

Abstract
Multipotent hematopoietic stem cells differentiate into an ensemble of committed progenitor cells that produce the diverse blood cells essential for life. Physiological mechanisms governing hematopoiesis, and mechanistic aberrations underlying non-malignant and malignant hematologic disorders, are often very similar in mouse and man. Thus, mouse models provide powerful systems for unraveling mechanisms that control hematopoietic stem/progenitor cell (HSPC) function in their resident microenvironments in vivo. Ex vivo systems, involving the culture of HSPCs generated in vivo, allow one to dissociate microenvironment-based and cell intrinsic mechanisms, and therefore have considerable utility. Dissecting mechanisms controlling cellular proliferation and differentiation is facilitated by the use of primary cells, since mutations and chromosome aberrations in immortalized and cancer cell lines corrupt normal mechanisms. Primary erythroid precursor cells can be expanded or differentiated in culture to yield large numbers of progeny at discrete maturation stages. We described a robust method for isolation, culture, and analysis of primary mouse erythroid precursor cells and their progeny.
Keywords: Hematopoiesis, Erythropoiesis, Ex vivo culture, Flow cytometry, Signaling
1 Introduction
Hematopoietic stem cells (HSCs), the essential stem cells responsible for generating and sustaining the blood system, are initially formed in the aorta-gonad-mesonephros region of the embryo (mice: embryonic day 10.5 or E10.5). Subsequently, the cells undergo a complex journey involving migration to and colonization of the fetal liver (mice: E11.5 onward). While the fetal liver is the primary site of hematopoiesis in late fetal development, after birth the HSCs migrate to and colonize the bone marrow [1–3]. Fetal liver-resident HSCs have a high proliferative capacity and generate large numbers of multi potent and committed progenitor cells [4, 5]. Therefore, the fetal liver provides a unique source of HSPCs for ex vivo analyses to elucidate mechanisms.
During hematopoiesis, HSCs generate progeny with unique signatures of cell surface proteins that are routinely used to distinguish distinct cell types. The signatures can be exploited using antibodies specific for the cell surface proteins and flow cytometry to analyze the composition of complex mixtures of cells and to isolate or enrich for unique cell types or populations. Alternatively, separations are also accomplished by incubating cell mixtures with antibody-bound beads, which can be isolated with a magnet, or based on other physical properties. Examples of instructive markers include: Ter119, erythroid [6]; CD45R (B220), B-cells [7]; and GR-1, myeloid [8]. The protocol presented in this chapter uses antibodies against cell-specific markers to bind to and remove differentiated myeloid, lymphoid, erythroid, and hepatic cells from a heterogeneous mixture of fetal liver cells, thus yielding an HSPC-enriched cell population deficient in terminally differentiated cells (Lineage− or Lin− cells). Lin− cells can be cultured in medium that supports rapid erythroid cell proliferation and differentiation, while suppressing the survival of other lineages [9].
Immortalized cell lines, which are often extremely easy to culture, have catalyzed many scientific advances and therefore can have high utility for certain experimental analysis. However, they often differ from primary cells in their growth, differentiation, and/or survival requirements, which is not surprising given mutations that may disrupt mechanisms governing fundamental cellular processes. The ability to culture, expand, and differentiate primary cells ex vivo is therefore a powerful approach for mechanistic analyses—either as an essential system to validate cell culture studies or as a front-line system for asking mechanistic questions. We describe herein a well-optimized method to isolate and culture primary murine fetal liver hematopoietic precursor cells, conduct loss-of-function analysis using retrovirus expressing an shRNA to target a specific mRNA, and quantitate molecular and cellular phenotypes (Fig. 1). The protocol can also be used for gain-of-function analyses, including rescue experiments, comparisons of mechanisms with cells from wild type and mutant mice, and pharmacological studies. The culture system can generate predominantly erythroid precursor cells (expansion culture) or mature erythroid cells (differentiation culture). This method can easily yield millions of the respective cells, consistent with requirements for diverse analytical approaches including genome-wide transcriptomics (RNA-seq), protein occupancy at chromatin sites (chromatin immunoprecipitation (ChIP), qChIP or ChIP-seq), protein analysis by mass spectrometry or Western blotting, and assessment of cellular attributes and/or signaling pathway status via flow cytometry. The application of these protocols has been described in multiple recent publications [10–16].
Fig. 1.
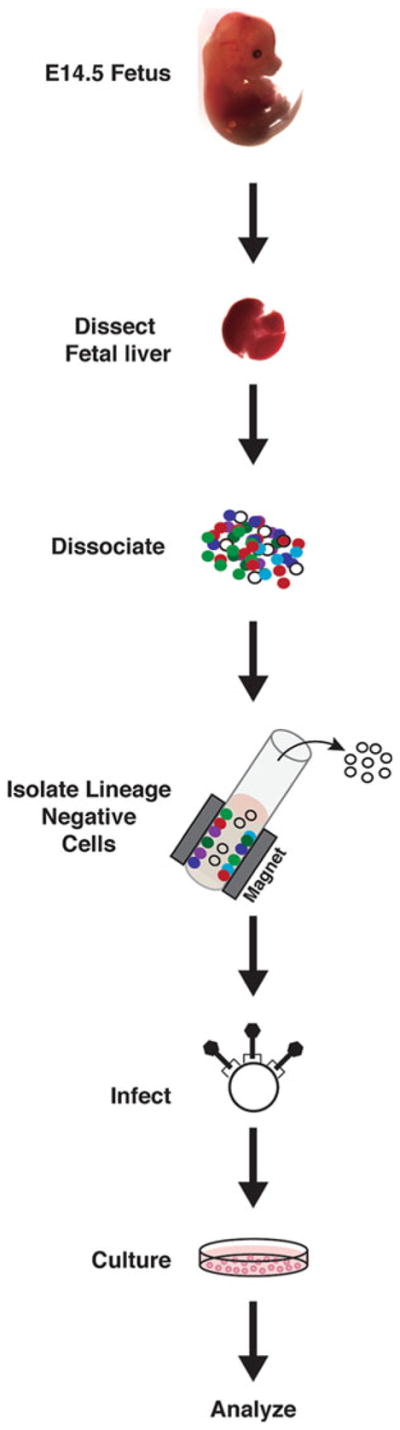
Isolation of lineage negative hematopoietic precursor cells
2 Materials
2.1 shRNA Cloning
Phusion high-fidelity DNA polymerase (ThermoFisher).
Restriction enzymes: BglII 10,000 U/ml, XhoI 20,000 U/ml.
T4 DNA ligase 400,000 U/ml.
Alkaline Phosphatase, Calf Intestinal (CIP) 10,000 U/ml.
Big Dye 3.1 Sequencing kit (ThermoFisher).
Primers: BglII-miR30sh-F: AGATCTAGATCTTGCTGTT-GACAGTGAGCG, XhoI-miR30sh-R: CTCGAGCTC-GAGTCCGAGGCAGTAGGC, PIG 5′Seq: CCCTTGAACCTCCTCGTTCGACC, PIG 3′Seq: GAGACGTGCTACTTCCATTTGTC.
PCR purification kit.
Gel purification kit.
Plasmid Miniprep kit.
Big Dye clean up kit.
Plasmid Endotoxin Free Maxiprep kit.
Expression vector: MSCV-PIG.
Apparatus and reagents to run DNA agarose gels.
Chemically competent bacteria, for example Jm109.
SOC medium.
LB broth and plates with 100 μg/ml Ampicillin.
15 ml round bottom snap top tubes.
2.2 Retrovirus Preparation
shRNA expression vector MSCV-PIG, purified via endotoxin-free maxiprep.
Retroviral packaging vector: pCL-Eco, purified via endotoxin-free maxiprep.
293T cells.
293T media: DMEM with glutamine and sodium pyruvate, 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin.
2× transfection buffer, 5 ml: 500 μl 0.5 M HEPES, 450 μl 2 M NaCl, 100 μl 100 mM Na2HPO4, 4.05 ml water.
10 cm culture plates.
10× NTE buffer: 1.5 M NaCl, 100 mM Tris-HCl (pH 7.4), 10 mM EDTA.
0.5 M Hepes (pH 7.1).
100 mM Na2HPO4 (pH 7.0 with phosphoric acid).
2 M CaCl2.
2 M NaCl.
2.3 Fetal Liver Isolation and Lineage Depletion
Dissection tools: forceps and scissors.
Dissection microscope.
12-well culture plates.
5 or 10 cm culture dishes.
5 mL round bottom tubes, solid and 40 μm filter cap.
PBS without calcium or magnesium.
Fetal Liver Juice (FLJ): 2% FBS, 2.5 mM EDTA and 10 mM Glucose in PBS.
Antibodies, all 0.5 mg/ml, anti-mouse and biotinylated: CD3e (clone 145-2C11), CD11b (clone M1/70), CD19 (clone 6D5), CD45R (B220) (clone RA3-6B2), GR-1 (clone RB6-8C5), CD71 (clone RI7217) and Ter119 (clone Ter119).
Streptavidin magnetic beads: MojoSort™ Streptavidin Nano-beads (BioLegend).
Magnet suitable for 5 ml tubes: MojoSort™ magnet (BioLegend) or EasySep™ magnet (StemCell Technologies).
2.4 Retroviral Infection
12-well culture plates.
Viral supernatant.
Polybrene: 2 mg/ml stock in water.
1 M HEPES.
Temperature-controlled centrifuge with swinging bucket rotor capable of spinning plates.
2.5 Culture of Lineage Negative Fetal Liver Cells
Expansion media: StemPro-34 (ThermoFisher) with 1× nutrient supplement, 2 mM L-Glutamine, 1% penicillin/streptomycin, 100 μM monothioglycerol, 1 μM dexamethasone, 0.5 U/ ml of erythropoietin (Epo), and 1% conditioned medium from a stem cell factor (SCF) producing CHO cell line (or 100 ng/ml recombinant SCF).
Differentiation media: IMDM (glutamine-free) with 10% FBS, 10% PDS (Plasma Derived Serum; Animal Technologies), 5% PFHM II, 2 mM L-glutamine, 1% penicillin/streptomycin, 100 μM monothioglycerol, and 6 U/ml erythropoietin.
2.6 Quantitating Erythroid Differentiation by Flow Cytometry
Antibodies: Fluorescent-conjugated anti-mouse CD71, Ter119, c-Kit.
Fetal Liver juice (FLJ): 2% FBS, 2.5 mM EDTA and 10 mM Glucose in PBS.
PBS without calcium or magnesium.
Cell-impermeable DNA dye, such as DAPI or PI, for live/dead cell discrimination.
5 mL tubes with 40 μm filter cap.
Multichannel analysis cytometer, e.g., BD LSR II or BD LSR Fortessa, or cell sorter, e.g., BD FACSAria III.
2.7 Quantitating Erythroid Differentiation by Morphology
50% FBS in PBS.
Glass slides.
Cytospin funnel.
Cytospin.
Giemsa stain.
2.8 Quantitating Colony-Forming Unit Activity
3 ml aliquots of MethoCult™ GF M3434 (StemCell Technologies).
16-gauge needles.
3 ml syringes.
Expansion media.
Sterile water.
35 mm × 10 mm culture dishes.
10 cm culture dishes.
Benzidine stain stock solution: 0.3 g benzidine, 9 ml acetic acid, 1 ml H2O. The stock is stable for 2 weeks.
Benzidine stain: 5 parts H2O, 0.1 part H2O2 (30%), 1 part benzidine stock (see item 8). The stain should be made just before use.
2.9 Quantitating Intracellular Signaling with Antibodies
Biotinylated anti-mouse Ter119.
Phospho-specific antibody against signing pathway components. For example, rabbit anti-mouse phospho-Akt, or rabbit anti-mouse phospho-ERK.
Fluorescent-conjugated secondary antibody. For example goat anti-rabbit-APC.
Fetal Liver juice (FLJ): 2% FBS, 2.5 mM EDTA and 10 mM Glucose in PBS.
Streptavidin magnetic beads: MojoSort™ Streptavidin Nano-beads (BioLegend).
Magnet suitable for 5 ml tubes: MojoSort™ magnet (BioLegend) or EasySep™ magnet (StemCell Technologies).
5 mL tubes, solid and 40 μm filter cap.
Cytokines of interest. For example SCF or Epo.
Fixable live/dead stain such as Zombie Dye (BioLegend).
1% BSA solution in IMDM.
PBS without calcium or magnesium.
4% FBS in HBSS.
16% paraformaldehyde.
Methanol.
3 Methods
3.1 Constructing shRNA-Expressing Retrovirus
Design shRNA sequences specific for your gene using a publically accessible database. For example: http://portals.broadinstitute.org/gpp/public/ is searchable using NCBI gene accession numbers. Select at least three sequences that target distinct regions of the gene of interest (see Note 1).
To plan how to integrate the shRNA into a miR-30 context, use the Ravi Lab resource: http://katahdin.cshl.org/html/scripts/resources.pl
Pick the RNAi project and siRNA/shRNA design and select shRNA psm2 design, then select mouse.
State the information you have available, for example, a 21mer. Input the sequence obtained in step 1 and the target gene NCBI accession number. If shRNA sequences are not available, the Ravi Lab resource will design shRNA from accession numbers. Order the 97mer oligonucleotide.
Add BglII and XhoI cut sites to the 97mer. To do this, set up 50 μl PCR reactions for each construct, according to the instructions provided with the high-fidelity DNA polymerase. Use a final concentration of 200 μM dNTPs, 150 μM of each primer (BglII-miR30sh-F and XhoI-miR30sh-R) and 300 μM of the shRNA 97mer oligonucleotide.
Run PCR: 98 °C 5 min, (98 °C 15 s, 72 °C 50 s) × 10, 72 °C 7 min.
Check for the presence of a ~100 bp band on a 1.5% agarose gel.
Purify the product with the PCR purification kit, and elute the shRNA construct with 30 μl water.
Cut the shRNA construct with BglII and XhoI, 28 μl of DNA from step 5, 1.5 μl each enzyme, 5 μl 10× restriction enzyme buffer, and water in 50 μl.
Cut 4 μg of MSCV-PIG vector with BglII and XhoI, 1.5 μl each enzyme, 5 μl 10× restriction enzyme buffer, and water in 50μl.
Incubate the restriction digest at 37 °C for 4 h (at 3.5 h, add 0.5 μl CIP to the MSCV-PIG vector).
Run the digested MSCV-PIG vector on a 1% agarose gel, visualize DNA, and excise the vector band (~7.5 kb).
Use the gel purification kit to purify the MSCV-PIG vector, and elute in 30 μl water.
Use the PCR purification kit to purify the shRNA products, and elute in 30 μl water.
Set up a ligation reaction with 16 μl of purified shRNA construct (from step 14), 1 μl purified cut MSCV-PIG vector (from step 13), 1 μl T4 DNA ligase, 2 μl 10× ligation buffer, in a 20 μl reaction. Incubate overnight at 16 °C.
Thaw competent bacteria on ice for 5 min (25–40 μl per reaction).
Add 7 μl of the ligation mix and incubate on ice for 20 min.
Heat-shock bacteria for 40 s at 42 °C, and then incubate on ice for 2 min.
Add 200 μl SOC medium and incubate for 1 h at 37 °C.
Plate 200 μl of bacteria on LB-Agar/Ampicillin plates and incubate overnight at 37 °C.
To screen colonies, make PCR mastermix sufficient for the number of colonies to test (25 μl per colony) according to the instructions provided with the DNA polymerase: Use a final concentration of 400 μM dNTPs, 400 μM each primer (PIG 5′Seq, PIG 3′Seq). Aliquot into PCR tubes.
Using a pipette tip, pick colonies of interest. First, dip tip into 2.5 ml LB broth/ampicillin and then into PCR master mix.
Perform PCR analysis: 95°C 5 min, (95°C 15 s, 54°C 30 s, 72°C 30 s) × 30, 72°C 7 min and analyze on a 1% agarose gel. Plasmids containing a single insert should have a 500 bp amplicon, plasmids with no insert will have a 400 bp amplicon.
Incubate LB broth samples from positive colonies at 37°C (shaking) for 10–24 h and perform minipreps to purify plasmids of interest.
To prepare the minipreps for sequencing, make a Big Dye reaction with 200 ng DNA template, 1.5 μl PIG 3′seq (10 mM stock), 3 μl Big Dye Buffer, 2 μl Big Dye Enzyme, water up to 20 μl. Amplify at 96 °C 3 min, (96 °C 10 s, 55 °C 15 s, 60 °C 3 min) × 34, 72 °C 7 min (see Note 2).
Purify product using a Big Dye clean-up kit and analyze by Sanger sequencing.
Once correct sequence is confirmed, inoculate 100 ml LB/ Ampicillin broth with bacteria, and incubate at 37 °C (shaking) for 16–18 h.
Purify plasmid using an endotoxin-free plasmid maxiprep kit.
3.2 Virus Preparation
Seed 3.5–4 million 293T cells in 10 ml medium in a 10 cm tissue culture dish, and incubate at 37 °C 5% CO2 overnight (see Note 3).
Make 2× transfection buffer (see Subheading 2.2, item 5).
Make DNA cocktail in eppendorf tubes: In this order, add water up to 500 μl, 62.5 μl 2 M CaCl2, 50 μl 10× NTE, 15 μg of shRNA expression plasmid (MSCV-PIG vector from Subheading 3.1, step 28), 15 μg retroviral packaging vector (pCL-Eco).
Aspirate the medium from the 10 cm 293T plates, and replace it with a 5 ml pre-warmed fresh medium (see Note 4).
Add 500 μl 2× transfection buffer to the DNA cocktail drop-wise. Bubble air through the liquid for 30 s to mix.
Slowly add transfection reaction to 293T cells on plate, and tilt to mix.
Incubate at 37°C, 5% CO2 for 6 h, and then replace the medium with a 6 ml pre-warmed fresh medium.
Collect viral supernatant by collecting medium from 293T at 24 h, replace the medium with 6 ml pre-warmed fresh medium, and repeat collection at 48 h.
Spin medium containing virus at 2000 rpm (670 × g) for 5 min at room temperature to pellet cell debris.
Aliquot the supernatant (0.5–1 ml). Snap freeze in liquid nitrogen and store at −80 °C.
3.3 Isolate Fetal Livers
Set up timed mating. Sacrifice a pregnant mouse 14 days after the observation of a vaginal plug. Embryos are considered E14.5 (see Note 5).
Place mouse on its back and grip the stomach, near the final set of nipples with forceps with the handle pointing toward the head, and pull upward gently.
Cut the skin and underlying muscular layer with a scissors in a V shape, starting underneath the region you are gripping with the forceps, until the uterus is easily accessible.
Grip the uterus between two fetuses and pull gently. Each of the two arms of the uterus will be attached at the ovary, near the ribs, and to the vagina near the legs with some blood vessels and fat in-between. Cut these connections and the uterus is extracted easily.
Place uteri containing the fetuses (usually around 9) into cold sterile PBS in a petri dish, wash once, and place in fresh PBS.
Isolate fetuses one at a time, using fine tipped forceps to tear uterus in the gap between the fetuses and squeeze out the fetus. If the yolk sac remains around the fetus, tease it off with two pairs of forceps. Immediately transfer fetus into cold FLJ in a new petri dish.
If working with mutant animals, obtain a portion of the tail and leg of each fetus and store separately to conduct genotyping.
Isolate livers by removing connective tissue surrounding the liver, under sterile conditions if possible. Place into a 12-well dish containing 1.5 ml FLJ and wash once. Intact fetal livers can be maintained on ice for several hours if required for genotyping, although minimizing this time is recommended. Wild-type fetal livers can be pooled for analysis.
Dissociate the fetal livers by pipetting in FLJ to generate a single-cell suspension (10–20 million cells per liver).
Pass the cell suspension through a 40 μm strainer top into a 5 ml tube.
Wash the strainer cap with cold FLJ until the tubes are full and replace the strainer cap with a solid cap.
Centrifuge for 5 min at 1200 rpm (250 × g) at 4°C, and proceed immediately to progenitor purification.
3.4 Lineage Depletion
This protocol is designed for the isolation of myelo-erythroid progenitors from three biological replicate cell suspensions, each containing three pooled wild-type fetal livers. Volumes can be scaled up or down depending on the desired cell number or number of replicates. The minimum volume per isolation is 100 μl, while the maximum volume is 1.5 ml.
Assemble master mix of all antibodies: 3 μl/ml each of CD3e, CD11b, CD19, CD45R, GR-1 and CD71, and 5 μl/ml of Ter119. For three replicates, each containing three fetal livers, use 2.25 ml FLJ, 6.75 μl each of CD3e, CD11b, CD19, CD45R, GR-1 and CD71, and 11.25 μl of Ter119 (see Note 6).
Resuspend fetal liver cells at 0.5–1 × 108 cells/ml (generally 250 μl/E14.5 liver) in antibody master mix (step 1).
Incubate on ice for 15 min, and then wash off unbound antibody by adding 3–4 ml FLJ, and centrifuge for 5 min at 1200 rpm (250 × g) at 4 °C. Discard the supernatant.
Assemble master mix of streptavidin beads: 75 μl/ml in FLJ. For three replicates each containing three fetal livers: 168.75 μl streptavidin beads in 2.25 ml FLJ.
Resuspend fetal liver cells in the bead master mix. Use a volume equal to that used in step 2.
Incubate on ice for 15 min, wash by adding 3–4 ml FLJ, and centrifuge for 5 min at 1200 rpm (250 × g) at 4 °C. Discard the supernatant.
Resuspend cells in 3 ml of FLJ using a P1000 or other pipetteman.
Remove the cap and incubate each tube in the magnet for 5 min.
Pour out cells of interest into a new tube in one continuous motion without removing the tube from the magnet. Do not shake/touch tube to the new tube etc. to obtain the last drop. This will cause the beads to contaminate your sample.
It is recommended that steps 7–9 are repeated once to increase yield.
Pass the cell suspension through a 40 μm strainer.
Count cells, isolate by centrifugation for 5 min at 1200 rpm (250 × g) at 4 °C, and resuspend in an appropriate volume of expansion media. A typical yield from three wild-type E14.5 fetal livers is 1.5–2 × 106 cells.
3.5 Viral Infection
Infections should be conducted immediately after lineage depletion. Generally, this includes retroviruses harboring a control shRNA (see Note 7) or an experimental shRNA. For flow cytometry applications, culture uninfected cells for single-stain controls.
Add 1–3 × 105 cells in 400 μl expansion medium to each well of a 12-well plate.
It is recommended that more than one well be infected per condition. Cells can be used as technical replicates or pooled to increase yield.
Add 100 μl viral supernatant, 2 μl polybrene solution, and 5 μl 1 M HEPES to each well and mix (see Note 8).
Centrifuge in a swinging bucket rotor at 2800 rpm (1300 × g) at 30°C for 90 min.
Add 500 μl expansion medium to each well and incubate at 37°C and 5% CO2.
3.6 Expansion Culture
The expansion medium used in this protocol is formulated to promote rapid proliferation of early erythroid cells, while preventing their terminal differentiation. Cells cultured in the expansion medium rarely differentiate beyond the stage of basophilic erythroblasts. SCF (Kit receptor agonist) and dexamethasone (glucocorticoid receptor agonist) stimulate expansion of immature erythroid precursor cells (burst forming unit - erythroid (BFU-E), colony forming unit - erythroid (CFU-E) and proerythroblasts) (see Note 9) [17]. A small amount of Epo in the medium supports the growth of proerythroblasts and the slightly more mature erythroblasts termed polychromatophilic erythroblasts. The expansion phase of the culture system permits analysis of early erythroid precursor function, e.g., to identify mechanisms that promote or suppress erythroblast proliferation, maturation, or survival.
Plate the cells from Subheading 3.5, step 5 at a concentration of 0.25–1 × 106 cells/ml in expansion media (see Note 10).
Cells are cultured 37 °C and 5% CO2 and double every 6–12 h (see Note 11).
It is recommended that the volume of medium is doubled each morning, and each afternoon count and dilute the cells to 0.25 × 106 cells/ml to ensure the cell concentration does not exceed one million cells/ml (see Note 12).
3.7 Differentiation Culture
The differentiation medium used in this protocol supports the terminal differentiation of erythroid cells. Cells cultured in this medium mature into basophilic and orthochromatic erythroblasts and about 20–30% enucleate after 48 h. This medium does not contain SCF or dexamethasone, and accordingly, early erythroid cells stop proliferating and differentiate. The high Epo concentration supports the erythroid cells upon terminal differentiation. The differentiation culture system enables the analysis of factors and signals that promote or inhibit terminal erythroid differentiation.
Collect greater than 0.5 × 106 cells in a 15 ml tube.
Centrifuge the cells for 5 min at 1200 rpm (250 × g) at 4 °C, and wash twice with 5 ml of PBS.
Resuspend cells in an appropriate volume of differentiation media. Cells must be maintained at a concentration of 1–2 × 106 cells/ml. Cells cultured at 37 °C and 5% CO2 double every 12–24 h, and proliferation slows during the culture period.
Phenotypic analyses of the differentiated erythroid cells can include flow cytometry, morphology, colony forming unit (CFU) assay, cell signaling, and cell cycle analyses, as described in the sections below. The optimal number of days in the differentiation medium will vary depending on the assay.
3.8 Quantitation of Erythroid Maturation by Flow Cytometry
During erythroid maturation, major changes occur in the expression of cell surface markers, which can be exploited to establish the maturation stage. For example, the earliest erythroid cells express c-Kit and then acquire CD71 expression. As maturation progresses these cells gain Ter119 expression. Finally, these cells lose CD71 expression [1]. The dynamic expression of cell surface proteins, along with the availability of fluorescently-conjugated antibodies recognizing these proteins, allows the quantitation and sorting of populations enriched for specific erythroid cell types. A common nomenclature for the erythroid maturation series is as follows: R1 (BFU-E, CFU-E, proerythroblast), R2 (proerythroblasts and early basophilic erythroblasts), R3 (early and late basophilic erythroblasts), R4 (chromatophilic and orthochromatic erythroblasts), and R5 (late orthochromatic erythroblasts and reticulocytes) [6]. During erythroid maturation, the expression of hundreds of genes increases or decreases. Thus, phenotypic changes (molecular or cellular) occurring between cells infected with control shRNA vs. a specific shRNA must be carefully interpreted (including the quantitation of knockdown efficiency), as such changes might be an indirect consequence of alterations in maturation. Typically, we isolate R1–R4/5 cells, using the cell surface markers CD71 and Ter119 by fluorescence activated cell sorting (FACS), to purify RNA from the specific cell populations. This minimizes the possibility that changes in mRNA expression reflect differences in cell maturation between control and treatment samples.
shRNA-mediated downregulation of specific mRNAs occurs by 16 h post-infection. Perturbations in erythroid maturation are particularly prominent after 48–72 h culture in expansion media. After culturing for 24 h, cells reside predominantly in R1, R2, and R3 populations. Further culture (48 and 72 h) decreases the percentage of cells in R1 and increases the R3 population (Fig. 2). After culturing for 48 h in differentiation media, the erythroid cells are predominantly in the R3 population, ~20–30% are in the R4 and ~1–5% are in the R5 population. It is important to evaluate the impact of downregulating factors on erythroid maturation at various times, while carefully considering the relationship between kinetics for shRNA-mediated downregulation of the target mRNA and the respective protein, and potential phenotypic alterations (Fig. 2).
Fig. 2.

Cell analysis using flow cytometry and Wight-Giemsa staining. (a) Representative flow cytometry plots of lineage negative cells stained with CD71-PE and Ter119-APC after 72 h expansion culture and then 48 h differentiation culture. R1 through R5 gates are denoted. (b) Representative Wright-Giemsa stain after 72 h expansion culture and then 48 h differentiation culture
Harvest 0.5–1 × 106 cells for flow cytometry analysis (FCM) or 10–20 × 106 for flow sorting (FACS). Rinse in PBS.
Assemble antibody master mix with 100 μl FLJ per sample. For FCM, add 1:100 (or 0.2 μg (1 μl)) each antibody: CD71, Ter119, c-Kit. For FACS, add 1:25 (or 0.8 μg (4 μl)) each antibody: CD71, Ter119, c-Kit (see Notes 13 and 14).
For either FCM or FACS, do single-stain controls using uninfected cells and resuspend each control 100 μl FLJ. Add each antibody individually to separate tubes and leave some cells unstained for use as a no-stain compensation control (see Note 15).
Resuspend fetal liver cells in 100 μl of antibody master mix, and incubate in the dark at 4 °C for 30 min with occasional mixing.
Add 2 ml PBS and isolate cells by centrifugation at 1200 rpm (250 × g) for 5 min at 4 °C.
Resuspend cells in 500 μl of PBS containing cell-impermeable DNA dye for live/dead discrimination. For example 1 μg/ml DAPI.
Pass the cell suspension through a 40 μm strainer.
Analyze or sort with a flow cytometer.
3.9 Morphological Analyses
The identification and quantitation of cells at distinct stages of erythroid maturation is readily accomplished by applying cells to a slide with a Cytospin and staining with Wright-Giemsa. Immature erythroid cells are large with large purple nuclei and a dark blue cytoplasm. As the cells mature, the erythroblasts get progressively smaller and their nuclei condense and become darker. The cytoplasm becomes hemoglobinized, attaining a light blue and then pink color. Terminal erythroid maturation involves enucleation. Wright-Giemsa stain also has utility for distinguishing between erythroid and non-erythroid lineages. For example, neutrophils have a lobed or donut-shaped nucleus, while macrophages are very large multi-nucleated cells [18, 19].
Large immature erythroblasts are abundant during the expansion culture. In the differentiation culture, which supports erythroblast terminal differentiation, cell size decreases, cytoplasm becomes hemoglobinized, and a significant percentage of the cells enucleate (Fig. 2).
Harvest 1 × 105 cells per slide, isolate by centrifugation, and resuspend in 500 μl of 50% FBS/PBS.
Load slide into the cytospin funnel apparatus, and add the cells.
Spin for 10 min at 700 rpm (55 × g) in the cytospin.
Remove the slides as soon as possible after the spin, and air dry for 10 min.
Store in the dark (up to 1 week) until staining (see Note 16).
3.10 Hematopoietic Progenitor Quantitation Using a Colony-Forming Unit Assay
The competence of hematopoietic precursors to differentiate into myeloid and erythroid cell types can be quantitated using CFU assays. The number, size, and morphology of colonies formed in semisolid medium provides a highly instructive metric of their differentiation and proliferation capabilities [20, 21]. This assay is performed within 24 h of lineage negative cell isolation, and varies depending on application. The GFP selection marker on the MSCV-PIG vector is easily detectable by 16 h post-infection. FACS can be used to isolate the GFP+ infected population, or a more specific cell population for analysis. CD71 and Ter119 markers are used to identify R1–R5 populations. The R1 population contains a high proportion of BFU-E and CFU-E. Total cells or GFP+ cells after 24 h expansion culture produce CFU-GM, BFU-E, CFU-E, with only a few CFU-GEMM.
Make a 10× cell suspension in expansion medium with total or GFP+ cells at 2 × 105 cells/ml. If using sorted R1 cells, suspend at 2–5 × 104 cells/ml in expansion medium.
Mix 300 μl cells with 3 ml MethoCult™ and vortex 5 s (see Note 17).
Incubate for 5 min to allow any bubbles to settle.
Use a 16-gauge needle and 3 ml syringe to dispense 1.1 ml Methocult™ into 35 mm × 10 mm dishes each (make two technical replicates). Distribute evenly by tilting the dishes.
Place dishes in a 10 cm plate, and add a third uncovered 35 mm × 10 mm dish containing 3 ml sterile H2O (The use of a water dish maintains humidity).
Score for CFU-E colonies (groups of 8–30 cells that appear to be fused together) after 2 days of culture at 37 °C and 5% CO2.
Score for BFU-E colonies (multiple cell clusters consisting of bright red, tiny, irregularly shaped cells that may appear to be fused together), CFU-GM colonies (colorless colonies consisting of large, oval cells (macrophage) and smaller, round cells (granulocytes)) and CFU-GEMM (typically large colonies consisting of red erythroid cells mixed with colorless granulocytes, macrophages, and megakaryocytes) after 7–10 days of culture at 37°C and 5% CO2. Before scoring for BFU-Es, plates can be stained with 500–700 μl of benzidine stain, which stains hemoglobin. Hemoglobinized BFU-E colonies will instantly stain dark blue. Benzidine stain will also stain CFU-E colonies, but is not compatible with continued culture to subsequently generate BFU-E, or for optional morphological analyses in step 8.
If colonies are to be harvested for morphological analysis, incubate plates at 4 °C for 2–3 h and add 1 ml of cold PBS per dish, pipetting up and down gently to mix the methylcellulose-based medium with the liquid medium. Move to a 15 ml conical tube. Wash the culture plate, and add an additional 5 ml PBS to the 15 ml conical tube. Centrifuge cells at 1200 rpm (250 × g) for 5 min at 4 °C, and wash twice with 5 ml PBS. Incubate for 10 min on a shaker at room temperature before spinning down after each wash. Count cells, and proceed with the morphological analysis procedure (Subheading 3.9).
3.11 Quantitation of Signaling
Cytokines control the survival, growth, and differentiation of erythroid precursor cells. Proliferation of immature erythroid precursors requires SCF [22, 23], while intermediate erythroblasts respond to SCF and Epo [24, 25] and terminally differentiated erythroid cells require Epo for survival [26]. A variety of mechanisms are in place to control the erythroid cell response to cytokines, and it would not be surprising if major additional mechanisms remain to be discovered. As cellular responses to cytokines can depend on the maturation status of the cells [27–29], this protocol provides a sensitive assay to quantitate cellular responses to cytokine signals in specific cell types delineated by their ensemble of cell surface proteins detected by flow cytometry.
Cytokines rapidly activate kinase cascades, which at least in certain contexts have a very restricted duration. To measure cell responses to a particular cytokine, it is commonplace to serum-starve cells, which decreases their basal signaling. Quantitation of intracellular signaling molecules by flow cytometry requires cellular fixation and permeabilization to allow appropriate antibodies to access their target, while retaining the target epitope. We find that common fixation procedures result in loss of the erythroid cell surface antigen recognized by the anti-Ter119 antibody. Thus in a typical experiment, we separate the erythroid cell population into early (Ter119−) and late (Ter119+) erythroblasts using magnetic beads prior to cytokine stimulation [14]. Typically, we compare the capacity of a cytokine to stimulate a control cell versus a cell in which a particular factor has been downregulated using shRNA (or cells from wild-type vs. mutant animals).
Centrifuge 5–20 × 106 cells at 1200 rpm (250 × g) for 5 min at 4 °C, aspirate medium, wash in ice-cold PBS, and transfer to a FACS tube.
Separate sample into Ter119+ and Ter119- populations using the lineage depletion protocol described in Subheading 3.4. Use 500 μl FLJ/sample, 2 μl/ml anti-Ter119, and 50 μl/ml streptavidin beads.
Count cells, wash and resuspend in 1 ml IMDM/1% BSA with a cell concentration that does not exceed 5 × 106 cells/ml.
Serum Starve the cells by incubating in IMDM/1% BSA for 60 min at 37 °C.
Isolate cells by centrifugation, and wash cells in PBS to remove all serum proteins.
Stain with fixable live/dead marker. For example, resuspend cells in 100 μl of a 1:100 dilution of Zombie Dye (BioLegend) in PBS, incubate at room temperature for 15 min, and then inactivate live/dead stain by adding pre-warmed IMDM/1% BSA.
Centrifuge cells at 1200 rpm (250 × g) for 5 min at room temperature and resuspend in IMDM/1% BSA at 37 °C, 1 ml/ condition (vehicle and cytokine treated).
Incubate for 10 min at 37 °C.
Stimulate with vehicle or cytokine of interest for 10 min at 37 °C (for example, 10 ng/ml SCF or 2 U/ml Epo). For precise timing, add SCF or vehicle every 20 s to individual cell suspensions.
Fix cells by adding 16% fresh paraformaldehyde (PFA) to a final concentration of 2% (143 μl/ml), and incubate for 10 min at 37°C (see Note 18).
Add an equal volume of PBS, isolate cells by centrifugation at 2200 rpm (800 × g) for 5 min at room temperature and wash twice with 4 ml PBS.
While vortexing, slowly add 2 ml ice-cold 95% methanol drop-wise to each cell suspension. Place each sample on ice immediately thereafter (no longer than 30 min). Tighten the lid, and store samples at −20 °C for at least 4–6 h and preferably overnight.
Add an equal volume of PBS. Do not mix, and isolate cells by centrifugation at 2200 rpm (800 × g) for 5 min at 4 °C.
Aspirate the supernatant carefully, leaving ~100–200 μl, and wash twice with 3 ml PBS.
Resuspend samples in 1 ml of cold HBSS/4% FBS, vortex, and incubate at 4 °C for 60 min.
Isolate cells by centrifugation at 2200 rpm (800 × g) for 5 min at 4 °C.
Resuspend in 100 μl primary antibody master mix and incubate for 30 min at room temperature. For example, rabbit anti-mouse phospho-AKT or phospho-ERK at 1:200 or biotinylated antibodies 1:200 in FLJ.
Wash cells with 4 ml PBS, and isolate cells by centrifugation at 2200 rpm (800 × g) for 5 min.
Resuspend in 100 μl fluorescently conjugated antibody master mix, and incubate for 30 min at room temperature in the dark. This master mix will contain both secondary antibodies: anti-rabbit 1:200 (e.g., APC), Streptavidin 1:400 (or 0.05 μg (0.25 μl), e.g., PE-Cy7) and any pre-conjugated primary antibodies: 1:50 (or 0.4 μg (2 μl), e.g., CD71-PE) in FLJ.
For single-stain controls, use fixed uninfected cells, and add antibodies separately to different tubes leaving an aliquot of cells unstained for a no-stain compensation control.
Wash once with 4 ml PBS, and resuspend in 400 μl PBS.
Pass the cell suspension through a 40 μm strainer.
Analyze data on flow cytometer as soon as possible. Use low/ middle pressure to collect the data.
The intensity of the cellular response to the cytokine is measured via the change in Median Fluorescence Intensity (MFI) of the phosphorylated kinase, e.g., phospho-Akt (vehicle vs. cytokine).
Compared to the vehicle control, cytokine stimulation will shift the histogram of fluorescence intensity of phospho-Akt to the right (Fig. 3).
Fig. 3.

Phospho-flow cytometric analysis of cytokine signaling. Left: Flow cytometry plots of phospho-Akt after 10 min treatment with vehicle (black) or 10 ng/ml SCF (red) in control Ter119−/CD71high cells cultured in expansion medium for 24 h. Right: Phospho-Akt quantitation. MFI is expressed relative to unstimulated cells (mean ± SE, three biological replicates)
3.12 Cell Cycle Analyses
Erythroid maturation is intimately linked to the cell cycle [30], and therefore cell cycle perturbation can have a large impact on maturation.
Isolate 5–20 × 106 cells by centrifugation at 1200 rpm (250 × g) for 5 min at 4 °C, aspirate the medium, and wash in cold PBS. Transfer to a FACS tube.
Separate the sample into Ter119+ and Ter119− populations using the lineage depletion protocol in Subheading 3.4. Use 500 μl FLJ/sample, 2 μl/ml anti Ter119, and 50 μl/ml strep-tavidin beads.
Count cells, and wash in PBS.
Alternatively, use FACS to isolate R1–R4 populations.
Resuspend cells in 100 μl of PBS and add ice-cold 70% ethanol dropwise.
Add DAPI at 5 μg/ml to the cells.
Incubate cells overnight at −20 °C.
Wash twice with PBS.
Collect DAPI fluorescence intensity on a linear scale on a flow cytometer.
Use Modfit software to analyze data. This software will attempt to fit the signal intensity of DAPI into a cell cycle model based on the quantity of DNA in the cell nucleus. Typically, this data is seen as two peaks, with the low-intensity peak representing G1/G0, the high-intensity peak representing G2/M, and S- phase representing the region between the peaks (Fig. 4).
Fig. 4.

Cell cycle analysis of control Ter119+ cells. Flow cytometry plots of DAPI stained Ter119+ cells after 24 or 72 h culture in expansion media. G1, S and G2/ M populations denoted
Acknowledgments
The lineage negative cell isolation and culture protocols were adapted from those originally provided by M.J. Weiss. This work was supported by NIH grants DK50107 and DK68634.
Footnotes
The cloning procedure described in Subheading 3.1 can be adapted to overexpress a gene of interest, for example to conduct a rescue experiment. A MSCV-PIG vector map is available at https://www.addgene.org/21654/.
The 3′PIG sequencing primer is best for sequencing short products. For longer products, run two sequencing reactions using the 3′PIG sequencing primer and the 5′PIG sequencing primer to cover the entire insert.
Ensure that the 293T cells do not become fully confluent during maintenance culture, which may negatively impact virus generation.
293T cells do not adhere strongly to the bottom of flasks and plates. Thus, to avoid disrupting the cells, add the medium and the buffers to the side of the plate slowly and gently.
E13.5–15.5 fetuses can also be used for the isolation of lineage negative cells. E14.5 and E15.5 livers give a greater yield than E13.5.
There are several commercial kits for the negative selection of lineage negative hematopoietic precursor cells. These include EasySep™ Mouse Hematopoietic Progenitor Cell Enrichment Kit (StemCell Technologies) and MojoSort™ Mouse Hematopoietic Progenitor Cell Isolation Kit (BioLegend). However, the hematopoietic selection cocktail must be supplemented with anti-CD71 biotin.
We use shRNA against luciferase as a negative control in knockdown experiments.
Fetal liver lineage negative cells are very easy to infect with an infection efficiency of over 90%. Inhibitors/activators can be used in the culture as an alternative to gene knockdown or overexpression.
Expansion culture: Cells grow better in SCF-containing conditioned medium than in medium containing recombinant SCF.
Expansion culture: Cells do not grow well if the concentration is less than 1 × 105 cells/ml.
shRNA-mediated knockdown of factors is observed by 16 h of culture.
In general, we grow the lineage negative cells for 72 h in the expansion medium and then move them to the differentiation medium for 48 h. By the end of the 72 h expansion culture, 0.2 × 106 cells will expand to at least 2 × 106 cells.
In general, our flow cytometric analysis panel includes any or all of following fluorochromes, PE, APC, PE-cy7, APC-cy7. A live dead marker, DAPI (or Zombie UV) and an infection marker, GFP.
An alternative measure of erythroid maturation replaces CD71 with CD44 in the flow cytometry panel [31].
- It is very important to save uninfected cells for compensation controls.
We subject our slides to Wright-Giemsa staining. A detailed protocol can be found in ref. 32.
Colony assays can also be performed with Methocult™ GF M3334 for exclusive analysis of CFU-Es and BFU-Es.
Although Ter119 staining is lost during cell fixation, CD71 staining and GFP signals are generally maintained when cells are fixed in paraformaldehyde/methanol. Antigen-recognition sites for antibodies targeting Ter119 and CD71, as well as GFP signal, are lost when cells are fixed/permeabilized in one step using 70% ethanol.
References
- 1.Dzierzak E, Philipsen S. Erythropoiesis: development and differentiation. Cold Spring Harb Perspect Med. 2013;3(4):a011601. doi: 10.1101/cshperspect.a011601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dzierzak E, de Pater E. Regulation of blood stem cell development. Curr Top Dev Biol. 2016;118:1–20. doi: 10.1016/bs.ctdb.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 3.Tober J, Maijenburg MW, Speck NA. Taking the leap: Runx1 in the formation of blood from endothelium. Curr Top Dev Biol. 2016;118:113–162. doi: 10.1016/bs.ctdb.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 4.Morrison SJ, Hemmati HD, Wandycz AM, Weissman IL. The purification and characterization of fetal liver hematopoietic stem cells. Proc Natl Acad Sci U S A. 1995;92(22):10302–10306. doi: 10.1073/pnas.92.22.10302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rebel VI, Miller CL, Eaves CJ, Lansdorp PM. The repopulation potential of fetal liver hematopoietic stem cells in mice exceeds that of their liver adult bone marrow counterparts. Blood. 1996;87(8):3500–3507. [PubMed] [Google Scholar]
- 6.Zhang J, Socolovsky M, Gross AW, Lodish HF. Role of Ras signaling in erythroid differentiation of mouse fetal liver cells: functional analysis by a flow cytometry-based novel culture system. Blood. 2003;102(12):3938–3946. doi: 10.1182/blood-2003-05-1479. [DOI] [PubMed] [Google Scholar]
- 7.Coffman RL, Weissman IL. B220: a B cell-specific member of the T200 glycoprotein family. Nature. 1981;289(5799):681–683. doi: 10.1038/289681a0. [DOI] [PubMed] [Google Scholar]
- 8.Fleming TJ, Fleming ML, Malek TR. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J Immunol. 1993;151(5):2399–2408. [PubMed] [Google Scholar]
- 9.Khandros E, Thom CS, D’Souza J, Weiss MJ. Integrated protein quality-control pathways regulate free alpha-globin in murine beta-thalassemia. Blood. 2012;119(22):5265–5275. doi: 10.1182/blood-2011-12-397729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeVilbiss AW, Sanalkumar R, Hall BD, Katsumura KR, de Andrade IF, Bresnick EH. Epigenetic determinants of erythropoiesis: role of the histone methyltransferase SetD8 in promoting erythroid cell maturation and survival. Mol Cell Biol. 2015;35(12):2073–2087. doi: 10.1128/MCB.01422-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McIver SC, Kang YA, DeVilbiss AW, O’Driscoll CA, Ouellette JN, Pope NJ, Camprecios G, Chang CJ, Yang D, Bouhassira EE, Ghaffari S, Bresnick EH. The exosome complex establishes a barricade to erythroid maturation. Blood. 2014;124(14):2285–2297. doi: 10.1182/blood-2014-04-571083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hewitt KJ, Kim DH, Devadas P, Prathibha R, Zuo C, Sanalkumar R, Johnson KD, Kang YA, Kim JS, Dewey CN, Keles S, Bresnick EH. Hematopoietic signaling mechanism revealed from a stem/progenitor cell cistrome. Mol Cell. 2015;59(1):62–74. doi: 10.1016/j.molcel.2015.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao X, Wu T, Johnson KD, Lahvic JL, Ranheim EA, Zon LI, Bresnick EH. GATA factor-G-protein-coupled receptor circuit suppresses hematopoiesis. Stem Cell Rep. 2016;6(3):368–382. doi: 10.1016/j.stemcr.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McIver SC, Katsumura KR, Davids E, Liu P, Kang Y-A, Yang D, Bresnick EH. Exosome complex orchestrates developmental signaling to balance proliferation and differentiation during erythropoiesis. eLife. 2016;5:e17877. doi: 10.7554/eLife.17877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hewitt KJ, Katsumura KR, Matson DR, Devadas P, Tanimura N, Hebert AS, Coon JJ, Kim JS, Dewey CN, Keles S, Hao S, Paulson RF, Bresnick EH. GATA factor-regulated Samd14 enhancer confers red blood cell regeneration and survival in severe anemia. Dev Cell. 2017;42(3):213–225e4. doi: 10.1016/j.devcel.2017.07.009. https://doi.org/10.1016/j.devcel.2017.07.009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehta C, Johnson KD, Gao X, Ong IM, Katsumura KR, McIver SC, Ranheim EA, Bresnick EH. Integrating enhancer mechanisms to establish a hierarchical blood development program. Cell Rep. 2017 doi: 10.1016/j.celrep.2017.08.090. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee HY, Gao X, Barrasa MI, Li H, Elmes RR, Peters LL, Lodish HF. PPAR-alpha and glucocorticoid receptor synergize to promote erythroid progenitor self-renewal. Nature. 2015;522(7557):474–477. doi: 10.1038/nature14326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams DA, Bunn HF, Sieff C, Zon LI. Hematopoiesis. In: Handin RI, Lux SE, Stossel TP, editors. Blood: principles and practice of hematology. 2. Lippincott Williams and Wilkins; Philadelphia, PA: 2003. pp. 147–208. [Google Scholar]
- 19.McGarry MP, Protheroe CA, Lee JJ. Mouse hematology: a laboratory manual. Cold Spring Harbor Laboratory Press; Woodbury, NY: 2010. [Google Scholar]
- 20.Broxmeyer HE. Colony assays of hematopoietic progenitor cells and correlations to clinical situations. Crit Rev Oncol Hematol. 1984;1(3):227–257. doi: 10.1016/s1040-8428(84)80013-x. [DOI] [PubMed] [Google Scholar]
- 21.Gregory CJ, Eaves AC. Three stages of erythropoietic progenitor cell differentiation distinguished by a number of physical and biologic properties. Blood. 1978;51(3):527–537. [PubMed] [Google Scholar]
- 22.Munugalavadla V, Kapur R. Role of c-Kit and erythropoietin receptor in erythropoiesis. Crit Rev Oncol Hematol. 2005;54(1):63–75. doi: 10.1016/j.critrevonc.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Muta K, Krantz SB, Bondurant MC, Dai CH. Stem cell factor retards differentiation of normal human erythroid progenitor cells while stimulating proliferation. Blood. 1995;86(2):572–580. [PubMed] [Google Scholar]
- 24.Sui X, Krantz SB, You M, Zhao Z. Synergistic activation of MAP kinase (ERK1/ 2) by erythropoietin and stem cell factor is essential for expanded erythropoiesis. Blood. 1998;92(4):1142–1149. [PubMed] [Google Scholar]
- 25.Wu H, Klingmuller U, Acurio A, Hsiao JG, Lodish HF. Functional interaction of erythropoietin and stem cell factor receptors is essential for erythroid colony formation. Proc Natl Acad Sci U S A. 1997;94(5):1806–1810. doi: 10.1160/TH08-08-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell. 1995;83(1):59–67. doi: 10.1016/0092-8674(95)90234-1. [DOI] [PubMed] [Google Scholar]
- 27.Wu H, Klingmuller U, Besmer P, Lodish HF. Interaction of the erythropoietin and stem-cell-factor receptors. Nature. 1995;377(6546):242–246. doi: 10.1038/377242a0. [DOI] [PubMed] [Google Scholar]
- 28.Hattangadi SM, Wong P, Zhang L, Flygare J, Lodish HF. From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood. 2011;118(24):6258–6268. doi: 10.1182/blood-2011-07-356006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haas N, Riedt T, Labbaf Z, Bassler K, Gergis D, Frohlich H, Gutgemann I, Janzen V, Schorle H. Kit transduced signals counteract erythroid maturation by MAPK-dependent modulation of erythropoietin signaling and apoptosis induction in mouse fetal liver. Cell Death Differ. 2015;22(5):790–800. doi: 10.1038/cdd.2014.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pop R, Shearstone JR, Shen Q, Liu Y, Hallstrom K, Koulnis M, Gribnau J, Socolovsky M. A key commitment step in erythropoiesis is synchronized with the cell cycle clock through mutual inhibition between PU.1 and S-phase progression. PLoS Biol. 2010;8(9):e1000484. doi: 10.1371/journal.pbio.1000484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen K, Liu J, Heck S, Chasis JA, An X, Mohandas N. Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc Natl Acad Sci U S A. 2009;106(41):17413–17418. doi: 10.1073/pnas.0909296106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dunning K, Safo AO. The ultimate Wright-Giemsa stain: 60 years in the making. Biotech Histochem. 2011;86(2):69–75. doi: 10.3109/10520295.2010.515496. [DOI] [PubMed] [Google Scholar]