

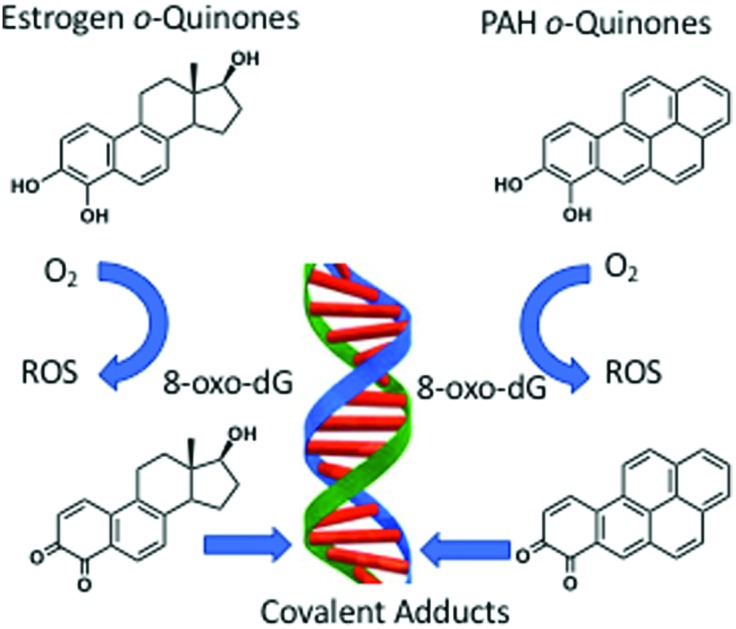
 o-Quinones are formed metabolically from natural and synthetic estrogens as well as upon exposure to polycyclic aromatic hydrocarbons (PAH) and contribute to estrogen and PAH carcinogenesis by genotoxic mechanisms.
o-Quinones are formed metabolically from natural and synthetic estrogens as well as upon exposure to polycyclic aromatic hydrocarbons (PAH) and contribute to estrogen and PAH carcinogenesis by genotoxic mechanisms.
Abstract
o-Quinones are formed metabolically from natural and synthetic estrogens as well as upon exposure to polycyclic aromatic hydrocarbons (PAH) and contribute to estrogen and PAH carcinogenesis by genotoxic mechanisms. These mechanisms include the production of reactive oxygen species to produce DNA strand breaks and oxidatively damaged nucleobases; and the formation of covalent depurinating and stable DNA adducts. Unrepaired DNA-lesions can lead to mutation in critical growth control genes and cellular transformation. The genotoxicity of the o-quinones is exacerbated by nuclear translocation of estrogen o-quinones by the estrogen receptor and by the nuclear translocation of PAH o-quinones by the aryl hydrocarbon receptor. The properties of o-quinones, their formation and detoxication mechanisms, quinone-mediated DNA lesions and their mutagenic properties support an important role in hormonal and chemical carcinogenesis.
1. Introduction
Quinones are aromatic compounds that contain conjugated diones within their structures. The dione may be either in the same ring (mononuclear quinones) or may be in different rings (polynuclear quinones). The arrangement of the dione in the mononuclear quinones can be p- or o- while the m-arrangement is less common.
Endogenous and exogenous estrogens can be metabolically activated to quinones which are involved in genotoxic mechanisms of estrogen carcinogenesis. The formation of quinones was first noted for the synthetic estrogen diethylstilbestrol in the Syrian hamster kidney, a site of estrogen carcinogenesis.1 Subsequently the formation of o-quinones was observed for estrone (E1) and 17β-estradiol (E2). Hydroxylation of E1 and E2 leads to the formation of the catechol estrogens: 2-hydroxy-estrone (E1)/estradiol (E2) and 4-hydroxy-E1/E22,3 which autoxidize to the o-quinones e.g. E1(E2)-2,3-dione and E1(E2)-3,4-dione. Xenoestrogens equilin and equilenin used in estrogen replacement therapy also exert their toxicological properties by metabolic activation to o-quinones,4,5 Fig. 1.
Fig. 1. Estrogen and PAH derived o-quinones.

Aromatic hydrocarbons are inert but can be converted to quinones in the atmosphere by oxy-radical attack of a carbon centered radical to form benzoquinone, naphthalene-1,2-dione, naphthalene-1,4-dione and phenanthrene-9,10-dione,6Fig. 2. Polycyclic aromatic hydrocarbons (PAH) that result from the incomplete combustion of fossil fuels and constituents of cigarette smoke, are suspect human carcinogens and are metabolically activated to quinones. The representative PAH, benzo[a]pyrene B[a]P is metabolized to mononuclear quinones e.g. B[a]P-7,8-dione7,8 and to polynuclear quinones e.g. B[a]P-1,6-dione, B[a]P-3,6-dione and B[a]P-6,12-dione.9,10
Fig. 2. Formation of PAH-quinones in the atmosphere.

Importantly, both E2 and B[a]P are classified as Group 1 known human carcinogens by the International Agency for Research on Cancer.11,12 The role of estrogen quinones in genotoxic mechanisms of breast and endometrial cancer and the role of PAH o-quinones in genotoxic mechanisms leading to lung cancer are thus of intense interest. This article will focus on the estrogen o-quinones and PAH o-quinones where distinct parallels exist in their physiochemical properties and genotoxicity.
2. Properties of o-quinones
A unique feature of the aromatic dione structure is its ability to exist in different redox states to shuttle electrons. o-Quinones can undergo two one electron reduction reactions. The first involves the formation of the o-semiquinone radical and the second involves the conversion of this radical to the catechol (hydroquinone). The reduction of the o-quinone to the catechol can be non-enzymatic and use reducing equivalents (NAD(P)H, Cu+, Fe2+) and or it can be enzymatic. Enzymatic reduction can involve two one electron steps catalyzed by NADPH-P450 oxidoreductase or it can involve a single two electron reduction step catalyzed by NAD(P)H quinone oxidoreductase (NQO1), aldo-keto reductases (AKRs) and by carbonyl reductases.13 The catechol if not intercepted by conjugation can autoxidize to the o-quinone in air with the concomitant generation of reactive oxygen species (ROS), Fig. 3. During autoxidation the initiating radical is superoxide anion radical and hydrogen peroxide is produced. In a second step, the o-semiquinone radical reacts with molecular oxygen to regenerate the o-quinone and superoxide anion radical to propagate subsequent steps of autoxidation.14 Hydrogen peroxide produced in this sequence undergoes Fenton chemistry in the presence of Fe2+ to produce the highly reactive hydroxyl radical (OH˙) and hydroxyl anion.
Fig. 3. Redox-cycling of quinones.

In an alternative sequence superoxide anion radical can react with nitric oxide to produce reactive nitrogen species (RNS). The initial product is peroxyntirite which in the presence of carbon dioxide can produce nitrosoperoxycarbonate which is a source of both nitrating and carbonylation radicals.15 Failure to intercept either the o-quinone or catechol leads to futile redox-cycles in which cellular reducing equivalents are depleted, ROS/RNS formation is amplified and the quinone/catechol are not consumed. Herein lies one major source of the toxicity of quinones.
Differences in the stability of estrogen catechols and PAH-catechols exist. Whereas estrogen catechols are stable and often require oxidants to produce estrogen o-quinones the reverse is true of the PAH-catechols where the PAH o-quinones are the more stable species. This difference is likely a reflection of the stabilization of the dione structure by the extensive aromatic system of the PAH.
The interconversion of the catechol via an o-semiquinone radical to produce a quinone, represents only one of three possible transitions. For example, the oxidation of a catechol to an o-quinone can occur at the level of the protonated catechol, the catechol itself or at the level of the catecholate anion, making nine possible transitions in all, Fig. 4. Which route occurs will be governed by the pH and pKa of the respective intermediates and the microenvironment which may alter the pKa of the intermediates. Furthermore, the o-semiquinone radical can often be stabilized by resonance of the unpaired electron around the aromatic ring system to give rise to a persistent organic radical.
Fig. 4. Catechol-o-quinone transitions.

o-Quinones also rely on their electrophilicity for their toxicity. As the dione is in extended conjugation they are Michael acceptors and that can undergo 1,4- and 1,6-Michael addition reactions. As a result they will react with cellular nucleophiles e.g. GSH, proteins (reaction with Cys and Lys residues),16,17 as well as nucleobases in RNA and DNA to from nucleoside-adducts.18,19 This can lead to loss or disruption of protein function and mutagenic lesions in DNA, respectively.
3. Metabolic formation of quinones and their detoxication
Endogenous estrogens (E1 and E2), as well as exogenous estrogens (equilin and equilenin) are activated by hydroxylation by P450 isozymes (P4501A1 and P4501B1) to the corresponding 2,3- and 3,4-catechols which then autoxidize to the corresponding o-quinones (see previous section).20 The major metabolite of equilin, 4-hydroxyequilin not only autoxidizes to the o-quinone but also isomerizes to yield 4-hydroxyequilenin.4
Compounds such as diethylstilbestrol can act as co-reductants of the peroxidase reaction to yield phenoxy radicals which upon further oxidation yield the corresponding polynuclear quinones.21 In this sequence compound I perferryl-oxygen (Fe–V O) requires two one electrons to return to the Fe(iii) resting state. By a similar mechanism B[a]P can also act as co-reductant of the peroxidase reaction to produce a C6-radical cation which is then the precursor of the polynuclear quinones, B[a]P-1,6-dione, B[a]P-3,6-dione and B[a]P-6,12-dione.9,22 Peroxidases capable of these reactions include P450 isozymes, prostaglandin H synthase, tyrosinase and horse-radish peroxidase.22–27
The formation of the PAH o-quinones is unique in that the precursors are the trans-dihydrodiol proximate carcinogens formed by the hydration of non-K-region arene oxides catalyzed by the sequential action of P4501A1/1B1 and epoxide hydrolase.10,28 trans-Dihydrodiols are oxidized by the NAD(P)+ dependent dihydrodiol dehydrogenase activity of human aldo-keto reductases (AKR1A1 and AKR1C1-AKR1C4) to yield ketols,29,30 which rearrange to the corresponding catechols, which autoxidize to the PAH o-quinone with the concomitant production of ROS.14
Catechol estrogens and PAH catechols can be detoxified by conjugation reactions to prevent their futile redox-cycling and ROS generation. This can be achieved by O-methylation catalyzed by catechol-O-methyl transferase,31–33 and by conjugation mediated by either sulfotransferases34,35 and by uridine diphosphate glucurosonyl transferases.36o-Quinones are also highly reactive with thiols and can be scavenged non-enzymatically by GSH16,17 and enzymatically by glutathione-S-transferases to form GSH conjugates.37 Even though 4-hydroxyequilenin is autoxidized to the o-quinone and readily reacts with GSH it can undergo a reverse Michael addition to regenerate the catechol to activate the estrogen receptor.38 Even in the case of B[a]P-7,8-dione, where a high second order rate constant for the formation of GSH conjugates exists,17 B[a]P-7,8-dione-GSH conjugates are not major metabolites observed in human lung cells.39
4. Oxidative and covalent DNA damage mediated by o-quinones
A large number of DNA adducts/lesions can arise from quinones which can be divided into those that come from ROS/RNS40–43 and those that result from covalent modification of the DNA by the quinone,18,19,44 Fig. 5. Each of these DNA lesions is discussed below with examples from the estrogen-o-quinone and PAH-quinone field.
Fig. 5. Spectrum of DNA adducts formed by o-quinones.

4.1. DNA strand breaks
DNA-strand breaks can occur when estrogen o-quinones and PAH o-quinones encounter DNA. This can be facilitated by the planar o-quinones which can intercalate between the bases in the double-helix of DNA. DNA strand scission could be mediated by either the o-semiquinone radical or ROS. PAH o-quinones in the presence of NADPH and Cu2+ cause DNA strand scission and produce an o-semiquinone radical detectable by EPR.45 Although the o-semiquinone radical could be quenched by the addition of DNA, strand scission was dependent upon a Cu(i)/Cu(ii) mediated redox cycle of the quinone that was prevented by Cu(i) chelators and OH˙ scavengers. Strand scission was accompanied by malondialdehyde formation which was also prevented by the same scavenging agents.45 These data are consistent with a hydroxyl radical initiated Criege rearrangement.46 In this sequence, a hydroxyl radical removes a hydrogen atom from the C4-position of the deoxyribose ring leading to the formation of a carbon centered radical which is attacked by molecular oxygen leading to a C4′-peroxy-deoxyribose intermediate, Fig. 6. Following ring expansion, and hydroxyl anion addition there is 3′ and 5′-cleavage of the phosphodiester linkage creating a strand break and the formation of a base propenal which hydrolyzes to yield malondialdehyde. Malondialdehyde is often cited in the literature as being a biomarker of lipid peroxidation but in this instance its source is from the deoxyribose moiety. A summary of oxidative DNA-lesions observed experimentally with o-quinones is given in Table 1.
Fig. 6. Criege rearrangement and DNA strand scission.

Table 1. Detection of oxidative DNA damage by o-quinones.
| Quinone | Treatment conditions | Lesion Type | Method of Detection | Reference |
| 4-OHEN | 4-OHEN + Sprague-Dawley rat mammary gland | DNA strand scission | Comet assay | Zhang et al.40 |
| 4-OHEN | 4-OHEN + Sprague-Dawley rat mammary gland | 8-Oxo-dG and 8-oxo-dA | SID-LC/MS/MS | Zhang et al.40 |
| 4-OHEN | 4-OHEN + S30 human breast cancer cells | 8-Oxo-dG | SID-LC/MS/MS and Comet assay coupled with Fpg | Wang et al.47 |
| 4-OHEN | 4-OHEN + Calf thymus DNA + NADPH + CuCl2 | 8-Oxo-dG and apurinic sites/apyridiminic sites | SID-LC/MS/MS and aldehyde reactive probe | Wang et al.47 |
| BPQ | BPQ + NADPH + CuCl2 + poly-dG:poly-dC or phage DNA | DNA strand scission | Electrophoresis | Flowers et al.45 |
| BPQ | BPQ + NADPH + CuCl2 + calf thymus DNA | Apurinic sites/apyridiminic sites | Aldehyde reactive probe | Park et al.41 |
| 8-Oxo-dG | EC-HPLC | |||
| BAQ | BAQ+ NADPH + CuCl2 + calf thymus DNA | Apurinic sites/apyridiminic sites | Aldehyde reactive probe | Park et al.41 |
| 8-Oxo-dG | EC-HPLC | Seike et al.58 | ||
| DMBAQ | DMBAQ + NADPH + CuCl2 + calf thymus DNA | Apurinic sites/apyridiminic sites | Aldehyde reactive probe | Park et al.41 |
| 8-Oxo-dG | EC-HPLC | |||
| BPQ | BPQ + NADPH + CuCl2 + p53 plasmid DNA | 8-Oxo-dG | EC-HPLC | Park et al.49 |
| BAQ | BAQ + NADPH + CuCl2 + p53 plasmid DNA | 8-Oxo-dG | EC-HPLC | Park et al.49 |
| DMBAQ | DMBAQ + NADPH + CuCl2 + p53 plasmid DNA | 8-Oxo-dG | EC-HPLC | Park et al.49 |
| BPQ | BPQ + H358 human lung epithelial cells | 8-Oxo-dG | SID-LC/MS/MS | Mangal et al.82 |
| Comet assay coupled to OGG1 | ||||
| BPQ | B[a]P-7,8-trans-dihydrodiol + A549 cells ± COMT inhibitor | 8-Oxo-dG | SID-LC/MS/MS | Park et al.96 |
| Comet assay coupled to OGG1 |
4.2. Oxidatively damaged nucleobases
At least 22 different oxidatively damaged DNA bases can result from ROS generated in the redox-cycling of quinones.46 The most common of these is 8′-hydroxy-dG or 8-oxo-dG (8-oxo-7,8-dihydro-2′-deoxygauosine). Evidence exists that estrogen o-quinones47,48 and a variety of PAH-o-quinones produce 8-oxo-dG41,49,50 in variety of in vitro and cell-based systems, see Table 1.
8-Oxo-dG can exist in a number of tautomeric forms. Of these the 6-keto-8-enol is the most common and is formed as follows. Initial attack of the C8 carbon of dG by OH˙ produces a reducing species which after losing an electron gives rise to the 6-keto-8-enol form. Tautomerization of the initial reducing adduct can also lead to ring opening to yield the formamidopyrimidine (FAPY)G adduct, Fig. 7. This creates the concept that 8-oxo-dG is a “fleeting” species since it is an intermediate to other adducts. Apart from FAPYG adducts, 8-oxo-dG can be the precursor of spiroiminohydantoin and 5-guandionhydantoin lesions,51Fig. 8. Although there is no evidence that quinone exposure under redox cycling conditions will lead to the formation of spiroiminohydantoin and 5-guandiohydantoin DNA lesions this is anticipated.
Fig. 7. Formation of 8-oxo-dG by hydroxyl radical or singlet oxygen.

Fig. 8. Spiroiminohydantoin and 5-guandinohydantoin lesions.

While OH˙ radical is the species that gives rise to 8-oxo-dG via Fenton chemistry, other oxidants can give rise to this lesion when the transition metal is different from Fe2+.50 For example, when Cu2+ is used a Cu(i)(OOH) can form which is the precursor of singlet oxygen. Cadet has shown that a 4,8-endoperoxide is a key-intermediate in 8-oxo-dG formation from singlet oxygen, Fig. 6.52 The distinction between Fenton chemistry and singlet oxygen mediated 8-oxo-dG formation was made using B[a]P-7,8-dione using different scavenging agents. For example, e.g. bathcuproine (Cu(i) scavenger); catalase, and methional (a Cu(i)OOH scavenger) abolished 8-oxo-dG formation in the presence of B[a]P-7,8-dione, NADPH and CuCl2. By contrast mannitol, sodium benzoate and sodium formate (OH˙ scavengers) had no effect. Sodium azide (a scavenger of both OH˙ and O21) abolished 8-oxo-dG formation confirming the role of singlet oxygen.50
Not all “G's” are equally prone to oxidation. Studies show that runs of “GG” residues are more prone to 8-oxo-dG formation and this has been documented on a genome-wide basis using whole genome next generating sequencing in OGG1 (oxoguanine glycosylase) knockout cells.53 The formation of 8-oxo-dG can also have epigenetic outcomes when they form on 5′-CpG′′ islands. When the cytosine is present as 5′-methylcytosine the 5′-MeCp8-oxo-G′′ can disrupt the binding of methyl-DNA binding proteins to enhance gene transcription.54
An additional consequence of ROS formation by o-quinones is the generation of lipid peroxides which decompose to reactive lipid aldehydes. Lipid aldehydes such as 4-hydroperoxy-2-nonenal can form etheno-dG adducts and 4-oxo-2-nonenal can form heptanone-etheno-dG and hetanone-etheno-dC adducts.55 The heptanone-etheno-dC adduct has a high frequency of miscoding upon translesional synthesis.56 Evidence that etheno- or haptanone-dG adducts from as a result of o-quinone mediated ROS production remains to be reported.
The major lesions produced by peroxynitrite reactions with dG include 8-nitro-2′-deoxyguanosine (8-nitro-dG) which readily depurinates to release 8-nitro-G, 5-guanidino-4-nitroimidazole, and 2,2-diamino4-[2(2-deoxy-B-d-erythro-pentofuranosy)amino]-5(2H)-oxazolone, however, no attempts have been made to detect these lesions following DNA exposure to o-quinones.57
Oxidatively generated DNA lesions mediated by o-quinones have been observed with 4-hydroxyequilenin in vitro in calf thymus DNA, in estrogen receptor positive breast cancer cells and in the mammary gland of Sprague-Dawley rats.40,47 Similarly, oxidatively damaged DNA lesions caused by B[a]P-7,8-dione have been observed in ds-oligonucleotides, in calf thymus DNA, plasmid DNA and human lung epithelial cells.41,49,50,58
4.3. Repair mechanisms for oxidatively damaged nucleobases
ROS-derived lesions are repaired by base-excision repair (BER). 8-Oxo-G mis-pairs with A during replication because of Hoogstein-base pair formation. In this instance, the 8-oxo-G base rotates from its anti- to syn-conformation to from hydrogen bonds with adenine. Monofunctional glycosylases (MutY) reverse the adenine mismatch to give rise to an abasic site.59 The abasic site is repaired by apurinic endonuclease (AP1). Whereas translesional synthesis by DNA polymerase alpha inserts an “A” opposite the abasic site.60 In a second mechanism, the bifunctional glycosylase MutM (OGG1 in humans), removes 8-oxo-dGuo leading to DNA strand scission that is repaired by AP1 endonuclease, DNA polymerase and DNA ligase.61 A separate group of glycosylases, NEIL 1 and NEIL 2 repair the spiroiminohydantoin and 5-guandionhydantoin lesions.62,63
4.4. Mutations due to oxidative DNA damage
8-Oxo-dGuo if unrepaired, is a mutagenic lesion64,65 As a result of mis-pairing with A during replication the daughter-strand, will give rise to a G to T transversion. Alternatively, when the A mismatch is repaired by MutY to give rise to an abasic site failure to repair this damage will result in the preferential insertion of A opposite the abasic site so that upon replication a C to T transition would result.60
5. Covalent quinone-DNA adducts repair and mutagenesis
Two forms of covalent DNA adducts can result from the reaction of either estrogen-o-quinones66–68 or PAH o-quinones with DNA.69,70 The first involves attack by the N7 position of dG or dA resulting in a labile N-glycosidic bond which is hydrolyzed to yield a depurinating quinone-N7 guanine or quinone-N7-adenine adduct, Fig. 9. Depurinating N7-guanine and N3-adenine adducts have been reported for the E2-2,3-dione and the E2-3,4-dione.71–73 The resultant abasic site that arises from the N7 gaunine depurinating adduct can give rise to a G to T transversion and the abasic site that arises from the N3 adenine depurinating adduct can give rise to an A to T transversion60 The formation of the estrogen o-quinone and PAH o-quinone depurinating DNA adducts has been readily observed in in vitro reactions performed in acetic acid and water which favors hydrolysis of the N-glycosidic bond. However, it remains doubtful as to whether depurinating DNA adducts are the major lesions responsible for quinone mediated mutation.74 A summary of the covalent o-quinone DNA adducts detected in various experimental systems is given in Table 2.
Fig. 9. Formation of depurinating quinone adducts.

Table 2. Formation of covalent o-quinone-DNA adducts.
| Quinone | Treatment Conditions | Structure of Adduct | Method of Detection | Reference |
| Estrone-2,3-dione | 2-OHE1 + MnO2 + dG + acetic acid/water | 2-OH-E1-6-N2-dG | NMR | Stack et al.72 |
| 2-OHE1 + MnO2 + dA + acetic acid/water | 2-OH-E1-6-N6-dA | |||
| Estradiol-2.3-dione | 2-OHE2 + MnO2 + Ade + acetic acid/water | 2-OH-E2-6-N3-adenine | NMR | Zahid et al.71 |
| 2-OHE2 + peroxidase + Calf thymus DNA | 2-OH-E2-6-N3-adenine | HPLC | ||
| 32P-post-labeling | ||||
| Estrogen-3,4-dione | 4-OHE1/E2 + MnO2 dG + acetic acid/water | 4-OH-E1-1-N7-guanine | NMR | Stack et al.72 |
| 4-OH-E2-1-N7-guanine | ||||
| Estrogen-3,4-dione | 4-OHHE2/E1 + MnO2 + Ade + acetic acid/water | 4-OH-E1(E2)-1-N3-adenine | NMR | Zahid et al.71 |
| 4-OHE2/E1 + Peroxidase + Calf thymus DNA | 4-OH-E1(E2)-1-N3-adenine | NMR | Li et al.73 | |
| 4-OHE2 or E-3,4-dione | 4-OH-E1(E2)-1-N7-guanine | LC-MS | ||
| ACI Rat Mammary Gland | 4-OH-E2-1-N3-adenine | HPLC | ||
| 4-OH-E2-1-N7-guanine | 32P-post-labeling | |||
| 4-OHEN | 4-OHEN in MetOH | 2-N1,3-(N2-dG)-1,3-Dihydroxy-5,7,9(10)-estratriene-4,17-dione. | 1H-NMR, 13C-NMR and LC/MS | Shen et al.18 |
| dG, dA or dC | 1-N6,3-C2-dA-2,3-Dihydroxy-5,7,9(10)-estratriene-4,17-dione | |||
| KPO4 buffer pH 7.4, 37 °C for 7 h or Calf-thymus DNA | 1-N3,3-(N4-dC)-2,3-Dihydroxy-5,7,9(10)-estratriene-4,17-dione. | |||
| 4-OHEN | 4-OHEN or 4-OHEN-diacetate + MCF7 cells | 1-N6,3-C2-dA-2,3-Dihydroxy-5,7,9(10)-estratriene-4,17-dione | SID-LC-MS/MS | Wang76 |
| N3,3-(N4-dC)-2,3-Dihydroxy-5,7,9(10)-estratriene-4,17-dione. | ||||
| 4-OHEN | 4-OHEN + SD rat mammary gland injection | 2-N1,3-(N2-dG)-1,3-Dihydroxy-5,7,9(10)-estratriene-4,17-dione | LC-MS/MS | Zhang et al.40 |
| 1-N6,3-C2-dA-2,3-Dihydroxy-5,7,9(10)-estratriene-4,17-dione | ||||
| NQ | dG/dA acetic acid/water/DMF | 1,2-Dihydroxy-naphthalene-4-N7-gaunine | UPLC/MS | Saeed et al.69 |
| dG/acetic acid : water | 1,2-Dihydroxy-naphthalene-4-N3-adenine | UPLC/MS | Saeed et al69 | |
| Naphthalene-1,2-dion-4-yl-N7-guanine | NMR/MS | McCoull et al.70 | ||
| PQ | dG/acetic acid : water or Calf Thymus DNA | Phenanthrene-1,2-dion-4-yl-N7-guanine | NMR/MS | McCoull et al.70 |
| BPQ | dG /acetic acid : water | Benzo[a]pyrene-7,8-dion-10-yl-N7-guanine | NMR/MS | McCoull et al.70 |
| BPQ | dG or dA/DMF/55 °C/pH 7.0 | 10-(N2-dG)-9,10-dihydro-9-hydroxy-B[a]P-7,8-dione | NMR | Balu et al.19 |
| 8-N1,9-(N2-dG)-8,10-Dihydroxy-9,10-dihydro-B[a]P-7(8H)-one | ||||
| 8-N6,10-N1-dA-8,9-Dihydroxy-9,10-dihydro-B[a]P-7(8H)-one | ||||
| BPQ | A549 cells and HBEC-tk cells incubated with 2 μM B [a]P-7,8-dione in HBSS + pyruvate | 6-(N2-dG)-9,10-Dihydro-10-hydroxy-B[a]P-7,8-dione | LC-MS/MS | Huang et al.44 |
| 10-(N1-dG)-9,10-Dihydro-9-hydroxy-B[a]P-7,8-dione | LC-MS/MS | Huang et al.44 | ||
| 10-(N6-dGA)-9,10-Dihydro-9-hydroxy-B[a]P-7,8-dione | LC-MS/MS | Huang et al.44 | ||
| 10-(N3-dA)-9,10-Dihydro-9-hydroxy-B[a]P-7,8-dione | LC-MS/MS | Huang et al.44 | ||
| 10-(N3-dA)-B[a]P-7,8-dione | LC-MS/MS | Huang et al.44 |
The second form of covalent adducts that form from o-quinones are the stable bulky adducts that result from either o-semiquinone radical attack of nucelobases or 1,4- or 1,6-Michael addition of the N2, N6, or N4-exocyclic amino group of dG, dA or dC, respectively.18 Many of these adducts undergo subsequent facile reactions involving hydration and cyclization.
Stable covalent DNA adducts have been described for equilenin-3,4-dione involving addition of the N4-, N6- and N2-exocyclic groups of dC, dA and dG, respectively where subsequent cyclization and hydration of the adducts was observed, Fig. 10.18,75 These stable adducts have been observed in calf-thymus DNA, in MCF7 human breast cancer cells and the mammary gland of Sprague-Dawley rats.18,40,76
Fig. 10. Formation of stable cyclic estrogen o-quinone DNA adducts: 4-OHEN-dC = N3,3-(N4-dC)-2,3-dihydroxy-5,7,9(10)-estratriene-4,17-dione; 4-OHEN-dA = 1-N6,3-C2-dA-2,3-dihydroxy-5,7,9(10)-estratriene-4,17-dione; 4-OHEN-dG = N1,3-(N2-dG)-1,3-dihydroxy-5,7,9(10)-estratriene-4,17-dione.

Stable covalent [3H]-B[a]P-7,8-dione-DNA adducts have been observed in reactions with deoxyribonucleosides, calf thymus and plasmid DNA, where a single dG adduct was detected but a structure was not assigned.77 In reactions of B[a]P-7,8-dione with deoxyribonucleosides at physiologic pH, in the presence of 50% DMF and 50 °C heat, formation of 1,4- and 1,6-Michael addition adducts occurred which subsequently underwent hydration and cyclization reactions to yield complex adducts, 1–6 19 similar to those observed with 4-hydroxyequilenin,18Fig. 11. However, a different set of stable B[a]P-7,8-dione-DNA adducts were observed in human lung cells, 7–11.44 In each case the stereochemistry of the adducts observed remains unresolved. In the A/J mouse model of lung carcinogenesis, stable covalent B[a]P-7,8-dione-DNA adducts could not be detected using [32P]-post-labeling when either the B[a]P-7,8-trans-dihydrodiol precursor or B[a]P-7,8-dione was administered in vivo.78,79 However, it is unknown if the A/J mouse lung contains the relevant AKRs to metabolically convert B[a]P-7,8-trans-dihydrodiol to B[a]P-7,8-dione. Also, since B[a]P-7,8-dione has poor bioavailability the studies should have employed B[a]P-7,8-catechol diacetate. Thus this negative outcome is not conclusive. Stable B[a]P-7,8-dione adducts have been observed in reactions with dG, calf thymus DNA, and in A549 and human bronchial epithelial cells.19,44,77
Fig. 11. Formation of stable cyclic PAH o-quinone DNA adducts.

5.1. Repair of covalent quinone-DNA adducts
The bulky quinone DNA adducts are anticipated to be repaired by nucleotide excision repair (NER) using global genomic repair or transcription coupled repair. If unrepaired the stable, bulky DNA lesions can lead to mutation by aberrant translesional bypass synthesis.80 NER and translesional bypass polymerase reactions have yet to be performed with either estrogen-o-quinone DNA adducts or PAH o-quinone–DNA adducts. This work has lagged behind because of either the lack of elucidation of the relevant stereoisomeric structures of the adducts or the difficulty in synthesizing adducts in oligonucleotides that match those formed in cells. For PAH o-quinones, overwhelming evidence suggests that oxidative DNA damage produced under redox-cycling conditions are the major DNA lesions formed.81
6. Challenges in measuring DNA adducts derived from quinone exposure
6.1. Detection of oxidative-DNA damage
Challenges exist in the detection and quantitation of 8-oxo-dG and the covalent quinone DNA adducts. State-of-the art approaches should employ stable-isotope dilution liquid chromatography tandem mass spectrometry, in which there is quantitative digestion of the DNA to the constituent deoxyribonucleosides so that adducts can be reported as adducts/107 deoxyribonucleosides. The method also entails the use of an internal standard preferably labeled with 15N5 so that both the adduct and the internal standard are chemically identical except for the incorporation of the stable isotope. The presence of the internal standard permits losses during the work up of the sample and recovery to be accounted for. Adduct identity is verified by correct retention time on liquid chromatography, correct MH+, and correct precursor-product mass transition, (usually the loss of deoxyribose m/z = 116) and quantified with respect to the internal standard recovered.
The quantitation of 8-oxo-dG can present additional challenges since dG undergoes adventitious oxidation to 8-oxo-dG during work up of the DNA sample, and unless precautions are taken to exclude transition metals from the buffers, values as high as 37.0 8-oxo-dG/107 dG can be observed when the true value should be closer to 3.0 8-oxo-dG/107-dG. A SID-LC-MS/MS method has been described which gives the expected baseline level of 8-oxo-dG and involves immunoaffinity purification of the adduct prior to LC-MS/MS.82 The method was subsequently modified to include two internal standards; 15N5-8-oxo-dG and 13C5-15N5-dG.83 The latter standard if converted to 8-oxo-dGuo during work up would give an accurate assessment of 8-oxo-dG formed adventitiously.
Other methods to detect 8-oxo-dG include the Comet assay coupled with either hOOG or FAPYG glycosylase, where DNA strand breaks are due to 8-oxo-dG formation.82 In addition, EC-HPLC,5032P-post-labeling, and immunohistochemistry approaches have been used.84 In each instance, these methods often lack the requisite specificity to ensure that 8-oxo-dG is being measured.
Apurinic/apyrimidinic sites that result from the repair of 8-oxo-dG or formation of covalent depurinating-DNA adducts, can be detected using the aldehydic probe (ARP).85 The ARP forms a Schiff's base with the aldehydic site and the biotin-tag binds to a streptavidin horseradish peroxidase conjugate. This approach has been used in several studies using 4-hydroxyequilenin and PAH o-quinones to detect oxidative DNA damage, see Table 1. However, this method will detect any aldehydic site in DNA.
6.2. Detection of covalent quinone DNA adducts
Stable-isotope dilution liquid chromatography tandem mass spectrometric assays for 4-hydroxyequilenin-DNA adducts have been established to detect covalent adducts in MCF7 cells. Using either 4-hydroxyequilenin or its catechol diacetate, four stereoisomeric isomers of cyclic hydrated 4-hydroxyequilenin N6-dA and 4-hydroxyequilenin N4-dC adducts were observed.76
Bulky Stable adducts of B[a]P-7,8-dione were synthesized with deoxyribonucleotides as standards for the 32P-post-labeling assay,79 however, using this approach the adduct standards only identified a fraction of the B[a]P-7,8-dione adducts formed in calf thymus DNA79 These adducts were not detected in A/J mouse lung treated with B[a]P-7,8-dione or its precursor B[a]P-7,8-trans-dihydrodiol.78 Stable-isotope dilution liquid chromatography tandem mass spectrometric assays for quantitation of B[a]P-7,8-dione DNA adducts have yet to be established.
7. Quinones as mutagens
Few studies have been performed to examine the mutagenicity of o-quinones in the Ames test. No studies have been performed with the catechol estrogens. Seven PAH o-quinones were tested as direct-acting mutagens in Salmonella typhimurium tester strains TA97a, TA98, TA100, TA102 and TA104. They were found to be more mutagenic than the test mutagens used for each tester strain, and were predominantly frameshift mutagens. The presence of an activating system (Aroclor-induced rat liver S9 plus NADPH) did not increase the mutagenicity of o-quinones in tester strains that were sensitive to oxidative mutagens (TA102 and TA104) suggesting that NADPH-P450 oxidoreductase activation of the o-quinone to an o-semiquinone radical did not occur.86
Using a yeast p53 reporter gene assay studies on the mutagenic potency of PAH o-quinones revealed that they were only mutagenic under redox-cycling conditions (NADPH and CuCl2) that would favor 8-oxo-dG formation.81 PAH o-quinones were not mutagenic in the absence of redox cycling suggesting that the major mutagenic lesions were related to oxidative DNA damage. Under redox-cycling conditions PAH o-quinones were effective at sub-micromolar concentrations and produced a large number of revertant p53 colonies. Sequencing of the mutated p53 showed that the mutation pattern was predominately G to T transversions consistent with 8-oxo-dG formation. Detection of 8-oxo-dG in the treated plasmid DNA showed a linear correlation between the number of 8-oxo-dG/dG and the number of revertants, Fig. 12.49 Examination of the p53 mutational spectrum showed that the codons mutated were randomly distributed through the DNA-binding domain of p53. However, when the revertant colonies were selected for dominance, the mutational spectrum showed that the codons mutated were those now seen in the tumors of lung cancer patients.49 These results suggest that 8-oxo-dGuo lesions derived from PAH-o-quinones can account for the mutational spectrum seen in lung cancer and that biological selection plays a more important role that sequence context for this lesion.
Fig. 12. p53 mutation by PAH o-quinones.

8. Nuclear translocation of o-quinones
Estrogen o-quinones and PAH o-quinones are products of microsomal and cytosolic metabolism, respectively but to exert their genotoxicity they must enter the nucleus. Bolton and coworkers were the first to show that estrogen o-quinones can be shuttled to the nucleus to form DNA-adducts by the ER, and this was referred to as the “Trojan-horse” hypothesis.48 Subsequently, it was found that planar PAH o-quinones can act as ligands for the aryl-hydrocarbon receptor and this can shuttle PAH o-quinones to the nucleus to cause 8-oxo-dG formation. This hypothesis was verified by demonstrating that the amount of 8-oxo-dG detected in murine Hepa1 cells by B[a]P-7,8-dione was dramatically reduced in Hepa1 cells deficient in either AhR or ARNT.87
9. Quinones and the initiation of cancer
Quinones derived from endogenous and exogenous estrogens likely play a role in the initiation of breast cancer by the formation of oxidative and stable covalent DNA adducts. Oxidized DNA and stable covalent lesions due to 4-hydroxyequilein have been detected in the mammary glands of Sprague-Dawley rats40 and in human mammary MCF7 cells,76 and cause anchorage independent growth in these cells.88 Detection of repaired estrogen-o-quinone DNA adducts could be valuable intermediate biomarkers of cancer risk. Depurinating catechol-estrogen DNA adducts in human serum have been used as biomarkers to discriminate between women of average risk of getting breast cancer versus those at high risk for breast cancer in women with newly diagnosed breast cancer.89 However, this study requires replication and validation. Collectively, these studies support the role of a genotoxic mechanism for estrogen carcinogenesis in the breast cancer.
PAH o-quinones likely play an important role in lung carcinogenesis. AKRs responsible for their formation are among the mostly highly expressed genes in non-small cell lung cancer90 and are overexpressed in human lung epithelial cells and buccal cells exposed to cigarette smoke and belong to the smoking gene battery.91–94 The contribution of AKRs to the metabolic activation of PAH has been assessed using SID-LC-MS/MS methods to measure the B[a]P metabolome. It was estimated that formation of PAH o-quinones and PAH diol-epoxides contribute equally to B[a]P metabolism.95 The pathway of B[a]P-7,8-trans-dihydrodiol oxidation, B[a]P-7,8-dione formation and consequential production of 8-oxo-dG has been demonstrated in human lung A459 adenocarcinoma cells.96 Additionally, B[a]P-7,8-dione under redox-cycling conditions mutated p53 to give a mutational pattern and spectrum reminiscent of that seen in human lung cancer.49,81 Collectively, these studies support the role of PAH o-quinones in human lung chemical carcinogenesis.
10. Conclusions
Understanding mechanisms of estrogen and PAH-induced hormonal and lung induced carcinogenesis can lead to new approaches to determining cancer risk. Identification of the genes involved in the formation and detoxication of estrogen o-quinones and PAH o-quinones allows assessment of the role of polymorphic variants of these genes in cancer susceptibility. In addition, understanding how these genes are regulated could provide routes to chemoprevention strategies. Identification of the DNA-lesions that are specific for the estrogen o-quinones and the PAH o-quinones can lead to the development of assays to detect the repaired adducts in serum or urine as intermediate cancer biomarkers.
Conflicts of interest
Dr Penning is a member of the Expert Panel for the Research Institute of Fragrance Materials.
Note added after first publication
This article replaces the version published on 13th September 2017, which contained errors in Table 1.
Acknowledgments
TMP is supported in part by grant P30-ES013508 from the National Institute of Environmental Health Sciences.
Biography

Trevor M. Penning
Trevor M. Penning, PhD, is the Thelma Brown and Henry Charles Molinoff Professor of Pharmacology, and Director of the Center of Excellence in Environmental Toxicology at the University of Pennsylvania. He obtained his PhD in Biochemistry with Professor M. Akhtar F.R.S. at Southampton University, UK and conducted postdoctoral training with Professor Paul Talalay at Johns Hopkins University. He joined the faculty at UPenn in 1982. He is internationally recognized for his research on how hormones and chemicals cause cancer. He is a member of The Johns Hopkins Society of Scholars, and Fellow of the American Chemical Society.
References
- Roy D., Liehr J. G. Carcinogenesis. 1989;10:1241–1245. doi: 10.1093/carcin/10.7.1241. [DOI] [PubMed] [Google Scholar]
- Purdy R. H., Moore Jr. P. H., Williams M. C., Goldzieher J. W., Paul S. M. FEBS Lett. 1982;138:40–44. doi: 10.1016/0014-5793(82)80390-6. [DOI] [PubMed] [Google Scholar]
- Ball P., Knuppen R. J. Clin. Endocrinol. Metab. 1978;47:732–737. doi: 10.1210/jcem-47-4-732. [DOI] [PubMed] [Google Scholar]
- Zhang F., Chen Y., Pisha E., Shen L., Xiong Y., van Breemen R. B., Bolton J. L. Chem. Res. Toxicol. 1999;12:204–213. doi: 10.1021/tx980217v. [DOI] [PubMed] [Google Scholar]
- Shen L., Pisha E., Huang Z., Pezzuto J. M., Krol E., Alam Z., van Breemen R. B., Bolton J. L. Carcinogenesis. 1997;18:1093–1101. doi: 10.1093/carcin/18.5.1093. [DOI] [PubMed] [Google Scholar]
- Pitts J. N., Cauwenberghe V. K. A., Grosjean D., Schmid J. P., Fitz D. R., Belser W. L., Knudson G. P., Hynds P. M. Science. 1978;202:515–519. doi: 10.1126/science.705341. [DOI] [PubMed] [Google Scholar]
- Smithgall T. E., Harvey R. G., Penning T. M. J. Biol. Chem. 1988;263:1814–1820. [PubMed] [Google Scholar]
- Smithgall T. E., Harvey R. G., Penning T. M. J. Biol. Chem. 1986;261:6184–6191. [PubMed] [Google Scholar]
- Lorentzen R. J., Ts'o P. O. Biochemistry. 1977;16:1467–1467. doi: 10.1021/bi00626a035. [DOI] [PubMed] [Google Scholar]
- Gelboin H. V. Physiol. Rev. 1980;60:1107–1166. doi: 10.1152/physrev.1980.60.4.1107. [DOI] [PubMed] [Google Scholar]
- IARC, Some Non-Heterocyclic Polycyclic Aromatic Hydrocarbons and Some Related Exposures, IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, World Health Organization, Lyon France, 2010, vol. 92. [PMC free article] [PubMed] [Google Scholar]
- IARC, Hormonal Contraception and Post-menopausal Hormonal Therapy, IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, World Health Organization, Lyon, France, 1999, vol. 72. [Google Scholar]
- Shultz C. A., Quinn A. M., Park J. H., Harvey R. G., Bolton J. L., Maser E., Penning T. M. Chem. Res. Toxicol. 2011;24:2153–2166. doi: 10.1021/tx200294c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penning T. M., Ohnishi S. T., Ohnishi T., Harvey R. G. Chem. Res. Toxicol. 1996;9:84–92. doi: 10.1021/tx950055s. [DOI] [PubMed] [Google Scholar]
- Uppu R. M., Pryor W. A. Biochem. Biophys. Res. Commun. 1996;229:764–769. doi: 10.1006/bbrc.1996.1878. [DOI] [PubMed] [Google Scholar]
- Murty V. S., Penning T. M. Bioconjugate Chem. 1992;3:218–224. doi: 10.1021/bc00015a003. [DOI] [PubMed] [Google Scholar]
- Murty V. S., Penning T. M. Chem.-Biol. Interact. 1992;84:169–188. doi: 10.1016/0009-2797(92)90077-x. [DOI] [PubMed] [Google Scholar]
- Shen L., Qiu S., Chen Y., Zhang F., van Breemen R. B., Nikolic D., Bolton J. L. Chem. Res. Toxicol. 1998;11:94–101. doi: 10.1021/tx970181r. [DOI] [PubMed] [Google Scholar]
- Balu N., Padgett W. T., Lambert G. R., Swank A. E., Richard A. M., Nesnow S. Chem. Res. Toxicol. 2004;17:827–838. doi: 10.1021/tx034207s. [DOI] [PubMed] [Google Scholar]
- Niwa T., Murayama N., Imagawa Y., Yamazaki H. Drug Metab. Rev. 2015;47:89–110. doi: 10.3109/03602532.2015.1011658. [DOI] [PubMed] [Google Scholar]
- Ross D., Mehlhorn R. J., Moldeus P., Smith M. T. J. Biol. Chem. 1985;260:16210–16214. [PubMed] [Google Scholar]
- Cavalieri E. L., Rogan E. G. Xenobiotica. 1995;25:677–688. doi: 10.3109/00498259509061885. [DOI] [PubMed] [Google Scholar]
- Cavalieri E. L., Li K. M., Balu N., Saeed M., Devanesan P., Higginbotham S., Zhao J., Gross M. L., Rogan E. G. Carcinogenesis. 2002;23:1071–1077. doi: 10.1093/carcin/23.6.1071. [DOI] [PubMed] [Google Scholar]
- Dwivedy I., Devanesan P., Cremonesi P., Rogan E., Cavalieri E. Chem. Res. Toxicol. 1992;5:828–833. doi: 10.1021/tx00030a016. [DOI] [PubMed] [Google Scholar]
- Metzler M., Zeeh J. J. Steroid Biochem. Mol. Biol. 1986;24:653–655. doi: 10.1016/0022-4731(86)90133-0. [DOI] [PubMed] [Google Scholar]
- Degen G. H., McLachlan J. A., Eling T. E., Sivarajah K. J. Steroid Biochem. Mol. Biol. 1987;26:679–685. doi: 10.1016/0022-4731(87)91039-9. [DOI] [PubMed] [Google Scholar]
- Cavalieri E. L., Devanesan P. D., Rogan E. G. Biochem. Pharmacol. 1988;37:2183–2187. doi: 10.1016/0006-2952(88)90579-5. [DOI] [PubMed] [Google Scholar]
- Shimada T., Martin M. V., Pruess-Schwartz D., Marnett L. J., Guengerich F. P. Cancer Res. 1989;49:6304–6312. [PubMed] [Google Scholar]
- Palackal N. T., Lee S. H., Harvey R. G., Blair I. A., Penning T. M. J. Biol. Chem. 2002;277:24799–24808. doi: 10.1074/jbc.M112424200. [DOI] [PubMed] [Google Scholar]
- Palackal N. T., Burczynski M. E., Harvey R. G., Penning T. M. Biochemistry. 2001;40:10901–10910. doi: 10.1021/bi010872t. [DOI] [PubMed] [Google Scholar]
- Zhang L., Jin Y., Chen M., Huang M., Harvey R. G., Blair I. A., Penning T. M. J. Biol. Chem. 2011;286:25644–25654. doi: 10.1074/jbc.M111.240739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J., Li Y., Chang M., Wu H., Yang X., Goodman J. E., Liu X., Liu H., Mesecar A. D., Van Breemen R. B., Yager J. D., Bolton J. L. Chem. Res. Toxicol. 2003;16:668–675. doi: 10.1021/tx0340549. [DOI] [PubMed] [Google Scholar]
- Yager J. D. Drug Discovery Today: Dis. Mech. 2012;9:e41–e46. doi: 10.1016/j.ddmec.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Huang M., Blair I. A., Penning T. M. J. Biol. Chem. 2012;287:29909–29920. doi: 10.1074/jbc.M112.386052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui Y., Yasuda S., Liu M. Y., Wu Y. Y., Liu M. C. Biol. Pharm. Bull. 2008;31:769–773. doi: 10.1248/bpb.31.769. [DOI] [PubMed] [Google Scholar]
- Zhang L., Huang M., Blair I. A., Penning T. M. Chem. Res. Toxicol. 2013;26:1570–1578. doi: 10.1021/tx400268q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterworth M., Lau S. S., Monks T. Carcinogenesis. 1997;18:561–567. doi: 10.1093/carcin/18.3.561. [DOI] [PubMed] [Google Scholar]
- Peng K. W., Chang M., Wang Y. T., Wang Z., Qin Z., Bolton J. L., Thatcher G. R. Chem. Res. Toxicol. 2010;23:1374–1383. doi: 10.1021/tx100129h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M., Liu X., Basu S. S., Zhang L., Kushman M. E., Harvey R. G., Blair I. A., Penning T. M. Chem. Res. Toxicol. 2012;25:993–1003. doi: 10.1021/tx200463s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Swanson S. M., van Breemen R. B., Liu X., Yang Y., Gu C., Bolton J. L. Chem. Res. Toxicol. 2001;14:1654–1659. doi: 10.1021/tx010158c. [DOI] [PubMed] [Google Scholar]
- Park J.-H., Troxel A. B., Harvey R. G., Penning T. M. Chem. Res. Toxicol. 2006;19:719–728. doi: 10.1021/tx0600245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Shen L., Zhang F., Lau S. S., van Breemen R. B., Nikolic D., Bolton J. L. Chem. Res. Toxicol. 1998;11:1105–1111. doi: 10.1021/tx980083l. [DOI] [PubMed] [Google Scholar]
- Chen Y., Liu X., Pisha E., Constantinou A. I., Hua Y., Shen L., van Breemen R. B., Elguindi E. C., Blond S. Y., Zhang F., Bolton J. L. Chem. Res. Toxicol. 2000;13:342–350. doi: 10.1021/tx990186j. [DOI] [PubMed] [Google Scholar]
- Huang M., Blair I. A., Penning T. M. Chem. Res. Toxicol. 2013;26:685–692. doi: 10.1021/tx300476m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flowers L., Ohnishi S. T., Penning T. M. Biochemistry. 1997;36:8640–8648. doi: 10.1021/bi970367p. [DOI] [PubMed] [Google Scholar]
- Breen A. P., Murphy J. A. Free Radicals Biol. Med. 1995;18:1033–1077. doi: 10.1016/0891-5849(94)00209-3. [DOI] [PubMed] [Google Scholar]
- Wang Z., Chandrasena E. R., Yuan Y., Peng K.-w., van Breeman R. B., Thatcher G. R. J., Bolton J. L. Chem. Res. Toxicol. 2010;23:1365–1373. doi: 10.1021/tx1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Yao J., Pisha E., Yang Y., Hua Y., van Breeman R. B., Bolton J. L. Chem. Res. Toxicol. 2002;15:512–519. doi: 10.1021/tx0101649. [DOI] [PubMed] [Google Scholar]
- Park J. H., Gelhaus S., Vedantam S., Oliva A. L., Batra A., Blair I. A., Troxel A. B., Field J., Penning T. M. Chem. Res. Toxicol. 2008;21:1039–1049. doi: 10.1021/tx700404a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.-H., Gopishetty S., Szewczuk L. M., Troxel A. B., Harvey R. G., Penning T. M. Chem. Res. Toxicol. 2005;18:1026–1037. doi: 10.1021/tx050001a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munk B. H., Burrows C. J., Schlegel B. J. Am. Chem. Soc. 2007;130:5245–5256. doi: 10.1021/ja7104448. [DOI] [PubMed] [Google Scholar]
- Ravanat J.-L., Cadet J. Chem. Res. Toxicol. 1995;8:379–388. doi: 10.1021/tx00045a009. [DOI] [PubMed] [Google Scholar]
- Ding Y., Fleming A. M., Burrows C. J. Am. Chem. Soc. 2017;139:2569–2572. doi: 10.1021/jacs.6b12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valinluck V., Tsai H. H., Rogstad D. K., Burdzy A., Bird A., Sowers L. Nucleic Acids. 2004;32:4100–4106. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair I. A. J. Biol. Chem. 2008;283:15524–15529. doi: 10.1074/jbc.R700051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack M., Yang I. Y., Kim H. Y., Blair I. A., Moriya M. Chem. Res. Toxicol. 2006;19:1074–1079. doi: 10.1021/tx0600503. [DOI] [PubMed] [Google Scholar]
- DeMott M. S. and Dedon P. C., in Chemical Biology of DNA Damage, ed. N. E. Geactinov, and S. Broyde, Wiley-VCH, Weinheim, Germany, 2010, pp. 21–52. [Google Scholar]
- Seike K., Murata M., Oikawa S., Hiraku Y., Hirakawa K., Kawanishi S. Chem. Res. Toxicol. 2003;16:1470–1476. doi: 10.1021/tx034103h. [DOI] [PubMed] [Google Scholar]
- Fromme J. C., Banerjee A., Huang S. J., Verdine G. L. Nature. 2004;427:652–656. doi: 10.1038/nature02306. [DOI] [PubMed] [Google Scholar]
- Shibutani S., Takeshita M., Grollman A. P. J. Biol. Chem. 1997;272:13916–13922. doi: 10.1074/jbc.272.21.13916. [DOI] [PubMed] [Google Scholar]
- Bruner S. D., Norman D. P., Verdine G. L. Nature. 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- Haller M. K., Slade P. G., Martin B. D., Rosenquist T. A., Sugden K. D. DNA Repair. 2005;4:41–50. doi: 10.1016/j.dnarep.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Fleming A. M., Burrows C. J. Free Radicals Biol. Med. 2017;107:35–52. doi: 10.1016/j.freeradbiomed.2016.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood M. L., Esteve A., Morningstar M. L., Kuziemko G. M., Essigmann J. M. Nucleic Acids Res. 1992;20:6023–6032. doi: 10.1093/nar/20.22.6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya M. Proc. Natl. Acad. Sci. U. S. A. 1993;90:1122–1126. doi: 10.1073/pnas.90.3.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed M., Gunselman S. J., Higginbotham S., Rogan E., Cavalieri E. Steroids. 2005;70:37–45. doi: 10.1016/j.steroids.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Markushin Y., Zhong W., Cavalieri E. L., Rogan E. G., Small G. J., Yeung E. S., Jankowiak R. Chem. Res. Toxicol. 2003;16:1107–1117. doi: 10.1021/tx0340854. [DOI] [PubMed] [Google Scholar]
- Cavalieri E. L., Stack D. E., Devanesan P. D., Todorovic R., Dwivedy I., Higginbotham S., Johansson S. L., Patil K. D., Gross M. L., Gooden J. K., Ramanathan R., Cerny R. L., Rogan E. G. Proc. Natl. Acad. Sci. U. S. A. 1997;94:10937–10942. doi: 10.1073/pnas.94.20.10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed M., Higginbotham S., Rogan E., Cavalieri E. Chem.-Biol. Interact. 2007;165:175–188. doi: 10.1016/j.cbi.2006.12.007. [DOI] [PubMed] [Google Scholar]
- McCoull K. D., Rindgen D., Blair I. A., Penning T. M. Chem. Res. Toxicol. 1999;12:237–246. doi: 10.1021/tx980182z. [DOI] [PubMed] [Google Scholar]
- Zahid M., Kohli E., Saeed M., Rogan E., Cavalieri E. Chem. Res. Toxicol. 2006;19:164–172. doi: 10.1021/tx050229y. [DOI] [PubMed] [Google Scholar]
- Stack D. E., Byun J., Gross M. L., Rogan E. G., Cavalieri E. L. Chem. Res. Toxicol. 1996;9:851–859. doi: 10.1021/tx960002q. [DOI] [PubMed] [Google Scholar]
- Li K. M., Todorovic R., Devanesan P., Higginbotham S., Köfeler H., Ramanathan R., Gross M. L., Rogan E. G., Cavalieri E. L. Carcinogenesis. 2004;25:289–294. doi: 10.1093/carcin/bgg191. [DOI] [PubMed] [Google Scholar]
- Boysen G., Pachkowski B. F., Nakamura J., Swenberg J. A. Mutat. Res. 2009;678:76–94. doi: 10.1016/j.mrgentox.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolbanovskiy A., Kuzmin V., Shastry A., Kolbanovskaya M., Chen D., Chang M., Bolton J. L., Geacintov N. E. Chem. Res. Toxicol. 2005;18:1737–1747. doi: 10.1021/tx050190x. [DOI] [PubMed] [Google Scholar]
- Wang Z., Edirisinghe P., Sohn J., Qin Z., Geacintov N. E., Thatcher G. R., Bolton J. L. Chem. Res. Toxicol. 2009;22:1129–1136. doi: 10.1021/tx900063g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou M., Harvey R. G., Penning T. M. Carcinogenesis. 1993;14:475–482. doi: 10.1093/carcin/14.3.475. [DOI] [PubMed] [Google Scholar]
- Nesnow S., Nelson G., Padegtt W. T., George M. H., Moore T., King L. C., Adams L. D., Ross J. A. Chem.-Biol. Interact. 2010;186:157–165. doi: 10.1016/j.cbi.2010.03.037. [DOI] [PubMed] [Google Scholar]
- Balu N., Padgett W. T., Nelson G. B., Lambert G. R., Ross J. A., Nesnow S. Anal. Biochem. 2006;355:213–223. doi: 10.1016/j.ab.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Cai Y., Kropachev K., Kolbanovskiy M., Kolbanovskiy A., Broyde S., Patel D. and Geactinov N., in The Chemial Biology of DNA Damage, ed. N. Geacitiov, and S. Broyde, Wiley-VCH, Weinheim, 2010, pp. 261–298. [Google Scholar]
- Yu D., Berlin J. A., Penning T. M., Field J. Chem. Res. Toxicol. 2002;15:832–842. doi: 10.1021/tx010177m. [DOI] [PubMed] [Google Scholar]
- Mangal D., Vudathala D., Park J. H., Lee S. H., Penning T. M., Blair I. A. Chem. Res. Toxicol. 2009;22:788–797. doi: 10.1021/tx800343c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesaros C., Arora J. S., Wholer A., Vachani A., Blair I. A. Free Radicals Biol. Med. 2012;53:610–617. doi: 10.1016/j.freeradbiomed.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E. M., Shigenaga M. K., Degan P., Korn T. S., Kitzler J. W., Wehr C. M., Kolachana P., Ames B. N. Proc. Natl. Acad. Sci. U. S. A. 1992;89:3375–3379. doi: 10.1073/pnas.89.8.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura J., Walker V. E., Upton P. B., Chiang S. Y., Kow Y. W., Swenberg J. A. Cancer Res. 1998;58:222–225. [PubMed] [Google Scholar]
- Flowers-Geary L., Bleczinki W., Harvey R. G., Penning T. M. Chem. -Biol. Interact. 1996;59:55–72. doi: 10.1016/0009-2797(95)03660-1. [DOI] [PubMed] [Google Scholar]
- Park J. H., Mangal D., Frey A. J., Harvey R. G., Blair I. A., Penning T. M. J. Biol. Chem. 2009;284:29725–29734. doi: 10.1074/jbc.M109.042143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuendet M., Liu X., Pisha E., Li Y., Yao J., Yu L., Bolton J. L. Mutat. Res. 2004;550:109–121. doi: 10.1016/j.mrfmmm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Pruthi S., Yang L., Sandhu N. P., Ingle J. N., Beseler C. L., Suman V. J., Cavalieri E. L., Rogan E. G. J. Steroid Biochem. Mol. Biol. 2012;132:73–79. doi: 10.1016/j.jsbmb.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto S.-I., Yamauchi N., Moriguchi H., Hippo Y., Watanbe A., Shibahara J., Taniguichi H., Ishikawa S., Ito H., Yamamato S., Iwanari H., Horonaka M., Ishikawa H., Niki T., Sohara Y., Kodama T., Mishimura M., Fukayama M., Doska-Akita H., Auratani H. Clin. Cancer Res. 2005;11:1776–1785. doi: 10.1158/1078-0432.CCR-04-1238. [DOI] [PubMed] [Google Scholar]
- Zhang L., Lee J. J., Tang H., Fan Y.-H., Xiao L., Ren H., Kurie J., Morice R. C., Hong W. K., Mao L. Cancer Prev. Res. 2008;1:112. doi: 10.1158/1940-6207.CAPR-07-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woenckhaus M., Klein-Hitpass L., Grepmeier U., Merk J., Pfeifer M., Wild P. J., Bettstetter M., Wuensch P., Blaszk H., Harrmann A., Hofstaedter F., Dietmaier W. J. Pathol. 2006;210:192–204. doi: 10.1002/path.2039. [DOI] [PubMed] [Google Scholar]
- Penning T. M., Lerman C. Cancer Prev. Res. 2008;1:80–83. doi: 10.1158/1940-6207.CAPR-08-0047. [DOI] [PubMed] [Google Scholar]
- Gumus Z. H., Du B., Kacker A., Boyle J. O., Bocker J. M., Mukherjee P., Subbaramaiah K., Dannenberg A. J., Weinstein H. Cancer Prev. Res. 2008;1:100–111. doi: 10.1158/1940-6207.CAPR-08-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D., Harvey R. G., Blair I. A., Penning T. M. Chem. Res. Toxicol. 2011;24:1905–1914. doi: 10.1021/tx2002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.-H., Mangal D., Tacka K. A., Quinn A. M., Harvey R. G., Blair I. A., Penning T. M. Proc. Natl. Acad. Sci. U. S. A. 2008;105:6846–6851. doi: 10.1073/pnas.0802776105. [DOI] [PMC free article] [PubMed] [Google Scholar]