Bertini I. DNP-Enhanced MAS NMR of Bovine Serum Albumin Sediments and Solutions. J Phys Chem B. 2014;118:2957–2965. doi: 10.1021/jp500016f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagaraj M, Franks TW, Saeidpour S, Schubeis T, Oschkinat H, Ritter C, van Rossum BJ. Surface Binding of TOTAPOL Assists Structural Investigations of Amyloid Fibrils by Dynamic Nuclear Polarization NMR Spectroscopy. ChemBioChem. 2016;17:1308–1311. doi: 10.1002/cbic.201600185. [DOI] [PubMed] [Google Scholar]
- 45.Wang TY, Friedman LJ, Gelles J, Min W, Hoskins AA, Cornish VW. The Covalent Trimethoprim Chemical Tag Facilitates Single Molecule Imaging with Organic Fluorophores. Biophys J. 2014;106:272–278. doi: 10.1016/j.bpj.2013.11.4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tugarinov V, Kanelis V, Kay LE. Isotope Labeling Strategies for the Study of High-Molecular-Weight Proteins by Solution NMR Spectroscopy. Nat Protoc. 2006;1:749–754. doi: 10.1038/nprot.2006.101. [DOI] [PubMed] [Google Scholar]
- 47.Otwinowski Z, Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 48.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser Crystallographic Software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: Building New Software for Automated Crystallographic Structure Determination. Acta Crystallogr, Sect D: Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 50.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and Development of Coot. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salzmann M, Wider G, Pervushin K, Senn H, Wüthrich K. TROSY-Type Triple-Resonance Experiments for Sequential NMR Assignments of Large Proteins. J Am Chem Soc. 1999;121:844–848. [Google Scholar]
- 52.Pines A, Gabby MG, Waugh JS. Proton-Enhanced NMR of Dilute Spins in Solids. J Chem Phys. 1973;59:569–590. [Google Scholar]
- 53.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN Data Model for NMR Spectroscopy: Development of a Software Pipeline. Proteins: Struct, Funct Genet. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 54.Battiste JL, Wagner G. Utilization of Site-Directed Spin Labeling and High-Resolution Heteronuclear Nuclear Magnetic Resonance for Global Fold Determination of Large Proteins with Limited Nuclear Overhauser Effect Data †. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 55.Gillespie JR, Shortle D. Characterization of Long-Range Structure in the Denatured State of Staphylococcal Nuclease. I. Paramagnetic Relaxation Enhancement by Nitroxide Spin Labels. J Mol Biol. 1997;268:158–169. doi: 10.1006/jmbi.1997.0954. [DOI] [PubMed] [Google Scholar]
- 56.Chang G, Guida WC, Still WC. An Internal Coordinate Monte Carlo Method for Searching Conformational Space. J Am Chem Soc. 1989;111:4379–4386. [Google Scholar]
- 57.Kolossváry I, Guida WC. Low-Mode Conformational Search Elucidated: Application to C39H80 and Flexible Docking of 9-Deazaguanine Inhibitors into PNP. J Comput Chem. 1999;20:1671–1684. [Google Scholar]
- 58.Borrelli KW, Cossins B, Guallar V. Exploring Hierarchical Refinement Techniques for Induced Fit Docking with Protein and Ligand Flexibility. J Comput Chem. 2010;31:2967–2970. doi: 10.1002/jcc.21409. [DOI] [PubMed] [Google Scholar]
- 59.Li Y, Berthold DA, Frericks HL, Gennis RB, Rienstra CM. Partial 13C and 15N Chemical-Shift Assignments of the Disulfide-Bond-Forming Enzyme DsbB by 3D Magic-Angle Spinning NMR Spectroscopy. ChemBioChem. 2007;8:434–442. doi: 10.1002/cbic.200600484. [DOI] [PubMed] [Google Scholar]
- 60.Stevens TJ, Fogh RH, Boucher W, Higman VA, Eisenmenger F, Bardiaux B, Van Rossum BJ, Oschkinat H, Laue ED. A Software Framework for Analysing Solid-State MAS NMR Data. J Biomol NMR. 2011;51:437–447. doi: 10.1007/s10858-011-9569-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fritzsching KJ, Yang Y, Schmidt-Rohr K, Hong M. Practical Use of Chemical Shift Databases for Protein Solid-State NMR: 2D Chemical Shift Maps and Amino-Acid Assignment with Secondary-Structure Information. J Biomol NMR. 2013;56:155–167. doi: 10.1007/s10858-013-9732-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sawaya MR, Kraut J. Loop and Subdomain Movements in the Mechanism of Escherichia Coli Dihydrofolate Reductase: Crystallographic Evidence. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 63.Bystroff C, Kraut J. Crystal Structure of Unliganded Escherichia Coli Dihydrofolate Reductase. Ligand-Induced Conformational Changes and Cooperativity in Binding. Biochemistry. 1991;30:2227–2239. doi: 10.1021/bi00222a028. [DOI] [PubMed] [Google Scholar]
- 64.Langen R, Oh KJ, Cascio D, Hubbell WL. Crystal Structures of Spin Labeled T4 Lysozyme Mutants: Implications for the Interpretation of EPR Spectra in Terms of Structure †. Biochemistry. 2000;39:8396–8405. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]
- 65.Iwahara J, Schwieters CD, Clore GM. Ensemble Approach for NMR Structure Refinement against (1){H} Paramagnetic Relaxation Enhancement Data Arising from a Flexible Paramagnetic Group Attached to a Macromolecule. J Am Chem Soc. 2004;126:5879–5896. doi: 10.1021/ja031580d. [DOI] [PubMed] [Google Scholar]
- 66.Kosen PA. Spin Labeling of Proteins. Methods Enzymol. 1989;177:86–121. doi: 10.1016/0076-6879(89)77007-5. [DOI] [PubMed] [Google Scholar]
- 67.Osborne MJ, Schnell J, Benkovic SJ, Dyson HJ, Wright PE. Backbone Dynamics in Dihydrofolate Reductase Complexes: Role of Loop Flexibility in the Catalytic Mechanism. Biochemistry. 2001;40:9846–9859. doi: 10.1021/bi010621k. [DOI] [PubMed] [Google Scholar]
- 68.Schilder J, Liu W-M, Kumar P, Overhand M, Huber M, Ubbink M. Protein Docking Using an Ensemble of Spin Labels Optimized by Intra-Molecular Paramagnetic Relaxation Enhancement. Phys Chem Chem Phys. 2016;18:5729–5742. doi: 10.1039/c5cp03781f. [DOI] [PubMed] [Google Scholar]
- 69.Bertini I, Luchinat C, Nagulapalli M, Parigi G, Ravera E. Paramagnetic Relaxation Enhancement for the Characterization of the Conformational Heterogeneity in Two-Domain Proteins. Phys Chem Chem Phys. 2012;14:9149. doi: 10.1039/c2cp40139h. [DOI] [PubMed] [Google Scholar]
- 70.Silvestre-Ryan J, Bertoncini CW, Fenwick RB, Esteban-Martin S, Salvatella X. Average Conformations Determined from PRE Data Provide High-Resolution Maps of Transient Tertiary Interactions in Disordered Proteins. Biophys J. 2013;104:1740–1751. doi: 10.1016/j.bpj.2013.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu K-N, Song C, Yu H-H, Swager TM, Griffin RG. High-Frequency Dynamic Nuclear Polarization Using Biradicals: A Multifrequency EPR Lineshape Analysis. J Chem Phys. 2008;128:052302. doi: 10.1063/1.2816783. [DOI] [PubMed] [Google Scholar]
- 72.Osborne MJ, Venkitakrishnan RP, Dyson HJ, Wright PE. Diagnostic Chemical Shift Markers for Loop Conformation and Substrate and Cofactor Binding in Dihydrofolate Reductase Complexes. Protein Sci. 2003;12:2230–2238. doi: 10.1110/ps.03219603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Linden AH, Franks WT, Akbey Ü, Lange S, van Rossum B-J, Oschkinat H. Cryogenic Temperature Effects and Resolution upon Slow Cooling of Protein Preparations in Solid State NMR. J Biomol NMR. 2011;51:283–292. doi: 10.1007/s10858-011-9535-z. [DOI] [PubMed] [Google Scholar]
- 74.Siemer AB, Huang K-Y, McDermott AE. Protein Linewidth and Solvent Dynamics in Frozen Solution NMR. PLoS One. 2012;7:e47242. doi: 10.1371/journal.pone.0047242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bauer T, Dotta C, Balacescu L, Gath J, Hunkeler A, Böckmann A, Meier BH. Line-Broadening in Low-Temperature Solid-State NMR Spectra of Fibrils. J Biomol NMR. 2017;67:51–61. doi: 10.1007/s10858-016-0083-4. [DOI] [PubMed] [Google Scholar]
- 76.Sergeyev IV, Itin B, Rogawski R, Day LA, McDermott AE. Efficient Assignment and NMR Analysis of an Intact Virus Using Sequential Side-Chain Correlations and DNP Sensitization. Proc Natl Acad Sci USA. 2017;114:5171–5176. doi: 10.1073/pnas.1701484114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koers EJ, Van Der Cruijsen EAW, Rosay M, Weingarth M, Prokofyev A, Sauvee C, Ouari O, Van Der Zwan J, Pongs O, Tordo P, et al. NMR-Based Structural Biology Enhanced by Dynamic Nuclear Polarization at High Magnetic Field. J Biomol NMR. 2014;60:157–168. doi: 10.1007/s10858-014-9865-8. [DOI] [PubMed] [Google Scholar]
- 78.Fricke P, Mance D, Chevelkov V, Giller K, Becker S, Baldus M, Lange A. High Resolution Observed in 800 MHz DNP Spectra of Extremely Rigid Type III Secretion Needles. J Biomol NMR. 2016;65:121–126. doi: 10.1007/s10858-016-0044-y. [DOI] [PubMed] [Google Scholar]
- 79.O’Grady C, Rempel BL, Sokaribo A, Nokhrin S, Dmitriev OY. One-Step Amino Acid Selective Isotope Labeling of Proteins in Prototrophic Escherichia Coli Strains. Anal Biochem. 2012;426:126–128. doi: 10.1016/j.ab.2012.04.019. [DOI] [PubMed] [Google Scholar]
- 80.Verardi R, Traaseth NJ, Masterson LR, Vostrikov VV, Veglia G. Isotope Labeling in Biomolecular NMR. Adv Exp Med Biol. 2012;992:35–62. doi: 10.1007/978-94-007-4954-2_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hoogstraten CG, J EJ., Jr Metabolic Labeling: Taking Advantage of Bacterial Pathways to Prepare Spectroscopically Useful Isotope Patterns in Proteins and Nucleic Acids. Concepts Magn Reson, Part A. 2008;32 A:34–55. [Google Scholar]
- 82.Takegoshi K, Nakamura S, Terao T. 13C-1H Dipolar-Assisted Rotational Resonance in Magic-Angle Spinning NMR. Chem Phys Lett. 2001;344:631–637. [Google Scholar]
- 83.Jaroniec CP, Filip C, Griffin RG. 3D TEDOR NMR Experiments for the Simultaneous Measurement of Multiple Carbon-Nitrogen Distances in Uniformly 13C, 15N Labeled Solids. J Am Chem Soc. 2002;124:10728–10742. doi: 10.1021/ja026385y. [DOI] [PubMed] [Google Scholar]
- 84.Baldus M. Cross Polarization in the Tilted Frame: Assignment and Spectral Simplification in Heteronuclear Spin Systems. Mol Phys. 1998;95:1197–1207. [Google Scholar]
- 85.Bobko AA, Efimova OV, Voinov MA, Khramtsov VV. Unique Oxidation of Imidazolidine Nitroxides by Potassium Ferricyanide: Strategy for Designing Paramagnetic Probes with Enhanced Sensitivity to Oxidative Stress. Free Radical Res. 2012;46:1115–1122. doi: 10.3109/10715762.2012.692785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bräuniger T, Wormald P, Hodgkinson P. Improved Proton Decoupling in NMR Spectroscopy of Crystalline Solids Using the SPINAL −64 Sequence. Monatsh Chem. 2002;133:1549–1554. [Google Scholar]
- 87.Sato H, Bottle SE, Blinco JP, Micallef AS, Eaton GR, Eaton SS. Electron Spin-Lattice Relaxation of Nitroxyl Radicals in Temperature Ranges That Span Glassy Solutions to Low-Viscosity Liquids. J Magn Reson. 2008;191:66–77. doi: 10.1016/j.jmr.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jaroniec CP. Solid-State Nuclear Magnetic Resonance Structural Studies of Proteins Using Paramagnetic Probes. Solid State Nucl Magn Reson. 2012;43–44:1–13. doi: 10.1016/j.ssnmr.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 89.Ravera E, Shimon D, Feintuch A, Goldfarb D, Vega S, Flori A, Luchinat C, Menichetti L, Parigi G. The Effect of Gd on Trityl-Based Dynamic Nuclear Polarisation in Solids. Phys Chem Chem Phys. 2015;17:26969–26978. doi: 10.1039/c5cp04138d. [DOI] [PubMed] [Google Scholar]
- 90.Leavesley A, Shimon D, Siaw T-A, Feintuch A, Goldfarb D, Vega S, Kaminker I, Han S. Effect of Electron Spectral Diffusion on Static Dynamic Nuclear Polarization at 7 T. Phys Chem Chem Phys. 2017;19:3596–3605. doi: 10.1039/c6cp06893f. [DOI] [PubMed] [Google Scholar]
- 91.Mentink-Vigier F, Paul S, Lee D, Feintuch A, Hediger S, Vega S, De Paepe G. Nuclear Depolarization and Absolute Sensitivity in Magic-Angle Spinning Cross-Effect Dynamic Nuclear Polarization. Phys Chem Chem Phys. 2015;17:21824–21836. doi: 10.1039/c5cp03457d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.