

Abstract
The efficacy of vaccine adjuvants such as Toll-like receptor agonists (TLRa) can be improved through formulation and delivery approaches. Here, we attached small molecule TLR-7/8a to polymer scaffolds (polymer-TLR-7/8a) and evaluated how varying physicochemical properties of the TLR-7/8a and polymer carrier influenced the location, magnitude and duration of innate immune activation in vivo. Particle formation by polymer-TLR-7/8a was critical for restricting adjuvant distribution and prolonging activity in draining lymph nodes. The improved pharmacokinetic profile by particulate polymer-TLR-7/8a was also associated with reduced morbidity and enhanced vaccine immunogenicity for inducing antibodies and T cell immunity. We extended these findings to the development of a modular platform in which protein antigens are site-specifically linked to temperature-responsive polymer-TLR-7/8a adjuvants that self-assemble into immunogenic particles at physiologic temperatures in vivo. Our findings provide a chemical and structural basis for optimizing adjuvant design to elicit broad-based antibody and T cell responses with protein antigens.
INTRODUCTION
While vaccines that mediate protection through antibodies are in routine clinical use1, vaccines that generate robust and durable T cell immunity are still needed for protection against certain infections (e.g., malaria2 and tuberculosis3) and as therapies for cancer4. One means of improving T cell immunity to vaccines is through the rational choice of adjuvants formulated with defined protein or peptide antigens5. Some of the most effective adjuvants for promoting T cell immunity are Toll-like receptor agonists (TLRa), which stimulate distinct populations of antigen-presenting cells (APCs), particularly dendritic cells (DCs), to present antigen, express costimulatory molecules and produce selective cytokines that drive T cell responses6.
Importantly, the formulation and delivery of TLRa play a critical role for influencing their in vivo activity when used with protein-based vaccines7–9 Accordingly, linking TLRa directly to protein antigen10–12, or co-administering protein with particle-based carriers9, 13–17 of TLRa have been shown to markedly improve both T cell and antibody responses. Recent reports show that the ability of formulations to spatially restrict TLRa activity to the sites of vaccine administration and draining lymph nodes are critical for enhancing antibody and T cell mediated protection18, 19. The capacity of particulate delivery platforms to target distinct APC populations in lymph nodes based on size has been studied20–23, but it is not clear how these and other physical and chemical properties of TLRa delivery influence the location, magnitude and duration of innate immune activation to enhance T cell immunity to protein vaccines in vivo.
Here, we investigated how delivery of TLRa on biocompatible polymer scaffolds can modulate innate immune activation in vivo to enhance T cell immunity to protein antigens. Small molecule agonists of endosomally localized TLRs 7 and 8 (TLR-7/8a) were selected as model TLRa for these studies because they have been shown in both humans24 and mice6, 25 to activate all major DC subsets and macrophages and induce the production of specific cytokines (i.e., IL-12 and Type I IFNs) that promote T cell immunity26. While FDA approval of two imidazoquinoline-based TLR-7/8a (i.e., Imiquimod and Resiquimod (R848)) for treating skin pathologies27, 28 provides evidence to support their safety and efficacy as immunotherapies in humans, poor pharmacokinetic properties that give rise to systemic toxicity29, 30 presently limit TLR-7/8a to topical uses.
Thus, to improve their pharmacokinetics and evaluate how various properties of delivery influence their activity for use in vaccines, we linked TLR-7/8a to polymers (Poly-7/8a) in a combinatorial process and evaluated how different physicochemical properties, such as the density of TLRa arrayed on polymers and inducible particle formation, influenced innate immune activation, adjuvant distribution, kinetics and uptake by different APCs in vivo. The data show that particle formation by Poly-7/8a is critical for enhancing the magnitude and duration of innate immune activation in draining lymph nodes while reducing systemic distribution and toxicity. The improved activity by particle-forming Poly-7/8a was associated with Th1 CD4 and CD8 T cell mediated-protection in two infectious challenge models, as well as Th1-skewed antibody responses. Finally, to extend these findings and develop an approach that combines the benefits of soluble antigens and adjuvants—high chemical definition and stability—with the superior activity of particulate vaccines, we developed temperature-responsive polymers co-delivering TLR-7/8a and protein antigen that exist as water soluble molecules in vitro at room temperature but that undergo temperature-dependent particle formation in vivo.
RESULTS
In vivo screening of Polymer-TLR-7/8a conjugates
The first studies focused on optimizing the delivery of small molecule TLR-7/8a (Supplementary Fig. 1a) on polymer carriers. We chose HPMA-based polymers (Supplementary Fig. 1b) as scaffolds based on their demonstrated safety in humans and because they provide a modular platform for delivering bioactive molecules31, 32. To evaluate how different properties, such as the density of TLR-7/8a arrayed on the polymer, or chemical composition of linkers (hydrophobic/hydrophilic, length) anchoring the agonist to the polymer backbone (Supplementary Fig. 1a) influence biological activity, a library of different Poly-7/8a (Fig. 1a and b, and Supplementary Fig. 1b) were generated through combinatorial synthesis (Supplementary Fig. 2) and then screened in vivo. As the hydrocarbon backbone of HPMA polymers is non-biodegradable, Poly-7/8a conjugates were kept at or below the previously reported glomerular filtration limit (~ 45 kDa33) to facilitate renal excretion.
Figure 1. Increasing densities of TLR-7/8a arrayed on polymer carriers is associated with particle formation and enhanced lymph node cytokine production.
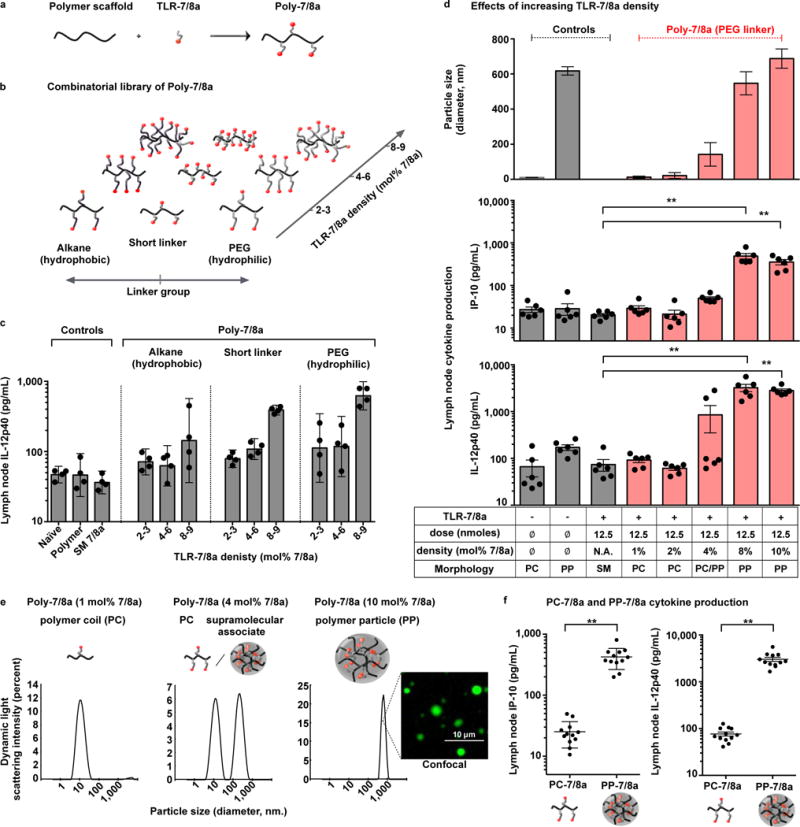
(a) Poly-7/8a were generated by reacting linear biocompatible polymers with nucleophilic TLR-7/8a. (b) Combinatorial synthesis was used to generate Poly-7/8a with varying linker group composition and TLR-7/8a density. The density of agonist arrayed on the polymers (mol% 7/8a) is reported as the percentage of monomers that are linked to TLR-7/8a (e.g., 10 mol% 7/8a indicates 10 out of 100 monomers are linked to TLR-7/8a). (c) A combinatorial library of Poly-7/8a (12.5 nmoles) were subcutaneously administered into hind footpads of mice. After 24 h, lymph nodes draining the site of immunization were harvested and processed to generate a cell suspension that was cultured for 8 h and then evaluated for the presence of IL-12p40 (n = 4). (d) Controls or Poly-7/8a with increasing densities of TLR-7/8a (normalized to 12.5 nmoles TLR-7/8A) were evaluated for particle formation by dynamic light scattering (n = 3) and the capacity to induce IP-10 (n = 6) and IL-12p40 (n = 6) cytokine production in draining lymph nodes at 24 h and 96 h, respectively, after administration. (e) Size distribution plots from dynamic light scattering are shown for selected samples; a confocal microscopy image is shown for a Poly-7/8a with 10 mol% 7/8a that forms particles in aqueous conditions. (f) Data from panel (d) for which Poly-7/8a with 1–2 mol% 7/8a that exist as unimolecular polymer coils (PC-7/8a) or Poly-7/8a with 8–10 mol% 7/8a that exist as submicron polymer particles (PP-7/8a) are grouped together to correlate the effect of Poly-7/8a morphology with lymph node IP-10 (n = 12) and IL-12p40 (n = 12). In vivo screens are representative of two independent experiments. Data on linear axes are reported as mean ± SEM. Data on log scale are reported as geometric mean with 95% Confidence Interval (CI). Comparison of multiple groups for statistical significance was determined using Kruskal-Wallis ANOVA with Dunn’s post test; Student’s T test was used for comparison of 2 groups; ns, not significant (P > 0.05); *, P < 0.05; **, P < 0.01. SM = small molecule; PC = polymer coil; PP = polymer particle.
We found that increasing the density of TLR-7/8a arrayed on polymers resulted in substantially higher lymph node production of cytokines critical for priming Th1 and CD8 T cell immunity (IL-12, IP-10 and IFNs), even though the same total dose of TLR-7/8a (12.5 nmoles) was administered (Fig. 1c and d, and Supplementary Fig. 3 and 4a–d). Notably, the enhanced in vivo activity observed for increasing densities of TLR-7/8a was associated with Poly-7/8a assembling into particles in aqueous conditions (Fig. 1d and e). Whereas Poly-7/8a with low to intermediate agonist densities (1–4 mol% TLR-7/8a) exist predominantly as unimolecular polymer coils (PC, ~ 10–20 nm diameter) and induce no measurable cytokine production in vivo, Poly-7/8a with high agonist density (8–10 mol% TLR-7/8a) assemble into submicron polymer particles (PP, ~ 700 nm diameter) and induce substantially higher lymph node cytokine production compared with the unformulated small molecule TLR-7/8a (SM 7/8a) and Poly-7/8a with low agonist density (1–2 mol% 7/8a) that exist as unimolecular polymer coils (Fig. 1d–f and Supplementary Fig. 4a–d). These data show that particle formation, increasing densities of TLR-7/8a on the polymers, or both, are critical for in vivo cytokine responses by Poly-7/8a. Such innate cytokine production by particle-forming Poly-7/8a is dependent on TLR-7 and independent of caspase 1/11 signaling, a major Inflammasome pathway that is activated by certain particulate delivery systems34 (Supplementary Fig. 4e and f).
Particle formation by Poly-7/8a promotes local retention and uptake by APCs
Cytokine production by lymph node APCs depends on the capacity of TLR-7/8a to access endosomally localized receptors within those cells35. However, the relative efficiency of TLR-7/8a delivery to target cells by different carrier morphologies is presently unknown. Thus, we assessed how morphology of the TLR-7/8a carrier, either small molecule (SM 7/8a), polymer coil (PC-7/8a) or particle-forming polymer (PP-7/8a), influences uptake by immune cells in lymph nodes draining the site of immunization following their subcutaneous administration in the hind footpads of mice. All TLR-7/8a constructs were fluorescently labeled (Supplementary Fig. 5a) to facilitate in vivo tracking. We prepared PC-7/8a and PP-7/8a with the same density of TLR-7/8a, while an inert hydrophobic ligand was attached to PP-7/8a to induce particle formation (Supplementary Fig. 4a–b and 5a). While the unformulated SM 7/8a exhibited systemic distribution (Fig. 2a) and rapid clearance (Fig. 2b), both PC-7/8a and PP-7/8a were primarily focused at the injection site (Fig. 2a) and persisted in draining lymph nodes for up to 20 days (Fig. 2b). Moreover, PP-7/8a led to ~400- and 4-fold higher levels of TLR-7/8a (AUC) in draining lymph nodes as compared with SM 7/8a and PC-7/8a, respectively (Fig. 2b). Analysis of lymph node sections by confocal microscopy (Fig. 2c) provided additional spatiotemporal resolution by revealing that PP-7/8a primarily localized within APCs situated in or near the medullary and cortical lymphatic sinuses of draining lymph nodes on day 1, but was also associated with a large influx of migratory APCs in the T cell zone at later time points (days 4 and 8). In contrast, only a limited amount of PC-7/8a was detectable and localized with APCs in the lymph node periphery. The SM 7/8a was undetectable in all lymph node sections (Fig. 2c), substantiating that the low molecular weight agonist was rapidly eliminated (Fig. 2b).
Figure 2. Particle formation by Poly-7/8a enhances local retention and promotes uptake by migratory APCs.
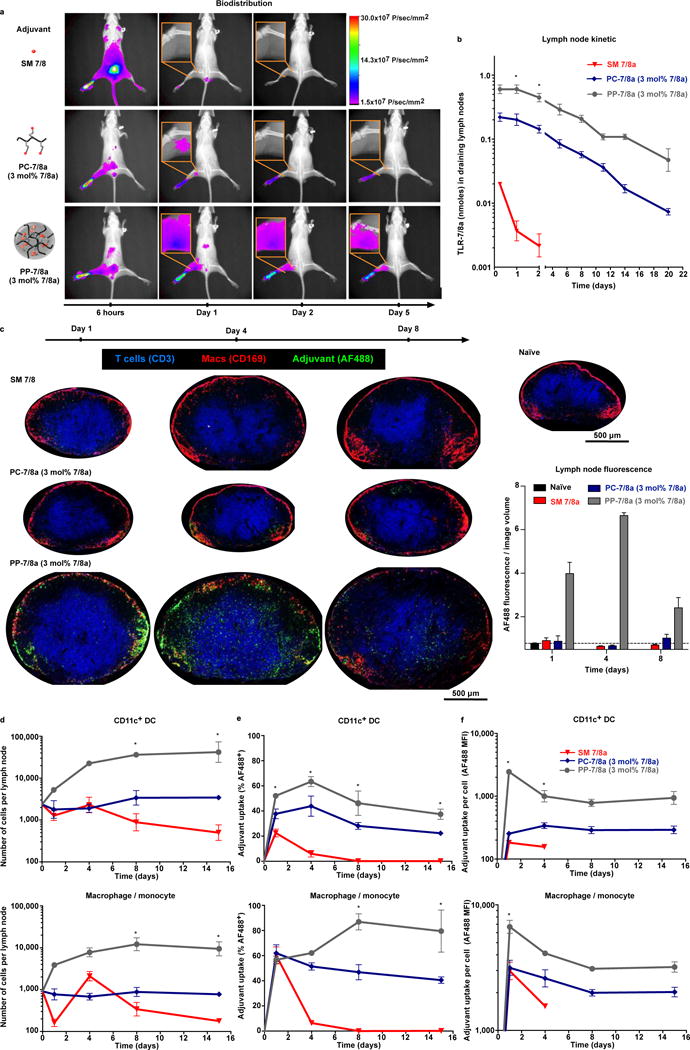
Dye-labeled particle-forming Poly-7/8a (PP-7/8a), polymer coil Poly-7/8a (PC-7/8a) and small molecule TLR-7/8a (SM 7/8a) were normalized for TLR-7/8a dose (62.5 nmoles) and administered subcutaneously to the left hind footpad of mice (see supplementary Fig. 5a for material composition and doses). (a, b) mice (n = 3) received Infrared (IR) dye-labeled materials and were imaged by X-ray and epifluorescence. (a) Representative images illustrate biodistribution over the first 5 days; (b) amount of TLR-7/8a (nmoles) in draining lymph nodes (n = 3) was estimated from epifluorescence images. (c–f) For mice that received Alexa Fluorophore 488 (AF488) labeled materials, draining lymph nodes were harvested at serial timepoints and evaluated by confocal microscopy (n = 3) or were processed to generate cell suspensions that were evaluated by flow cytometry (n = 3). (c) Representative confocal microscopy images are shown for the groups at 1, 4 and 8 days after administration of adjuvant, with the total fluorescence per imaged section volume quantified and compared across all samples (n = 3). (d) Flow cytometry analysis of the number of total CD11c+ DCs and macrophages/monocytes in lymph nodes (n = 3), as well as (e) percent adjuvant uptake (% AF488+, n = 3) and (f) the relative amount of adjuvant taken up on a per cell basis (AF488 MFI, n = 3). Whole animal imaging and flow cytometry data are representative of two independent experiments and confocal images from each group are representative of 3 replicates. All data are reported as mean ± SEM; statistical significance is shown for comparisons between PP-7/8a and SM 7/8a and was determined using Kruskal-Wallis ANOVA with Dunn’s post test; ns, not significant (P > 0.05); *, P < 0.05; **, P < 0.01.
Further assessment of specific lymph node APC populations by flow cytometry (Supplementary Fig. 5b and c) showed that PP-7/8a leads to a substantially greater (>10-fold) influx of CD11c+ DCs and macrophages/monocytes (CD11bhiF4/80+) as compared with SM 7/8a and PC-7/8a (Fig. 2d). Furthermore, while PC-7/8a and PP-7/8a localized in draining lymph nodes with similar proportions of total DCs (~40–60%) and macrophages/monocytes (~60–80%) (Fig. 2e), the relative amount of material taken up by APCs on a per cell basis was markedly higher (~10–20 fold) for PP-7/8 as compared with PC-7/8a (Fig. 2f). Further stratification of DC subsets revealed uptake of PP-7/8a by monocyte-derived DCs (CD11c+CD11bhiCD8−B220−) as well as lymph node resident CD8− and CD8+ DCs (CD11c+CD11bloCD8−B220− and CD11c+CD8+, respectively) populations at day 1, but the CD11c+CD8−B220− population enriched for migratory APCs accounted for the majority of uptake at later time points (Supplementary Fig. 5d). These findings are consistent with earlier reports that submicron sized particles (>100 nm), such as PP-7/8a, are primarily trafficked to lymph nodes by migratory APCs21.
TLR 7/8a density potentiates innate and adaptive immunity
We then assessed how carrier morphology and properties of the linked TLR-7/8a influence the magnitude and spatiotemporal characteristics of innate immune activation in vivo. The locally restricted PP-7/8a was associated with significantly increased costimulatory molecule expression by DCs and macrophages/monocytes (Fig. 3a and b and Supplementary Fig. 6a and b), and enhanced cytokine production (Fig. 3c), in draining lymph nodes. Analysis of APC populations in the spleen showed that only the systemically distributed SM 7/8a (see Fig. 2a) induced splenic DC activation (Supplementary Fig. 6c) and serum cytokines (Supplementary Fig. 6d). Notably, PC-7/8a was largely inactive in vivo, despite efficient local retention. We hypothesized that increasing the molecular weight of PC-7/8a may lead to improved activity in vivo; however, PC-7/8a across a range of molecular weights (5–300 kDa) were still inactive (data not shown). Altogether, these results show that particle formation by Poly-7/8a is critical for mediating local DC activation and cytokine production.
Figure 3. Particle forming Poly-7/8a induce high magnitude and persistent local innate immune activation that is associated with enhanced CD8 T cell responses and Th1 skewed antibody responses.
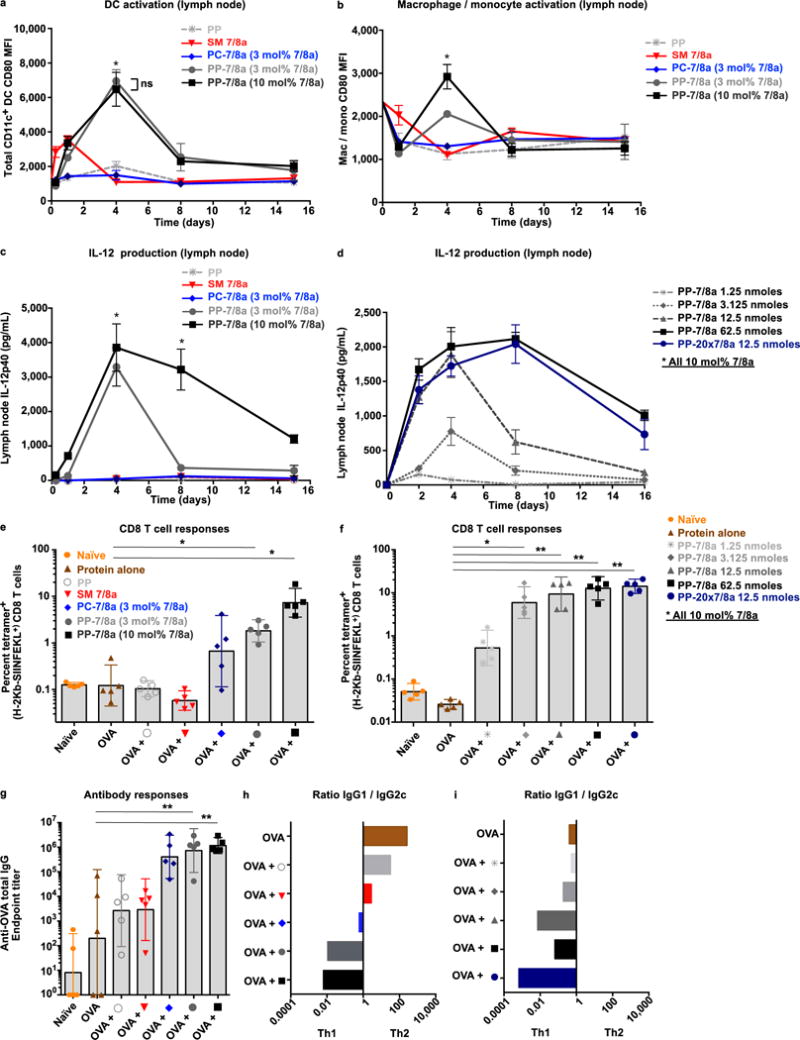
Poly-7/8a or controls normalized for TLR-7/8a dose (62.5 nmoles), or Poly-7/8a delivered at different doses (1.25 to 62.5 nmoles), were administered subcutaneously to the left hind footpad of mice. (a–b) Draining lymph nodes were harvested at serial timepoints thereafter and were processed to generate cell suspensions that were evaluated by flow cytometry. (a) CD11c+ DCs (n = 3) and (b) macrophages/monocytes (n = 3) were evaluated for their expression of the co-stimulatory molecule CD80. (c, d) Lymph nodes draining the site of administration were harvested at serial timepoints after administration and were processed to generate a cell suspension that was cultured 8 h and then evaluated for the presence of IL-12p40 (n = 4). (e–i) Poly-7/8a, SM 7/8a and controls were formulated with 50 μg of OVA in PBS and given subcutaneously to C57BL/6 mice at days 0 and 14 (see supplementary figure 8a for doses). At day 28, tetramer+ CD8 T cell responses were evaluated from whole blood of mice that received either (e) different formulations of TLR-7/8a (n = 5) or (f) different doses or potencies of particle-forming Poly-7/8a (PP-7/8a) admixed with OVA (n = 5). Serum from vaccinated mice was evaluated for (g) anti-OVA total IgG antibody titers (n = 5), as well as (h, i) the ratio of Ig1/IgG2c isotypes (n = 5). All data are representative of two or more independent experiments. Data on log scale are reported as geometric mean with 95% CI. Comparison of multiple groups for statistical significance was determined using Kruskal-Wallis ANOVA with Dunn’s post test; ns, not significant (P > 0.05); *, P < 0.05; **, P < 0.01.
Increasing agonist density (3 to 10 mol% 7/8a) on PP-7/8a administered at the same dose of TLR-7/8a (62.5 nmoles) led to higher costimulatory molecule expression by macrophages/monocytes (Fig. 3b) and certain DC subsets (Supplementary Fig. 6b), as well as more persistent cytokine production (Fig. 3c). Furthermore, using either PP-7/8a at different doses of TLR-7/8a (1–62.5 nmoles), or with a ~ 20-fold more potent agonist (PP-20x7/8a, Supplementary Fig. 2a and b), led to higher magnitude and more persistent (> 8 days) cytokine responses (Fig. 3d). Consistent with recent in vitro findings that the C4 amine can be blocked to delay onset of immune activity36, we also found that reversing the orientation of TLR-7/8a on the polymers delayed the onset and reduced the magnitude of lymph node cytokine responses (Supplementary Fig. 7a–c).
We next assessed CD8 T cell and antibody responses following co-administration of the different adjuvant formulations with a model protein antigen, Ovalbumin (OVA). After two immunizations, the magnitude of CD8 T cell responses (Fig. 3e and f) was concordant with local lymph node cytokine responses (Fig. 3c and d). PP-7/8a with high TLR-7/8a density (10 mol% TLR-7/8a) elicited substantially higher CD8 T cell responses as compared with all other groups (Fig. 3e). Moreover, increasing either the dose of TLR-7/8a delivered with PP-7/8a or the potency of the agonists attached to PP-7/8a resulted in significantly increased magnitude of CD8 T cell responses (Fig. 3f).
We also assessed the influence of the Poly-7/8a formulations on antibody responses. While the magnitude of anti-OVA total IgG antibody responses was comparable between the different Poly-7/8a formulations (Fig. 3g), increasing density, potency and dose of TLR-7/8a delivered on polymer particles was associated with strikingly more biased antibody class switching to IgG2c (Figure 3h and i and Supplementary Fig. 8a–d), consistent with the persistence of local IL-12 and IFN production driving the skewing of antibody responses to isotypes associated with Th1 immunity.
Particulate formulation of other TLRa limits systemic toxicity
We assessed whether different adjuvants that induce innate immune activation locally, systemically or both lead to protective T cell immunity as well as morbidity (weight loss) in mice. Responses induced by PP-7/8a were benchmarked against two commercially available TLRa, a small molecule TLR-7/8a, R848 (Resiquimod), and a TLR-9 agonist, CpG (ODN 1826). Whereas the low molecular weight agonist, R848, induced only systemic (serum) cytokines (Fig. 4a and Supplementary Fig. 9a), the PP-7/8a induced predominantly local (lymph node) cytokine production (Fig. 4b and Supplementary Fig. 9b). In contrast, CpG induced high levels of both local and systemic cytokine production (Fig. 4a and b and Supplementary Fig. 9a and b). The systemic cytokine production induced by R848 and CpG was associated with transient decreases in body weight (Fig. 4c), which was not observed for locally retained PP-7/8a (Fig. 4c and d).
Figure 4. Persistent, locally restricted innate immune activation is necessary and sufficient for eliciting protective CD8 and Th1-type CD4 T cell responses.
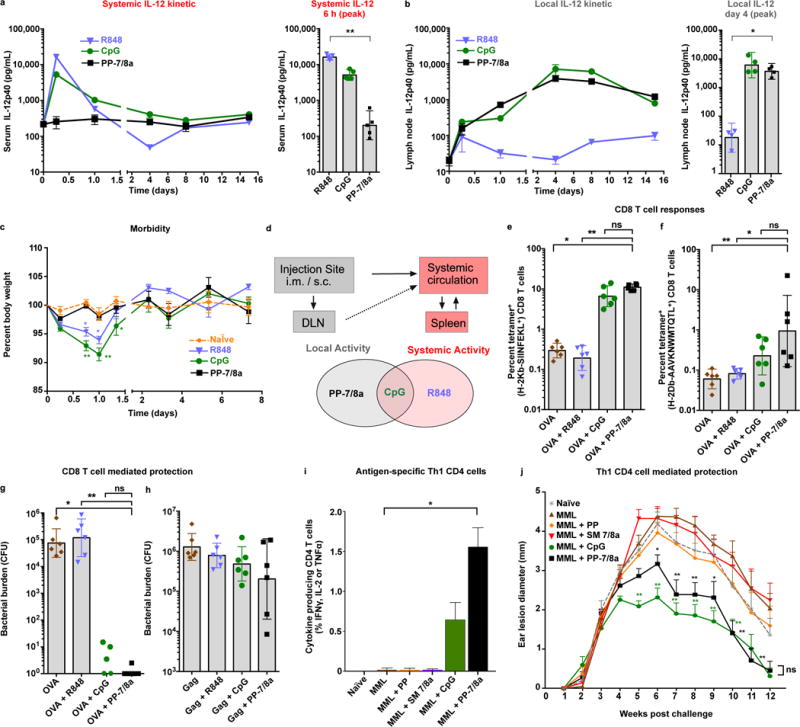
(a–c) CpG ODN 1826 (3.1 nmoles, 20 μg), R848 (62.5 nmoles, 20 μg) or PP-7/8a (62.5 nmoles, 120 μg) were delivered subcutaneously into both hind footpads of C57BL/6 mice. (a) Supernatant of ex vivo cultured lymph node cell suspensions (n = 4) and (b) serum (n = 5) were assessed for IL-12p40 by ELISA at serial timepoints. (c) Percent body weight change (n = 3) following subcutaneous administration of different vaccine adjuvants (significance is shown for comparison with naïve; two-way ANOVA with Bonferroni correction). (d) Relationship between biodistribution and local and systemic innate immune activation. (e, f) C57BL/6 mice received subcutaneous administration of protein antigen (either 50 μg of OVA, or 20 μg of SIV Gag p41) formulated with adjuvant at days 0 and 14. At day 24, tetramer+ CD8 T cell responses were assessed from whole blood by flow cytometry (n = 6). (g, h) Mice were challenged intravenously at day 28 with either (g) LM-OVA or (h) LM-Gag, and bacterial burden in spleens (n = 6) was evaluated on day 31 and 30, respectively. (i–l) C57BL/6 mice received subcutaneous immunizations of 20 μg of MML with or without adjuvant on days 0, 21 and 42. (i) Splenocytes were isolated on day 70 and stimulated in vitro with an MML peptide pool. CD4 T cells in the mixed splenocyte cultures were evaluated for Th1 characteristic cytokine (IFNγ, IL-2 and TNFα) production (n = 4). (j) Mice were challenged intradermally in both ears with L. major at day 70. Ear lesion diameters (n = 6) were measured for 12 weeks (significance is shown for comparison with protein alone). All data are representative of two or more independent experiments, except the Leishmania ear lesion kinetic is from a single study. Data on log scale are reported as geometric mean with 95% CI. Unless stated otherwise, comparison of multiple groups for statistical significance was determined using Kruskal-Wallis ANOVA with Dunn’s post test; ns, not significant (P > 0.05); *, P < 0.05; **, P < 0.01.
To extend these findings to other TLRa, particulate carriers of TLR-2/6a, TLR-4a and TLR-9a (Supplementary Fig. 10 and Supplementary Methods) were compared with their respective unconjugated TLRa and evaluated for local and systemic innate immune activation and morbidity (Supplementary Fig. 11a–d). Delivery of the various TLRa on particulate carriers largely resulted in enhanced DC activation and lymph node cytokine production while reducing systemic cytokine production and morbidity relative to the unconjugated TLRa. Grouping data from several studies in a meta-analysis shows that the systemic cytokine production induced by unconjugated TLRa is associated with acute TLRa toxicity and morbidity (Supplementary Fig. 11e).
Particulate TLRa enhances protective T cell immunity
The adjuvant capacity of PP-7/8a, R848 and CpG to elicit protective CD8 T cell responses was next determined using OVA and SIV Gag protein as immunogens and Listeria monocytogenes (LM) expressing either OVA (LM-OVA) or SIV Gag (LM-Gag) for challenge to assess vaccine efficacy (Fig. 4e–h and Supplementary Fig. 12a–c). After two immunizations with OVA protein, PP-7/8a and CpG (Fig. 4b) were associated with increased OVA-specific CD8 T cell responses (12% and 8%, respectively) (Fig. 4e) and significant protection (5- and 4-log reductions in bacterial burden, respectively) against LM challenge (Fig. 4g). Of note, the magnitude of CD8 T cell responses observed with PP-7/8a co-administered with OVA was ~10-fold higher than with SIV Gag (Fig. 5f) and this was associated with far less protection against LM-Gag challenge (Fig. 4h). Finally, these studies were extended to show that PP-7/8a conferred Th-1 mediated protection in the mouse model of Leishmania major when used as an adjuvant (Fig 4i and j, and Supplementary Fig. 13).
Figure 5. Thermo-responsive polymer particles (TRPP) permit temperature-dependent particle assembly that leads to persistent innate immune activation and protective CD8 T cell responses.
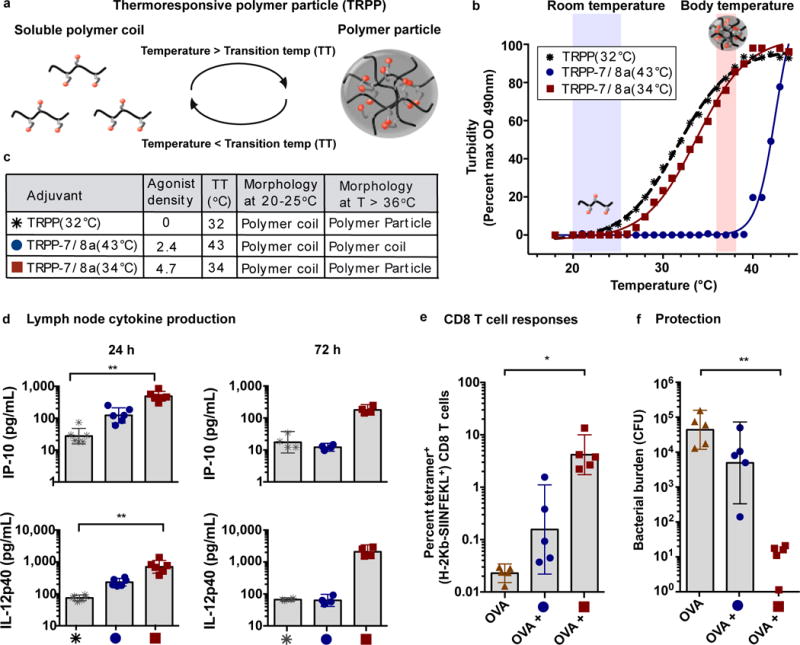
(a) Schematic of TRPP shown reversibly assembling into particles. (b) Transition temperatures (TT) were empirically determined by measuring the turbidity (OD at 490 nm) of solutions of TRPP in PBS over a range of temperatures. (c) Table summarizing the thermo-responsive properties of select TRPP. (d) TRPP-7/8a and a TRPP control were delivered subcutaneously into both hind footpads of C57BL/6 mice. Popliteal lymph nodes draining the vaccination site were harvested at 24 h (n = 6) and 72 h (n = 4) and then processed to create a cell suspension that was cultured ex vivo for 8h and then evaluated for the presence of IL-12p40 and IP-10 by ELISA. (e, f) C57BL/6 mice (n = 5) received subcutaneous administration of 50 μg of OVA alone or admixed with adjuvant at days 0 and 14. (e) Tetramer+ CD8 T cell responses were evaluated at day 24 (n = 5). (f) Mice were challenged intravenously at day 28 with LM-OVA and bacterial burden in spleens was evaluated on day 31 (n = 5). All data are representative of two independent experiments. Data on log scale are reported as geometric mean with 95% CI. Comparison of multiple groups for statistical significance was determined using Kruskal-Wallis ANOVA with Dunn’s post test; ns, not significant (P > 0.05); *, P < 0.05; **, P < 0.01.
In vivo particle formation with thermo-responsive Poly-7/8a
To substantiate that particle scaffolds are critical for optimizing TLR-7/8a activity as an adjuvant and provide a potentially more flexible commercial platform for manufacturing and storage, we developed temperature-responsive polymer particle (TRPP)-7/8a conjugates (Supplementary Fig. 14a) that exist as water-soluble macromolecules during manufacturing and storage (T < 30°C) but that undergo temperature-driven (T > 36°C) particle assembly in vivo (Fig. 5a). While temperature-sensitive polymers have been used as adjuvants alone37 or to physically entrap admixed (i.e., non-linked) Immunomodulators38, 39, herein, we evaluated how varying densities of TLR-7/8a linked directly to the polymer backbone though covalent interactions influenced immune activity in vivo.
Our data show that the transition temperature of TRPP-7/8a is tunable by modulating the density and the hydrophilic or hydrophobic character of ligands attached to the polymer backbones (Supplementary Fig. 14b), allowing for production of TRPP-7/8a that form particles at precisely defined temperatures either in vitro or at body temperature in vivo (Fig. 5b and c). Consistent with our earlier findings with PP-7/8a, only TRPP-7/8a capable of forming particles in vivo lead to persistent and high magnitude local cytokine production that is associated with protective CD8 T cell responses and Th1-skewed antibodies (Fig. 5d–f and Supplementary Fig. 14c–g).
Steps were taken to further refine the structure of TRPP-7/8a to promote biodegradability and improve generalizability of the approach. First, as bioaccumulation of polymers is a potential safety concern, a di-block copolymer with esterase sensitive side chains was used to promote degradation of the particles to individual polymer chains that can be excreted by the kidneys. Secondly, as prior studies by others and us have shown that synchronous delivery of protein antigen with innate immune stimulation is a highly efficient approach for optimizing T cell priming10, 40, 41, a TRPP-7/8a was prepared with coil peptides to provide a generalizable strategy for site-specifically linking antigen-coil fusion proteins to polymer carriers through coiled-coil interactions as described in previous reports42, 43. To demonstrate the utility of this approach for ensuring co-delivery of antigen and adjuvant, a recombinant HIV Gag-coil fusion protein was site-specifically linked to a TRPP-7/8a through self-assembly using peptide-based coiled coil interactions (Fig. 6a and Supplementary Fig. 15a–d). Mixing aqueous solutions of the HIV Gag-coil protein with a TRPP-7/8a modified with a complementary coil peptide results in self-assembly of a TRPP-7/8a-(coil-coil)-Gag heterodimer that undergoes particle formation (Fig. 6b and c) at temperatures greater than 34°C and ensures co-delivery of Gag with TRPP-7/8a (Fig. 6d and e). Co-delivery of Gag with TRPP-7/8a (TRPP-7/8a-(coil-coil)-Gag) resulted in enhanced T cell and antibody responses as compared with Gag-coil admixed with either the free TLR-7/8a or TRPP-7/8a (Fig. 6f–i).
Figure 6. Co-delivery of TLR-7/8a and protein antigen on a self-assembling thermo-responsive vaccine particle.
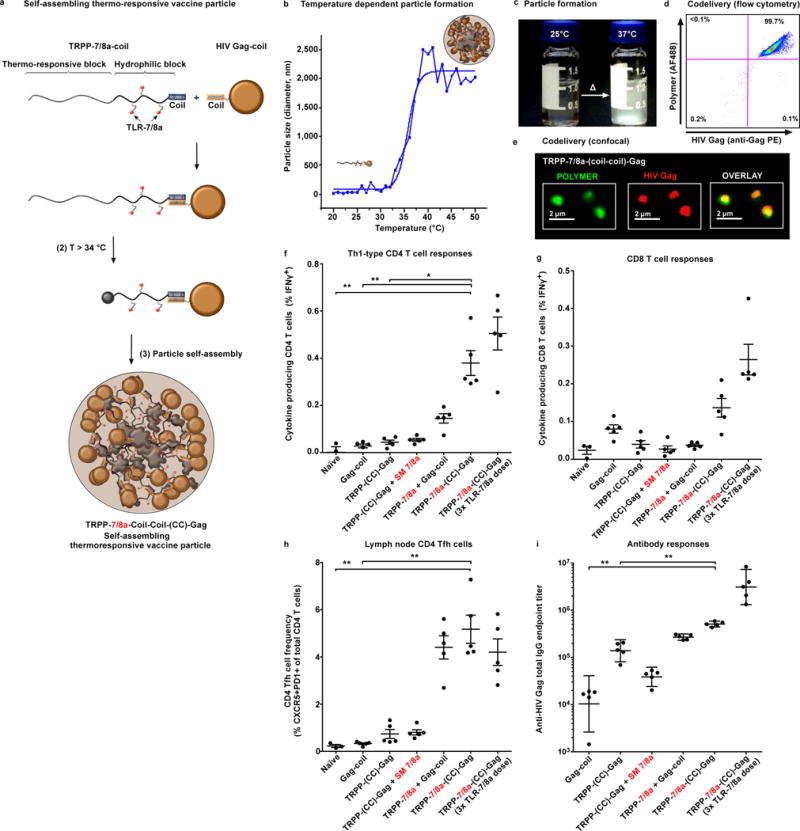
(a) Cartoon schematic of a thermo-responsive Poly-7/8a (TRPP-7/8a) modified with a coil peptide that forms heterodimers with a recombinant HIV Gag-coil fusion protein to form TRPP-7/8a-(CC)-Gag. Heterodimerization occurs at room temperature and particle formation results at temperatures greater than 33°C. (b) Temperature-dependent particle formation illustrated by dynamic light scattering. (c) Aqueous solutions of TRPP-7/8a-(CC)-Gag at 25°C and 37°C. (d, e) Co-localization of HIV Gag (labeled with anti-Gag PE) with TRPP-7/8a (labeled with carboxyrhodamine 110) was confirmed by (d) flow cytometry and (e) confocal microscopy. (f–i) BALB/c mice received subcutaneous administration of 50 μg of HIV-Gag coil formulated with either a control or TRPP-7/8a normalized for TLR-7/8a dose (1x dose = 2.5 nmoles, or 3x dose = 7.5 nmoles) at days 0 and 14. At day 28, DLN, spleen and serum from vaccinated mice were collected for analysis. Splenocytes were stimulated in vitro with an HIV Gag peptide pool. Antigen-specific IFNγ-producing (f) CD4 T cells (n = 5) and (g) CD8 T cells (n = 5) in the mixed splenocyte cultures, as well as (h) Tfh cells (n = 5) in draining lymph nodes were quantified by flow cytometry. (i) Serum was evaluated for anti-HIV Gag total IgG antibody titers (n = 5). In vivo studies are representative of two independent experiments. Data on linear axes are reported as mean ± SEM. Data on log scale are reported as geometric mean with 95% CI. Comparison of multiple groups for statistical significance was determined using Kruskal-Wallis ANOVA with Dunn’s post test; ns, not significant (P > 0.05); *, P < 0.05; **, P < 0.01. CC = coiled-coil interaction.
DISCUSSION
Herein, we systematically refined properties of polymer-TLR-7/8a to elicit robust T cell and antibody responses to protein antigen. These studies established that particulate carriers (PP-7/8a) displaying high densities and potencies of TLR-7/8a are critical properties for promoting high magnitude and persistent (> 8 days) innate immune activation restricted to draining lymph nodes that is necessary for eliciting protective CD4 and CD8 T cell responses and high antibody titers.
Our results extend earlier findings on particulate delivery of TLRa15, 16 to additional TLRa (i.e., TLR-2/6a, TLR-4a, TLR-7/8a and TLR-9a) and show that, in the context of TLR-7/8a delivery, improved retention is necessary but not sufficient for enhancing T cell immunity. We find that despite improved retention at the injection site and draining lymph nodes by all polymer carriers of TLR-7/8a, only the particles (PP-7/8a) are taken up efficiently by APCs and induce persistent innate immune activation in draining lymph nodes. These findings provide additional mechanistic insights from studies by others and us that TLR-7/8a attached to protein antigens are markedly more immunogenic upon aggregation44 or when adsorbed to alum microparticles45.
The patterns of DC uptake observed for the particulate Poly-7/8a (~ 700 nm) used in this study are consistent with earlier findings that ~ 100–1000 nm particles are primarily trafficked to lymph nodes through uptake by migratory and monocyte-derived DCs21, 22, however, it will be important to determine whether particulate carriers of TLR-7/8a that passively traffic to lymph nodes, or larger particles that require uptake by DCs in the periphery, are preferred for generating T cell immunity. Notably, most existing vaccines use Alum microparticles that form depots to promote DC activation and antigen uptake in the periphery8. Moreover, direct intranodal injection of microparticle carriers of TLR-3a have been shown to be more immunogenic than intranodal injection of nanoparticles14. Thus, additional studies are needed to carefully characterize which sizes of particulate TLRa carriers are optimal for enhancing vaccine immunogenicity.
While this report used linear polymers based on HPMA and NIPAM as scaffolds for TLR-7/8a delivery, other particle-based delivery platforms have been used in formulations with TLRa (e.g., poly(lactic-co-glycolic acid) (PLGA)13–15, liposomes9, 17 and polymersomes46) and can likely be adapted to satisfy the key physicochemical requirements for TLR-7/8a adjuvant activity defined herein. However, particulate delivery systems used in vaccines can be unstable during long-term storage and often require specialized manufacturing and storage conditions that contribute significantly to cost8. The thermo-responsive Poly-7/8 (TRPP-7/8a) we report herein could potentially overcome the manufacturing and storage limitations of preformed particles and may allow for the use of single-vial water-soluble preparations of both antigen and adjuvant.
Finally, it should be noted that other delivery approaches, such as albumin hitchhiking18 or linear polymers that do not form particles but can promote local retention and efficient uptake by APCs in draining lymph nodes may also provide the persistence of lymph node restricted innate immune activity that is needed to drive antibody and T cell responses. Indeed, as shown here, phosphorothioate-modified CpG oligonucleotides can induce protective T cell responses to protein antigen despite being a relatively low molecular weight oligomer. This may be accounted for by local retention of CpG47 through extracellular matrix protein binding48; durability through endonuclease resistance; and uptake by APCs through lectin receptors49. Even so, numerous studies do demonstrate that the activity of CpG can be improved through formulation and delivery approaches18, 50.
In conclusion, polymer carriers of TLRa represent a diverse and versatile class of adjuvant that can be systematically tuned to achieve the optimal magnitude and spatiotemporal characteristics of innate immune activity required for eliciting antibody and T cell immunity for applications in preventive and therapeutic vaccines for infections and tumors.
ONLINE METHODS
Synthesis of small molecule TLR-7/8a (SM 7/8a)
A detailed description of the synthesis, purification and chemical characterization of the small molecules used in this study is provided in the Supplementary Materials and Methods. The conjugatable imidazoquinoline-based small molecule TLR-7/8a (SM 7/8a, SM-7/8a-Alkane, SM-7/8a-PEG, SM 20x7/8a and SM R20x7/8a) and TLR-7/8a dye conjugates were prepared according to previous reports27, 35, 51.
Synthesis of polymer-TLR-7/8a (Poly-7/8a) conjugates
A detailed description of the synthesis, purification and characterization of the Poly-7/8a is provided in the Supplementary Materials and Methods. Briefly, polymer-TLR-7/8a (Poly-7/8a) conjugates were synthesized in a combinatorial approach by reacting varying amounts of nucleophilic TLR-7/8a (e.g., SM 7/8a) with amine reactive copolymers based on N-(2-hydroxypropyl)methacrylamide (HPMA) and N-isopropylacrylamide (NIPAM) that have been previously described52, 53. For dye-labeled polymers, Alexa Fluor 488 (AF488) Cadaverine (Life Technologies, Carlsbad, CA) or Cruz Fluor 8 amine (Santa Cruz Biotechnology, Dallas, TX) were reacted with the amine reactive copolymers for 2 h prior to the addition of the nucleophilic TLR-7/8a. Poly-7/8a were purified by dialysis against methanol using Spectra/Por7 Standard Regenerated Cellulose dialysis tubing with a molecular weight cut-off (MWCO) of 25 kDa (Spectrum Labs, Rancho Dominguez, CA). The solution of methanol was changed twice each day for 3 days to ensure efficient removal of the unreacted small molecules. Poly-7/8a were collected following evaporation of the solvent and characterized according to the methods described below.
Determination of polymer molecular weights
Molecular weights and polydispersities of polymers and copolymers were measured by gel permeation chromatography using an HPLC system (Shimadzu, Japan) equipped with refractive index (RI), UV and multiangle light scattering (MALS) DAWN 8 EOS detectors (Wyatt Technology, USA). Either a Superose 6 column (GE Healthcare, USA was used with a 0.3 M acetate buffer (pH 6.5) eluent at 0.5 mL min−1, or a MicroSuperose 6 column was used with PBS (pH 7.4) as the eluent at 0.1 mL min−1. Weight-average molecular weights (Mw) were calculated from the light-scattering detector based on the known injected mass while assuming 100% mass recovery. Number-average molecular weights (Mn) were determined by refractive index measurements and were calculated assuming a dn/dc value of 0.167 mL/g. Polydispersity is defined as (Mw/Mn).
Determination of TLR-7/8a and fluorophore content on polymers
The amount of TLR-7/8a or fluorophore attached to the copolymers was determined by UV-Vis spectroscopy. Briefly, samples were suspended in solutions of 1% triethylamine/methanol at known concentrations and added to quartz cuvettes with a path length of 1 cm. Absorption was recorded over a spectrum from 250 – 775 nm using a Lambda25 UV-Vis spectrophotometer from Perkin Elmer (Waltham, MA). The amount of TLR-7/8a or fluorophore in solution was calculated from the Beer-Lambert law relationship. Example calculations are provided in the Supplementary Materials and Methods.
Particle size determination by dynamic light scattering
Z-average diameter (nm) of copolymers in PBS (pH 7.4) was determined by dynamic light scattering using a Zetasizer Nano ZS purchased from Malvern (Malvern Instruments, Malvern, United Kingdom).
Transition temperature determination for thermo-responsive polymers
Thermo-responsive polymers were suspended at 0.1 mg/mL PBS (pH 7.4) in 200 μL volumes in Costar polystyrene 96 well flat bottom culture plates (Corning, NY). Absorption at 490 nm was evaluated as a function of temperature (1°C/5 min steps) on a SpectraMax Plus spectrophotometer (Molecular Devices, Sunnyvale, CA). The Data was analyzed using Prism 6 (GraphPad, La Jolla, CA) and non-linear regression fit was used to calculate transition temperature (~ temperature at ½ max absorbance).
Cloning and expression of the Gag-coil fusion protein
The HIV Gag-KSK coil fusion protein was produced using an expression construct encoding the p41 region of the HIV Gag (HXB2) gene fused to the KSK coil peptide sequence IAALKSKIAALKSEIAALKSKIAALKSK. Briefly, a 1,131 bp fragment from HIV Gag p41 was prepared with the following additions: an N-terminal His8 tag followed by the Tobacco Etch Virus endopeptidase (TEV) cleavage site ENLYFQS, and a C-terminal Gly10 spacer followed by the KSK coil. This sequence was codon optimized for expression in E. coli and synthesised commercially (DNA2.0, USA) before sub-cloning into a T7-driven expression plasmid (OG37) (Oxford Genetics, UK) for expression and purification from E. coli. The His-tagged Gag-coil fusion protein was purified from cell lysates and Sigma Protease Inhibitors (P8849, Sigma) were added to prevent proteolysis. Cell lysate was loaded onto a 10 mL IMAC column (Thermo Scientific) and eluted with Imidazole. Fractions containing the Gag-coil fusion protein were extracted with Triton X-114 (Sigma) at 1% to remove endotoxin and then buffer exchanged into 20 mM HEPES, pH 7.3, 300 mM NaCl and 2 mM β-mercaptoethanol to a final concentration of 1.7 mg/mL HIV Gag-coil (< 2.9 EU/mg protein).
Animal protocols
All animal experiments were conducted at the Vaccine Research Center (VRC) at the National Institutes of Health (Bethesda, MD) and were in compliance with the guidelines set by the Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC) and the Institutional Animal Care and Use Committee (ACUC). All experimental animal protocols underwent review and were approved by the ACUC prior to the start of experiments.
Animals
BALB/c mice, C57BL/6 (B6) mice, and B6N.129S2-Casp1tm1Flv/J (Caspase 1/11, Inflammasome KO) mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and maintained at the Vaccine Research Center’s (VRC) Animal Care Facility (Bethesda, MD) under pathogen-free conditions. TLR7 KO mice were kindly provided by Ross Kedl at the University of Colorado (Denver, CO, USA) and were bred and maintained at the VRC. BALB/c and B6 mice used in this study were female and were between 8 and 12 weeks old at the start of experiments. Both female and male Caspase 1/11 and TLR-7 KO mice were used in this study and were between 6 and 20 weeks old at the start of experiments. Animals were randomly assigned to either control or experimental groups.
Immunizations
Vaccines were prepared in sterile, endotoxin-free (<0.05 EU/mL) PBS (Gibco, Life Technologies) and administered subcutaneously in a total volume of 50 μL. All immunogens were certified endotoxin free (<1 EU/mg) by the manufacturer or were prepared in-house with <5 EU/mg as determined by LAL assay (Genscript, Piscataway, NJ). EndoFit Ovalbumin was obtained from Invivogen (San Diego, CA). SIV Gag p55 was obtained from Protein Sciences Corporation (Meriden, CT) and MML (also known as Leish-111f) was produced as previously described54 by the Protein Expression Laboratory at the National Cancer Institute (Frederick, MD). Adjuvants had < 1EU/mg endotoxin and were either prepared in-house as described elsewhere or were acquired from commercial sources. CpG ODN 1826 and R848 were obtained from Invivogen (San Diego, CA).
Ex vivo lymph node cultures for cytokine determination
Proximal draining lymph nodes were harvested at various time points following subcutaneous administration of different adjuvants or controls and placed in 300 μL of RPMI supplemented with 10% (v/v) fetal calf serum, 50 U/mL penicillin, 50 μg/mL streptomycin and 2 mM L-glutamine in 1.5 mL DNase, RNase, Pyrogen free Kontes Pellet Pestle Grinders (Kimble-Chase, Vineland, NJ) sitting on ice. Lymph nodes were gently mechanically disrupted using sterile pestles and the resulting suspensions were vortexed for 5 seconds and added to a 96 well round bottom culture plate that was incubated at 37°C/5% CO2 for 8 h. Supernatant was collected and stored at -80°C until analyzed by ELISA or cytokine bead array.
Cytokine measurements
Cytokines in lymph node culture supernatants and sera were determined using ELISA and cytokine bead array kits according to the manufacturer’s recommended guidelines. ELISA kits for murine IL-12p40, IP-10 and IFNγ were obtained from Peprotech (Rocky Hill, NJ), and an ELISA kit for the detection of murine IFNα (Verikine Mouse IFNA Alpha ELISA Kit) was purchased from PBL Assay Science (Piscataway, NJ). Customized mouse cytokine/chemokine magnetic bead panels were purchased from EMD Millipore (Billerica, MA) and were run on a FLEX MAP3D multiplex system from Luminex (Austin, TX) according to the manufacturer’s guidelines. Concentrations of individual cytokines in the supernatants were determined from standard curves.
Whole animal imaging
Biodistribution and kinetics of IR Dye-labeled materials was evaluated using a Bruker In Vivo Xtreme (Bruker, Billerica, MA) combined optical and x-ray small animal imaging system. Following subcutaneous administration of IR dye-labeled materials, mice were imaged at serial timepoints using a two-step imaging protocol: (1) epifluorescence (excitation = 760 nm; emission = 830 nm; 0.915 second exposure); followed by (2) X-ray (45 kVp; 1.0 second exposure). Amount of TLR-7/8a (nmoles) within a region of interest (ROI) was calculated using a standard curve of (log(photons/mm2 × seconds) versus TLR-7/8a amount (nmoles).
Confocal microscopy of lymph node sections
20 μm lymph node sections were prepared and imaged as previously described, with minor modifications55. In brief, lymph nodes were fixed with 0.05 M phosphate buffer containing 0.1 M L-lysine (pH 7.4), 2 mg/mL NaIO4, and 10 mg/mL paraformaldehyde over night at 4°C, equilibrated in 30% sucrose solution for 24 h, fixed and sectioned using a Leica cryostat. Tissue sections were then blocked for 1–2 h with 1% normal mouse serum and bovine serum albumin solution containing 0.3% Triton X-100 and stained with directly conjugated antibodies for 6–10 h at 4°C. Sections were then imaged using a Leica SP8 confocal microscope equipped with a 1.3NA 40x objective and a motorized tiling stage. Potential fluorescence spillover was removed using Channel Dye Separation analysis module in the Leica LAS AF software. Final image analysis was conducted using Imaris software (Bitplane). For quantification, 3D isosurfaces of whole imaged lymph node sections were first generated using the Imaris surface creation module and statistics for the total surface volume and sum of AF488 fluorescence signal were exported into Excel (Microsoft, Redmond, WA) for further calculations.
Flow cytometry analysis of innate immune cells from lymph nodes and spleen
The magnitude, activation status and adjuvant uptake of innate immune cells in draining lymph nodes and spleen were evaluated as previously described44, with slight modifications. Briefly, lymph nodes were harvested and added to pestle tubes for mechanical disruption as described above. Resulting lymph node cell suspensions were spun down, supernatant was removed and cells were re-suspended in 1 mL of an enzyme cocktail comprised of 1 mg/mL collagenase D (Roche, Basel, Switzerland) and 100 U/mL recombinant DNase I (Roche) in RPMI for 30 minutes at 37°C. Lymph node cells were then washed and resuspended in PBS and added to 96 well plates for staining. To isolate splenocytes, 5 mL of enzyme cocktail was injected directly into spleens that were then incubated at 37°C for 30 minutes. Spleens were mechanically disrupted over 70 μm nylon strainers (Corning) and collected in 50 mL Falcon tubes (Corning). Splenocytes were washed with HBSS and resuspended in 2 mL of ACK lysing buffer (Life Technologies) followed by incubation for 4 minutes at room temperature. Splenocytes were then washed with HBSS and then resuspended with PBS and added to 96 well plates for staining. Cells were stained with LIVE/DEAD cell stain (Aqua, Life Technologies) for 10 minutes at room temperature. Without washing, cells were stained for 15 minutes with FcR-Block, anti-CD16/CD32 (clone 2.4G2, BD Biosciences, Franklin Lakes New Jersey), followed by the addition of Brilliant Violet (BV) 510-anti-CD3e (145-2C11, BD), BV421-anti-CD19 (1D3, BD), BV605-anti-Ly-6G (1A8, BD), BV786-anti-CD8 (53-6.7, BD), BV510-anti-NK-1.1 (PK136, BD), Cy7-PE-anti-B220 (RA3-6B2, BD), PE-anti-CD11c (HL3, BD), Ax700-anti-CD11b (M1/70, BioLegend, San Diego, CA), Cy5-PE-anti-F4/80 (BM8, eBioscience, San Diego, CA), and CF594-PE-anti-CD80 (16-10A1, BD). Following incubation in the antibody cocktail for 20 minutes, cells were washed with PBS, resuspended in 0.5% paraformaldehyde/PBS and then evaluated by flow cytometry.
Tetramer staining of CD8 T cells from whole blood
Tetramer+ CD8 T cell responses were characterized from whole blood as previously described56. Briefly, 200 μL whole blood was collected from immunized mice in heparinized 1.5 mL polypropylene tubes at various time points described in figure legends. Following red blood cell lysis using ACK Lysing buffer (Life Technologies), cells were washed with PBS and then added to 96 well plates for staining. Cells were stained with the viability dye LIVE/DEAD Fixable Orange (OrViD, Life Technologies) for 10 minutes at room temperature. After washing, cells were stained for 15 minutes with either PE-H-2Kb OVA (SIINFEKL) tetramer (Beckman Coulter, Brea, California) or PE –H-2Db SIV Gag (AAVKNWMTQTL) tetramer (Beckman Coulter). Cells were then blocked with anti-CD16/CD32 (clone 2.4G2, BD) for 10 minutes, followed by the addition of APC-Cy7-anti-CD8 (53-6.7, Biolegend), PE-Cy7-anti-CD62L (MEL-14, Abcam, Cambridge, England), eFluor-660-anti-CD127 (A7R34, eBioscience) and FITC-anti-KLRG1 (2F1, Southern Biotech, Birmingham, Alabama). After incubating for 20 minutes at room temperature, cells were washed and then incubated with Fix/Perm solution (BD) for 20 minutes at 4°C. After washing, cells were suspended in Perm/Wash buffer containing PerCP-Cy5.5-anti-CD3 (145-2C11, BD) and incubated at 4°C for 30 minutes. Cells were washed and suspended in Perm/Wash buffer and then evaluated by flow cytometry.
Analysis of polyfunctional CD4 T cell responses by flow cytometry
Evaluation of polyfunctional CD4 T cell responses was determined as previously described57, 58. Briefly, splenocytes were isolated from vaccinated mice at several timepoints and 1.5×106 splenocytes were cultured in 96-well plates with 2 μg/mL anti-CD28 (37.51, eBioscience) alone (background response), or in combination with 20 μg/mL MML protein (Leish-111f, derived from Leishmania spp.) or a 2 μg/mL solution of MML peptides (Mimotopes Pty Ltd; 15-mers overlapping by 11). Cells were incubated for 2 h before brefeldin A (BFA, Sigma-Aldrich) was added to a final concentration of 10 μg/ml and cells were incubated for an additional 4 h. After washing with PBS, cells were stained with Violet Dead Cell Stain (ViViD, Life Technologies), then washed and blocked with anti-CD16/CD32 (clone 2.4G2, BD) for 10 minutes at room temperature. After blocking, the following surface antibodies were added for 30 minutes at room temperature: APC-Cy7-anti-CD8 (clone 53-6.7, Biolegend) and Ax700-anti-CD4 (RM4-5). Cells were then fixed and permeabilized using Fix/Perm solution (BD) and incubated at 4°C for 30 minutes. Cells were washed and then suspended in Perm/Wash buffer containing PE-Cy5-anti-CD3 (145-2C11, BD), APC-anti-IFNγ (XMG1.2, BD), PE-anti-IL-2 (JES6-5H4, BD), PE-Cy7-anti-TNFα (MP6-XT22, BD) for 30 minutes at room temperature. Cells were washed and suspended in Perm/Wash buffer and then evaluated by flow cytometry.
Flow cytometry
Samples were acquired on a modified LSR II flow cytometer (BD). Results were analyzed using FlowJo version 9.3, Pestle version 1.6.2, and SPICE version 5.22 software (Mario Roederer, Vaccine Research Center, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD).
Listeria monocytogenes
Recombinant Listeria monocytogenes expressing-OVA (LM-OVA) was a gift from H. Shen (University of Pennsylvania, Philadelphia, PA) and attenuated (ΔactA, ΔintB) Listeria monocytogenes expressing-SIV Gag (LM-Gag) from SIV strain mac239 was provided by ANZA Therapeutics (Concord, CA). Both strains were maintained at -80°C as a stock in brain-heart infusion/50% glycerol (BHI/50% glycerol).
Listeria monocytogenes infectious challenge
Evaluation of CD8 T cell mediated protection using Listeria monocytogenes was carried out as previously described56. For infection with recombinant LM-OVA and LM-Gag, mice were infected with 3×104 and 2×107 CFU, respectively, in 0.3 mL PBS that was delivered intravenously into the tail vein. Spleens were harvested 48 or 72 h after infection and mechanically homogenized in 1 mLPBS using a Tissue Ruptor (Qiagen, Valencia, CA). Spleen homogenate was plated in duplicate as a 10-fold dilution series on brain heart infusion agar (Difco, Detroit, MI). Plates were incubated at 37°C for 24 h and colonies were counted by personnel blinded to group assignment to determine total CFU per spleen (bacterial burden).
Leishmania major challenge
L. major clone V1 (MHOM/IL/80/Friedlin) promastigotes were grown as previously described57. Briefly, infectious-stage metacyclic promastigotes of L. major were isolated from stationary cultures (4–5 days old) by negative selection using peanut agglutinin (Vector Laboratories). Mice were infected 4 weeks after boost by injecting 750 metacyclic promastigotes intradermally within both ears. Personnel blinded to the vaccination assignments of the infected mice recorded weekly ear measurements using a metric calliper.
Serum antibody measurements
Immulon 4HBX plates (Thermo Scientific) were coated overnight at 4°C with HIV Gag protein diluted in 0.1 M Carbonate Buffer (pH 9.8). Plates were then blocked with 1% FBS/PBS, and plasma from Gag immunized mice was applied in serial 10-fold dilutions in ELISA diluent (0.1% FBS/PBS) and incubated for 1 h at 37°C. Biotinylated Goat-anti-mouse total IgG (Jackson Immuno-Research, Westgrove, Pennsylvania) was added and incubated for 1 h at 37°C. For detection, Avidin-Horse Radish Peroxidase (BD) was added and incubated for 30 minutes at room temperature, followed by the addition of TMB substrate-chromogen (Dako, Glostrup, Denmark) and a 2N sulfuric acid stop solution. Washing was performed between steps with PBS + 0.05% Tween 20. Plates were read on a SpectraMax Plus spectrophotometer (Molecular Devices) at 450 nm. Data was analyzed in Prism to generate a non-linear regression fit to estimate EC50 (midpoint titer) or EC05 (endpoint titers).
Evaluation of TLR-7 activity in vitro using HEK293 reporter cells
HEK293 expressing human TLR-7 (HEK-TLR7) were purchased from Invivogen (San Diego, CA) and used according to the manufacturer’s guidelines.
Statistics and graphs
Sample sizes for biological studies were chosen based on calculations using JMP statistical analysis software (Cary, NC); standard deviations and pre-specified differences in groups (“differences to detect”) were based on historical data and type I and type II error rates were set at 0.05 and 0.2, respectively. Data on linear axes are reported as mean ± SEM. Data on log scale are reported as geometric mean with 95% CI. Statistical analyses were carried out using Prism software (GraphPad). Unless stated otherwise within the figure legends, Kruskal-Wallis one-way ANOVA with Dunn’s post test was used to calculate p values for comparisons between > 2 groups. Differences were found to be significant when P was less than 0.05 or 0.01, as indicated by single (*) or double asterisks (**) within the figures. Most graphs were produced using Prism. Flow cytometry data was processed using FlowJo (Tree Star). To evaluate the multifunctional cytokine response for antigen-specific T cells, Boolean gating was performed in Flow Jo and the resulting data was processed in Pestle (M. Roederer, VRC, NIAID, NIH, USA) and graphed using SPICE (http://exon.niaid.nih.gov/spice/; provided by Mario Roederer).
Supplementary Material
Acknowledgments
The authors wish to acknowledge M. Dillon, K. Wuddie, and C. Chiedi at the Vaccine Research Center and B. Klaunberg and V. Diaz at the Mouse Imagine Facility (MIF) for their valuable support and assistance with the animal studies. We would also like to thank K. Ulbrich, R. Swenson and G. Griffiths for their support and helpful insights. This work was supported in part by the BIOPOL project (Grant of the Ministry of Education, Youth and Sports of the Czech Republic, no. EE2.3.30.0029); by the Czech Science Foundation (15-15181S); Charles University (UNCE 204025/2012) a Cancer Research UK grant (C552/A17720); the Office of AIDS Research; and NIAID at the NIH.
Footnotes
AUTHOR CONTRIBUTIONS
G.M.L., R.L., K.D.F, L.W.S, and R.A.S were involved in experimental planning, interpreting data and writing the manuscript. G.M.L, R.L., A.E.D., O. Vasalatiy, J.T., L.S., K.A.R., and A.P.E-K planned and carried out the synthesis, purification and characterization of small molecules. R.L., R.P., M.P., T.E., O. Vanek and G.M.L planned and completed the synthesis, purification and characterization of the polymer precursors and polymer conjugates. P.A.D, A.S.I., A.B., A.Y., K.M.Q., K.K. and J.R.F. planned and conducted many of the biological studies. M.Y.G. and M.G.S. carried out the confocal microscopy studies on lymph node sections and polymer particles, respectively. T.H. and R.C. developed the plasmids to express the HIV Gag-coil fusion protein. R.L., M.P., R.P. and T.E. devised the coil-coil strategy. A.P.E-K., T.E., O.V., K.D.F, L.W.S, and R.A.S. are principal investigators who advised the studies.
COMPETING FINANCIAL INTERESTS
G.M.L., R.L., K.D.F, L.W.S. and R.A.S. are listed as inventors on patents describing polymer-based vaccines. K.D.F and L.W.S. are scientific founders and equity holders in PsiOxus Therapeutics, Ltd. (Oxford, UK). G.M.L. and J.R.F. are scientific founders and equity holders in Avidea Technologies, Inc., which is developing polymer-based technologies for immunotherapeutic applications (Baltimore, MD, USA).
References
- 1.Plotkin SA. Correlates of protection induced by vaccination. Clinical and vaccine immunology: CVI. 2010;17:1055–1065. doi: 10.1128/CVI.00131-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Epstein JE, et al. Live attenuated malaria vaccine designed to protect through hepatic CD8(+) T cell immunity. Science. 2011;334:475–480. doi: 10.1126/science.1211548. [DOI] [PubMed] [Google Scholar]
- 3.Andersen P, Woodworth JS. Tuberculosis vaccines–rethinking the current paradigm. Trends Immunol. 2014;35:387–395. doi: 10.1016/j.it.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Arens R, van Hall T, van der Burg SH, Ossendorp F, Melief CJ. Prospects of combinatorial synthetic peptide vaccine-based immunotherapy against cancer. Seminars in immunology. 2013;25:182–190. doi: 10.1016/j.smim.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 5.Guy B. The perfect mix: recent progress in adjuvant research. Nature reviews Microbiology. 2007;5:505–517. doi: 10.1038/nrmicro1681. [DOI] [PubMed] [Google Scholar]
- 6.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 7.Nordly P, Madsen HB, Nielsen HM, Foged C. Status and future prospects of lipid-based particulate delivery systems as vaccine adjuvants and their combination with immunostimulators. Expert opinion on drug delivery. 2009;6:657–672. doi: 10.1517/17425240903018863. [DOI] [PubMed] [Google Scholar]
- 8.Brito LA, O’Hagan DT. Designing and building the next generation of improved vaccine adjuvants. Journal of controlled release: official journal of the Controlled Release Society. 2014;190C:563–579. doi: 10.1016/j.jconrel.2014.06.027. [DOI] [PubMed] [Google Scholar]
- 9.Fox CB, et al. TLR4 ligand formulation causes distinct effects on antigen-specific cell-mediated and humoral immune responses. Vaccine. 2013;31:5848–5855. doi: 10.1016/j.vaccine.2013.09.069. [DOI] [PubMed] [Google Scholar]
- 10.Wille-Reece U, et al. HIV Gag protein conjugated to a Toll-like receptor 7/8 agonist improves the magnitude and quality of Th1 and CD8+ T cell responses in nonhuman primates. Proc Natl Acad Sci U S A. 2005;102:15190–15194. doi: 10.1073/pnas.0507484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shukla NM, et al. Toward self-adjuvanting subunit vaccines: model peptide and protein antigens incorporating covalently bound toll-like receptor-7 agonistic imidazoquinolines. Bioorg Med Chem Lett. 2011;21:3232–3236. doi: 10.1016/j.bmcl.2011.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu CC, et al. Immunotherapeutic activity of a conjugate of a Toll-like receptor 7 ligand. Proc Natl Acad Sci U S A. 2007;104:3990–3995. doi: 10.1073/pnas.0611624104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kasturi SP, et al. Programming the magnitude and persistence of antibody responses with innate immunity. Nature. 2011;470:543–547. doi: 10.1038/nature09737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jewell CM, Lopez SC, Irvine DJ. In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles. Proc Natl Acad Sci U S A. 2011;108:15745–15750. doi: 10.1073/pnas.1105200108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ilyinskii PO, et al. Adjuvant-carrying synthetic vaccine particles augment the immune response to encapsulated antigen and exhibit strong local immune activation without inducing systemic cytokine release. Vaccine. 2014;32:2882–2895. doi: 10.1016/j.vaccine.2014.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moon JJ, et al. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc Natl Acad Sci U S A. 2012;109:1080–1085. doi: 10.1073/pnas.1112648109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smirnov D, Schmidt JJ, Capecchi JT, Wightman PD. Vaccine adjuvant activity of 3M-052: an imidazoquinoline designed for local activity without systemic cytokine induction. Vaccine. 2011;29:5434–5442. doi: 10.1016/j.vaccine.2011.05.061. [DOI] [PubMed] [Google Scholar]
- 18.Liu H, et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 2014;507:519–522. doi: 10.1038/nature12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu TY, et al. Rational design of small molecules as vaccine adjuvants. Sci Transl Med. 2014;6:263ra160. doi: 10.1126/scitranslmed.3009980. [DOI] [PubMed] [Google Scholar]
- 20.Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10:787–796. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 21.Manolova V, et al. Nanoparticles target distinct dendritic cell populations according to their size. Eur J Immunol. 2008;38:1404–1413. doi: 10.1002/eji.200737984. [DOI] [PubMed] [Google Scholar]
- 22.Reddy ST, Rehor A, Schmoekel HG, Hubbell JA, Swartz MA. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. Journal of controlled release: official journal of the Controlled Release Society. 2006;112:26–34. doi: 10.1016/j.jconrel.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 23.Kourtis IC, et al. Peripherally administered nanoparticles target monocytic myeloid cells, secondary lymphoid organs and tumors in mice. PLoS One. 2013;8:e61646. doi: 10.1371/journal.pone.0061646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorden KB, et al. Synthetic TLR Agonists reveal functional differences between human TLR7 and TLR8. Journal of Immunology. 2005;174:1259–1268. doi: 10.4049/jimmunol.174.3.1259. [DOI] [PubMed] [Google Scholar]
- 25.Oh JZ, Kurche JS, Burchill MA, Kedl RM. TLR7 enables cross-presentation by multiple dendritic cell subsets through a type I IFN-dependent pathway. Blood. 2011;118:3028–3038. doi: 10.1182/blood-2011-04-348839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity. 2010;33:492–503. doi: 10.1016/j.immuni.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerster JF, et al. Synthesis and structure-activity-relationships of 1H-imidazo[4,5-c]quinolines that induce interferon production. J Med Chem. 2005;48:3481–3491. doi: 10.1021/jm049211v. [DOI] [PubMed] [Google Scholar]
- 28.A AG, Tyring SK, Rosen T. Beyond a decade of 5% imiquimod topical therapy. Journal of drugs in dermatology: JDD. 2009;8:467–474. [PubMed] [Google Scholar]
- 29.Savage P, et al. A phase I clinical trial of imiquimod, an oral interferon inducer, administered daily. British journal of cancer. 1996;74:1482–1486. doi: 10.1038/bjc.1996.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pockros PJ, et al. Oral resiquimod in chronic HCV infection: safety and efficacy in 2 placebo-controlled, double-blind phase IIa studies. Journal of hepatology. 2007;47:174–182. doi: 10.1016/j.jhep.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 31.Seymour LW, et al. Hepatic drug targeting: phase I evaluation of polymer-bound doxorubicin. J Clin Oncol. 2002;20:1668–1676. doi: 10.1200/JCO.2002.20.6.1668. [DOI] [PubMed] [Google Scholar]
- 32.Liu XM, Miller SC, Wang D. Beyond oncology–application of HPMA copolymers in non-cancerous diseases. Advanced drug delivery reviews. 2010;62:258–271. doi: 10.1016/j.addr.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seymour LW, Duncan R, Strohalm J, Kopecek J. Effect of Molecular-Weight (Mbarw) of N-(2-Hydroxypropyl)Methacrylamide Copolymers on Body Distribution and Rate of Excretion after Subcutaneous, Intraperitoneal, and Intravenous Administration to Rats. J Biomed Mater Res. 1987;21:1341–1358. doi: 10.1002/jbm.820211106. [DOI] [PubMed] [Google Scholar]
- 34.Sharp FA, et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci U S A. 2009;106:870–875. doi: 10.1073/pnas.0804897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Russo C, et al. Small molecule Toll-like receptor 7 agonists localize to the MHC class II loading compartment of human plasmacytoid dendritic cells. Blood. 2011;117:5683–5691. doi: 10.1182/blood-2010-12-328138. [DOI] [PubMed] [Google Scholar]
- 36.Ryu KA, Stutts L, Tom JK, Mancini RJ, Esser-Kahn AP. Stimulation of Innate Immune Cells by Light-Activated TLR7/8 Agonists. Journal of the American Chemical Society. 2014;136:10823–10825. doi: 10.1021/ja412314j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shakya AK, Holmdahl R, Nandakumar KS, Kumar A. Characterization of chemically defined poly-N-isopropylacrylamide based copolymeric adjuvants. Vaccine. 2013;31:3519–3527. doi: 10.1016/j.vaccine.2013.05.084. [DOI] [PubMed] [Google Scholar]
- 38.Shi HS, et al. Novel vaccine adjuvant LPS-Hydrogel for truncated basic fibroblast growth factor to induce antitumor immunity. Carbohydr Polym. 2012;89:1101–1109. doi: 10.1016/j.carbpol.2012.03.073. [DOI] [PubMed] [Google Scholar]
- 39.Chou HY, et al. Hydrogel-delivered GM-CSF overcomes nonresponsiveness to hepatitis B vaccine through the recruitment and activation of dendritic cells. J Immunol. 2010;185:5468–5475. doi: 10.4049/jimmunol.1001875. [DOI] [PubMed] [Google Scholar]
- 40.Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440:808–812. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 41.Nair-Gupta P, et al. TLR signals induce phagosomal MHC-I delivery from the endosomal recycling compartment to allow cross-presentation. Cell. 2014;158:506–521. doi: 10.1016/j.cell.2014.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jing P, Rudra JS, Herr AB, Collier JH. Self-assembling peptide-polymer hydrogels designed from the coiled coil region of fibrin. Biomacromolecules. 2008;9:2438–2446. doi: 10.1021/bm800459v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pechar M, Pola R. The coiled coil motif in polymer drug delivery systems. Biotechnology advances. 2013;31:90–96. doi: 10.1016/j.biotechadv.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 44.Kastenmuller K, et al. Protective T cell immunity in mice following protein-TLR7/8 agonist-conjugate immunization requires aggregation, type I IFN, and multiple DC subsets. J Clin Invest. 2011;121:1782–1796. doi: 10.1172/JCI45416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vecchi S, et al. Conjugation of a TLR7 agonist and antigen enhances protection in the S. pneumoniae murine infection model. European journal of pharmaceutics and biopharmaceutics: official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik e.V. 2014;87:310–317. doi: 10.1016/j.ejpb.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 46.Scott EA, et al. Dendritic cell activation and T cell priming with adjuvant- and antigen-loaded oxidation-sensitive polymersomes. Biomaterials. 2012;33:6211–6219. doi: 10.1016/j.biomaterials.2012.04.060. [DOI] [PubMed] [Google Scholar]
- 47.Liu H, Kwong B, Irvine DJ. Membrane anchored immunostimulatory oligonucleotides for in vivo cell modification and localized immunotherapy. Angewandte Chemie. 2011;50:7052–7055. doi: 10.1002/anie.201101266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krieg AM, Stein CA. Phosphorothioate oligodeoxynucleotides: antisense or anti-protein? Antisense research and development. 1995;5:241. doi: 10.1089/ard.1995.5.241. [DOI] [PubMed] [Google Scholar]
- 49.Lahoud MH, et al. DEC-205 is a cell surface receptor for CpG oligonucleotides. Proc Natl Acad Sci U S A. 2012;109:16270–16275. doi: 10.1073/pnas.1208796109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Titta A, et al. Nanoparticle conjugation of CpG enhances adjuvancy for cellular immunity and memory recall at low dose. Proc Natl Acad Sci U S A. 2013;110:19902–19907. doi: 10.1073/pnas.1313152110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shukla NM, Malladi SS, Mutz CA, Balakrishna R, David SA. Structure-activity relationships in human toll-like receptor 7-active imidazoquinoline analogues. J Med Chem. 2010;53:4450–4465. doi: 10.1021/jm100358c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hruby M, et al. New bioerodable thermoresponsive polymers for possible radiotherapeutic applications. Journal of controlled release: official journal of the Controlled Release Society. 2007;119:25–33. doi: 10.1016/j.jconrel.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 53.Subr V, Ulbrich K. Synthesis and properties of new N-(2-hydroxypropyl)-methacrylamide copolymers containing thiazolidine-2-thione reactive groups. React Funct Polym. 2006;66:1525–1538. [Google Scholar]
- 54.Skeiky YA, et al. Protective efficacy of a tandemly linked, multi-subunit recombinant leishmanial vaccine (Leish-111f) formulated in MPL adjuvant. Vaccine. 2002;20:3292–3303. doi: 10.1016/s0264-410x(02)00302-x. [DOI] [PubMed] [Google Scholar]
- 55.Gerner MY, Kastenmuller W, Ifrim I, Kabat J, Germain RN. Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity. 2012;37:364–376. doi: 10.1016/j.immuni.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quinn KM, et al. Comparative analysis of the magnitude, quality, phenotype, and protective capacity of simian immunodeficiency virus gag-specific CD8+ T cells following human-, simian-, and chimpanzee-derived recombinant adenoviral vector immunization. J Immunol. 2013;190:2720–2735. doi: 10.4049/jimmunol.1202861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Darrah PA, et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nature medicine. 2007;13:843–850. doi: 10.1038/nm1592. [DOI] [PubMed] [Google Scholar]
- 58.Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8:247–258. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.