

Abstract
Recently, a crystal structure has been reported of a new catalytic RNA, the TS ribozyme, that has been identified through comparative genomics and is believed to be a metalloribozyme having novel mechanistic features. Although this data provides invaluable structural information, analysis suggests a conformational change is required to arrive at a catalytically relevant state. We report results of molecular simulations that predict a spontaneous local rearrangement of the active site, leading to solution structures consistent with available functional data and providing competing mechanistic hypotheses that can be experimentally tested. The two competing hypotheses differ in the proposed identity of the catalytic general acid: either a water molecule coordinating a Mg2+ ion bound at the Watson–Crick edge of residue C7, or the N3 position of residue C7 itself.
Keywords: biocatalysis, general acids, molecular dynamics, ribozymes, RNA
Communications
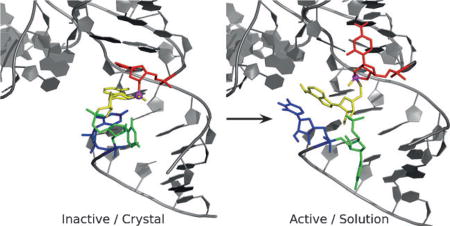
A new catalytic RNA, the TS ribozyme, is believed to be a metalloribozyme having novel mechanistic features. Molecular dynamic simulations predict a spontaneous local rearrangement of the active site, leading to solution structures consistent with available functional data and providing competing mechanistic hypotheses that can be experimentally tested.
Small nucleolytic ribozymes are important model systems in the study of RNA catalysis.[1] Recently, there has been a surge of progress in the identification of new classes of self-cleaving nucleolytic ribozymes that have been revealed by comparative genomics analysis.[2,3] Following in the wake of this discovery have been intense efforts to determine crystal structures of these new ribozymes and gain insight into the origin of their function.[4–10] However, crystal structures often are not representative of the active states in solution. In the case of the recently discovered TS ribozyme, the crystal structure[4] does not provide a mechanistic explanation of available functional data, leading to the assertion that a local rearrangement of the active site must occur to form a catalytically active state.
Herein, we use molecular dynamics (MD) simulations and molecular solvation theory to develop two distinct models for the active state of the TSrz in solution that are consistent with the currently available functional data.[3, 4] The “Mg Acid” model (Supplementary Dataset 1) positions a divalent metal ion to play the role of the general acid, similar to what has been suggested as a plausible mechanism in the hammerhead ribozyme.[12–14] The “C7 Acid” model (Supplementary Dataset 2), points to a highly conserved cytosine residue that may play the role of the general acid, similar to that proposed for the HDV ribozyme.[11] Both models require local rearrangements of the active site from the crystal and, unlike the crystal structure, are able to both provide a plausible rationale for the current body of experimental functional data[3, 4] and to suggest experimentally testable predictions that would distinguish pathways.
The TSrz crystal structure revealed several divalent metal ion binding sites (labeled M1–M7 in the original work)[4] that presumably stabilize tertiary contacts in the crystal. Initial simulations departing from the crystallographic structure that preserved these divalent metal ion binding modes did not lead to any significant rearrangement in solution (Supporting Information, Figure S1). We then hypothesized that the crystal packing environment may have created divalent metal ion binding sites that differ from those present in solution. To explore this possibility, we used a 3D reference site interaction model (3D-RISM)[18] to predict cation occupation sites in TSrz both in a crystalline environment and in solution. Crystal packing models with 3D-RISM indicated cation binding sites consistent with the crystallographic data. However, calculations of the solution TSrz model revealed a shift in the predicted cation binding (Figure 1) from direct coordination of G5 proR and C54 proR (M4 site) in the crystal to a distinct position at the nearby Watson–Crick edge of residue C7, referred to as the M4′ site.
Figure 1.

Difference map of cation number density from 3D-RISM calculations of the solution structure and crystallographic coordinates. The green mesh surfaces indicate that there is a predicted increase in cation density at both the crystallographic M4 site (where a Mg2+ ion is observed to directly coordinate G5 proR and C52 proR), as well as the Watson–Crick edge of residue C7 (shown in a colored stick representation) labeled as the M4′ site.
We therefore placed a Mg2+ ion at this alternative position, and re-ran three independent simulations of a transition state mimic in solution. Within 2 ns, these simulations all produced a spontaneous rearrangement of the active site residues (C54, A55, U56, and U57) leaving the global fold, inferred from the original crystallographic work,[4] largely unaffected (Supporting Information, Figures S2 and S3). In this model (Figure 2), a Mg2+ ion at residue C7 is poised to act as a general acid, another interacting with residue A9 supports general base catalysis, and the rearrangement of residue A55 promotes in-line fitness and provides electrostatic stabilization of the transition state.
Figure 2.

MD average structure for the “Mg Acid” model. Key nucleotides are shown in stick form, with hydrogen bonds indicated by broken lines. a) Local environment of C7, highlighting hydrogen bonding between C7:N4 and G51:O2′ as well as the positioning of the Mg2+ ion at the M4′ site, directly coordinating C7:O2. b) View of the M1 site Mg2+, outer-sphere coordinating the sugar edge of A9 as well as the O2′ nucleophile. c) Close up view of the hydrogen-bonding scaffold that is proposed to anchor residue A55 in a splayed-out conformation relative to residue C54, in order to promote in-line fitness.
Table 1 summarizes the current body of available mutagenesis data and compares structural and functional interpretations to the experimental crystal structure and to the computational solution simulation models reported. The “Mg Acid” model predicts that the four most severe mutations involve disruptions to interactions that position C7 within the active site and to Mg2+ binding at the O2 position. It should be noted that this Mg2+ ion remained stably bound at C7:O2 throughout all MD simulations and 3D-RISM calculations of the average structure for the “Mg Acid” model predict a higher fractional occupancy at M4′ than at the M4 site (Supporting Information, Figure S4). It follows that these mutations would be the most detrimental as they would directly impact the general acid step whereby a water molecule inner-sphere coordinated to the Mg2+ bound at C7:O2 is poised to act as a proton donor to stabilize the negatively charged O5′ leaving group. Furthermore, this model suggests that the C7 5F mutation would be the least impactful of this group as it only weakens H-bonding and Mg2+ binding rather than eliminating the key H-bonding interaction between C7:N4 and G51:O2′.
Table 1.
Comparison of structural and functional interpretations of the available mutational data. Decrease in rate is shown as a fold decrease due to the mutation relative to the wild type sequence; N/A indicates no observable activity. The structural interpretation listed is the apparent interaction(s) disrupted by the mutation in the crystal or simulation model. A…B represents the disruption of a hydrogen bond, while A◦B indicates outer-sphere coordination of a metal and A•B indicates direct coordination. All rate measurements from Liu et al. 2017.
| Mutation | Fold Decr. |
Crystal Structure (PDBID: 5T5A) | Solution Simulation (Mg Acid) | Solution Simulation (C7 Acid) | |||
|---|---|---|---|---|---|---|---|
| Structural | Functional | Structural | Functional | Structural | Functional | ||
| C7U | N/A | C7:N4…G51:O2′ | Folding | C7:N4…G51:O2′ | General Acid Mg2+ Binding at C7:O2 | C7:N4…A55:O5′ | General Acid |
| C7Z | N/A | C7:N3H+ | |||||
| G51 O2′H | 1600 | G51:O2′…G6:OP2 | Folding+ | ||||
| C7 5F | 940 | C7:N4…A55:O5′ | General Acid C7:N3H | ||||
| A9 N3CH O2′H | 890 | Mg2+◦A9:N3 | General Base Mg2+ Binding | Mg2+◦A9:N3 | General Base Mg2+ Binding | Mg2+◦A9:N3 | General Base Mg2+ Binding |
| A9 O2′H | 3.2 | X | X | Mg2+◦A9:O2′ | Mg2+◦A9:O2′ | ||
| A55 N3CH O2′H | 270 | X | X | A55:N3•Na+•A55:OP1 A55:O2′…U56:O3′ | Positioning of A55 → Inline Fitness | A55:O2′…U56:O5′ | Positioning of A55 → Inline Fitness |
| A55 N7CH | 59[a] | X | X | G6:N1/N2…A55:N7 | G6:N1/N2…A55:N7 | ||
| A55 I | 30[a] | A55:N6…C7:O2 | X | A55:N6…A25:N1 C24:N4…A55:N1 | A55:N6…A25:N1 C24:N4…A55:N1 | ||
| A55 O2′H | 9.5 | X | X | A55:O2′…U56:O3′ | A55:O2′…U56:O5′ | ||
| A55 P | 6.3[a] | A55:N6…C7:O2 | X | A55:N6…A25:N1 | A55:N6…A25:N1 | ||
| A25 I | 3.5 | X | X | A55:N6…A25:N1 | A55:N6…A25:N1 | ||
The crystallographic structure and the computational model are in close agreement for both the structural and functional explanations of the mutations to A9, with the sugar edge providing hydrogen-bond acceptors for outer-sphere coordination of the Mg2+ implicated in the general base step. The structural rearrangement in the computational model, however, allows for additional hydrogen bonding to A9:O2′ for which there is a small observed mutational effect.
Finally, the mutational data suggests that A55 is involved in numerous H-bonding interactions not observed in the crystal structure. The anomalously high B-values of residue A55 in the crystal (>60) suggest this residue may have a low barrier to local rearrangement.[4] The spontaneous structural rearrangement observed in our simulations positions residue A55 to form a constellation of hydrogen bonds that promotes an active in-line conformation of the nucleophile. The hydrogen bond between residues A55 and G6 along with the interaction of the Watson–Crick edge of A55 and residues C24 and A25 in the L4 loop provides a scaffold for positioning A55 in a splayed out conformation relative to residue C54, a common structural feature in ribozymes that favors in-line attack.[19]
However, an alternative explanation for the mutagenesis data is that residue C7 may itself act as the general acid, similar to what has been proposed in the HDV ribozyme.[12–14] Starting from the previously discussed structural model with local rearrangement of residue A55, we explored simulations with residue C7 protonated while donating a hydrogen bond to the leaving group, as would be required to function as a general acid, and the Mg2+ ion previously at the M4′ site is returned to the M4 site. 3D-RISM calculations predict significant cation density centered around the Mg2+ ion bound at the M4 site, consistent with the position of metal ions in the crystal structure (Supporting Information, Figure S5). The system was equilibrated and relaxed in this state and simulated without restraints for 50 ns. The unrestrained simulation showed that this new structure was stable on that timescale, maintaining all of the key hydrogen bonding interactions representative of the model.
From these simulations, we present an alternative “C7 Acid” model that can be used as an alternative to the “Mg Acid” model to interpret the experimental functional data in Table 1. The only notable differences between the two computational models involve residue C7 (Figure 3). In the “C7 Acid” model, C7:N4 is hydrogen bonding to the leaving group instead of G51:O2′ in the crystal and the “Mg Acid” model. The interpretation of the complete loss of activity with C7U and C7Z mutants is then both the shifting of the pKa of C7:N3 away from neutrality and the loss of the H-bond from C7:N4. The C7 5F mutation is now explained by the shift in the C7:N3 pKa down 2 units (cytosine: pKa=4.2, 5-fluorocytidine: pKa=2.3), lowering the probability of residue C7 being protonated and the ribozyme being in an active protonation state at neutral pH, in addition to weakening of the hydrogen bonding between C7:N4 and the O5′ leaving group. A shift in pKa down by 2 units would presumably disfavor the protonated state by 2.6 kcal mol−1 corresponding to an approximately 80-fold decrease in the observed rate. This is an order of magnitude less than the observed decrease (940-fold) in rate, and it could be the weakening of the hydrogen bonding between C7:N4 and the leaving group, due to the 5F substitution, that partially accounts for the remainder of the rate decrease. This supposition of the catalytic importance of hydrogen bonding between residue C7 and the leaving group is supported by the elimination of activity observed with the C7Z mutation since the pKa of zebularine and 5-fluorocytidine are approximately the same, but zebularine lacks the exocyclic amine entirely. Finally, the proposed “C7 Acid” model presents a hypothesis that the G51:O2′H mutation is detrimental because of the importance of residue G51 in folding, rather than a direct interaction with residue C7.
Figure 3.

MD average structure for “C7 Acid” model. Key nucleotides are shown in stick form, with hydrogen bonds indicated by broken lines. a) Close up view of C7:N3H+ donating hydrogen bonds to the O5′ leaving group and thus poised to act as the general acid. b) Interactions between the first solvation shell of the Mg2+ ion at the M1 site and both the sugar edge of A9 and the O2′ nucleophile. c) Hydrogen-bonding scaffold supporting the positioning of residue A55 as splayed-out from residue C54, proposed to support in-line fitness.
To test these competing mechanistic hypotheses, the activity of a TSrz ribozyme mutant with C7 replaced by N3-methylcytosine or N3-deazacytosine (both eliminate the ability of residue C7 to donate a proton from the N3 position) could be measured experimentally. If these mutants had only a moderate effect on catalysis, the most straightforward interpretation would be that C7:N3 most likely does not act as the general acid. However, if this mutation eliminated catalytic activity, the result would be consistent with the role of residue C7 as a general acid and could be further validated by testing whether introduction of an enhanced leaving group, such as a 5′ thio substitution, would have a rescue effect. It is clear that while a crystal structure is an essential starting point for the study of the TS ribozyme, computational modelling can provide important insights that not only aid in the interpretation of the available experimental data but also drive future studies. Additional computational work must be done to explore the relevant chemical steps along the reaction pathway. Likewise, additional biochemical experiments are needed to refine the working hypothesis for the mechanism of this catalytic RNA.
Supplementary Material
Acknowledgments
We thank Dr. David M. J. Lilley and Dr. Timothy J. Wilson for valuable discussion and sharing of experimental data. The authors are grateful for financial support provided by the National Institutes of Health (Grant GM62248 to D.M.Y.). Computational resources were provided by the National Institutes of Health under Grant No. S10OD012346 and by the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant No. OCI-1053575 (Project No. TGMCB110101). This work also benefited from access to RDI2 Research Instruments.
Footnotes
Supporting information, including molecular dynamics simulation and 3D-RISM calculation methods, RMSD time series for the transition state mimic simulations, and a comparison between the average structure from the simulation where the spontaneous rearrangement was observed and the crystallographic structure, and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201705608.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Wilson TJ, Liu Y, Lilley DM. Front. J. Chem. Sci. Eng. 2016;10:178–185. [Google Scholar]
- 2.Roth A, Weinberg Z, Chen AG, Kim PB, Ames TD, Breaker RR. Nat. Chem. Biol. 2014;10:56–62. doi: 10.1038/nchembio.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weinberg Z, Kim PB, Chen TH, Li S, Harris KA, Lünse CE, Breaker RR. Nat. Chem. Biol. 2015;11:606–610. doi: 10.1038/nchembio.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Y, Wilson TJ, Lilley DM. J. Nat. Chem. Biol. 2017;13:508–513. doi: 10.1038/nchembio.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y, Wilson TJ, McPhee SA, Lilley DM. J. Nat. Chem. Biol. 2014;10:739–744. doi: 10.1038/nchembio.1587. [DOI] [PubMed] [Google Scholar]
- 6.Ren A, Košutic M, Rajashankar KR, Frener M, Santner T, Westhof E, Micura R, Patel DJ. Nat. Commun. 2014;5:5534–5544. doi: 10.1038/ncomms6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eiler D, Wang J, Steitz TA. Proc. Natl. Acad. Sci. USA. 2014;111:13028–13033. doi: 10.1073/pnas.1414571111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kosutic M, Neuner S, Ren A, Flür S, Wunderlich C, Mairhofer E, Vusurovic N, Seikowski J, Breuker K, Höbartner C, Patel DJ, Kreutz C, Micura R. Angew. Chem. Int. Ed. 2015;54:15128–15133. doi: 10.1002/anie.201506601. Angew. Chem.2015, 127, 15343–15348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ren A, Vusurovic N, Gebetsberger J, Gao P, Juen M, Kreutz C, Micura R, Patel D. Nat. Chem. Biol. 2016;12:702–708. doi: 10.1038/nchembio.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen LA, Wang J, Steitz TA. Proc. Natl. Acad. Sci. USA. 2017;114:1021–1026. doi: 10.1073/pnas.1611191114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mir A, Golden BL. Biochemistry. 2016;55:633–636. doi: 10.1021/acs.biochem.5b01139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakano S, Proctor DJ, Bevilacqua PC. Biochemistry. 2001;40:12022–12038. doi: 10.1021/bi011253n. [DOI] [PubMed] [Google Scholar]
- 13.Das S, Piccirilli J. Nat. Chem. Biol. 2005;1:45–52. doi: 10.1038/nchembio703. [DOI] [PubMed] [Google Scholar]
- 14.Chen J-H, Yajima R, Chadalavada DM, Chase E, Bevilacqua PC, Golden BL. Biochemistry. 2010;49:6508–6518. doi: 10.1021/bi100670p. [DOI] [PubMed] [Google Scholar]
- 15.Emilsson GM, Nakamura S, Roth A, Breaker RR. RNA. 2003;9:907–918. doi: 10.1261/rna.5680603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breaker RR, Emilsson GM, Lazarev D, Nakamura S, Puskarz IJ, Roth A, Sudarsan N. RNA. 2003;9:949–957. doi: 10.1261/rna.5670703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breaker RR. ACS Chem. Biol. 2017;12:886–891. doi: 10.1021/acschembio.7b00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luchko T, Gusarov S, Roe DR, Simmerling C, Case DA, Tuszynski J, Kovalenko A. J. Chem. Theory Comput. 2010;6:607–624. doi: 10.1021/ct900460m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cochrane JC, Strobel SA. Acc. Chem. Res. 2008;41:1027–1035. doi: 10.1021/ar800050c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.