

Abstract
Background
The etiology of idiopathic dilated cardiomyopathy (DCM) is unknown by definition, but its familial subtype is considered to have a genetic component. We hypothesize that most idiopathic DCM, whether familial or non-familial, has a genetic basis, in which case a genetics-driven approach to identifying at-risk family members for clinical screening and early intervention could reduce morbidity and mortality.
Methods
Based on this hypothesis, we have launched the NHLBI- and NHGRI-funded DCM Precision Medicine Study, which aims to enroll 1,300 individuals (600 non-Hispanic African ancestry, 600 non-Hispanic European ancestry, and 100 Hispanic) who meet rigorous clinical criteria for idiopathic DCM along with 2,600 of their relatives. Enrolled relatives will undergo clinical cardiovascular screening to identify asymptomatic disease, and all individuals with idiopathic DCM will undergo exome sequencing to identify relevant variants in genes previously implicated in DCM. Results will be returned by genetic counselors 12-14 months after enrollment. The data obtained will be used to describe the prevalence of familial DCM among idiopathic DCM cases and the genetic architecture of idiopathic DCM in multiple ethnicity-ancestry groups. We will also conduct a randomized controlled trial to test the effectiveness of Family Heart Talk, an intervention to aid family communication, for improving uptake of preventive screening and surveillance in at-risk first-degree relatives.
Conclusions
We anticipate this study will demonstrate that idiopathic DCM has a genetic basis and guide best practices for a genetics-driven approach to early intervention in at-risk relatives.
Keywords: dilated cardiomyopathy, genetics, idiopathic, familial, communication
Journal Subject Terms: Cardiomyopathy, Genetics, Clinical Studies, Behavioral/Psychosocial Treatment, Compliance/Adherence
Introduction
Dilated cardiomyopathy (DCM), defined as the presence of both left-ventricular enlargement and systolic dysfunction, is a leading cause of heart failure (HF), with 30-50% of enrolled patients in many HF registries and clinical trials having non-ischemic DCM.1 Most of these patients have idiopathic DCM, for which usual clinical investigations to understand etiology, excluding genetic evaluation, have been completed and are negative.2 Because these individuals may have had asymptomatic left-ventricular enlargement, systolic dysfunction, or idiopathic DCM for many years before presenting with HF, arrhythmia, or embolus,3,4 there is a significant opportunity to delay or stop progression to HF with early intervention.2 The potential benefits are compelling: HF has a 5-year survival of approximately 50%1 and is expected to affect >8 million US adults and cost $69.7 billion, or approximately $244 per US adult, by 2030.5
A meta-analysis of single-center studies suggests that there is evidence of familial disease in 23% of idiopathic DCM cases.6 Such familial DCM cases are considered to have a genetic etiology,7 which has led us to hypothesize that all idiopathic DCM, whether familial or non-familial, has a genetic basis. If this hypothesis is correct, a genetics-driven, family-based approach to early intervention in at-risk individuals could be fruitful for idiopathic DCM more generally.8 However, the available evidence regarding the prevalence of familial DCM among idiopathic DCM cases and the genetic architecture of idiopathic DCM is currently insufficient to make a compelling case for a family-centric model of care. Moreover, there are a number of unresolved challenges in implementing a family-centric model of care for any disease. These issues, considered in greater detail below, led to the design and launch of the DCM Precision Medicine Study.
Prevalence of Familial DCM
While the occurrence of familial DCM in a significant proportion of idiopathic DCM cases has been established,6,9 uncertainty among cardiovascular practitioners regarding its prevalence persists. Because DCM is clinically silent until late in its course,7 self-reported family history has low sensitivity, and clinical screening of relatives with echocardiography is necessary to discover familial disease.10 As a result, cardiovascular practitioners may systematically underestimate the prevalence of familial DCM based on their own clinical experience. Unfortunately, the scientific literature adds to this uncertainty. Published estimates of the prevalence of familial DCM among idiopathic DCM cases vary widely, ranging from 2% to 65% in 23 single-center studies.6,9 This marked heterogeneity stems in part from the variable definitions of familial DCM across studies, which range from positive family history alone to DCM in at least 2 relatives.6
Genetic Architecture
The genetic architecture of a phenotype is a description of how genetic factors influence its manifestation.11,12 These factors include the loci affecting the phenotype, their interactions with each other and the environment, the effects of particular alleles at these loci, and the frequencies of these alleles/genotypes in the population.11,12 For DCM, the genetic architecture is still being characterized.2,7,8,13 While a consensus has emerged that genetic cause underlies familial DCM,13 both US and European consensus documents consider the role of genetics as an open question for idiopathic DCM cases with negative family histories. The most recent US consensus document classified hypertrophic (HCM) and arrhythmogenic right ventricular (ARVC) cardiomyopathies and channelopathies as “genetic” but classified DCM as “mixed”.14 The 2013 ACC/AHA Heart Failure Guidelines were silent on this question, citing the US consensus document and noting that classification is challenging.1 The European consensus document classified familial DCM, along with HCM and ARVC, as genetic, but also noted that DCM could result from a variety of environmental impacts (e.g., hypertension, viral infections).15 A more recent European position statement has discussed genetic causes for DCM but noted that the inheritance model is still being elucidated and that environmental causes, either alone or in combination with genetic predisposition, may underlie disease.16
In targeted resequencing studies of 11 candidate genes in a series of 312 individuals with idiopathic DCM, we observed similar frequencies of variants in familial and non-familial cases.17 Our finding suggests that non-familial idiopathic DCM may also have a genetic basis. However, non-familial cases were categorized mostly on the basis of a negative family history,18,19 potentially missing cases that would have been identified by systematic clinical screening of family members.10
Racial and Ethnic Minorities
Because almost all genetic investigations to date have been conducted in individuals of European ancestry (EA), data on individuals of African ancestry (AA) and Hispanic ethnicity (HE) are lacking. The paucity of data on individuals of African ancestry (AA) is particularly problematic; non-ischemic DCM is considered to be more prevalent in AA, with earlier age of onset and worse outcomes in most,20–23 but not all,24 studies. Although environmental risk factors, such as hypertension20 or adverse socioeconomic status,25 are known to be more prevalent in AA, an analysis of cardiovascular mortality from the Studies of Left Ventricular Dysfunction that controlled for these issues suggested that individuals of AA still had increased mortality compared to EA.26 A natural question, then, is whether the genetic architecture of DCM differs in AA, but such questions cannot be answered without additional data on minority populations.
Challenges of Family-Centric Early Intervention
A diagnosis of idiopathic DCM should trigger family-based evaluation,27 as early intervention with DCM may forestall the onset of advanced heart failure. However, a family-centric model for early intervention presents formidable implementation challenges not encountered in conventional patient-centric treatment of symptomatic DCM. Chief among these is the fact that the clinician often cannot communicate directly with at-risk family members and so must transmit complex genetic information and recommendations through the patient. Research on high risk breast and colon cancer cases and their first-degree relatives (FDRs) has found that communication about risk does not flow seamlessly among family members28,29 and often does not motivate clinical screening or genetic testing.30 In cardiomyopathies, retrospective single-center studies of mostly hypertrophic cardiomyopathy cases have also demonstrated incomplete uptake of genetic counseling (40%), genetic testing (39-51%) and cardiovascular screening (76%) among FDRs for whom these interventions are indicated.31,32 Moreover, best practices for communication of genetic test results may also depend on cultural or social context. For example, studies have found that uptake of BRCA1/2 tests to identify inherited risk for breast/ovarian cancer was lower among AA compared to EA.33,34 At present, methods for addressing these implementation challenges in DCM have not been studied.
Methods
Design
The DCM Precision Medicine Study is a cross-sectional observational study of families with an embedded open-label randomized controlled trial. An overview of the study flow is provided in the Figure 1. We aim to recruit 1,300 individuals (600 non-Hispanic AA (NHE-AA), 600 NHE-EA, and 100 HE; 50% female) meeting rigorous diagnostic criteria for idiopathic DCM (Table 1) and 2,600 of their relatives. These probands (Table 2) are inpatients or outpatients identified by heart failure and cardiac transplant cardiovascular physicians and clinical research personnel in heart failure/heart transplant programs at sites in the DCM Consortium (Appendix). The DCM phenotype was selected for this study because of its prevalence and its prior inclusion in the development of virtually all pharmacotherapy for non-ischemic heart failure from reduced ejection fraction. Hence, any genetic insights gained should be able to be broadly generalized for the greatest public health impact.
Figure 1.
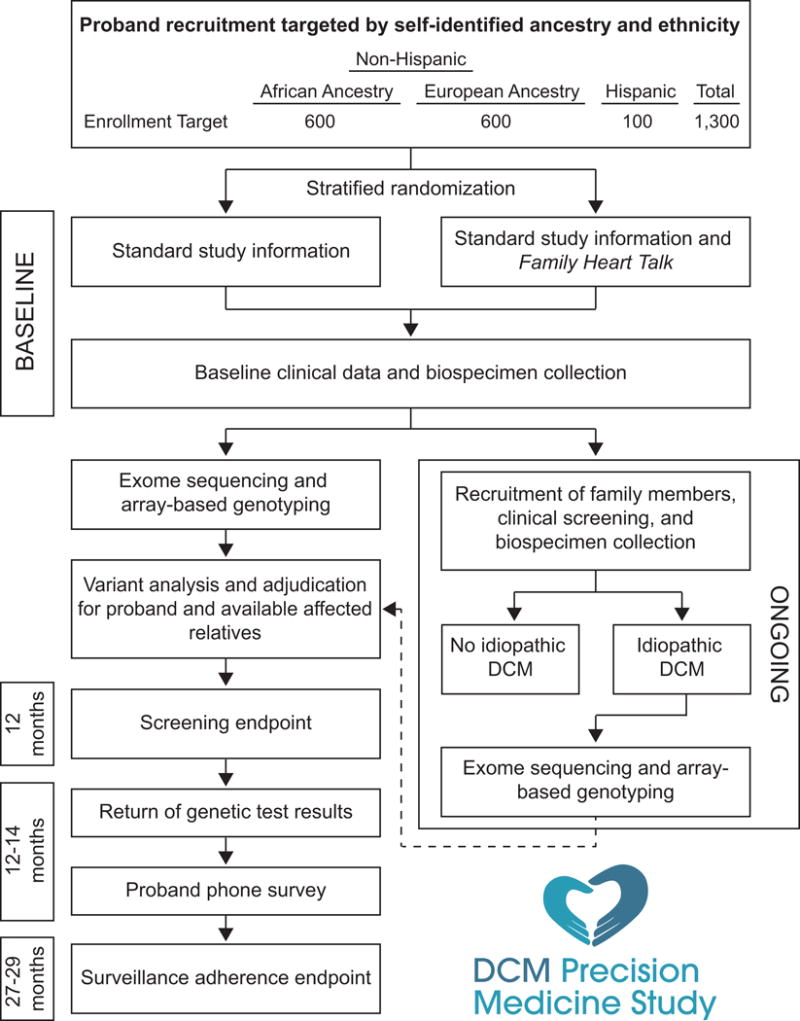
Flow of study processes for a single family. DCM = Dilated cardiomyopathy.
Table 1.
Diagnostic criteria for idiopathic dilated cardiomyopathy.
| Criterion |
|---|
| Left ventricular ejection fraction <50% |
| Left ventricular enlargement (echo-derived left ventricular end-diastolic dimension ≥95th percentile for gender/height)35 |
|
|
| Exclusion of all of the following at the time of primary DCM diagnosis: |
|
|
| Coronary artery disease (CAD) causing ischemic cardiomyopathy (>50% narrowing, any major epicardial coronary artery; clinical testing is indicated to exclude CAD routinely in (1) males >40 years or females >45 years without risk factors and (2) males or females >30 years with risk factors) |
| Primary valvular disease |
| Cardiotoxic drug exposure, including Adriamycin and other cancer chemotherapeutics |
| Other forms of cardiomyopathy (e.g., hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, restrictive cardiomyopathy, Chagas cardiomyopathy) |
| Congenital/structural heart disease |
| Sarcoid |
| Amyloid |
| Iron overload |
| Other active multisystem disease that may plausibly cause DCM (e.g., hypereosinophilic syndrome, cardiac involvement with connective tissue disease, Loeffler’s endocarditis, endomyocardial fibrosis) |
| Severe and untreated or untreatable hypertension* |
| Exclusion beyond a reasonable doubt of all other detectable causes of cardiomyopathy (other than genetic) at the time of primary DCM diagnosis using a rigorous clinical standard congruent with an expert approach rendered by a board-certified heart failure/transplant cardiologist |
DCM = Dilated cardiomyopathy.
Severe hypertension is defined as systolic blood pressures routinely >180 mm Hg and/or diastolic blood pressures >120 mm Hg. Untreated hypertension is an individual receiving no medications, and untreatable hypertension is an individual with severe hypertension persisting despite multidrug regimens and/or associated with other multisystem disease (e.g., scleroderma, other vasculitides, etc).
Table 2.
Definitions of terms.
| Term | Definition |
|---|---|
| Proband | The first individual from a particular family who meets diagnostic criteria for idiopathic DCM (Table 1) and enrolls in the study. |
| Familial DCM | Idiopathic DCM along with at least one first- or second-degree relative with confirmed idiopathic DCM. |
| Non-familial DCM | Idiopathic DCM not meeting the definition of familial, including when no first- or second-degree relatives complete clinical screening. |
| Plausible genetic cause | At least one variant in one or more of the DCM-relevant genes selected for this study adjudicated pathogenic or likely pathogenic without using familial genetic data. |
| Hispanic ethnicity (HE) | An individual who self-identifies as Cuban, Mexican, Puerto Rican, South or Central American, or another culture of Spanish origin. |
| Self-Identified Ancestry Categories | |
| African (AA) | An individual who self-identifies as “Black or African American” either alone or in combination with “White.” |
| European (EA) | An individual who self-identifies as only “White.” |
| Native American (NAA) | An individual who self-identifies as an “American Indian or Alaska Native” either alone or in combination with “White.” |
| Genomic Ancestry Categories | |
| African (AA) | An individual with more than 50% of ancestry estimated to derive from the populations represented in the African reference samples, regardless of the estimated ancestry proportions for other continental groups. |
| European (EA) | An individual with at least 87.5% of ancestry estimated to derive from the populations represented in European reference samples, regardless of the estimated ancestry proportions for other continental groups. |
| Native American (NAA) | An individual with more than 50% of ancestry estimated to derive from the Native American reference population(s) or apparent ancestral population, regardless of the estimated ancestry proportions for other continental groups. |
DCM = Dilated cardiomyopathy.
Accrual began in June 2016 and is currently underway. To ensure attainment of the overall recruitment targets above, the study has implemented site-specific race, ethnicity and gender enrollment expectations and provides site-specific and overall study reporting of enrollment data to all active sites every two weeks.
Objectives
The observational study will yield estimates of the prevalence of familial DCM (Table 2) among probands in multiple ethnicity-ancestry groups. It will also provide estimates of the probability of finding a plausible genetic cause (Table 2) in a proband of a particular ethnicity-ancestry and familial/non-familial DCM classification. The randomized controlled trial will test the effectiveness of an intervention, Family Heart Talk, for improving the uptake of preventive screening and surveillance in at-risk first-degree relatives.
Organizational Structure
The DCM Precision Medicine Study is executed under the auspices of the multi-site DCM Consortium (Appendix; see also http://www.dcmproject.com). The purpose of the DCM Consortium, created and directed by the study Principal Investigator (REH), is to conduct DCM genetics and genomics research. The Ohio State University (OSU) is the lead site of the DCM Consortium, the coordinating site for this study, and a clinical site responsible for recruiting and enrolling probands and relatives. The remaining sites are clinical sites that are also responsible for recruiting and enrolling probands and relatives. DCM Consortium contracts negotiated between the lead site and each clinical site established research relationships for regulatory and related items. Following local Institutional Review Board applications and approvals for a preliminary version of the DCM Precision Medicine Study, patient protected health information and biological materials could be transferred to the coordinating site to demonstrate feasibility and to obtain preliminary data for an NIH R01 funding application. Following funding, the Institutional Review Boards at The Ohio State University and all clinical sites approved the DCM Precision Medicine Study. Written informed consent is obtained from all subjects.
Inclusion/Exclusion Criteria
Probands must meet diagnostic criteria for idiopathic DCM (Table 1), be in one of the target ethnicity-ancestry groups (Figure 1), be able to communicate in English (or in Spanish at sites approved to recruit individuals of HE), be able to give informed consent (or assent and parental consent for children), and be willing and able to participate in a family-based study. Relatives must satisfy these same requirements except for meeting diagnostic criteria for idiopathic DCM. Risk factors considered conventional for DCM, such as obesity, routinely treated hypertension, alcohol use/abuse, peripartum cardiomyopathy, or the presence of left-ventricular non-compaction, are not used as exclusion criteria.
Recruitment of Relatives
Probands are asked at the time of enrollment to refer all living FDRs and any affected family member with heart disease, including idiopathic DCM, to the study. Study staff at the coordinating and clinical sites work with the probands to follow up on these referrals and enroll the relatives. Relatives known or found to be affected with idiopathic DCM are asked to refer their family members to expand the family’s pedigree. Relatives are not contacted by study personnel without oral consent for contact. This permission to contact is transmitted to study personnel in most cases via the proband; the family member may also contact study staff directly. At enrollment, the proband is requested to document, in writing, that contact information will only be provided with the family member’s consent.
Family Heart Talk Intervention
Family Heart Talk was developed to help probands communicate DCM risk and stimulate clinical screening of their family members. It is based on Leventhal’s Self-Regulation Model of Health Behavior36 and is modeled after a previously developed web-based family communication intervention (DJB, WB) for melanoma survivors that resulted in increased family communication about shared risk.37 Family Heart Talk has been vetted by a focus group of cardiovascular and genetics experts and in structured interviews with DCM patients.
The Family Heart Talk intervention consists of a guide to family communication about DCM provided in both print and web-based forms. It includes visuals and lay language explanations of the evaluation and care of individuals with DCM, including the necessity of a clinical cardiac evaluation in family members because DCM can be asymptomatic. It also provides guidance on talking with family members about DCM risk and includes samples of emails and letters.
Randomization
Probands receive either standard study information (control group) or Family Heart Talk along with this information (intervention group) at the time of enrollment. Standard study information includes a study brochure with information for family members, a “Dear Family Member” letter, and a letter to physicians of family members. Proband enrollment kits for each ethnicity-ancestry stratum (NHE-AA, NHE-EA, or HE) at each clinical site are assembled at the coordinating site and are marked with accession codes specifying the clinical site, the ethnicity-ancestry stratum, and the order of use that can be seen by personnel enrolling probands. If the randomization assignment corresponding to the kit’s accession code is to the intervention group, printed Family Heart Talk materials as well as a login and password for accessing an electronic version of these materials on our website (http://www.dcmproject.com) are inserted. After assembly is complete, the opaque shipping envelope containing the kit is sealed. Recruiting staff enrolling a proband at a clinical site select the kit with the next sequential accession code for the proband’s self-identified ethnicity-ancestry stratum, and the intervention assignment is revealed to the recruiting staff only after the kit is opened. This assignment is recorded on study documents returned to the coordinating site and compared against the actual randomization assignment for the kit in the database.
The randomization assignment for each accession code is based on randomization schedules that were generated independently for each ethnicity-ancestry stratum within each recruitment site. For each stratum, the randomization list was generated using randomly permuted blocks with equal treatment allocations and an equiprobable random block size of 2, 4, or 6. The randomization schedule for each stratum has more than enough randomization assignments to accommodate projected recruitment, and additional randomization schedules can be generated for new DCM Consortium sites using the same procedure.
Baseline Clinical Data Collection
Upon enrollment, all probands and relatives are interviewed by study personnel. A cardiovascular history questionnaire is collected, and a questionnaire probing quality of life, health behaviors, and family dynamics is administered. All probands are also provided a comprehensive family history questionnaire, which is completed following enrollment. Study personnel from OSU contact probands by telephone within 1-3 days following enrollment and expand the family history and construct a pedigree that is maintained in the study database. The pedigree is further augmented with the return of the family history questionnaire to OSU.
Following enrollment, study personnel complete a medical record questionnaire for probands, which details all aspects of their disease onset, evaluation, and past and current treatment. All information is supported by medical records, which are electronically transmitted to OSU. An OSU collaborating cardiologist reviews all data submitted for each proband to ensure study inclusion/exclusion criteria have been met, and, if so, a diagnosis of idiopathic DCM is entered into the study database. A similar process is applied to family members with known or suspected DCM or other cardiovascular disease; medical record questionnaires are completed by study personnel, and cardiovascular medical records are obtained and systematically reviewed. Family members must meet the same stringent requirements as probands to be diagnosed with DCM. All information is stored in the study database.
OSU clinical study personnel have been trained by a study genetic counselor in taking a cardiovascular family history and preparing pedigrees. Site personnel are trained in all study procedures, including taking and recording family histories and proper completion of all study documents, by the study PI, the study clinical research manager, and the study genetic counselor at site activation events.
Baseline Biospecimen Collection
Peripheral blood is drawn and sent to the DCM Consortium biobank at Nationwide Children’s Hospital for production of DNA (all individuals) and preservation of viable lymphocytes (individuals with an idiopathic DCM diagnosis only). All steps of phelebotomy, blood handling and shipment, blood receipt, DNA preparation, and storage at the study biobank follow CLIA guidelines and preserve CLIA chain of custody. CLIA chain of custody is also maintained when DNA specimens are shipped to the CLIA clinical sequencing laboratory at the University of Washington, where a portion of the DNA samples are transferred to the non-CLIA research laboratory at the University of Washington for exome sequencing. Variants identified at exome sequence analysis and considered clinically relevant undergo Sanger sequencing in the University of Washington CLIA-compliant clinical sequencing laboratory.
Clinical Screening of Relatives
For relatives who have not yet had clinical screening but are presumed free of cardiovascular disease, clinical (phenotype) screening, consisting of a cardiovascular-directed history and exam, ECG, and a trans-thoracic echocardiogram, is conducted at a nearby DCM Consortium site (if geographically feasible) or at the relative’s physician as part of recommended clinical care.27,38 Screening results are transmitted to OSU and reviewed to exclude DCM. Abnormal clinical results obtained from study-initiated research-based screening are returned to participants by study personnel at the site where the family member was enrolled with recommendations for additional (non-research) clinical cardiovascular evaluation. For all such participants who undergo additional testing, their subsequent clinical cardiovascular medical records are requested and reviewed to confirm the presence or absence of idiopathic DCM.
Genotyping
Research exome sequencing and array-based genotyping of individuals with idiopathic DCM diagnoses are conducted at the University of Washington Genome Sciences laboratory, and genomic data files are transferred to the Division of Human Genetics Data Management Platform at the Ohio Supercomputer Center for further analysis of a panel of genes considered clinically relevant for DCM (Supplemental Table 1) by staff at the coordinating site. Exome sequencing, rather than sequencing a large DCM gene panel, was considered the safest remedy should a novel high-impact gene not yet on any clinical panel be discovered during the study. Exome sequence also provides a basis for future discovery studies; funding limitations permitted neither discovery studies nor genome sequencing to be included in the current study.
Variant Analysis and Interpretation
Our approach to adjudicating variants is tailored after that used by the ClinGen initiative.39 Each variant passing quality control in these genes is assigned to one of the American College of Medical Genetics variant categories: pathogenic, likely pathogenic, variant of uncertain significance, likely benign, or benign.40 A Variant Adjudication Oversight Committee is charged with approving the criteria used for these assignments. Variants considered pathogenic or likely pathogenic and variants of uncertain significance that may be reclassified based on assessment of segregation are resequenced in the DNAs from probands and their available informative family members with CLIA-compliant Sanger sequencing at the University of Washington. These data are used for variant confirmation and, along with exome sequence data from affected family members, assessment of segregation in adjudication.
Return of Genetic Results
Genetic test results for DCM-relevant genes will be returned to probands 12-14 months after proband enrollment. If pathogenic or likely pathogenic variants are found in the proband, at-risk relatives who have DNAs available for CLIA-compliant resequencing will also receive their genetic test results for these variants at this time. Exome sequence data will not be analyzed for secondary genetic findings outside of DCM-relevant genes. Results will be returned to consented individuals by study genetic counselors via telephone calls along with a report and counseling letter. Recommendations with regard to follow-up surveillance will be tailored according to Figure 2.
Figure 2.
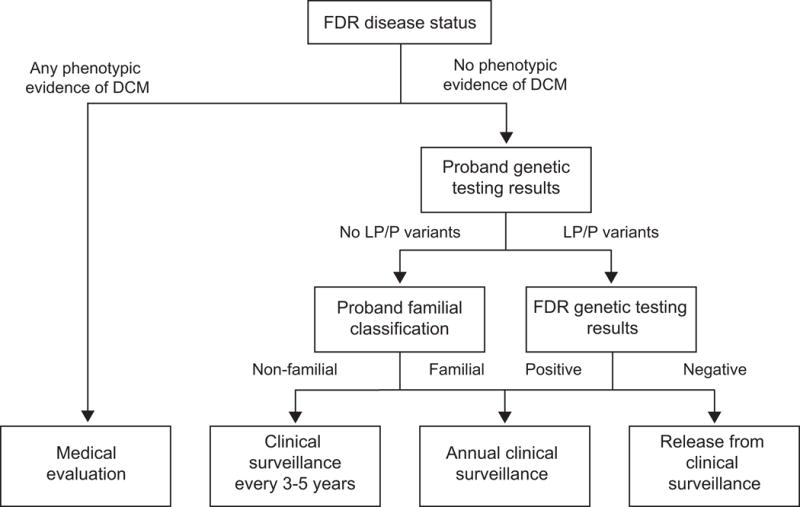
Surveillance recommendations for first-degree relatives. DCM = Dilated cardiomyopathy; FDR = First-degree relative; LP = Likely pathogenic; P = Pathogenic.
Annual Follow-up
Study staff will recontact probands by telephone after return of genetic results and annually thereafter to administer questionnaires probing quality of life, health behaviors, and family dynamics.
Statistical Analysis
Observational Study
We are interested in whether genomic background, cultural and social background (e.g., through greater or lesser exposure to environmental risk factors), or both influence the prevalence of familial DCM or the probability of finding a plausible genetic cause in probands. Ancestry can be measured by self-identification or genomic analysis; the two measures are often highly, although not perfectly, correlated41,42 and so would both reflect genomic as well as cultural and social background. Moreover, self-identified ancestry is most likely to be used in clinical care. Therefore, we will perform separate analyses for observational study outcomes using both self-identified and genomic ancestry categories (Table 2). Genomic ancestry will be determined based on global ancestry proportions inferred from array-based genotypes using established techniques, such as the widely vetted STRUCTURE model43 implemented in ADMIXTURE software.44 Probands not falling into one of these genomic ancestry categories will be excluded from that analysis. Ethnicity, which is a cultural construct, will be based on self-identification in both analyses.
Because of the diverse referral populations across the centers in this study and the previously observed heterogeneity in single-site estimates of the prevalence of familial DCM in probands,6 we expect to observe variation in observational study outcomes between sites and ethnicity-ancestry groups beyond what can be explained by sampling error alone. For this reason, our statistical analyses for all observational study outcomes will use generalized linear mixed models (GLMMs),45 which allow modeling of this heterogeneity. We will include normally distributed random effects for site and ethnicity-ancestry group (NHE-AA, NHE-EA, HE-AA, HE-EA, and HE Native American ancestry (HE-NAA)) within site for all observational study outcomes. The site-level random effects are designed to reflect site-specific characteristics that would affect all ethnicity-ancestry groups in the same way, and the site-ethnicity-ancestry random effects are designed to reflect factors that may cause variation among ethnicity-ancestry groups at the same site.
The prevalence of familial DCM among probands will be analyzed using a logistic-normal GLMM with the proband’s familial classification as the outcome. The linear predictor will include fixed effects for ethnicity-ancestry group, age quartile, and sex along with the random effects previously described. We will estimate the marginally standardized predicted probabilities of familial DCM in populations of AA or EA probands at a typical site as well as their difference.45–47
Genetic architecture will be explored using a logistic-normal GLMM with the presence or absence of a plausible genetic cause as the outcome. The linear predictor will include fixed effects for ethnicity-ancestry group interacted with familial classification, age quartile, and sex along with the random effects previously described. We will estimate the marginally standardized predicted probabilities for populations of familial or non-familial probands, for populations of AA or EA probands, and their differences at a typical site. Note that, because variant adjudication criteria often use segregation among multiple affected family members as a criterion for determining pathogenicity, we ignore the evidence provided by familial genetic data in our definition of plausible genetic cause (Table 2) to avoid bias in comparisons between familial and non-familial probands.
Randomized Trial
There are two primary endpoints for measuring effectiveness of the intervention: (1) whether a living FDR without a previously confirmed idiopathic DCM diagnosis completes clinical screening within 12 months after proband recruitment; and (2) whether a living FDR adheres to appropriate surveillance recommendations for his or her situation (Figure 2) during the 15 months after genetic testing results are first released to the family. For the first endpoint, living FDRs without a previously confirmed idiopathic DCM diagnosis will be considered; the second endpoint will consider all living FDRs. Living FDRs who do not enroll in the study will be considered non-completers for the screening endpoint and non-adherent for the surveillance endpoint. For the surveillance endpoint, failure to complete clinical screening (if not previously completed), failure to obtain a recommended surveillance procedure (hypovigilance), or obtaining a surveillance procedure that was not recommended (hypervigilance) within the appropriate timeframe will be counted as non-adherence. Endpoints have been defined using only FDRs rather than including more distantly related relatives because the proband is more likely to communicate regularly with FDRs and family information commonly flows through next of kin.
Because randomization is performed within ethnicity-ancestry strata at each site and the intervention is administered at the family level via the proband, this trial is stratified and cluster-randomized,48 with each family defining a cluster. For each primary endpoint, we will fit a logistic-normal GEE-type GLMM45 to the outcomes in individual FDRs. The linear predictor will include fixed effects for randomization assignment (Family Heart Talk or control), age quartile, sex, and proband-reported family history of DCM (present or absent). Stratified randomization will be accounted for by including random effects for site and ethnicity-ancestry stratum (NHE-AA, NHE-EA, or HE) within site. Note that stratum effects are modeled in this manner rather than with fixed effects to avoid estimation problems arising from relatively large number of planned strata. Correlation between the outcomes of FDRs of the same proband will be modeled with a compound symmetry working correlation structure. The null hypothesis for each endpoint is that the odds ratio between the Family Heart Talk and control arms is ≤1, and the alternative hypothesis is that it is >1. The alpha level for each primary endpoint will be 0.05.
Sample Size Justifications
Monte Carlo simulations were used to estimate the precision of estimates for observational study outcomes and the power to detect an effective intervention for the randomized trial. Results are summarized below; full details are provided in the Supplemental Methods.
Observational Study
Under our assumptions, this study has at least an 80% chance of ruling out the following at an alpha of 0.05: 1) a prevalence of familial DCM below 0.249 in self-identified AA and EA probands and differences between these populations of more than 0.121 at a typical site; 2) probabilities of plausible genetic cause below 0.290 in non-familial probands and differences between familial and non-familial probands of more than 0.136 at a typical site; and 3) probabilities of plausible genetic cause below 0.287 for self-identified AA and EA probands and differences between these populations of more than 0.137 at a typical site.
Randomized Trial
The randomized trial has at least 99% power to detect an effective intervention at an alpha of 0.05 for each endpoint under our assumptions.
Discussion
The DCM Precision Medicine Study will rigorously obtain phenotype and pedigree data on a large, multi-racial cohort of idiopathic DCM cases and their family members, identify relevant genetic variants through exome sequencing, and return the results from known DCM genes to probands and affected family members. With the data from this cohort, we will obtain precise estimates of the prevalence of familial DCM among AA and EA probands as well as the proportion of probands with plausible genetic cause by ancestry and familial/non-familial classification. If our hypothesis is correct, this study is likely to establish a higher prevalence of familial DCM than is indicated by the extant literature as well as a genetic etiology for non-familial idiopathic DCM. We will also obtain valuable knowledge regarding family dynamics and interactions around return of genetic results and rigorously test the effectiveness of a family communication tool with a well-powered randomized trial. The results of this study could hasten the adoption of a genetics-driven approach to early intervention in at-risk relatives of individuals with idiopathic DCM while also guiding best practices for its implementation.
Supplementary Material
Acknowledgments
The DCM Precision Medicine Study is supported by computational infrastructure provided by The Ohio State University Division of Human Genetics Data Management Platform and the Ohio Supercomputer Center.
Sources of Funding: Research reported in this publication was supported by the National Heart, Lung, and Blood Institute and National Human Genome Research Institute of the National Institutes of Health under award number R01HL128857. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Appendix: DCM Consortium institutions and personnel participating in the DCM Precision Medicine Study
Study Principal Investigator and Co-Investigators
The Ohio State University
Ray E. Hershberger (Principal Investigator)
Daniel D. Kinnamon
Ana Morales
Nationwide Children’s Hospital
Julie M. Gastier-Foster
University of Washington
Wylie Burke
Deborah J. Bowen
Deborah A. Nickerson
Michael O. Dorschner
DCM Consortium Clinical Site Principal Investigators and Clinical Site Other Significant Contributors
The following clinical sites and individuals contributed to the submission of R01HL128857 as Site Principal Investigators (Site PI) or as Other Significant Contributors (OSC):
The Ohio State University
Garrie Haas (Site PI)
William Abraham (OSC)
Philip Binkley (OSC)
Ayesha Hasan (OSC)
Jennifer Host (OSC)
Brent Lampert (OSC)
Sakima Smith (OSC)
Tufts Medical Center
Gordon Huggins (Site PI)
David DeNofrio (OSC)
Michael Kiernan (OSC)
University of Washington
Daniel Fishbein (Site PI)
Richard Cheng (OSC)
Todd Dardas (OSC)
Wayne Levy (OSC)
Claudius Mahr (OSC)
Sofia Masri (OSC)
April Stempien-Otero (OSC)
University of Maryland, Baltimore
Stephen Gottlieb (Site PI)
Stanford University
Matthew Wheeler (Site PI)
Euan Ashley (OSC)
Julia Platt (Co-Investigator)
Medstar Washington Hospital Center
Mark Hofmeyer (Site PI)
Cleveland Clinic
Wilson Tang (Site PI)
Randall Starling (OSC)
Rocio Moran (OSC)
University of Pennsylvania
Anjali Owens (Site PI)
Kenneth Marguilies (OSC)
Thomas Cappola (OSC)
Lee Goldberg (OSC)
Susan Brozena (OSC)
J. Rame (OSC)
Rhondalyn McLean (OSC)
University of Mississippi Medical Center
Charles Moore (Site PI)
Matthew deShazo (OSC)
Robert Long (OSC)
Baptist Health South Florida – Miami Cardiac and Vascular Institute
Francisco Jimenez Carcamo (Site PI)
Hakop Hrachian-Haftevani (OSC)
Houston Methodist Hospital
Barry Trachtenberg (Site PI)
Guhu Ashrith (OSC)
Arvind Bhimarahj (OSC)
Jerry Estep (OSC)
An updated list of DCM Consortium sites currently participating in the DCM Precision Medicine Study is available at http://www.dcmproject.com.
The following clinical site was added following study approval but prior to NHGRI supplemental funding:
University of Arizona Sarver Heart Center
Nancy Sweitzer (Site PI)
Study Consultants
Carlos D. Bustamante, Stanford University School of Medicine
Gail P. Jarvik, University of Washington
Eden R. Martin, University of Miami Miller School of Medicine
Heidi Rehm, Harvard Medical School
National Institutes of Health Program Personnel
National Heart, Lung, and Blood Institute
Patrice Desvigne-Nickens
James Troendle
Yi-Ping Fu
National Human Genome Research Institute
Lucia Hindorff
Footnotes
Clinical Trial Registration: www.clinicaltrials.gov, Unique Identifier: NCT03037632.
Disclosures: None.
References
- 1.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, et al. 2013 ACCF/AHA guideline for the management of heart failure. Circulation. 2013;128:e240–e327. doi: 10.1161/CIR.0b013e31829e8776. [DOI] [PubMed] [Google Scholar]
- 2.Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: A review for genetics professionals. Genet Med. 2010;12:655–667. doi: 10.1097/GIM.0b013e3181f2481f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Japp AG, Gulati A, Cook SA, Cowie MR, Prasad SK. The diagnosis and evaluation of dilated cardiomyopathy. J Am Coll Cardiol. 2016;67:2996–3010. doi: 10.1016/j.jacc.2016.03.590. [DOI] [PubMed] [Google Scholar]
- 4.Mahon NG, Murphy RT, MacRae CA, Caforio ALP, Elliott PM, McKenna WJ. Echocardiographic evaluation in asymptomatic relatives of patients with dilated cardiomyopathy reveals preclinical disease. Ann Intern Med. 2005;143:108–115. doi: 10.7326/0003-4819-143-2-200507190-00009. [DOI] [PubMed] [Google Scholar]
- 5.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics—2016 update. Circulation. 2016;133:e38–360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 6.Petretta M, Pirozzi F, Sasso L, Paglia A, Bonaduce D. Review and metaanalysis of the frequency of familial dilated cardiomyopathy. Am J Cardiol. 2011;108:1171–1176. doi: 10.1016/j.amjcard.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 7.Piran S, Liu P, Morales A, Hershberger RE. Where genome meets phenome: Rationale for integrating genetic and protein biomarkers in the diagnosis and management of dilated cardiomyopathy and heart failure. J Am Coll Cardiol. 2012;60:283–289. doi: 10.1016/j.jacc.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Hershberger RE, Siegfried JD. Update 2011: Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2011;57:1641–1649. doi: 10.1016/j.jacc.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005;45:969–981. doi: 10.1016/j.jacc.2004.11.066. [DOI] [PubMed] [Google Scholar]
- 10.Michels VV, Moll PP, Miller FA, Tajik AJ, Chu JS, Driscoll DJ, et al. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med. 1992;326:77–82. doi: 10.1056/NEJM199201093260201. [DOI] [PubMed] [Google Scholar]
- 11.Weiss KM. Genetic variation and human disease: principles and evolutionary approaches. Cambridge; New York: Cambridge University Press; 1993. [Google Scholar]
- 12.Hansen TF. The evolution of genetic architecture. Annu Rev Ecol Evol Syst. 2006;37:123–157. [Google Scholar]
- 13.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–547. doi: 10.1038/nrcardio.2013.105. [DOI] [PubMed] [Google Scholar]
- 14.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, et al. Contemporary definitions and classification of the cardiomyopathies. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 15.Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: A position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur Heart J. 2008;29:270–6. doi: 10.1093/eurheartj/ehm342. [DOI] [PubMed] [Google Scholar]
- 16.Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Böhm M, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016;37:1850–1858. doi: 10.1093/eurheartj/ehv727. [DOI] [PubMed] [Google Scholar]
- 17.Hershberger RE, Norton N, Morales A, Li D, Siegfried JD, Gonzalez-Quintana J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3:155–161. doi: 10.1161/CIRCGENETICS.109.912345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kushner JD, Nauman D, Burgess D, Ludwigsen S, Parks SB, Pantely G, et al. Clinical characteristics of 304 kindreds evaluated for familial dilated cardiomyopathy. J Card Fail. 2006;12:422–429. doi: 10.1016/j.cardfail.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 19.Siegfried JD, Morales A, Kushner JD, Burkett E, Cowan J, Mauro AC, et al. Return of genetic results in the familial dilated cardiomyopathy research project. J Genet Couns. 2013;22:164–174. doi: 10.1007/s10897-012-9532-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics—2013 update. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yancy CW. Heart failure in African Americans. Am J Cardiol. 2005;96:3–12. doi: 10.1016/j.amjcard.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 22.Bibbins-Domingo K, Pletcher MJ, Lin F, Vittinghoff E, Gardin JM, Arynchyn A, et al. Racial differences in incident heart failure among young adults. N Engl J Med. 2009;360:1179–1190. doi: 10.1056/NEJMoa0807265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franciosa JA, Ferdinand KC, Yancy CW, On behalf of the Consensus Statement on Heart Failure in African Americans Writing Group. Treatment of heart failure in African Americans: A consensus statement. Congest Heart Fail. 2010;16:27–38. doi: 10.1111/j.1751-7133.2009.00118.x. [DOI] [PubMed] [Google Scholar]
- 24.Markham DW, Dries DL, King LP, Leonard D, Yancy CW, Peshock RM, et al. Blacks and whites have a similar prevalence of reduced left ventricular ejection fraction in the general population: The Dallas Heart Study (DHS) Am Heart J. 2008;155:876–882. doi: 10.1016/j.ahj.2007.11.038. [DOI] [PubMed] [Google Scholar]
- 25.Coughlin SS, Myers L, Michaels RK. What explains black-white differences in survival in idiopathic dilated cardiomyopathy? The Washington, DC, Dilated Cardiomyopathy Study. J Natl Med Assoc. 1997;89:277–282. [PMC free article] [PubMed] [Google Scholar]
- 26.Dries DL, Exner DV, Gersh BJ, Cooper HA, Carson PE, Domanski MJ. Racial differences in the outcome of left ventricular dysfunction. N Engl J Med. 1999;340:609–616. doi: 10.1056/NEJM199902253400804. [DOI] [PubMed] [Google Scholar]
- 27.Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MRG, Towbin JA. Genetic evaluation of cardiomyopathy—A Heart Failure Society of America practice guideline. J Card Fail. 2009;15:83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Schroy PC, Lal SK, Wilson S, Heeren T, Farraye FA. Deficiencies in knowledge and familial risk communication among colorectal adenoma patients. J Clin Gastroenterol. 2005;39:298–302. doi: 10.1097/01.mcg.0000155129.31208.0d. [DOI] [PubMed] [Google Scholar]
- 29.Pho K, Geller A, Schroy PC. Lack of communication about familial colorectal cancer risk associated with colorectal adenomas. Cancer Causes Control. 11:543–546. doi: 10.1023/a:1008932417421. [DOI] [PubMed] [Google Scholar]
- 30.Boehmer U, Harris J, Bowen DJ, Schroy PC. Surveillance after colorectal cancer diagnosis in a safety net hospital. J Health Care Poor Underserved. 2010;21:1138–1151. doi: 10.1353/hpu.2010.0918. [DOI] [PubMed] [Google Scholar]
- 31.Christiaans I, Birnie E, Bonsel GJ, Wilde AAM, van Langen IM. Uptake of genetic counselling and predictive DNA testing in hypertrophic cardiomyopathy. Eur J Hum Genet. 2008;16:1201–1207. doi: 10.1038/ejhg.2008.92. [DOI] [PubMed] [Google Scholar]
- 32.Miller EM, Wang Y, Ware SM. Uptake of cardiac screening and genetic testing among hypertrophic and dilated cardiomyopathy families. J Genet Couns. 2013;22:258–267. doi: 10.1007/s10897-012-9544-4. [DOI] [PubMed] [Google Scholar]
- 33.Levy DE, Byfield SD, Comstock CB, Garber JE, Syngal S, Crown WH, et al. Underutilization of BRCA1/2 testing to guide breast cancer treatment: Black and Hispanic women particularly at risk. Genet Med. 2011;13:349–355. doi: 10.1097/GIM.0b013e3182091ba4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sherman KA, Miller SM, Shaw L-K, Cavanagh K, Gorin SS. Psychosocial approaches to participation in BRCA1/2 genetic risk assessment among African American women: a systematic review. J Community Genet. 2013;5:89–98. doi: 10.1007/s12687-013-0164-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vasan RS, Larson MG, Levy D, Evans JC, Benjamin EJ. Distribution and categorization of echocardiographic measurements in relation to reference limits. Circulation. 1997;96:1863–1873. doi: 10.1161/01.cir.96.6.1863. [DOI] [PubMed] [Google Scholar]
- 36.Cameron L, Leventhal EA, Leventhal H. Symptom representations and affect as determinants of care seeking in a community-dwelling, adult sample population. Health Psychol. 1993;12:171–179. doi: 10.1037//0278-6133.12.3.171. [DOI] [PubMed] [Google Scholar]
- 37.Bowen DJ, Albrecht T, Hay J, Eggly S, Harris-Wei J, Meischke H, et al. Communication among melanoma family members. J Health Commun. 2017;22:198–204. doi: 10.1080/10810730.2016.1259374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Europace. 2011;13:1077–1109. doi: 10.1093/europace/eur245. [DOI] [PubMed] [Google Scholar]
- 39.Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, et al. ClinGen — The Clinical Genome Resource. N Engl J Med. 2015;372:2235–2242. doi: 10.1056/NEJMsr1406261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–423. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruiz-Linares A, Adhikari K, Acuña-Alonzo V, Quinto-Sanchez M, Jaramillo C, Arias W, et al. Admixture in Latin America: geographic structure, phenotypic diversity and self-perception of ancestry based on 7,342 individuals. PLoS Genet. 2014;10:e1004572. doi: 10.1371/journal.pgen.1004572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bryc K, Durand EY, Macpherson JM, Reich D, Mountain JL. The genetic ancestry of African Americans, Latinos, and European Americans across the United States. Am J Hum Genet. 2015;96:37–53. doi: 10.1016/j.ajhg.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stroup WW. Generalized linear mixed models: modern concepts, methods and applications. Boca Raton: CRC Press; 2012. [Google Scholar]
- 46.Muller CJ, MacLehose RF. Estimating predicted probabilities from logistic regression: different methods correspond to different target populations. Int J Epidemiol. 2014;43:962–970. doi: 10.1093/ije/dyu029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Molenberghs G, Verbeke G. Models for discrete longitudinal data. New York: Springer; 2005. [Google Scholar]
- 48.Hayes RJ, Moulton LH. Cluster randomised trials. Boca Raton: CRC Press; 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.